

Abstract
BACKGROUND
Women demonstrate a memory advantage when cognitively healthy yet lose this advantage to men in Alzheimer's disease. However, the genetic underpinnings of this sex difference in memory performance remain unclear.
METHODS
We conducted the largest sex‐aware genetic study on late‐life memory to date (N males = 11,942; N females = 15,641). Leveraging harmonized memory composite scores from four cohorts of cognitive aging and AD, we performed sex‐stratified and sex‐interaction genome‐wide association studies in 24,216 non‐Hispanic White and 3367 non‐Hispanic Black participants.
RESULTS
We identified three sex‐specific loci (rs67099044—CBLN2, rs719070—SCHIP1/IQCJ‐SCHIP), including an X‐chromosome locus (rs5935633—EGL6/TCEANC/OFD1), that associated with memory. Additionally, we identified heparan sulfate signaling as a sex‐specific pathway and found sex‐specific genetic correlations between memory and cardiovascular, immune, and education traits.
DISCUSSION
This study showed memory is highly and comparably heritable across sexes, as well as highlighted novel sex‐specific genes, pathways, and genetic correlations that related to late‐life memory.
Highlights
Demonstrated the heritable component of late‐life memory is similar across sexes.
Identified two genetic loci with a sex‐interaction with baseline memory.
Identified an X‐chromosome locus associated with memory decline in females.
Highlighted sex‐specific candidate genes and pathways associated with memory.
Revealed sex‐specific shared genetic architecture between memory and complex traits.
Keywords: aging, Alzheimer's disease, endophenotypes, cognition, Genomics, GWAS, memory, sex differences, sex‐specific
1. BACKGROUND
Alzheimer's disease (AD) is a global health crisis, affecting more than 50 million people worldwide, with AD cases expected to steeply increase to over 150 million people by 2050. 1 Notably, AD is characterized by a neuropathological cascade lasting decades, resulting in neurodegeneration and cognitive impairment. 2 One of the earliest and most detectable clinical manifestations of both aging and AD is changes in memory performance. Those with AD demonstrate impairment for verbal and non‐verbal memory in a pattern that is distinguishable from both cognitively unimpaired controls and other memory‐related disorders. 3 , 4 , 5 , 6 Individuals in prodromal AD exhibit difficulties with acquisition of new information, which can be detected with immediate and delayed recall memory tasks. 3 Furthermore, episodic memory tests can isolate distinct cognitive trajectories for those who will go on to develop AD up to 12 years before clinical AD diagnosis, and these changes track with neuropathological changes. 4 , 5 , 6
Recent literature has highlighted robust sex/gender differences in memory performance throughout the life course including in aging and AD. In clinically healthy individuals, women (women—based on self‐report) on average have an advantage on episodic and verbal memory tasks, measured by such tests as the California Verbal Learning Test and the Weschler Memory Scale. 7 , 8 These memory advantages emerge at a young age in women and persist into adulthood, 7 , 8 and this advantage is consistent across the literature. 9 In contrast, men (men—based on self‐report), on average perform better at tasks requiring visuospatial ability, such as analysis of figure tasks. 7 , 8 A meta‐analysis on visuospatial ability highlighted the consistency of the advantage in men across studies, beginning at adolescence and increasing in magnitude with age. 10 Furthermore, sex differences in cognitive performance change with the onset of disease. While women with AD lose their episodic and verbal memory advantages, 11 men with AD appear to maintain their visuospatial advantage. 12 Taken together, sex/gender differences in memory performance are complex, with contributions from factors such as age, type of memory task, and disease stage.
Genetic studies have started to uncover the genetic architecture of memory performance in older adults, identifying loci that may contribute to variability in memory. One major challenge surrounding large‐scale genomic studies of memory performance is that different neuropsychological test batteries are administered across studies. 13 , 14 Even if the same neuropsychological battery is administered, variability may exist in the way a test item is assessed or coded across studies. 13 , 14 These complexities surrounding studying memory result in a lower generalizability of findings across studies, and prevents large, systematic meta‐analyses on memory. 13 , 14 Our group has addressed this challenge by leveraging modern, psychometric techniques to harmonize memory scores across cohort studies. As a result, we have robust memory composite scores across many time‐points that are on the same metric across multiple cohorts of cognitive aging and AD, as well as genotype data for nearly 27,000 of these individuals. Additionally, we have a published pipeline for the genetic study of sex differences in AD endophenotypes. 15 Together, this uniquely positions our group to conduct large, sex‐aware genetic analyses on memory performance in older adults. We hypothesize that a large genetic component of memory performance in older adults will be shared across sexes, but we believe that some genetic drivers will differ across sexes.
2. METHODS
Please see Figure 1 for an overview of the harmonized memory endophenotype and of our analytical workflow.
FIGURE 1.
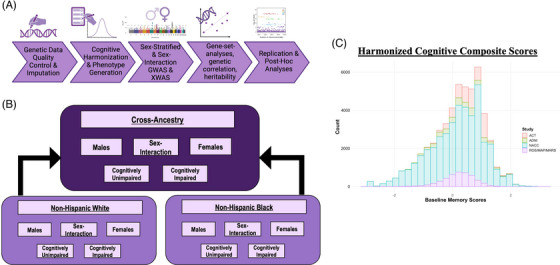
Overview of this study's genome‐wide sex‐aware cross‐ancestral analysis on late‐life memory performance. (A) Analytical workflow overview. (B) Ancestry, diagnosis, and sex strata workflow. (C) Histogram highlighting the harmonized memory endophenotype at baseline cognitive visit for each cohort. ACT, Adult Changes in Thought; ADNI, Alzheimer's Disease Neuroimaging Initiative; GWAS, genome‐wide association studies; ROS/MAP/MARS, Religious Orders Study/Memory and Aging Project/Minority Aging Research Study; XWAS, X‐wide association studies.
2.1. Participants
We leveraged four well‐characterized cognitive aging and AD cohorts: Adult Changes in Thought (ACT), Alzheimer's Disease Neuroimaging Initiative (ADNI), National Alzheimer's Coordinating Center (NACC), and Religious Orders Study (ROS)/Rush Memory and Aging Project (MAP)/Minority Aging Research Study (MARS). ACT 16 began in Seattle in 1994, recruiting cognitively unimpaired individuals in the area. The current number of enrolled participants is now at 4960. ADNI (https://adni.loni.usc.edu) began in 2004 and includes four phases: ADNI‐1, ADNI‐2, ADNI‐GO, and ADNI‐3, all of which were included in this study. Participants recruited for ADNI were a mix of cognitively unimpaired, mild cognitive impairment (MCI), and AD dementia, and were followed to track pathological and disease progression over time. NACC 17 , 18 , 19 , 20 , 21 began in 1999 and is comprised of data measured by 42 Alzheimer's Disease Research Centers (ADRCs); 15 ADRCs were leveraged for this analysis. The goal of NACC is to standardize a large database of clinical and neuropathological research data. ROS 22 began in 1994 and recruited older Catholic nuns, priests, and bothers in orders without known dementia. MAP 22 started in 1997 and recruited older individuals without known dementia in the Chicago area. Both studies are longitudinal, epidemiological clinical‐pathological studies of aging and AD. MARS 23 began in 2004 and recruited older African American individuals without known dementia. All ROS/MAP/MARS participants completed informed and repository consents. ROS, MAP, and MARS studies share a large common core of data that can be merged at the item level. Please see Table 1 for participant characteristics.
TABLE 1.
Participant characteristics.
| Non‐Hispanic White (N = 24,216) | Non‐Hispanic Black (N = 3367) | Cross‐ancestry (N = 27,583) | ||||
|---|---|---|---|---|---|---|
| Men | Women | Men | Women | Men | Women | |
| N (total) | 11,062 | 13,154 | 880 | 2487 | 11,942 | 15,641 |
| Baseline characteristics | ||||||
| Age at first cognitive visit (mean years ± SD) | 74.35 ± 7.73 | 74.56 ± 8.13 | 72.79 ± 7.27 | 73.39 ± 7.62 | 74.24 ± 7.71 | 74.37 ± 8.06 |
| Education (mean years ± SD) | 16.36 ± 3.06 | 15.39 ± 2.85 | 14.33 ± 3.43 | 14.41 ± 3.16 | 16.21 ± 3.13 | 15.23 ± 2.92 |
| Memory score (mean score ± SD) | 0.16 ± 0.83 | 0.36 ± 0.90 | 0.04 ± 0.79 | 0.24 ± 0.80 | 0.15 ± 0.82 | 0.34 ± 0.88 |
| Longitudinal characteristics | ||||||
| Number of visits (mean ± SD) | 7.40 ± 4.29 | 8.29 ± 4.67 | 6.93 ± 3.50 | 7.80 ± 4.07 | 7.37 ± 4.25 | 8.22 ± 4.59 |
| Years of follow‐up (mean years ± SD) | 3.84 ± 3.95 | 4.42 ± 4.32 | 3.52 ± 3.38 | 3.98 ± 3.74 | 3.82 ± 3.93 | 4.36 ± 4.24 |
| Memory decline score (mean slope ± SD) | −0.09 ± 0.10 | −0.07 ± 0.10 | −0.08 ± 0.09 | −0.06 ± 0.08 | −0.09 ± 0.10 | −0.07 ± 0.10 |
| Clinical diagnosis at first cognitive visit | ||||||
| N (%) normal cognition (out of those diagnosed) | 4950 (44.75%) | 7839 (59.59%) | 391 (44.43%) | 1384 (55.65%) | 5341 (44.72%) | 9223 (58.97%) |
| N (%) MCI/AD (out of those diagnosed) | 4478 (40.48%) | 3987 (30.31%) | 373 (42.39%) | 869 (34.94%) | 4851 (40.62%) | 4856 (31.05%) |
| APOE carrier status | ||||||
| N (%) ɛ2 carrier (out of those that have APOE data) | 1329 (12.01%) | 1623 (12.34%) | 173 (19.66%) | 457 (18.38%) | 1502 (12.58%) | 2080 (13.30%) |
| N (%) ɛ4 carrier (out of those that have APOE data) | 4154 (39.35%) | 4766 (36.23%) | 415 (47.16%) | 1047 (42.10%) | 4569 (38.26%) | 5813 (37.17%) |
Abbreviation: APOE, apolipoprotein E; MCI/AD, mild cognitive impairment.
2.2. Clinical diagnosis determination
ACT, ADNI, NACC, and ROS/MAP/MARS have their own enrollment criteria (see Section 2.1 Participants), but in all studies a clinical diagnosis of dementia and AD dementia followed standard criteria of the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association. ADNI, ROS/MAP, and NACC provide an MCI designation for those who have signs of cognitive impairment on neuropsychological testing but do not meet full criteria for dementia. ACT does not identify individuals with MCI. To harmonize diagnostic coding across studies, we mapped study‐specific diagnostic codes to either (1) cognitively unimpaired, (2) MCI, or (3) AD. For this study, baseline visit was selected to make diagnostic determinations for diagnosis‐stratified genetic analyses (see Section 2.5.4). Given the variable coverage of MCI across studies, we also mapped a binary classifier as either cognitively unimpaired or cognitively impaired (ie, either MCI or AD) that was used in the diagnosis‐stratified analyses.
RESEARCH IN CONTEXT
Systematic review: Women have a memory advantage that they lose to men with Alzheimer's disease (AD), but the genetic architecture of this sex difference has yet to be elucidated. This study leveraged harmonized memory scores and genetic data from four cohorts of aging and AD to conduct the most comprehensive sex‐aware genetic analysis on late‐life memory to date.
Interpretation: We showed that memory performance is comparably heritable across sexes, and we identified three sex‐specific genetic loci that relate to memory. Additionally, we identified sex‐specific candidate genes and pathways relating to memory, including targets previously associated with sex hormones, the X‐chromosome, and neurodevelopment.
Future directions: Although the genetic contribution to late‐life memory may be similar across sexes, some of the genetic drivers appear to differ by sex. Our study emphasizes the importance of investigating AD cognitive endophenotypes in a sex‐specific manner, to identify novel targets that may best suit each sex.
2.3. Cognitive harmonization
Qualified neuropsychologists and behavioral neurologists (authors A.J.S., J.M., E.H.T.) categorized test item‐level data into memory, executive functioning, language, or visuospatial functioning domains (or none of these). , These experts evaluated each test item to ensure that scoring was equivalent across studies, and recoded scoring to match when necessary (eg, higher score = always better performance, irrespective of study). Additionally, the recoding process involved collapsing missing data into as few categories as possible. Then, a confirmatory factor analysis was performed in each cohort for each domain, leveraging the last visit for each individual, to determine a composite score that best captured domain variance. The best single factor or bi‐factor model was chosen for each domain based on acceptable ranges for three fit statistics: confirmatory fit index, Tucker‐Lewis Index, and root mean square error of approximation. Supplemental materials for our published harmonization methods paper include study‐specific fit statistics for each domain, all of which are in the “good” to “excellent” range. 14
Next, in order to facilitate co‐calibration, common items across test protocols were selected, and placed into a confirmatory factor analysis to ensure equivalency across studies (and confirmed equivalent by author E.H.T.). Once confirmed, these common items, denoted as “anchor items,” were selected as anchors across studies. Co‐calibration was performed using a confirmatory factor analysis model where the anchor items were restricted to have the same parameters across studies. Parameters were allowed to vary by study for all items that were specific to a particular study. Finally, all visits were included in one main model with all estimated item parameters fixed to generate factor scores for each participant at each visit. The resulting domain composite scores were on the same metric across studies (Figure 1C). Thus, the final distribution of factor scores for each study was on a z‐score scale with a mean = 0 and standard deviation = 1, where +1 or −1 point meant the same across studies. For more information on cognitive harmonization, see a recent paper by Mukherjee and colleagues describing the harmonization protocol at length. 14
2.4. Autosomal and X‐chromosome genetic data quality control and imputation
Raw genetic data were collected on the genotyping arrays outlined to follow: ACT data were collected on the Infinium Global Screening Array‐24 BeadChip. ADNI data were collected on four arrays: Illumina Human610‐Quad BeadChip, Illumina HumanOmniExpress BeadChip, Illumina Omni 2.5 M, and Illumnia Global Screening Array v2. NACC genetic data acquisition is described on their website: https://naccdata.org/nacc‐collaborations/partnerships. ROS/MAP/MARS data were collected on three arrays: Global Screening Array‐24 v3.0 BeadChip, Affymetrix GeneChip 6.0, and some data were obtained from the Alzheimer's Disease Genetics Consortium genome‐wide association studies (GWAS) datasets. We performed a standardized quality control and imputation pipeline on raw genotypes, which we will outline below. For quality control, we made non‐Hispanic White (NHW) and non‐Hispanic Black (NHB) determinations based on self‐reported race/ethnicity and based on a principal component analysis, which also facilitated ancestry‐specific outlier removal.
First, we applied a series of variant‐level filters, removing variants that had >5% missingness or a minor allele frequency (MAF) of <1%. Second, we applied a series of sample‐level filtering, removing samples that had >1% missingness, related individuals, individuals with mismatched self‐reported and genetically determined sex, and individuals with excessive heterozygosity (p < 10−7). For X‐chromosome variants, genetic data were compared between sexes, and variants with differential missingness were filtered (p < 10−7). We conducted a Hardy‐Weinberg equilibrium (HWE) exact test, filtering out variants with p < 10−6. We phased and imputed the cleaned genetic data on the Trans‐Omics for Precision Medicine imputation server (hg38). 24 , 25 , 26 Raw imputed data were filtered for an imputed r2 < 0.8 and duplicated/multi‐allelic variants. Then original genotypes were merged back into the imputed data and another HWE exact test was performed in the respective datasets. All variants were subsequently filtered for MAF < 1%.
All imputed, cleaned chip‐specific datasets from each cohort were merged into one final dataset. First, we compared samples across chips for duplicate samples, and checked concordance across those duplicated samples. If concordance was >99%, the sample on the chip with greater coverage was typically retained. Each chip was then compared and filtered for reference allele mismatches and MAF differences of >10%. Then, clean imputed data were merged across chips for each cohort. We next assessed the merged datasets for cryptic relatedness, removing related individuals, and for genetic ancestry outliers, filtering out ancestry‐specific outliers.
2.5. Statistical analyses
2.5.1. Single nucleotide polymorphism‐heritability tests
We next performed single nucleotide polymorphism (SNP)‐heritability analyses (with GCTA 27 ) in a diagnosis‐ and sex‐stratified manner, restricting the sample first to cognitively unimpaired and then to cognitively impaired participants. This exploratory analysis was completed to deconvolve some of the phenotypic heterogeneity present in the overall sample that includes individuals with and without dementia. While it is well established that memory performance is a heritable trait, and AD dementia is a heritable trait, we were interested in deconvolving the late‐life memory performance data to better understand whether there are any sex differences in the heritability of memory performance (leveraging a published formula 28 ) in the absence of baseline cognitive impairment (to simplify the sample to individuals at a similar diagnostic starting point) and whether there are sex differences in the heritability of memory performance following a clinical diagnosis of dementia (acting as a marker of disease progression).
2.5.2. Memory endophenotype selection
In this study, we were interested in leveraging memory performance as an endophenotype for AD. Memory was the selected endophenotype because it (1) increases precision by focusing on a single, consistently measured quantitative metric; and (2) allows for increased statistical power due to longitudinal data that more accurately represent the continuum of change across the diagnostic spectrum. In particular, we were interested in “late‐life memory,” defined in this study as memory performance after age 60. Thus, our target population was older adults, who as a subgroup are more likely to experience preclinical memory changes or AD‐related decline.
To study memory performance, we selected both baseline memory and memory decline measures outlined as follows. First, we extracted baseline memory scores for each participant, based on their first cognitive visit. This served as our baseline memory phenotype for subsequent GWAS and post‐GWAS analyses. Second, we calculated memory slopes for each participant who had at least two cognitive visits, using all datapoints available for each of these participants. Slopes were calculated with a null linear mixed effects model, letting slope and intercept vary for each participant. The interval term used in our linear mixed effects regression model is quantified as years from baseline for each observation for each participant, so the extracted slope from the model represents a standardized metric of annual change in memory performance for each participant. The individual memory slopes served as our memory decline phenotype for subsequent GWAS and post‐GWAS analyses.
2.5.3. Genome‐wide and X‐wide association studies among all participants
Prior to performing GWAS, cryptic relatedness was assessed across all four cohorts, and related individuals were typically removed from the larger cohort. Baseline memory and memory decline GWAS were performed with PLINK's 29 linear association tool with additive variant coding. All GWAS described were performed in males, in females, and with a sex‐interaction. Covariates included baseline age and the first five genetic ancestry principal components, and additionally the sex‐interaction GWAS contained an SNP‐by‐sex interaction term. We performed GWAS in ACT, ADNI, NACC, and ROS/MAP/MARS separately. Within each cohort, GWAS were performed among NHW and among NHB participants separately. X‐wide association studies (XWAS) were performed identically to the GWAS, except that male genotypes were coded as 0/2 (instead of 0/1) to account for X‐chromosome dosage differences between males and females.
2.5.4. GWAS and XWAS subgroup analyses
Within each ancestry group, we additionally performed sex‐stratified and sex‐interaction GWAS and XWAS subgroup analyses, by first limiting the sample to cognitively unimpaired and then limiting to cognitively impaired participants. Clinical diagnosis determinations are outlined above in Section 2.2, and the distributions of memory scores within each diagnosis group are displayed in Figures S1 and S2. These stratified analyses allowed us to explore genetic effects contributing to memory performance in the absence of cognitive impairment as well as in the context of MCI/AD.
2.5.5. Sensitivity analyses
For all genome‐wide significant variants, we performed a two‐part sensitivity analysis. First, we conducted post hoc SNP‐interaction analyses. Thus, for each significant variant, we had a series of three linear models that had SNP‐by‐ancestry‐by‐sex, SNP‐by‐diagnosis‐by‐sex, and SNP‐by‐ancestry‐by‐diagnosis‐by‐sex interaction terms, respectively. Second, we checked the association of genome‐wide significant variants in the diagnosis‐stratified subgroup analyses (please see Section 2.5.4).
2.5.6. Within‐ancestry genome‐wide meta‐analyses
Male, female, and sex‐interaction individual cohort GWAS were meta‐analyzed implementing a fixed‐effects model with beta and standard error input (eg, GWAMA v2.2.2.). 30 Meta‐analyses were performed within each ancestry group and furthermore within each diagnostic category mentioned above. Meta‐analysis results were restricted to SNPs present in three or four (out of four) cohorts. Results were further filtered to retain SNPs with a stratum specific MAF of >1%. These filtered meta‐analysis results were leveraged for the cross‐ancestry meta‐analyses (see next section).
2.5.7. Cross‐ancestry genome‐wide meta‐analyses
We additionally performed cross‐ancestry meta‐analyses. To do this, we took each within‐ancestry genome‐wide meta‐analysis described above (ie, for NHW and NHB) and meta‐analyzed those with a fixed‐effects approach. 30 SNPs were retained if they were present in both ancestry groups.
2.5.8. Expression quantitative trait locus analysis
All genome‐wide significant variants were queried in the following expression quantitative trait loci (eQTL) databases: GTEx (https://gtexportal.org/), BRAINEAC (http://www.braineac.org), eQTLGen Consortium (whole blood; https://www.eqtlgen.org), Brain xQTLServe (http://mostafavilab.stat.ubc.ca/xqtl/), BrainSeq (dorsolateral prefrontal cortex [DLPFC] and hippocampus; http://eqtl.brainseq.org), and MetaBrain (https://www.metabrain.nl). The eQTL significance threshold was set a priori at p < 0.05. For each eQTL, p‐values were determined by the given p‐values in each database for the given tissue(s).
2.5.9. Functional annotation of top genetic loci
When applicable, we conducted variant mapping with the Functional Mapping and Annotation of Genome‐Wide Association Studies online software tool (FUMA; https://fuma.ctglab.nl/). 31 , 32 All SNPs in linkage disequilibrium (LD) with the top SNP in the locus, irrespective of their p‐values, were also considered for annotation. In brief, FUMA performs three types of mapping: (1) positional, (2) eQTL, and (3) chromatin. The chromatin mapping looks for regions in the locus that are enriched in topologically associated domains, measured by Hi‐C assays. Furthermore, FUMA checks to see if these enriched regions in the topologically associated domains overlap with promoters or enhancers of genes across multiple cell types and tissues.
2.5.10. Gene‐set analyses
Prior to conducting gene‐set analyses, we removed a 1Mb region up‐ and downstream of the apolipoprotein E [APOE] gene from our meta‐analysis results, to allow for investigation of genes and pathways contributing to memory beyond APOE. Next, we performed male, female, and sex‐interaction gene‐ and pathway‐based tests with the Multi‐marker Analysis of GenoMic Annotation (MAGMA v1.09) analytical tool 33 on both the within‐ancestry and the cross‐ancestry filtered meta‐analysis results. MAGMA gene‐set analyses consist of permutation‐like tests to determine if a set of SNPs or a gene‐set is associated with a gene or a biological pathway, respectively, more than would be expected by random chance. Gene annotations were curated leveraging our genetic data files and Genome Reference Consortium Human Build 38 gene locations. Pathway annotations were compiled from gene sets from the Molecular Signatures Database 34 v.7.0. All gene‐ and pathway‐based tests were adjusted for multiple comparisons with the false discovery rate (FDR) procedure with an a priori significance threshold set at FDR < 0.05.
2.5.11. Genetic correlation tests
Prior to conducting genetic correlation analyses, we removed a 1Mb region up‐ and downstream of APOE from our meta‐analysis results, to allow us to investigate the sex‐specific genetic architecture of memory beyond the APOE locus. We leveraged our male, female, and sex‐interaction NHW meta‐analysis filtered results to perform genetic correlation tests between our GWAS and GWAS of other complex traits. Genetic correlation estimates were calculated with the GENOVA. 35 When correlation estimates were unstable due to sample size limitations of the original GWAS, genetic covariance estimates were used instead. Genetic correlations/covariances were adjusted for LD inflation with ancestry‐specific LD‐scores, calculated based on the 1000 Genomes (European) reference genetic dataset. All genetic correlation (or covariance) values were adjusted for multiple comparisons with the FDR procedure with significance set a priori as FDR < 0.05. We calculated genetic correlations with a curated set of 65 complex traits.
3. RESULTS
Our study included a total of 27,583 participants, comprised of 24,216 NHW individuals (54.3% female) and 3367 NHB individuals (73.4% female). Participant characteristics are presented in Table 1. Females were 59% cognitively unimpaired, 37% APOE ɛ4 carriers, with a mean age of 74 years at baseline. Males were 45% cognitively unimpaired, 38% APOE ɛ4 carriers, with a mean age of 74 years at baseline.
3.1. Sex‐aware heritability calculations of memory performance by ancestry and by sex
We calculated heritability estimates for memory performance in each sex as well as in both sexes combined, and we did this among NHW individuals and among NHB individuals. All heritability estimates can be found in Table 2. Heritability estimates in both sexes for NHW participants ranged from 0.14 to 0.17 for baseline memory and 0.10 to 0.15 for memory decline. Across sexes for NHW individuals, estimates trended a bit higher for males, but there were no statistically significant differences between sexes. Additionally, among NHW participants of each sex, baseline memory heritability estimates were higher when estimated among cognitively unimpaired individuals, and memory decline heritability estimates were higher when estimated among cognitively impaired individuals. Heritability estimates for NHB participants in both sexes (and sex‐stratified) appeared a bit higher than NHW for baseline memory, albeit the standard errors were quite large suggesting that estimates were less stable. For memory decline among NHB individuals, several of the heritability estimates were too small (and unstable) to interpret, likely due to small sample sizes. In contrast with the NHW results, heritability estimates appeared to trend higher among NHB females compared to NHB males, but no estimates significantly differed by sex.
TABLE 2.
Sex‐specific heritability of memory performance.
| Non‐Hispanic White | Sex differences test | |||||
|---|---|---|---|---|---|---|
| Phenotype | diagnosis | Both sexes | Men | Women | z‐score | p‐value |
| Baseline memory | All diagnoses |
β = 0.17 (SE = 0.01) p < < < 0.01 |
β = 0.16 (SE = 0.03) p = 8.93 × 10–12 |
β = 0.17 (SE = 0.03) p = 1.94 × 10–14 |
0.24 | 0.81 |
| Cognitively unimpaired |
β = 0.17 (SE = 0.03) p < < < 0.01 |
β = 0.25 (SE = 0.06) p = 6.74 × 10–9 |
β = 0.21 (SE = 0.04) p = 2.65 × 10–13 |
−0.55 | 0.58 | |
| Cognitively impaired |
β = 0.14 (SE = 0.04) p = 9.44 × 10–9 |
β = 0.20 (SE = 0.07) p = 6.87 × 10–6 |
β = 0.11 (SE = 0.07) p = 8.73 × 10–3 |
−0.91 | 0.36 | |
| Memory decline | All diagnoses |
β = 0.14 (SE = 0.02) p < < < 0.01 |
β = 0.20 (SE = 0.04) p = 2.26 × 10–10 |
β = 0.12 (SE = 0.03) p = 2.12 × 10–6 |
−1.60 | 0.11 |
| Cognitively unimpaired |
β = 0.10 (SE = 0.03) p = 1.01 × 10–9 |
β = 0.14 (SE = 0.06) p = 2.23 × 10–5 |
β = 0.09 (SE = 0.04) p = 2.83 × 10–4 |
−0.69 | 0.49 | |
| Cognitively impaired |
β = 0.15 (SE = 0.05) p = 1.92 × 10–7 |
β = 0.31 (SE = 0.09) p = 1.54 × 10–5 |
β = 0.21 (SE = 0.09) p = 5.04 × 10–5 |
–0.79 | 0.43 | |
| Non‐Hispanic Black | Sex differences test | |||||
|---|---|---|---|---|---|---|
| Phenotype | diagnosis | Both sexes | Men | Women | z‐score | p‐value |
| Baseline memory | All diagnoses |
β = 0.27 (SE = 0.09) p = 4.36 × 10–5 |
β = 0.19 (SE = 0.18) p = 6.81 × 10–2 |
β = 0.21 (SE = 0.12) p = 1.75 × 10–2 |
0.09 | 0.93 |
| Cognitively unimpaired |
β = 0.28 (SE = 0.15) p = 2.64 × 10–3 |
β = 0.14 (SE = 0.33) p = 2.96 × 10–1 |
β = 0.37 (SE = 0.20) p = 8.29 × 10–3 |
0.60 | 0.55 | |
| Cognitively Impaired |
β = 0.07 (SE = 0.21) p = 3.96 × 10–1 |
β = 1 × 10–6 (SE = 0.41) p = 5.00 × 10–1 |
β = 0.15 (SE = 0.26) p = 3.08 × 10–1 |
0.31 | 0.76 | |
| Memory decline | All diagnoses |
β = 0.19 (SE = 0.11) p = 2.61 × 10–2 |
β = 0.14 (SE = 0.32) p = 3.50 × 10–1 |
β = 0.19 (SE = 0.14) p = 4.47 × 10–2 |
0.14 | 0.89 |
| Cognitively unimpaired |
β = 0.15 (SE = 0.17) p = 1.37 × 10–1 |
β = 1 × 10–6 (SE = 0.52) p = 5.00 × 10–1 |
β = 0.24 (SE = 0.21) p = 8.61 × 10–2 |
0.43 | 0.67 | |
| Cognitively impaired |
β = 1E‐06 (SE = 0.26) p = 5.00 × 10–1 |
β = 1 × 10–6 (SE = 0.48) p = 5.00 × 10–1 |
β = 1 × 10–6 (SE = 0.34) p = 5.00 × 10–1 |
0.00 | 1.00 | |
3.2. Sex‐stratified and sex‐interaction genome‐wide significant loci among a cross‐ancestry sample of individuals spanning the AD diagnostic spectrum
All top variant associations (p < 1 × 10−5) from the NHW, NHB, and cross‐ancestry meta‐analyses can be found in Tables S1–S6.
In the cross‐ancestry meta‐analysis among individuals across the AD diagnostic spectrum, we identified a significant sex‐interaction at a chromosome 18 locus whereby the minor allele was associated with baseline memory (Figure 2A; top SNP: rs67099044; MAF = 0.16; βinteraction = 0.10; p interaction = 2.15 × 10−8). Notably, this SNP was also nominally significant in each sex and had a flipped effect across sexes (βmales = −0.05, p males = 2.66 × 10−4; βfemales = 0.04, p females = 2.20 × 10−4). The top SNPs in this locus (rs67099044, rs34049053) are intronic variants within a long, non‐coding RNA, LOC107985179. When stratifying by ancestry, this locus significantly interacted with sex in NHW individuals (βinteraction = 0.16, p interaction = 1.47 × 10−8). Similar to the cross‐ancestry analysis, the locus was nominally significant in each sex, and had a flipped effect across sexes (βmales = −0.05, p males = 2.42 × 10−4; βfemales = 0.04, p females = 3.08 × 10−4). Sensitivity analyses revealed that there was a significant three‐way SNP‐by‐sex‐by‐diagnosis interaction (p interaction = 0.04), whereby cognitively unimpaired individuals seemed to drive the observed sex‐interaction with baseline memory (cognitively unimpaired: βsex‐interaction = 0.08, p sex‐interaction = 4.38 × 10−7; cognitively impaired: βsex‐interaction = 0.03, p sex‐interaction = 0.41). In addition, variant annotation revealed that this locus is enriched in Hi‐C chromatin loops in multiple cell types/tissues, one of which is neural progenitor cells, and that the Hi‐C chromatin loops overlap with promoter regions for a wide‐variety of genes, including CBLN2.
FIGURE 2.
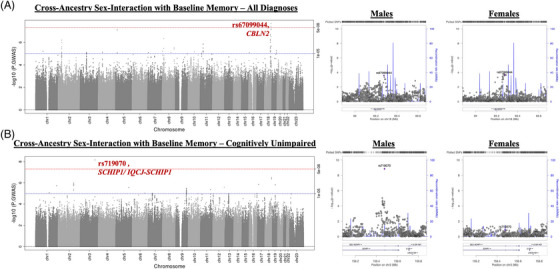
Sex‐specific genetic loci associated with late‐life memory performance. (A) Genome‐wide meta‐analysis on baseline memory in a cross‐ancestral sample, highlighting a significant locus with a sex‐interaction on chromosome 18 (rs67099044); the right plots are regional plots in males (left) and in females (right) surrounding rs67099044. (B) Genome‐wide meta‐analysis on baseline memory among cognitively unimpaired, cross‐ancestral individuals, highlighting a significant locus with a sex‐interaction on chromosome 3 (rs719070); the right plots are regional plots in males (left) and in females (right) surrounding rs719070.
For memory decline among a cross‐ancestry sample spanning the AD diagnostic spectrum, we identified a female‐specific association with a previously published AD risk locus on chromosome 2, BIN1 (top SNP rs6733839; MAF = 0.40; βfemales = −0.01, p females = 3.70 × 10−9; βmales = −3.60 × 10−3, p males = 6.73 × 10−4), but there was no evidence of a sex‐interaction (p interaction = 0.17). Additionally, sensitivity analyses did not reveal any additional effect modifications by diagnosis or by ancestry.
3.3. Sex‐stratified and sex‐interaction genome‐wide significant loci within diagnosis strata
In our cross‐ancestry meta‐analysis among individuals without cognitive impairment, we identified a significant sex‐interaction with baseline memory in an intronic variant within SCHIP1/IQCJ‐SCHIP1 (Figure 2B; rs719070 on chromosome 3; MAF = 0.18; βinteraction = 0.09; p interaction = 7.03 × 10−9). In the sex‐stratified GWAS, this SNP was also significant genome‐wide among cognitively unimpaired males (βmales = −0.07, p males = 1.41 × 10−9), but not among female counterparts (βfemales = 0.01, p females = 0.14). Sensitivity analyses revealed that there was a significant three‐way SNP‐by‐diagnosis‐by‐sex interaction (p interaction = 0.03), and a significant three‐way SNP‐by‐ancestry‐by‐sex interaction (p interaction = 1.80 × 10−3), whereby cognitively unimpaired NHW and NHB males each appeared to drive the observed memory association. In addition to being an intron variant within SCHIP1, functional annotation (eg, FUMA) showed that the top variant in this locus is enriched in Hi‐C chromatin loops in multiple cell types/tissues for SCHIP1.
Furthermore, we identified a locus of nominal significance in the cross‐ancestry X‐wide meta‐analysis (rs5935633: MAF = 0.07; βfemales = −0.01, p females = 2.77 × 10−3; βmales = −3.46E x10−3, p males = 0.26) that reached genome‐wide significance among cognitively impaired NHB participants, whereby this X‐chromosome locus was significantly associated with memory decline among females (Figure 3; top SNP rs5935633; MAF = 0.12; βfemales = −0.04, p females = 3.55 × 10−8) but not among males (βmales = −2.29 × 10−3, p males = 0.74). The female‐specific association did not reach genome‐wide significance in the cross‐ancestry meta‐analysis of cognitively impaired participants, likely because the association was null among NHW participants. Sensitivity analyses showed that the four‐way SNP‐by‐ancestry‐by‐diagnosis‐by‐sex interaction was significant (p interaction = 0.03), whereby the observed association seemed to be specific to cognitively impaired NHB females. Interestingly, the top variant in this locus, rs5935633, is an intron variant within EGFL6 and is an eQTL for TCEANC in the DLPFC and for OFD1 in the hippocampus.
FIGURE 3.
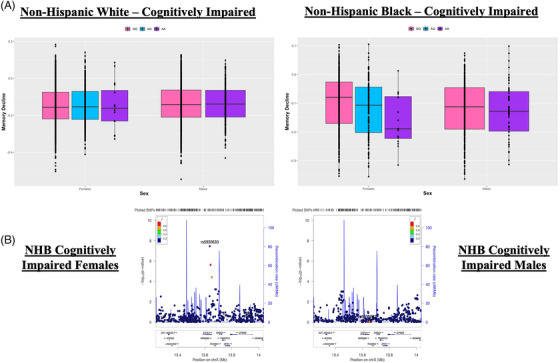
X‐chromosome locus associated with late‐life memory decline. (A) Nominally significant cross‐ancestry X‐chromosome locus that reached genome‐wide significance among cognitively impaired non‐Hispanic Black (NHB) females. Top variant, rs5935633, is plotted by sex among non‐Hispanic White (NHW) participants (left) and NHB participants (right). (B) Regional plot in NHB males (left) and in NHB females (right) surrounding rs5935633.
3.4. Gene‐set analyses identify candidate genes and biological pathways significantly associated with memory performance in a sex‐specific manner
All top genes and pathways (p < 0.05) from the NHW, NHB, and cross‐ancestry gene‐set analyses can be found in Tables S7–S18.
In the cross‐ancestry analysis, we identified Gene Ontology biological pathways (Figure 4) that were associated with baseline memory only among females: Heparan Sulfate Proteoglycan Biosynthetic Process Polysaccharide Chain Biosynthetic Process (p males = 0.86, p.FDRmales = 1.00; p females = 1.82 × 10−6, p.FDRfemales = 0.02); Heparan Sulfate N‐Acetylglucosaminyltransferase Activity (p males = 0.78, p.FDRmales = 0.99; p females = 8.87 × 10−6, p.FDRfemales = 0.04); Heparan Sulfate Sulfotransferase Activity (p males = 0.82, p.FDRmales = 0.99; p females = 9.39 × 10−6, p.FDRfemales = 0.04); and Heparin Biosynthetic Process (p males = 0.99, p.FDRmales = 1.00; p females = 1.48 × 10−5, p.FDRfemales = 0.045).
FIGURE 4.
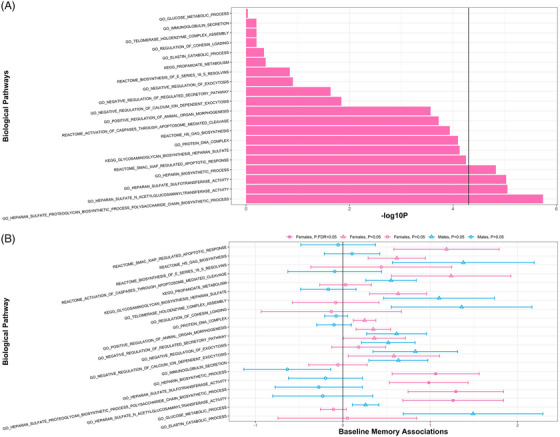
Sex‐specific biological pathways associated with late‐life memory performance. (A) Bar chart displaying the ‐log10 p‐values of the top biological pathways (p < 1E‐03) associated with baseline memory among cross‐ancestral females. (B) The 95% confidence intervals of the top biological pathways associated with baseline memory in cross‐ancestral females (pink) with corresponding confidence intervals in cross‐ancestral males (blue), with shapes denoting level of significance. GO, Gene Ontology.
We also identified one biological pathway with a sex‐interaction with baseline memory among cognitively unimpaired individuals—Reactome Interleukin‐6 Signaling (p interaction = 1.50 × 10−6, p.FDRinteraction = 0.02)—and this pathway was nominally significant among the cross‐ancestry sample of cognitively unimpaired males (p males = 4.10 × 10−4). Additionally, this pathway had a nominally significant sex‐interaction in both NHW (p interaction = 0.01) and NHB (p interaction = 0.01) cognitively unimpaired individuals.
3.5. Some complex traits have sex‐specific shared genetic architecture with memory performance
All genetic correlations were conducted among NHW individuals only, and all genetic correlation results can be found in Tables S19–S24.
We evaluated shared genetic architecture between a curated set of 65 GWAS of complex traits and our sex‐stratified meta‐analyses on memory performance. All correlations reported below are for individuals spanning the AD diagnostic spectrum and survived adjustment for multiple comparisons with the FDR procedure (FDR < 0.05). Five traits were significantly associated with baseline memory among males and 16 traits were significantly associated among females. Of traits surviving FDR correction in at least one sex, 95% confidence intervals did not overlap between sexes for five traits: anxiety disorder, educational attainment, insomnia, risky behavior, and heart rate variability (HRV), suggestive of true sex effects (Figure 5A). Anxiety disorder was significantly and negatively correlated among females and not correlated among males; educational attainment was significantly and positively correlated in both sexes, but stronger among females; insomnia was significantly and negatively correlated among females and nominally significant and positively correlated among males; risky behavior was significantly and positively correlated in males and not correlated in females; and finally, HRV was nominally significant and negatively correlated among males and significantly and positively correlated among females.
FIGURE 5.
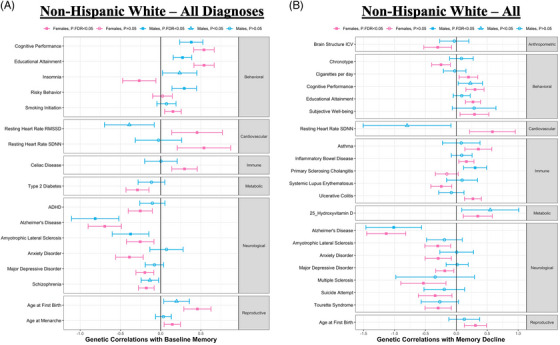
Sex‐specific genetic correlations with late‐life memory performance. (A) Genetic correlations between baseline memory and complex traits by sex (among non‐Hispanic White [NHW] participants), with males in blue, females in pink, and shapes denoting level of significance. All traits displayed are false discovery rate (FDR)‐significant in at least one sex. (B) Genetic correlations between memory decline and complex traits by sex (among NHW participants), with males in blue, females in pink, and shapes denoting level of significance. All traits displayed are FDR‐significant in at least one sex. ADHD, attention‐deficit/hyperactivity disorder; ICV, total intracranial volume; RMSSD, root mean square of successive R‐R interval differences; SDNN, standard deviation of the N‐N intervals.
We observed two traits associated with memory decline among males and 20 traits associated with memory decline among females. Of traits surviving FDR correction in at least one sex, 95% confidence intervals did not overlap between sexes for three traits—primary sclerosing cholangitis, HRV, and ulcerative colitis—suggestive of true sex effects (Figure 5B). Primary sclerosing cholangitis was not correlated among females and significantly and positively correlated among males; HRV was nominally significant and negatively corelated among males and significantly and positively correlated among females; and ulcerative colitis was not correlated among males and significantly and positively correlated among females.
4. DISCUSSION
We completed the largest and most comprehensive analysis of sex‐specific genetic predictors of late‐life memory performance to date. We observed that memory is comparably and highly heritable across ancestry and sex, and we highlighted novel genetic variants, candidate genes, molecular pathways, and genetic correlations that relate to memory in a sex‐specific manner. Our results suggest that the genetic contribution to memory is comparable across sexes, but the molecular drivers of memory vary substantially. In particular, genetic drivers of the X‐chromosome and sex hormones appear to be relevant among females, while genetic drivers relevant to neurodevelopment appear to be relevant among males.
4.1. Memory performance is comparably and highly heritable across sexes
We calculated SNP‐heritability estimates stratified by ancestry and by sex (Table 2). Although estimates trended higher in males among NHW and higher in females among NHB, estimates did not significantly differ by sex, consistent with a previous twin study evaluating sex differences in memory heritability. 36 In our study, narrow‐sense heritability estimates were slightly lower (≈0.10 to 0.25 compared to 0.30 to 0.81 in twin studies), 37 , 38 , 39 perhaps due to the older age and high phenotypic heterogeneity in our sample. 40 In support of that possibility, we noticed that our heritability estimates for baseline memory were higher when estimated among the more homogenous sample of cognitively unimpaired individuals. We have observed this in prior AD endophenotype GWAS by our group. 15 , 41 Overall, our results add strong evidence that memory performance is highly heritable with a comparable genetic component across sex and ancestry.
4.2. Heparan sulfate signaling and the X‐chromosome are implicated as sex‐specific genetic drivers of memory
We found that heparan sulfate signaling pathways associated with baseline memory among a cross‐ancestry sample of females (Figure 4). Heparan sulfate signaling has been implicated in the AD neuropathological cascade and in female‐related disorders. First, heparan sulfate signaling has a hypothesized role in amyloidosis, 42 , 43 and genes that contribute to the heparan pathways are upregulated in the AD brain. 44 Second, heparan sulfate signaling is implicated in female sex biology. Heparan sulfate is related to the menstrual cycle 45 and the onset of labor. 46 Increased heparan sulfate proteoglycans are linked to polycystic ovary syndrome 47 and preeclampsia. 48 Lastly, heparan signaling is involved with embryo implantation 49 and oocyte patterning. 50 In our results, upregulation of these heparan signaling pathways were associated with better memory performance among females but not among males (Figure 4). One notable significant gene‐level association, NDST3, contributed to the heparan sulfate pathway signal among females. NDST3 is involved in microtubule acetylation, and notably microtubule deacetylation is a hallmark of neurodegenerative disorders. 51 Indeed, atypical expression of NDST3 is observed in multiple neurodegenerative disorders, including AD. 51
In a recent sex‐aware AD endophenotype GWAS study by our group, we hypothesized that female‐specific genomic findings may be driven by (1) X‐chromosome, (2) sex hormones, or (3) both. 15 Thus, it is noteworthy that we identified a hormone‐related target (heparan sulfate signaling) and an X‐chromosome candidate (EGFL6; Figure 3). EGFL6 is involved in cell‐cycle regulation, craniofacial development, 52 and female cancers. 53 , 54 The top variant in the locus, rs5935633, was a hippocampal eQTL for OFD1 and a DLPFC eQTL for TCEANC. TCEANC and OFD1 are additional sex‐specific candidate genes, as one study showed that TCEANC escapes X‐inactivation across multiple species, 55 and OFD1 mutations lead to neurological‐related phenotypes. 56
Overall, our results again underscore the importance of analyzing the X‐chromosome when looking for sex‐specific effects and highlight the heparan sulfate signaling pathway as a potential novel female‐specific genetic driver of memory performance. While we highlight some evidence that the heparan sulfate effect may be modulated by sex hormones, further work will be needed to replicate and evaluate this hypothesis.
4.3. Genetic correlations between complex traits and memory performance differ by sex
We examined the shared genetic architecture between memory performance and complex traits (Figure 5) and identified traits that significantly differed by sex. We observed evidence of female‐specific genetic correlations between memory and immune traits, consistent with our previous findings, 15 and a slightly stronger genetic correlation between memory and educational attainment among females, consistent with the stronger association between educational attainment and dementia among women in the literature. 57 , 58
One sex‐specific genetic correlation we found interesting was the correlation between memory decline and HRV. Among females, we saw an expected genetic correlation (Figure 5B) such that genetic susceptibility towards worse HRV was associated with a faster rate of memory decline, a direction of effect consistent with the literature, where reduced HRV is associated with worse cognition, 59 particularly among women. 60 In contrast, among males we observed a counter‐intuitive genetic correlation such that genetic susceptibility towards worse HRV was associated with a slower rate of memory decline, although the male‐stratified correlation did not survive correction for multiple comparisons (Figure 5B). The male‐stratified results are also in contrast to our previously published male‐specific association with cognitive resilience. 15
Upon further investigation, local genetic correlations 61 highlighted that the regions driving this study's sex‐specific association were RGS6 (chromosome 14), which is implicated in HRV 62 and is dysregulated in AD, 63 as well as CD33 (chromosome 19), which is a known AD risk gene. 64 The present results suggest that genetic drivers of HRV in females may be particularly relevant to memory decline in cognitive aging and AD, but more work is needed to deconvolve the complex interplay between sex, HRV, neuropathology, and cognition.
4.4. Sex‐specific variant associations with baseline memory performance
We identified a cross‐ancestry, genome‐wide significant locus (chromosome 18; top variant, rs67099044) that had a sex‐interaction with baseline memory (Figure 2A). Functional annotation of this locus revealed a candidate gene, CBLN2, which is involved in synaptogenesis. There was also nominal eQTL evidence for CBLN2 in the brain. CBLN2 has been previously implicated in neurodevelopmental disorders, such as autism and Tourette's syndrome, 65 , 66 and a prior sex‐specific association was observed with bilirubin concentrations. 67 While there was evidence of this locus's involvement in the brain and potential sex‐specific effects of CBLN2, this locus needs to be replicated in future sex‐aware genetic studies.
We also identified a cross‐ancestry, genome‐wide significant chromosome 3 locus. The top variant in this locus was rs719070, an intronic variant within SCHIP1/IQCJ‐SCHIP1 that was significant among cognitively unimpaired males and had a significant sex‐interaction (Figure 2B). SCHIP1 and IQCJ‐SCHIP1 are both hypothesized to be involved with axon growth during brain development 68 and with the nodes of Ranvier in the adult brain. 69 , 70 , 71 Additionally, IQCJ‐SCHIP1 was previously implicated in neurodevelopmental disorders such as autism and language deficit disorders 71 , 72 and is highly expressed in the fetal brain. 69 Future work will need to be done to replicate our study's finding and confirm the causal gene in the region.
4.5. Strengths and limitations
Our study had many strengths but was not without limitations. We leveraged four deeply phenotyped cohorts, each with years of longitudinal follow‐up. Each participant had harmonized memory composite scores, although we were unable to evaluate subcomponents of memory, such as episodic memory. As Kremen and colleagues stated, 73 individual memory tasks may be driven by different genetic factors, which we were unable to fully address in this study. 73 Future studies will need larger sample sizes in order to harmonize memory subcomponents (eg, episodic memory) and investigate the genetic factors driving each of these individual subcomponents. Furthermore, most of our findings in this study were baseline memory associations driven by preclinical individuals. The relative lack of significant findings for longitudinal memory may be due, in part, to the substantial inter‐individual variability in cognitive trajectories, especially among those with MCI/AD. Furthermore, there is no gold standard for analyzing longitudinal phenotypes in the context of GWAS. Therefore, advanced modeling techniques and larger sample sizes with less phenotypic heterogeneity are needed to detect genetic factors contributing to memory change in AD. Additionally, in this analysis, we had data from NHW and NHB individuals, and leveraged a cross‐ancestral approach. Although we were somewhat underpowered to detect NHB‐specific effects, as sample sizes continually grow for more diverse cohorts, we will be able to include more ancestries and diverse samples. Furthermore, with few published genomic studies on the X‐chromosome and AD, a strength of this study was inclusion of the X‐chromosome, albeit we were a bit underpowered to detect its effects. Finally, we did not have a replication sample for our top sex‐specific variant associations.
5. CONCLUSIONS
We conducted the largest sex‐aware, cross‐ancestral genetic study on late‐life memory performance to date. From this study, we identified three sex‐specific loci, including one X‐chromosome locus. We identified multiple sex‐specific candidate genes and sex‐vulnerable biological pathways both with ties to neurodevelopment and female‐related disorders. Overall, our study suggests that although much of the genetic architecture of late‐life memory performance is shared across sexes, a sex‐specific genetic component to memory does exist, providing exciting targets for future intervention. Our findings highlight the benefits of characterizing sex‐specific genes and pathways and their relation to cognitive aging and AD.
Data collection and sharing for ADNI were supported by National Institutes of Health Grant U01‐AG024904 and Department of Defense (award number W81XWH‐12‐2‐0012). ADNI is also funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Ann McKee, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD).
The Alzheimer's Disease Genetics Consortium supported genotyping and data processing of samples through National Institute on Aging (NIA) grants U01‐AG032984. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689). The Adult Changes in Thought Study, based at Kaiser Permanente Health Research Institute of Washington and the University of Washington, is supported through U19‐AG066567.
Additional support includes R01‐AG073439, U24‐AG074855, K01‐AG049164, R01‐AG059716, R01‐AG061518, R21‐AG059941, K12‐HD043483, K24‐AG046373, HHSN311201600276P, S10‐OD023680, R01‐AG034962, R01‐NS100980, R01‐AG056534, P30‐AG010161, R01‐AG15819, R01‐AG17917, U01‐AG46152, R01‐AG048927, U19‐AG068753, U01 AG058654, R01‐AG062634, R01‐AG048927, U19‐AG068753, K23:AG045966, the Nancy and Buster Alvord Endowment (C. D. K.), the Vanderbilt Clinical Translational Science Award (UL1‐TR000445), the Vanderbilt Memory and Alzheimer's Center, and the Vanderbilt Alzheimer's Disease Research Center (P20‐AGAG068082). Part of Figure 1 was created with BioRender.com. Additional funding also includes F31‐AG077791.
The ADSP Phenotype Harmonization Consortium (ADSP‐PHC) is funded by NIA (U24 AG074855, U01 AG068057 and R01 AG059716). The ADSP‐PHC cohorts include: Adult Changes in Thought (ACT, U01 AG006781, U19 AG066567), the Alzheimer's Disease Centers (ADC, P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD), ADNI funded by U01 AG024904, and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). The Memory and Aging Project at the Knight‐ADRC, is supported by NIH grants R01AG064614, R01AG044546, RF1AG053303, RF1AG058501, U01AG058922 and R01AG064877 to Carlos Cruchaga. The recruitment and clinical characterization of research participants at Washington University was supported by NIH grants P30AG066444, P01AG03991, and P01AG026276. Data collection and sharing for this project was supported by NIH grants RF1AG054080, P30AG066462, R01AG064614 and U01AG052410.
This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the Neurogenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/) and the Departments of Neurology and Psychiatry at Washington University School of Medicine; the Minority Aging Research Study (MARS, R01 AG22018, R01 AG42210), the National Alzheimer's Coordinating Center (NACC, U01 AG016976, U24 AG072122),the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐ LOAD, U24 AG056270), the Religious Orders Study (ROS, P30 AG10161, P30 AG72975, R01 AG15819, R01 AG42210), the RUSH Memory and Aging Project (MAP, R01 AG017917, R01 AG42210), the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, and the Genome Center for Alzheimer's Disease (U54AG052427), funded by NIA.
The Alzheimer's Disease Genetics Consortium supported collection and genotyping of samples used in this study through National Institute on Aging (NIA) grants U01AG032984 and RC2AG036528. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689‐01), The Center for Applied Genomics at the Children's Hospital of Philadelphia Research Institute performed genotyping of samples. The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD). Samples from the National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. (www.fnih.org). Rush University grants P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG030146, R01 AG01101, RC2 AG036650, R01 AG22018, U01 AG06781, U01 HG004610.
CONFLICT OF INTEREST STATEMENT
T.J.H. is a member of the scientific advisory board for Vivid Genomics, and a Senior Associate Editor for Alzheimer's & Dementia. The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Written informed consent was obtained from all participants in the ACT, ADNI, NACC, and ROS/MAP/MARS cohorts, and research was carried out in accordance with Institutional Review Board‐approved protocols. Secondary analyses of all data were approved by the Vanderbilt University Medical Center Institutional Review Board. Please see Table 1 for an overview of the study cohort demographics separated by sex and by ancestry.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
Data collection was supported through funding by NIA grants P30AG10161 (ROS), P30AG72975, R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG22018, R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq) RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP‐AD, targeted proteomics), U01AG46161 (TMT proteomics), U01AG61356 (whole genome sequencing, targeted proteomics, ROSMAP AMP‐AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.synapse.org). Study data were provided by the Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. Additional phenotypic data can be requested at www.radc.rush.edu.
1.
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Eissman JM, Archer DB, Mukherjee S, et al. Sex‐specific genetic architecture of late‐life memory performance. Alzheimer's Dement. 2024;20:1250–1267. 10.1002/alz.13507
REFERENCES
- 1. GBD 2019 Dementia Forecasting Collaborators . Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of disease study 2019. Lancet Public Health. 2022;7(2):e105‐e125. doi: 10.1016/S2468-2667(21)00249-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. The Lancet Neurology. 2010;9(1):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albert MS. The ageing brain: normal and abnormal memory. Philos Trans R Soc Lond B Biol Sci. 1997;352(1362):1703‐1709. doi: 10.1098/rstb.1997.0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492‐498. doi: 10.1002/ana.21509 [DOI] [PubMed] [Google Scholar]
- 5. Bäckman L, Small BJ, Fratiglioni L. Stability of the preclinical episodic memory deficit in Alzheimer's disease. Brain. 2001;124(Pt 1):96‐102. doi: 10.1093/brain/124.1.96 [DOI] [PubMed] [Google Scholar]
- 6. Grober E, Lipton RB, Hall C, Crystal H. Memory impairment on free and cued selective reminding predicts dementia. Neurology. 2000;54(4):827‐832. doi: 10.1212/wnl.54.4.827 [DOI] [PubMed] [Google Scholar]
- 7. Munro CA, Winicki JM, Schretlen DJ, et al. Sex differences in cognition in healthy elderly individuals. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2012;19(6):759‐768. doi: 10.1080/13825585.2012.690366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herlitz A, Rehnman J. Sex differences in episodic memory. Curr Dir Psychol Sci. 2008;17(1):52‐56. [Google Scholar]
- 9. Hirnstein M, Stuebs J, Moè A, Hausmann M. Sex/Gender differences in verbal fluency and verbal‐episodic memory: a meta‐analysis. Perspect Psychol Sci. 2023;18(1):67‐90. doi: 10.1177/17456916221082116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voyer D, Voyer SD, Saint‐Aubin J. Sex differences in visual‐spatial working memory: a meta‐analysis. Psychon Bull Rev. 2017;24(2):307‐334. doi: 10.3758/s13423-016-1085-7 [DOI] [PubMed] [Google Scholar]
- 11. Brunet HE, Caldwell JZK, Brandt J, Miller JB. Influence of sex differences in interpreting learning and memory within a clinical sample of older adults. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2020;27(1):18‐39. doi: 10.1080/13825585.2019.1566433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Millet X, Raoux N, Le Carret N, Bouisson J, Dartigues JF, Amieva H. Gender‐related differences in visuospatial memory persist in Alzheimer's disease. Arch Clin Neuropsychol. 2009;24(8):783‐789. doi: 10.1093/arclin/acp086 [DOI] [PubMed] [Google Scholar]
- 13. Crane PK, Narasimhalu K, Gibbons LE, et al. Item response theory facilitated cocalibrating cognitive tests and reduced bias in estimated rates of decline. J Clin Epidemiol. 2008;61(10):1018‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukherjee S, Choi SE, Lee ML, et al. Cognitive domain harmonization and cocalibration in studies of older adults. Neuropsychology. 2022;37(4):409‐423. doi: 10.1037/neu0000835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eissman JM, Dumitrescu L, Mahoney ER, et al. Sex differences in the genetic architecture of cognitive resilience to Alzheimer's disease. Brain. 2022;145(7):2541‐2554. doi: 10.1093/brain/awac177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59(11):1737‐1746. doi: 10.1001/archneur.59.11.1737 [DOI] [PubMed] [Google Scholar]
- 17. Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91‐101. doi: 10.1097/WAD.0b013e318191c7dd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Besser L, Kukull W, Knopman DS, et al. Version 3 of the National Alzheimer's Coordinating Center's Uniform Data Set. Alzheimer Dis Assoc Disord. 2018;32(4):351‐358. doi: 10.1097/WAD.0000000000000279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18(4):270‐277. [PubMed] [Google Scholar]
- 20. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21(3):249‐258. doi: 10.1097/WAD.0b013e318142774e [DOI] [PubMed] [Google Scholar]
- 21. Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer Disease Centers’ Neuropsychological test battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10‐17. doi: 10.1097/WAD.0000000000000223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64(s1):S161‐S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barnes LL, Shah RC, Aggarwal NT, Bennett DA, Schneider JA. The Minority Aging Research Study: ongoing efforts to obtain brain donation in African Americans without dementia. Curr Alzheimer Res. 2012;9(6):734‐745. doi: 10.2174/156720512801322627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284‐1287. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv. 2019. [DOI] [PMC free article] [PubMed]
- 26. Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2014;31(5):782‐784. doi: 10.1093/bioinformatics/btu704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome‐wide complex trait analysis. Am J Hum Genet. 2011;88(1):76‐82. doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin J, Khramtsova EA, Goleva SB, et al. Examining Sex‐differentiated genetic effects across neuropsychiatric and behavioral traits. Biol Psychiatry. 2021;89(12):1127‐1137. doi: 10.1016/j.biopsych.2020.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4(1):7. doi: 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mägi R, Morris AP. GWAMA: software for genome‐wide association meta‐analysis. BMC Bioinf. 2010;11:288. doi: 10.1186/1471-2105-11-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi: 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watanabe K, Umićević Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D. Genetic mapping of cell type specificity for complex traits. Nat Commun. 2019;10(1):3222. doi: 10.1038/s41467-019-11181-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219‐e1004219. doi: 10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu Q, Li B, Ou D, et al. A powerful approach to estimating annotation‐stratified genetic covariance via GWAS summary statistics. Am Hum Genet. 2017;101(6):939‐964. doi: 10.1016/j.ajhg.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Read S, Pedersen NL, Gatz M, et al. Sex differences after all those years? Heritability of cognitive abilities in old age. J Gerontol B Psychol Sci Soc Sci. 2006;61(3):P137‐143. doi: 10.1093/geronb/61.3.p137 [DOI] [PubMed] [Google Scholar]
- 37. Wilson RS, Barral S, Lee JH, et al. Heritability of different forms of memory in the late onset Alzheimer's disease family study. J Alzheimers Dis. 2011;23(2):249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee JH, Flaquer A, Stern Y, Tycko B, Mayeux R. Genetic influences on memory performance in familial Alzheimer disease. Neurology. 2004;62(3):414‐421. doi: 10.1212/01.wnl.0000106461.96637.ac [DOI] [PubMed] [Google Scholar]
- 39. Finkel D, Pedersen NL, McGue M, McClearn GE. Heritability of cognitive abilities in adult twins: comparison of Minnesota and Swedish data. Behav Genet. 1995;25(5):421‐431. doi: 10.1007/BF02253371 [DOI] [PubMed] [Google Scholar]
- 40. Darst BF, Koscik RL, Hermann BP, et al. Heritability of cognitive traits among siblings with a parental history of Alzheimer's disease. J Alzheimers Dis. 2015;45(4):1149‐1155. doi: 10.3233/jad-142658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143(8):2561‐2575. doi: 10.1093/brain/awaa209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fu Y, Zhao J, Atagi Y, et al. Apolipoprotein E lipoprotein particles inhibit amyloid‐β uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener. 2016;11(1):37. doi: 10.1186/s13024-016-0099-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kunkle BW, Schmidt M, Klein HU, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta‐analysis. JAMA Neurol. 2021;78(1):102‐113. doi: 10.1001/jamaneurol.2020.3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pérez‐López N, Martín C, García B, et al. Alterations in the expression of the genes responsible for the synthesis of heparan sulfate in brains with Alzheimer disease. J Neuropathol Exp Neurol. 2021;80(5):446‐456. doi: 10.1093/jnen/nlab028 [DOI] [PubMed] [Google Scholar]
- 45. Maroclo MVO, Pereira SD, Sampaio FJB, Cardoso LEM. Urinary glycosaminoglycan excretion during the menstrual cycle in normal young women. J Urol. 2005;173(5):1789‐1792. doi: 10.1097/01.ju.0000154621.18695.b7 [DOI] [PubMed] [Google Scholar]
- 46. Hjelm AM, Barchan K, Malmström A, Ekman‐Ordeberg GE. Changes of the uterine proteoglycan distribution at term pregnancy and during labour. Eur J Obstet Gynecol Reprod Biol. 2002;100(2):146‐151. doi: 10.1016/s0301-2115(01)00476-6 [DOI] [PubMed] [Google Scholar]
- 47. Giordano MV, Giordano LA, Gomes RCT, et al. The evaluation of endometrial sulfate glycosaminoglycans in women with polycystic ovary syndrome. Gynecol Endocrinol. 2015;31(4):278‐281. doi: 10.3109/09513590.2014.989980 [DOI] [PubMed] [Google Scholar]
- 48. Khedun SM, Naicker T, Moodley J, Gathiram P. Urinary heparan sulfate proteoglycan excretion in black African women with pre‐eclampsia. Acta Obstet Gynecol Scand. 2002;81(4):308‐312. doi: 10.1034/j.1600-0412.2002.810405.x [DOI] [PubMed] [Google Scholar]
- 49. Yin Y, Wang A, Feng L, et al. Heparan sulfate proteoglycan sulfation regulates uterine differentiation and signaling during embryo implantation. Endocrinology. 2018;159(6):2459‐2472. doi: 10.1210/en.2018-00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Watson LN, Mottershead DG, Dunning KR, Robker RL, Gilchrist RB, Russell DL. Heparan sulfate proteoglycans regulate responses to oocyte paracrine signals in ovarian follicle morphogenesis. Endocrinology. 2012;153(9):4544‐4555. doi: 10.1210/en.2012-1181 [DOI] [PubMed] [Google Scholar]
- 51. Tang Q, Li X, Wang J. Tubulin deacetylase NDST3 modulates lysosomal acidification: implications in neurological diseases. Bioessays. 2022;44(11):e2200110. doi: 10.1002/bies.202200110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skare Ø, Lie RT, Haaland ØA, et al. Analysis of parent‐of‐origin effects on the X chromosome in Asian and European Orofacial Cleft Triads Identifies Associations with DMD, FGF13, EGFL6, and additional loci at Xp22.2. Front Genet. 2018;9:25. doi: 10.3389/fgene.2018.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. An J, Du Y, Fan X, et al. EGFL6 promotes breast cancer by simultaneously enhancing cancer cell metastasis and stimulating tumor angiogenesis. Oncogene. 2019;38(12):2123‐2134. doi: 10.1038/s41388-018-0565-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Y, Wang J, Wang F, Gao C, Cao Y, Wang J. Identification of specific cell subpopulations and marker genes in ovarian cancer using single‐cell RNA sequencing. Biomed Res Int. 2021;2021:1005793. doi: 10.1155/2021/1005793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Balaton BP, Fornes O, Wasserman WW, Brown CJ. Cross‐species examination of X‐chromosome inactivation highlights domains of escape from silencing. Epigenetics Chromatin. 2021;14(1):12. doi: 10.1186/s13072-021-00386-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Del Giudice E, Macca M, Imperati F, et al. CNS involvement in OFD1 syndrome: a clinical, molecular, and neuroimaging study. Orphanet J Rare Dis. 2014;9:74. doi: 10.1186/1750-1172-9-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reifegerste J, Veríssimo J, Rugg MD, et al. Early‐life education may help bolster declarative memory in old age, especially for women. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2021;28(2):218‐252. doi: 10.1080/13825585.2020.1736497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bloomberg M, Dugravot A, Dumurgier J, et al. Sex differences and the role of education in cognitive ageing: analysis of two UK‐based prospective cohort studies. Lancet Public Health. 2021;6(2):e106‐e115. doi: 10.1016/S2468-2667(20)30258-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schaich CL, Malaver D, Chen H, et al. Association of heart rate variability with cognitive performance: the multi‐ethnic study of atherosclerosis. J Am Heart Assoc. 2020;9(7):e013827. doi: 10.1161/JAHA.119.013827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morandi GN, Lin SH, Lin CW, et al. Heart rate variability is associated with memory in females. Appl Psychophysiol Biofeedback. 2019;44(2):117‐122. doi: 10.1007/s10484-018-9425-1 [DOI] [PubMed] [Google Scholar]
- 61. Zhang Y, Lu Q, Ye Y, et al. SUPERGNOVA: local genetic correlation analysis reveals heterogeneous etiologic sharing of complex traits. Genome Biol. 2021;22(1):262. doi: 10.1186/s13059-021-02478-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nolte IM, Munoz ML, Tragante V, et al. Genetic loci associated with heart rate variability and their effects on cardiac disease risk. Nat Commun. 2017;8:15805. doi: 10.1038/ncomms15805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arzouni N, Matloff W, Zhao L, Ning K, Toga AW. Identification of dysregulated genes for late‐onset Alzheimer's disease using gene expression data in brain. J Alzheimers Dis Parkinsonism. 2020;10(6):498. [PMC free article] [PubMed] [Google Scholar]
- 64. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4 /MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43(5):436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clarke RA, Lee S, Eapen V. Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including autism. Transl Psychiatry. 2012;2(9):e158. doi: 10.1038/tp.2012.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jones RM, Cadby G, Melton PE, Abraham LJ, Whitehouse AJ, Moses EK. Genome‐wide association study of autistic‐like traits in a general population study of young adults. Front Hum Neurosci. 2013;7:658. doi: 10.3389/fnhum.2013.00658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coltell O, Asensio EM, Sorlí JV, et al. Genome‐Wide Association Study (GWAS) on bilirubin concentrations in subjects with metabolic syndrome: sex‐Specific GWAS analysis and gene‐diet interactions in a Mediterranean population. Nutrients. 2019;11(1):E90. doi: 10.3390/nu11010090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Martin PM, Cifuentes‐Diaz C, Devaux J, et al. Schwannomin‐interacting protein 1 isoform IQCJ‐SCHIP1 is a multipartner Ankyrin‐ and Spectrin‐binding protein involved in the organization of nodes of Ranvier. J Biol Chem. 2017;292(6):2441‐2456. doi: 10.1074/jbc.M116.758029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Elsaid MF, Chalhoub N, Ben‐Omran T, et al. Homozygous nonsense mutation in SCHIP1/IQCJ‐SCHIP1 causes a neurodevelopmental brain malformation syndrome. Clin Genet. 2018;93(2):387‐391. doi: 10.1111/cge.13122 [DOI] [PubMed] [Google Scholar]
- 70. Klingler E, Martin PM, Garcia M, et al. The cytoskeleton‐associated protein SCHIP1 is involved in axon guidance, and is required for piriform cortex and anterior commissure development. Development. 2015;142(11):2026‐2036. doi: 10.1242/dev.119248 [DOI] [PubMed] [Google Scholar]
- 71. Martin PM, Carnaud M, Garcia del Caño G, et al. Schwannomin‐interacting protein‐1 isoform IQCJ‐SCHIP‐1 is a late component of nodes of Ranvier and axon initial segments. J Neurosci. 2008;28(24):6111‐6117. doi: 10.1523/JNEUROSCI.1044-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kwaśnicka‐Crawford DA, Carson AR, Scherer SW. IQCJ‐SCHIP1, a novel fusion transcript encoding a calmodulin‐binding IQ motif protein. Biochem Biophys Res Commun. 2006;350(4):890‐899. doi: 10.1016/j.bbrc.2006.09.136 [DOI] [PubMed] [Google Scholar]
- 73. Kremen WS, Panizzon MS, Franz CE, et al. Genetic complexity of episodic memory: a twin approach to studies of aging. Psychol Aging. 2014;29(2):404‐417. doi: 10.1037/a0035962 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information