

Abstract
This editorial summarizes advances from the Clearance of Interstitial Fluid and Cerebrospinal Fluid (CLIC) group, within the Vascular Professional Interest Area (PIA) of the Alzheimer's Association International Society to Advance Alzheimer's Research and Treatment (ISTAART). The overarching objectives of the CLIC group are to: (1) understand the age‐related physiology changes that underlie impaired clearance of interstitial fluid (ISF) and cerebrospinal fluid (CSF) (CLIC); (2) understand the cellular and molecular mechanisms underlying intramural periarterial drainage (IPAD) in the brain; (3) establish novel diagnostic tests for Alzheimer's disease (AD), cerebral amyloid angiopathy (CAA), retinal amyloid vasculopathy, amyloid‐related imaging abnormalities (ARIA) of spontaneous and iatrogenic CAA‐related inflammation (CAA‐ri), and vasomotion; and (4) establish novel therapies that facilitate IPAD to eliminate amyloid β (Aβ) from the aging brain and retina, to prevent or reduce AD and CAA pathology and ARIA side events associated with AD immunotherapy.
Keywords: amyloid, ARIA, cerebrospinal fluid, clearance, interstitial fluid, IPAD, ISTAART
1. AGING, ARIA, AND CAA‐RELATED INFLAMMATION
Fluid and solutes are eliminated from the brain along the basement membranes of capillaries and arteries as intramural periarterial drainage (IPAD). This clearance mechanism fails with increasing age, possession of apolipoprotein E4 (APOE ε4) genotype and cardiovascular risk factors such as a high fat diet. 1 , 2 Disturbances in the clearance of interstitial fluid (ISF) and cerebrospinal fluid (CSF) from the brain also is suggested to contribute to the development of amyloid‐related imaging abnormalities (ARIA). 3 , 4 , 5 Immune complexes can form after immunotherapy and may also block IPAD. 6 , 7 Increasing evidence suggests that this phenomenon may be causally linked to the occurrence of both ARIA associated with raised CSF concentrations of anti‐amyloid β (Aβ) autoantibodies spontaneously occurring in patients with cerebral amyloid angiopathy (CAA)‐related inflammation (CAA‐ri) 8 and ARIA associated with a class of anti‐Aβ disease‐modifying immunotherapies with monoclonal antibodies (mAbs) in for Alzheimer's disease (AD). 3 , 4 , 5 In this framework, therapy‐induced ARIA are becoming increasingly recognized as the iatrogenic manifestation of CAA‐ri. 8 , 9
ARIA is an umbrella term used to generally define the detection of two types of (sub)acute image abnormalities on magnetic resonance imaging (MRI): (i) ARIA‐edema (ARIA‐E), defined as cortical hyperintensities in the brain parenchyma or sulcal effusion in the leptomeninges/sulci, on T2 weighted fluid‐attenuated inversion recovery (FLAIR) sequences; (ii) ARIA‐hemorrhage (ARIA‐H), defined as microhemorrhages or cortical superficial siderosis, on T2* or susceptibility weighted imaging (SWI). 10 Notably, the term ARIA is not intended to provide any information concerning the associated manifestations of clinical symptoms.
Current data indicate that high immunotherapy drug dosing, APOE ε4, and background CAA burden at the start of treatment are the main risk factors for the occurrence of radiographic ARIA‐E events detected in 12% to 40% of recent clinical trial participants. 11 , 12 A recent meta‐analysis of phase III randomized control trials also showed that although the incidence of ARIA was high for all drugs, except for Solanezumab, only Aducanumab caused both the greatest brain Aβ reduction and the greatest risk for ARIA. 13 This finding is in accordance with the “ARIA paradox” ethiopathogenic model, which posits that Aβ mobilization achieved by mAbs targeting plaques may be causally linked to both efficacy and ARIA risk in a dose‐dependent fashion. 14
Although the optimal balance between dose regimens to minimize risk of ARIA and maximize efficacy has yet to be determined, this is generating concerns over the potential of mAbs to aggravate CAA at the sites of greater plaque removal 8 , 15 (Figure 1). Moving beyond ARIA incidence, describing individual ARIA cases based on longitudinal biomarker variations will be instrumental in solidifying our understanding of ARIA mechanisms and risk factors. With respect to CAA‐ri, the integration of multimodal and multiparametric longitudinal testing for CSF and imaging biomarkers revealed a specific regional and temporal association between focal areas of microglia activation and ARIA‐E only in patients with coexisting CAA and AD pathology, with subsequent in‐time presentation of ARIA‐H after the first ARIA‐E index event. 8
FIGURE 1.
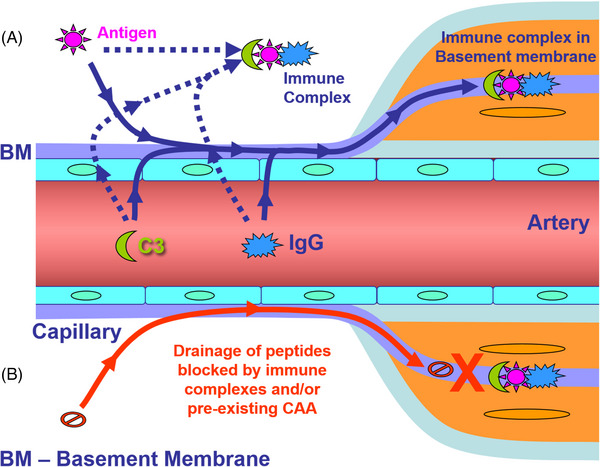
The possible mechanism by which immune complexes disrupt IPAD. In (A), antigens (peptides) in the extracellular space interact with IgG extravasated from the circulation resulting in immune complex formation and fixation of complement C3. The immune complexes formed block the arterial basement membranes that represent the IPAD pathway. In (B), the drainage of peptides such as Aβ is blocked by the presence of immune complexes or pre‐existing CAA in the arterial basement membranes
In clinical trials, the precise pathophysiology of ARIA is not fully understood, but several mechanisms have been postulated. 4 , 16 Therapy‐induced ARIA is associated with mAbs that rely on Fc receptor‐mediated phagocytosis of Aβ aggregates by immune cells. 17 Therefore, an exaggerated inflammatory response of vessel‐adjacent immune cells, such as microglia and perivascular macrophages, may locally perturb the integrity of the cerebrovascular wall, leading to leakage of fluid and/or blood products into the brain parenchyma or sulci. 18 In addition, the drug‐mediated breakdown of parenchymal Aβ aggregates and their subsequent mobilization along IPAD may further increase the Aβ vascular burden and exacerbate CAA‐related pathology in some individuals, 19 , 20 while also altering the integrity of the vascular wall. It has been suggested that the cerebral blood vessels affected by ARIA may be able to recover and withstand continued dosing. 21 It remains unclear to what extent the location and speed of Aβ removal as well as the degree of Aβ burden, in particular the severity of CAA, increase the risk of ARIA and its severity and why some individuals experience asymptomatic ARIA.
The aforementioned ARIA mechanisms were recently translated into the first semi‐mechanistic, in the silico model of ARIA‐E, the vascular wall disturbance (VWD) model. 22 The VWD model hypothesized that high local rates of Aβ removal (parenchymal or vascular) can trigger the ARIA‐E onset, as long as they are not counterbalanced by the rate of an intrinsic vascular repair process. The VWD model successfully described individual‐level time‐courses of ARIA‐E and highlighted the need to identify additional biomarkers and individual characteristics that could explain the variable presentation of ARIA‐E; for example, biomarkers able to (i) distinguish between the vascular and parenchymal Aβ burden or (ii) differentiate between individuals with fast and slow vascular repair processes.
With the first anti‐Aβ therapies in AD being recently granted accelerated approval by the US Food and Drug Administration (FDA) and possibly others on their path to approval, an unprecedented era of ARIA information is emerging. 5 , 11 , 15 , 23 The data collected within the setting of anti‐Aβ clinical trials should make it possible to evaluate the correlation between the location of ARIA events, the regional changes in Aβ burden and the regional presence of both hemorrhagic and non‐hemorrhagic markers of CAA. 8 , 24 , 25 Such an integrated analysis will help clarify the relative risk of ARIA associated with the local vascular load of Aβ and its removal. Although less common, ARIA‐E recurrent events, in particular their location will cast light on the intrinsic recovery ability of vessels affected by ARIA and ultimately increase confidence to restart therapy after the resolution of ARIA‐E. 8 Post mortem neuropathological follow‐up of individuals with and without ARIA will complement the image‐based assessments by increasing confidence in diagnosis and providing insights about ARIA‐induced neuropathological alterations of the blood vessels. 26 The lack of ARIA‐E animal models and human samples from clinical trials for independent research is still representing one of the main road blocks to fill current knowledge gaps in our understanding of ARIA.
RESEARCH IN CONTEXT
Systematic review: This work reviews (using traditional sources, such as PubMed) the current understanding and evidence for the clearance of interstitial and cerebrospinal fluids (CSFs) from the brain. The authors are part of an international group hosted by the vascular professional interest area of Alzheimer's Association.
Interpretation: Our findings lead to a series of working hypotheses regarding the pathogenesis of amyloid‐related imaging abnormalities (ARIA), biomarkers, and new therapeutic strategies for Alzheimer's disease.
Future directions: Future directions should concentrate on (a) clarifying the role of a novel PET tracer [11C]‐Butanol as an in vivo marker of drainage of cerebral interstitial fluid; (b) establishing an experimental model for ARIA in correlation with the human CAA‐ri; (c) clarifying the pathways for drainage of cerebral proteins into the nose; (d) implementing retinal biomarkers into clinical practice and (e) developing therapeutic strategies to facilitate intramural periarterial drainage.
The last 10 years of research has provided compelling evidence in the commonalities between CAA‐ri and therapy‐induced ARIA, suggesting the latter is the iatrogenic manifestation of CAA‐ri, a rare autoimmune encephalopathy characterized by raised CSF levels of anti‐Aβ autoantibodies, spontaneous ARIA‐E events and focal areas of activated microglia in patients with coexisting AD and CAA. 8 , 9 , 27
Large longitudinal cohorts registries and biorepositories of well‐defined patients with CAA‐ri, such as the iCAB International Network, as well as future registry of patients treated with mAbs, such as the ALZ‐NET, will be essential for gathering data from clinical practice, both to advance current recommendations limits for treatment decisions and outcome interpretations. 8
The priority and urgency in studying the mechanistic aspects of ARIA are further motivated by the fact that we are still searching for best‐in‐class biomarkers to fill current knowledge gaps and most clinicians still have little experience with the use of immunotherapeutic drugs in patients with AD and CAA.
New research studies aimed to advance current understanding on ARIA‐E pathophysiological mechanisms and biomarkers, including those disentangling potential associations between autoantibody levels, micro‐glial reactivity, Aβ deposition, and impairment of the IPAD clearance pathways, should be top research priorities.
Given the biological complexity of ARIA, a multimodal and multiparametric approach in large longitudinal cohorts of samples from clinical practice will be the only way to reach this aim, thus increasing generalizability and facilitating the validation of emerging candidate biomarkers, including:
MRI markers and rating systems for CAA and ARIA‐E
CSF and plasma testing for autoantibodies, APOE, astrocyte and microglial reactivity, and blood‐brain barrier damage biomarkers.
In this framework, CSF testing for anti‐Aβ autoantibodies may meet the specific requirement of companion diagnostic biomarkers to assess risk stratification, drug tailoring, and dose monitoring in order to maintain a therapeutic window for the safe and effective clearance of Aβ with minimal occurrence of ARIA. 8 , 9 , 15 , 28
2. FUTURE OF IMMUNOTHERAPY
Lessons learned from other complex diseases underscore the importance of starting immunotherapy in asymptomatic individuals in the initial stages of disease, ensuring that vascular health is optimal. This also opens the opportunity to use emerging techniques to harness the innate immune system. Examples include using different helper T‐cell peptide epitopes (UBITh technology), 29 , 30 to elicit an enhanced B‐cell response while avoiding harmful pro‐inflammatory T‐cell responses, or alternative adjuvants such as CpG 1018 made up of short, unmethylated cytosine‐phosphate‐guanine oligodeoxynucleotides targeting microglia. 31 , 32 However, both active and passive anti‐Aβ immunotherapies have their advantages and disadvantages, 33 , 34 , 35 and it is probable that both approaches will be utilized in the future. The use of novel and sensitive biomarker methods to detect signs of the neurodegenerative process long before the symptoms occur will certainly facilitate the advancement of immunotherapies through clinical development and potentially provide means to prevent or slow AD progression. There are now exciting promising trials for synucleinopathies. 36 There is a need for sensitive, specific biomarkers that detect the neuropathology early, before clinical signs and symptoms. In the case of AD, biomarkers need to be able to determine the health of the walls of cerebral blood vessels and, therefore, their ability to cope with IPAD and clearing immune complexes.
3. VESSEL‐WALL CELL DYSFUNCTION MECHANISMS AND NOVEL THERAPEUTIC STRATEGIES FOR CAA/AD DISCUSSED DURING CLIC 2022
Vascular cells are crucial to maintain neurovascular unit function and proper brain clearance of solutes, including Aβ and debris. Indeed, vascular dysfunction is one of the earliest events in AD pathogenesis, and contributes to the failure of clearance pathways such as IPAD. In particular, it has been shown that cells composing the blood‐brain barrier (such as endothelial and smooth muscle cells) are affected by Aβ deposits such as those characterizing CAA. This results in the activation of mitochondrial dysfunction pathways, with release of hydrogen peroxide, loss of mitochondrial membrane potential and cytochrome C release from the mitochondria, resulting in the instigation of pro‐apoptotic mechanisms. 37 , 38 , 39 Increases in blood‐brain barrier permeability and defects in angiogenesis have also been reported after challenge of cerebral endothelial cells with multiple Aβ peptides, including fragments and mutants. 40 However, the specific molecular mechanisms and possible strategies for prevention of endothelial and smooth muscle cell dysfunction in AD and CAA are not yet clear. Novel studies from the Fossati Group highlighted carbonic anhydrases as a possible therapeutic target to prevent mitochondrial dysfunction and cell death of cells composing the vessel wall. 41 , 42 , 43 , 44
4. CARBONIC ANHYDRASE INHIBITORS
Methazolamide, acetazolamide, and analogue carbonic anhydrase inhibitors are FDA‐approved agents for glaucoma, high altitude sickness and other indications (they are also legacy diuretics). Carbonic anhydrase inhibitors can cross the blood‐brain barrier and are known to stimulate cerebral blood flow. The group led by Silvia Fossati has been the first to test carbonic anhydrase inhibitors cellular and animal models of AD and CAA, showing that these drugs prevent the toxic effects of Aβ on endothelial, glial and neuronal cells and mitochondrial dysfunction, improving cerebrovascular function and glial cell clearance in TgSwDI mice. 45 These positive effects are due to the prevention of the loss of mitochondrial ΔΨ (membrane potential), production of mitochondrial reactive oxygen species and release of cytochrome C induced by Aβ.
Current work suggests that these compounds may also prevent smooth muscle cell death and mitochondrial dysfunction in vitro and in vivo. These properties, together with their known effects in increasing vasoreactivity and stimulating cerebral blood flow, 46 , 47 , 48 will possibly facilitate IPAD and other brain clearance pathways.
5. RIVASTIGMINE
Mathematical modeling and experimental work has demonstrated that the motive force for IPAD is derived from the spontaneous contractions of arterial smooth muscle cells, a process known as vasomotion. 49 , 50 Arterial smooth muscle cells have cholinergic receptors and in AD there is early degeneration and loss of the basal forebrain nuclei of cholinergic supply. 51 Cholinesterase inhibitors are administered late in the disease process and their use has therefore been limited to symptomatic treatment of neurodegenerative dementias. Unpublished data from the Carare group suggests that early treatment with dual cholinesterase inhibitor Rivastigmine results in an improvement of IPAD clearance of tracer molecules. This interpretation is also suggested by a study showing that AD patients treated with acetylcholinesterase inhibitors showed increased plasma levels of anti‐Aβ autoantibodies than untreated AD patients. 52 This is in accordance with the ARIA PARADOX model, which states that therapy‐induced ARIA are the iatrogenic exacerbation of the neuroinflammatory and IPAD mechanisms spontaneously occurring in CAA‐ri. 8 Efforts are now underway to characterize the cholinergic and adrenergic receptors of vascular smooth muscle cells and determine the effects of their selective inhibition or augmentation as disease modification strategies.
6. CEREBROSPINAL FLUID
From the time of its discovery, significant debate has existed regarding the major anatomical sites of CSF efflux from the central nervous system (CNS). 53 For the majority of the 20th century, it was understood that CSF drains to the dural venous sinuses through structures known as arachnoid villi or granulations. However, more recently, scientists and clinicians have recognized an important, perhaps even predominant, role for lymphatic vessels in draining CSF from the subarachnoid space. Recent tracer studies in rodents using magnetic resonance or fluorescence imaging have shown that bulk outflow pathways exist from the subarachnoid space around the brain to the lymphatic vessels draining to deep or superficial cervical lymph nodes. 54 , 55 , 56 The rapid clearance of even micron‐sized beads from the CSF to downstream lymph nodes 57 indicates that pathways through or around the arachnoid membrane (which is considered to be impermeable) must exist. Recent efforts have concentrated on elucidating potential fluid routes to the newly rediscovered dural lymphatic vessels 55 , 58 , 59 or to extracranial lymphatic vessels that are in close proximity to exiting cranial nerves. 54 With respect to the latter, much focus has been given to efflux routes at the cribriform plate of the ethmoid bone, where olfactory nerves exit through foramina to terminate at the olfactory epithelium. 60 , 61 , 62 It is currently debated whether lymphatic vessels traverse the cribriform plate to access the subarachnoid space 57 , 61 , 63 and/or if perineural routes around the olfactory nerves carry the fluid and solutes to lymphatic vessels within the submucosa. 64 , 65 , 66 Steven Proulx and colleagues are currently elucidating the drainage pathways at this location using CSF infusions of PEGylated microbeads into transgenic reporter mice expressing fluorescent proteins for the leptomeninges and lymphatic vessels followed by confocal imaging of decalcified sections. They show that open and direct pathways exist through breaches in the arachnoid barrier for CSF to drain to lymphatic vessels present on both the CNS and nasal mucosal sides of the cribriform plate. 67 Further studies utilizing these methods will elucidate CSF drainage pathways around other nerves to lymphatic vessels, including the optic, trigeminal, and spinal nerves. A diagrammatic summary of all the drainage routes is presented in Figure 2, adapted from. 68
FIGURE 2.
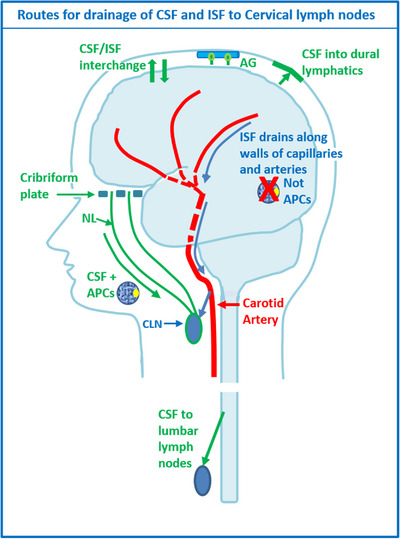
Diagram to illustrate the drainage pathways for CSF and interstitial fluid (ISF) to cervical lymph nodes. Reproduced with permission. 68 CSF and ISF drain to lymph nodes by different and distinct pathways. In humans, CSF drains into the blood of venous sinuses through well‐developed arachnoid villi and granulations (AG). Lymphatic drainage of CSF occurs via nasal and dural lymphatics and along cranial and spinal nerve roots (outlined in green). Channels that pass from the subarachnoid space through the cribriform plate allow passage of CSF (green line), T cells and antigen presenting cells (APC) into nasal lymphatics (NL) and cervical lymph nodes (CLN). CSF from the lumbar subarachnoid space drains to lumbar lymph nodes. ISF from the brain parenchyma drains along basement membranes in the walls of cerebral capillaries and arteries (blue arrows) to cervical lymph nodes adjacent to the internal carotid artery just below the base of the skull. This narrow intramural perivascular drainage pathway does not allow the traffic of APC. There is interchange between CSF and ISF (convective influx/glymphatic system) as CSF enters the surface of the brain alongside penetrating arteries
Mony de Leon's group previously reported using a tau PET tracer ([18F]‐THK5117) that reduced CSF clearance and was detected in the cribriform plate and nasal turbinates in subjects with AD. 69 Recently they tested a novel PET tracer [11C]‐Butanol that has an advantage of not binding to brain tissue, thus providing an unbiased estimate of tracer flow and clearance through ISF and CSF compartments. 70 In 26 cognitively normal individuals aged > 65years, they estimated the time for 75% of the tracer entering the cribriform plate and nasal turbinate region (AUC) to be cleared. The results show that tracer levels remain elevated in ventricle, brain, and CSF drainage pathways up to 45 min after IV administration. In contrast, arterial and venous blood levels peak and return to near asymptotic baseline levels within 5 min. The clinical data demonstrate that ventricular tracer clearance and turbinate clearance are highly correlated, p < 0.01. Importantly, individuals with brain Aβ deposits (n = 10) based on [11C]‐PiB PET scan show, relative to Aβ negative controls (n = 16), an increased time to clear the tracer from the turbinates (Figure 3). These data replicate the prior observation by the De Leon group that brain Aβ is associated with slower nasal CSF clearance. Current studies are underway testing the magnitude of these relationships in the brain and other CSF egress pathways.
FIGURE 3.
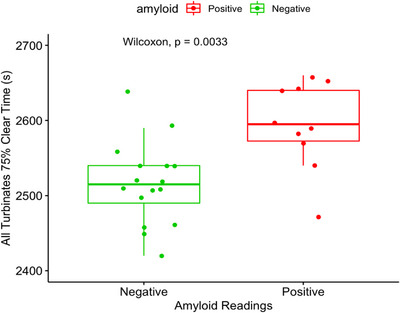
The PET tracer [11C]‐Butanol was used as a biomarker for CSF clearance. The data show in elderly individuals with brain amyloid deposits ([11C]‐PiB imaging) that compared with controls, nasal turbinate tracer drainage is slower. These data suggest that the slowed ventricular CSF drainage found in neurodegenerative diseases such as Alzheimer's can be detected peripherally
Douglas Ethell's group has described subarachnoid evaginations under the olfactory bulb that project into cribriform plate apertures, connecting with a cribrose watershed. 71 This watershed interconnects large cribriform plate apertures with conduits that run from the Crista Galli's cistern to a manifold structure within the olfactory fossa's back wall. Tiny tubules from the watershed project downward into the nasal mucosa, releasing CSF into the submucosal space where lymphatic vessels are present. 72 In a study of more than 600 human cribriform plates, 72 Dr Ethell's team also found age‐dependent declines in porosity that reduce CSF egress capacity and may explain why the loss of smell is a common early sign in AD. 73 The importance of CSF egress along other cranial and spinal nerves is also worthy of investigation. As the largest brainstem nerve, the trigeminal nerve (CN5) has afferent and efferent branches that could accommodate microchannels for CSF outflow.
7. IMPLICATIONS FOR NEW BIOMARKERS FOR AD AND CAA
7.1. Retinal biomarkers
The neural retina shares a common embryonic origin with the brain and is a structurally direct and physiologically intact extension of the brain. Since both CNS compartments display structural and functional resemblance and comprise similar neuronal, blood barrier, and glial cell types, 74 , 75 , 76 their susceptibility to common pathological processes is not surprising. The retina is easily accessible for direct, non‐invasive imaging at ultra‐high spatial resolution; hence, it may offer an unparalleled at affordable means to visualize and monitor CNS targets on the microvascular, cellular, and molecular levels in the clinical settings.
Growing evidence from multiple biochemical and histological studies indicated that the pathological features of AD manifest in the retina of AD patients, including mild cognitive impairment (MCI) and preclinical AD cases, with parallels between the pathology in the brain and retina. 77 , 78 , 79 , 80 , 81 Early research found degeneration of retinal ganglion cells and their axonal projections. 82 Koronyo‐Hamaoui and colleagues identified the pathological hallmarks, Aβ deposits and neurofibrillary tangles, in post mortem retinas of AD patients, including those with early‐stage disease. 83 , 84 Retinal Aβ42 accumulation seems to follow a similar trajectory to that of brain Aβ burden during disease progression, and significantly correlated with the ABC and BRAAK stages and the Mini‐Mental State Examination (MMSE) cognitive status. 84 , 85 , 86 Additional studies also demonstrated the existence of retinal Aβ and tau oligomers, various phosphorylated tau forms, microgliosis, astrogliosis, and associated retinal neurodegeneration in these patients. 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 , 100 , 101 , 102 The proteome signatures of the AD retina and brain were recently reported, with common changes especially found between changes in the superior‐ and inferior‐temporal retina and the temporal cortex, including elevated inflammatory and apoptotic markers and deficiencies in oxidative phosphorylation and mitochondrial markers. 85 Likewise, numerous studies in animal models of AD have demonstrated reported AD‐related pathologies in the retina of these models, including visual impairments, corresponding to the changes in the brain for example. 83 , 101 , 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 , 112 , 113 , 114 , 115 , 116 , 117 , 118 , 119 , 120
The first in vivo imaging of Aβ pathology in living AD patients was demonstrated by the Koronyo‐Hamaoui group together with NeuroVision Imaging LLC, utilizing a modified confocal scanning laser ophthalmoscope following the oral administration of highly bioavailable Longvida curcumin. 84 Since then, additional teams showed the feasibility of detecting retinal Aβ deposits in human AD trials by non‐invasive retinal optical and hyperspectral imaging, in which the Aβ load was greater in MCI and AD patients, as well as in brain Aβ‐positive preclinical patients, than in healthy control individuals. 84 , 121 , 122 , 123 , 124 , 125 , 126 , 127 Importantly, the extent of retinal Aβ burden correlated with cerebral Aβ–PET load, hippocampal and whole gray matter atrophy, and the cognitive deficit. 121 , 122 , 123 , 124 , 125 As both retinal Aβ accumulation and retinal vascular pathology 77 , 101 , 102 , 110 , 128 , 129 , 130 were reported in patients with MCI and AD patients, another imaging study explored the interplay between retinal vascular geometric measures and retinal Aβ burden using retinal fluorescence imaging. This pilot study found that a joint index of retinal venular tortuosity and retinal Aβ burden significantly correlated with verbal memory and Short Form‐36 (SF‐36) mental component scores in MCI patients. 124 This study suggests that a combined Aβ –vascular indexes are better discriminators of cognitive function, with the potential for use as outcome measures in AD and mixed dementia trials.
Prior reports demonstrated that retinal vascular abnormalities may be used as biomarkers of early or preclinical dementia, and retinal microvascular changes in MCI and dementia have been described applying various retinal vasculature imaging modalities, including optical coherence tomography angiography, retinal fundus photography, high‐frequency flicker‐light stimulation, and the retinal function imager. 77 , 79 , 124 , 131 , 132 , 133 , 134 , 135 , 136 Histologically, a wide range of vascular changes has been described in the retina of MCI and AD patients. 77 , 101 , 102 , 137 These changes include perivascular and vascular Aβ40 and Aβ42 accumulations associated with pericyte and capillary loss and tight junction damage. Early and progressive deficiency in vascular platelet–derived growth factor receptor–beta (vPDGFRβ) along with pericyte apoptosis were identified in the post mortem retinas of MCI and AD patients. 101 The vPDGFRβ significantly correlated with retinal vAβ40 and vAβ42 load, CAA severity, and cognitive scores. In the APPSWE/PS1ΔE9‐transgenic model mice, retinal capillary and PDGFRβ losses and vascular Aβ deposits were also detected and were associated with inner blood‐retinal barrier (BRB) tight‐junction changes and BRB leakage. 129 Notably, AD retinopathy was linked with color and contrast vision deficits. 107
Furthermore, Aβ40 was found to be deposited in the smooth muscle layer of retinal arteries 102 in a similar pattern to its deposition in CAA. This intense retinal Aβ40 accumulation in arterioles that was far less prevalent in retinal venules suggests a failure of Aβ clearance via the IPAD pathway in the retina of MCI and AD patients. IPAD failure in the brain is considered central to the pathogenesis of CAA, leading to neuronal, and homeostatic dysfunctions, and often associated with worsening cognitive impairments.
Tight junctions between endothelial cells are critical components of the blood‐brain barrier and inner BRB that are essential for maintaining both cerebral and retinal homeostasis. A substantial decrease in the markers of blood‐brain barrier, zonula occludens‐1 (ZO‐1), and claudin‐5 was observed in the retinal vessels of MCI and AD patients that was correlated with abundant arteriolar Aβ40 deposition as well as with the severity of CAA in these patients. 102 Similar to the findings in the retina, degeneration of critical tight junction molecules as claudin‐5 and ZO‐1 were described in cerebral capillaries of AD patients with CAA and in the 5xFAD AD‐model mice. 138 , 139 , 140 The early loss of both ZO‐1 and claudin‐5 in retinal blood vessels of MCI patients versus cognitively normal controls suggests that the BRB damage appears at the earliest stages of functional impairment in the AD continuum. Notably, almost no claudin‐5 expression in retinal blood vessels was demonstrated in AD patients with moderate to severe CAA. Such findings indicate a disrupted inner BRB during AD progression. Altogether, exploring the manifestations of AD in the retina and its relationship to brain pathology in order to develop non‐invasive retinal imaging to detect and monitor AD is thus a priority.
The retina is connected to the brain through bundles of neuronal axons forming the optic nerve and by retinal and cerebral blood vessels, which may facilitate transportation of abnormal Aβ and tau species and further lead to the spread of AD pathology throughout the CNS. 141 The blood‐brain barrier is established by endothelial cells firmly joined by tight junction proteins forming vessel walls, astrocyte end‐feet, and supporting pericytes in the basement membrane. Similarly, in the retina, the BRB is comprised of an inner barrier with vascular endothelial cells and an outer barrier with epithelial cells. Both include tight junctions and supporting pericytes. 142 , 143 , 144 It is well established that compromised blood‐brain barrier vascular networks in AD are likely an important cause of cerebral Aβ deposition due to impaired Aβ removal to the circulating blood. 145 , 146 , 147 , 148 , 149 , 150 The retina and the brain do not have traditional lymphatic vessels. It is believed that the ocular and brain glymphatic fluid transport systems share similar characteristics for waste clearance. These consist of a CSF influx along the periarterial space, an efflux path along the perivenous space, and a final collection site by the dural and cervical lymphatic vessels. 151 , 152 Recent findings by Wang et al., suggest that the glymphatic pathway, through astrocytic aquaporin‐4 water channels in the retina, facilitates the removal of metabolites and fluid from the intraocular space, as well as the efflux of Aβ tracers through the optic nerve and meningeal lymph vessels. 151 Radial Müller cells spanning the neuroretina, as well as fibrous astrocytes along the optic nerve, express abundant aquaporin‐4, and the expression of aquaporin‐4 in astrocyte endfeet is diminished in AD pathogenesis. 153 Furthermore, extensive Müller degeneration has been reported in the AD retina. 90 It has been speculated that Aβ could be cleared from the retina via perivascular transport, and pathological changes in AD may interfere with glymphatic flow, leading to neurotoxicity through Aβ accumulation. 154 Aβ build‐up along the blood vessels during AD pathogenesis may reduce the large, low‐resistance perivascular spaces necessary for glymphatic flux, 155 thus creating a vicious cycle of Aβ accumulation, neuronal death, and impaired glymphatic drainage. Koronyo‐Hamaoui and colleagues recently demonstrated substantial amounts of retinal vascular and perivascular Aβ deposition in AD transgenic mice, as well as in the retina of MCI and AD patients, compared to cognitively normal subjects. 156 Aβ deposits were found in perivascular regions near pericytes, in the lumen adjacent to an endothelial cell, inside pericytes, and in the vascular wall (tunica media, tunica adventitia, and intima). These findings suggest that Aβ deposition around and inside the blood vessels may impede glymphatic drainage, resulting in aberrant Aβ accumulation.
Overall, given that vascular amyloidosis and tight junction deficiencies have been detected in the AD retina, future studies should aim to determine whether BRB permeability is also altered in AD either as a cause or effect of vascular amyloidosis. Indeed, studies in AD patients have generally supported that these vascular abnormalities in the retina can predict cognitive decline. 157 , 158 , 159 Recent progress in retinal amyloid imaging, 122 , 123 , 137 , 160 the clinically available fundus fluorescein angiography and OCTA microvascular imaging 161 , 162 , 163 are promising tools to aid in the detection of pathological vascular features of AD in an outpatient clinical setting. In future practice, the recently developed OCT‐Leakage method to assess vascular circulation, BRB permeability, and edema, 164 , 165 together with the potential of pericyte imaging by adaptive optics 166 should allow for a comprehensive assessment of retinal Aβ and vascular pathology. These methodologies, which allow for a dynamic and in vivo assessment of evolving AD pathology in conjunction with validated cognitive testing and advanced cerebral imaging techniques, may revolutionize AD diagnosis and provide a novel way to track disease progression.
7.2. Biomarkers in the nose
Based on the observations from animal models and latest in vivo studies revealing that cerebral proteins can be identified in the nasal mucosa, there are now advanced efforts to develop sensitive and specific biomarker testing in the nose. As the olfactory bulb is one of the first brain regions affected by AD, measuring brain‐derived analytes closer to the area of neurodegeneration means more accurate and earlier detection. Such non‐invasive measurement also allows for longitudinal monitoring of therapeutic treatment. Noselab, founded in 2020, has already developed a method for detecting several biomarkers including phospho‐tau, total tau, Aβ−42 and Aβ−40 in nasal secretion for diagnosing AD. Arethusta technology from Leucadia Therapeutics claims to improve CSF flow across the cribriform plate as a therapeutic measure for AD. These are two of a number of technologies being applied to identify low cost and non‐invasive markers of disease‐related pathological measures with minimum barrier.
8. ZOOMING INTO PERIVASCULAR SPACES IN THE BRAIN
The prefix peri means “around” or “about” whereas para indicates “alongside”. Perivascular space (PVS) was first described by Johann Henrich Pestalozzi as a space that existed between the adventitia and the tunica media after observing hemorrhage collect in this space. Soon after, these spaces were described as specific channels in the subadventitial or intra‐adventitial by Rudolf Virchow and Charles Filippe Robin respectively. 167 However, these spaces were never illustrated and it is not clear which tissues and spaces the authors were referring to. Later works including those by Charles Weed established direct connections of PVS and the subarachnoid space (SAS). 168 Weed's experimental technique was challenged and subsequently Schaltenbrand and Bailey in reviewing the vast literature produced by the early 1930s, observed that blood vessels in the brain were covered by a “pia‐arachnoid” layer of connective tissue. They termed the outer pial boundary of blood vessels which is fused to the cortical glial membrane as “pialglialmembran”, now known as the pial‐glial membrane. 169 This space between the pial‐glial membrane and the tunica adventitia was considered to be the true perivascular space by Woollam, which terminated at the level of the capillaries. While controversies regarding the precise anatomy of the perivascular space still persist, current view holds that this space is bounded by the pial‐glial membrane on the outside and the tunica adventia of penetrating arteries on the inside and is not in direct communication with SAS surrounding the vessels. 170 The pial sheath reflects over the penetrating arterioles and is continuous over the arteries on the surface of the cortex within the subarachnoid space. The basement membranes of both the pia mater and the pial‐glial basement membrane and those of the outer vessel wall surround the underlying tunica media, which creates a pathway for centrifugal flow of ISF. This pathway also known as the IPAD, appears to drain waste products that later reach the cervical lymph nodes. 171 , 172 In contrast to a single layer of pia mater surrounding the cortical arteries and arterioles, a double layer of leptomeninges has been identified surrounding the penetrating lenticulostriate arteries in the basal ganglia enclosing a true CSF filled space which is in direct connection with the SAS. 173 , 174
MRI is currently the only in vivo technique that holds promise in illustrating anatomy of the perivascular spaces in the human brain but also in providing an understanding of the entry and exit pathways using approved tracers such as gadolinium‐based contrast agents (GBCA). The fluid within these spaces is isointense relative to CSF and appears bright on T2 weighted sequences but dark on FLAIR sequence and on T1 weighted images. MRI can clearly distinguish lacunar infarcts from dilated perivascular spaces or dilated Virchow‐Robin spaces within the basal ganglia. 175 In the past two decades, a clear correlation between the number, size, and the location of dilated perivascular spaces in the brain and several neurological diseases was found leading to the development of scoring methods that have been widely validated. 176 , 177 , 178 , 179 These scores are based on the appearance of PVS on T1, T2 weighted, and FLAIR images. Dilated PVS in the basal ganglia are linked to cerebrovascular pathologies such as stroke, cerebral haemorrhage, CAA, hypertensive encephalopathy and small vessel disease. 176 , 180 , 181 , 182 , 183 , 184 , 185 , 186 On the other hand, dilation of PVS in the white matter of centrum semiovale were reported to be more frequent in patients affected with CAA or dementia. More recently, dilation of PVS in the white matter has also been found relevant in paediatric adrenoleukodystrophy, epilepsy, multiple sclerosis, and cognitive impairment. 187 , 188 , 189 , 190 , 191 , 192 , 193
MRI of brain tissue after intravenous injection GBCAs has been employed in tracing CSF flow in the brain. GBCAs injected intravenously escape through fenestrated capillaries in the choroid plexi and are transported in the ventricles along with CSF flow towards the SAS. 194 Imaging using heavily T2 weighted sequence clearly demonstrates gadolinium induced changes in signal intensity within the CSF spaces in the ventricles, in the SAS, around the perioptical subrachnoid sheath and in the retina in rodents and humans. 194 , 195 , 196 , 197 However, in all these studies, post gadolinium imaging was performed at very long time points and, therefore, lack the capacity to clearly demonstrate whether gadolinium molecules enter via paravascular space or whether they enter the pial‐glial basement membrane (periarterial space). Dynamic contrast enhanced perfusion weighted imaging has been employed in rodents; however the lack of both temporal and spatial resolution does not allow the depiction of a precise anatomical entry and exit pathway of gadolinium ions. 198 Intrathecal administration of GBCAs in normal subjects and patients with normal pressure hydrocephalus also confirms the notion that GBCAs enter the brain parenchyma from the CSF‐filled SAS; however, the long imaging times do not allow for a precise determination of entry and exit pathways of GBCAs. 199
The percentage change in the signal intensity on post gadolinium imaging of fluid within enlarged PVS in the basal ganglia is similar to that obtained in the CSF filled SAS after 4 h of intravenous gadolinium injection. However, the same is not true for fluid within the PVS in the white matter of the centrum semiovale where signal intensity changes only slightly, suggesting that dilated PVS in the basal ganglia and those in the centrum semiovale contain fluids with different composition and represent different drainage pathways. 200 , 201 This difference is likely due to the known histologic differences in the leptomeningeal coverings around vessels in the two regions. 173 It is noteworthy that, although the capillary density is at least eight times higher in the cortical gray matter with respect to white matter, dilated PVS are not present in the cortex and are only seen within the white matter, again pointing to the differential significance of dilated PVS in the basal ganglia and the white matter.
While it is well‐established that dilated PVS in the white matter and the basal ganglia represent pathological changes in the brain, the underlying pathophysiology is still hypothesized as either being a result of a neuroinflammatory or a neurodegenerative process. We have demonstrated that IPAD blockage can result in dilated PVS within the white matter but not in the gray matter. 202 Dilated PVS in the white matter of the centrum semiovale can also result in a spectrum of protein elimination failure angiopathies characterized by accumulation of insoluble proteins within the arterial walls. 6 This difference is likely due to the known histologic differences in the leptomeningeal coverings around vessels in the two regions. 173
MR still lacks the spatial and temporal resolution necessary to visualize membranes and spaces surrounding the vessel wall at rapid time points and thereby to clearly distinguish Virchow‐Robin spaces from dilated periarterial spaces. Volumetric images are necessary to increase the sensitivity and accuracy in determining the presence of PVS. 179 , 203 The challenges in identifying subtle change in signal intensity within pathologically dilated PVS containing protein rich fluid need to be addressed. GBCA based imaging should be considered within minutes of its injection to identify the exact entry and exit pathway. 204
9. CONCLUSION
In summary, we provide an update on the latest findings and challenges for understanding the mechanistic sequela underlying ARIA and its relationship to clearance pathways, such as IPAD. We posit that the continued development of mechanistic models such a VWD and those afforded by more precise spatiotemporal resolution on MRI, biomarkers for clearance failure (e.g., retinal), and novel treatments for preclinical testing such as CAIs should all be prioritized as key research objectives for advancing AD and CAA disease‐modifying therapeutics.
CONFLICT OF INTEREST STATEMENT
R.A. is a full‐time employee and shareholder of F. Hoffmann‐La Roche Ltd. All other authors declare no conflicts of interest. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
Multiple authors are members of the ISTAART Vascular Cognitive Disorders PIA. This manuscript was facilitated by the Alzheimer's Association International Society to Advance Alzheimer's Research and Treatment (ISTAART), through the Vascular Professional Interest Area (PIA). The views and opinions expressed by authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer's Association. Award Grant Number: 23AARG‐1030214. Program: Alzheimer's Association Research Grant (AARG). Project Title: UncoveriNg Immune MechanIsms and Biomarkers of ARIA (UNIMIB‐ARIA Toolkit). Principal Investigator: Prof. Fabrizio Piazza, PhD.
Kelly L, Brown C, Michalik D, et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group‐part of Vascular Professional Interest Area (PIA), updates in 2022‐2023. Cerebrovascular disease and the failure of elimination of Amyloid‐β from the brain and retina with age and Alzheimer's disease: Opportunities for therapy. Alzheimer's Dement. 2024;20:1421–1435. 10.1002/alz.13512
REFERENCES
- 1. Albargothy NJ, Johnston DA, MacGregor‐Sharp M, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018; 136(1):139‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hawkes CA, Gentleman SM, Nicoll JA, Carare RO. Prenatal high‐fat diet alters the cerebrovasculature and clearance of β‐amyloid in adult offspring. J Pathol. 2015;235(4):619‐631. [DOI] [PubMed] [Google Scholar]
- 3. Sperling RA, Jack CR Jr, Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenberg SM, Bacskai BJ, Hernandez‐Guillamon M, Pruzin J, Sperling R, Van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cogswell PM, Barakos JA, Barkhof F, et al. Amyloid‐Related imaging abnormalities with emerging Alzheimer disease therapeutics: detection and reporting recommendations for clinical practice. AJNR Am J Neuroradiol. 2022;43(9):E19‐e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carare RO, Teeling JL, Hawkes CA, et al. Immune complex formation impairs the elimination of solutes from the brain: implications for immunotherapy in Alzheimer's disease. Acta Neuropathol Commun. 2013;1(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziliotto N, Bernardi F, Piazza F. Hemostasis components in cerebral amyloid angiopathy and Alzheimer's disease. Neurolog Sci. 2021;42(8):3177‐3188. [DOI] [PubMed] [Google Scholar]
- 8. Piazza F, Caminiti SP, Zedde M, et al. Association of microglial activation with spontaneous ARIA‐E and CSF levels of anti‐Aβ autoantibodies. Neurology. 2022;99(12):e1265‐e1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Antolini L, DiFrancesco JC, Zedde M, et al. Spontaneous ARIA‐like events in cerebral amyloid angiopathy–related inflammation: a multicenter prospective longitudinal cohort study. Neurology. 2021;97(18):e1809‐e1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barakos J, Purcell D, Suhy J, et al. Detection and management of amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with Anti‐Amyloid Beta therapy. J Prev Alzheimers Dis. 2022;9(2):211‐220. [DOI] [PubMed] [Google Scholar]
- 11. Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salloway S, Chalkias S, Barkhof F, et al. Amyloid‐related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79(1):13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid‐β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta‐analysis of phase III RCTs in Alzheimer's disease. Ageing Res Rev. 2021;68:101339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piazza F, Winblad B. Amyloid‐related imaging abnormalities (ARIA) in immunotherapy trials for Alzheimer's disease: need for prognostic biomarkers? J Alzheimer's Dis: JAD. 2016;52(2):417‐420. [DOI] [PubMed] [Google Scholar]
- 15. Zedde M, Pascarella R, Piazza F. CAA‐ri and ARIA: two faces of the same coin? Am J Neuroradiol. 2023;44(2):E13‐E14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sperling R, Salloway S, Brooks DJ, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ‐801‐the first wave of amyloid‐targeting drugs for Alzheimer's disease with potential for near term approval. Alzheimers Res Ther. 2020;12(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198‐207. [DOI] [PubMed] [Google Scholar]
- 19. Boche D, Zotova E, Weller RO, et al. Consequence of Aβ immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131(12):3299‐3310. [DOI] [PubMed] [Google Scholar]
- 20. Weller RO, Preston SD, Subash M, Carare RO. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimer's Res Ther. 2009;1(2):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zago W, Schroeter S, Guido T, et al. Vascular alterations in PDAPP mice after anti‐Aβ immunotherapy: implications for amyloid‐related imaging abnormalities. Alzheimers Dement. 2013;9(5 Suppl.):S105‐S115. [DOI] [PubMed] [Google Scholar]
- 22. Aldea R, Grimm HP, Gieschke R, et al. In silico exploration of amyloid‐related imaging abnormalities in the gantenerumab open‐label extension trials using a semi‐mechanistic model. Alzheimers Dement (N Y). 2022;8(1):e12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cummings J, Apostolova L, Rabinovici GD, et al. Lecanemab: appropriate use recommendations. J Prev Alzheimers Dis. 2023; 10(3):362‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klein G, Delmar P, Rehal S, et al. Consistently large amyloid reductions in patients with and without ARIA‐E in the gantenerumab SCarlet RoAD and Marguerite RoAD open‐label extension studies (S9.007). Neurology. 2019;92(15 Supplement):S9007. [Google Scholar]
- 25. Charidimou A, Boulouis G, Frosch MP, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI‐neuropathology diagnostic accuracy study. Lancet Neurol. 2022;21(8):714‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scherlek A, Kozberg M, Nicoll JAR, et al. MRI‐histopathology correlations of amyloid‐related imaging abnormalities (ARIA) in postmortem human brain samples. Alzheimer Dement. 2020;16(S2):e041579. [Google Scholar]
- 27. Piazza F, Greenberg SM, Savoiardo M, et al. Anti–amyloid β autoantibodies in cerebral amyloid angiopathy–related inflammation: implications for amyloid‐modifying therapies. Ann Neurol. 2013;73(4):449‐458. [DOI] [PubMed] [Google Scholar]
- 28. Piazza F, Frölich L, Padovani A. Large collaborative registries and real‐world data to manage amyloid‐related imaging abnormalities. JAMA Neurol. 2022;79(6):633‐634. [DOI] [PubMed] [Google Scholar]
- 29. Wang CY, Finstad CL, Walfield AM, et al. Site‐specific UBITh amyloid‐beta vaccine for immunotherapy of Alzheimer's disease. Vaccine. 2007;25(16):3041‐3052. [DOI] [PubMed] [Google Scholar]
- 30. Wang CY, Wang PN, Chiu MJ, et al. UB‐311, a novel UBITh(®) amyloid β peptide vaccine for mild Alzheimer's disease. Alzheimer Dement. 2017;3(2):262‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patel AG, Nehete PN, Krivoshik SR, et al. Innate immunity stimulation via CpG oligodeoxynucleotides ameliorates Alzheimer's disease pathology in aged squirrel monkeys. Brain. 2021;144(7):2146‐2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scholtzova H, Do E, Dhakal S, et al. Innate immunity stimulation via toll‐like receptor 9 ameliorates vascular amyloid pathology in Tg‐SwDI mice with associated cognitive benefits. J Neurosci. 2017;37(4):936‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song C, Shi J, Zhang P, et al. Immunotherapy for Alzheimer's disease: targeting β‐amyloid and beyond. Transl Neurodegener. 2022;11(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nimmo JT, Kelly L, Verma A, Carare RO, Nicoll JAR, Dodart J‐C. Amyloid‐β and α‐Synuclein immunotherapy: from experimental studies to clinical trials. Front Neurosci. 2021;15:733857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winblad B, Graf A, Riviere M‐E, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer's disease. Alzheimer's Res Ther. 2014;6(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nimmo JT, Smith H, Wang CY, et al. Immunisation with UB‐312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric alpha‐synuclein accumulation in the brain and gut. Acta Neuropathol. 2022;143(1):55‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parodi‐Rullan R, Sone JY, Fossati S. Endothelial mitochondrial dysfunction in cerebral amyloid angiopathy and Alzheimer's disease. J Alzheimers Dis. 2019;72(4):1019‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer's Aβ. Cell Death Dis. 2012;3:e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fossati S, Cam J, Meyerson J, et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid‐beta variants in cells composing the cerebral vessel walls. FASEB J. 2010;24(1):229‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parodi‐Rullan R, Ghiso J, Cabrera E, Rostagno A, Fossati S. Alzheimer's amyloid beta heterogeneous species differentially affect brain endothelial cell viability, blood‐brain barrier integrity, and angiogenesis. Aging Cell. 2020;19(11):e13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lemon N, Canepa E, Ilies MA. Fossati S. carbonic anhydrases as potential targets against neurovascular unit dysfunction in Alzheimer's disease and stroke. Front Aging Neurosci. 2021;13:772278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Provensi G, Carta F, Nocentini A, et al. A new kid on the block? carbonic anhydrases as possible new targets in Alzheimer's disease. Int J Mol Sci. 2019;20(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solesio ME, Peixoto PM, Debure L, et al. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell. 2018:e12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fossati S, Giannoni P, Solesio ME, et al. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta‐induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis. 2016;86:29‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Canepa E, Parodi‐Rullan R, Vazquez‐Torres R, et al. FDA‐approved carbonic anhydrase inhibitors reduce amyloid β pathology and improve cognition, by ameliorating cerebrovascular health and glial fitness. Alzheimers Dement. 2023. n/a(n/a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rasmussen JK, Boedtkjer E. Carbonic anhydrase inhibitors modify intracellular pH transients and contractions of rat middle cerebral arteries during CO2/HCO3(‐) fluctuations. J Cereb Blood Flow Metab. 2018;38(3):492‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang L, Yang Q, Zhang L, Chen X, Huang Q, Wang H. Acetazolamide improves cerebral hemodynamics in CADASIL. J Neurol Sci. 2010;292(1‐2):77‐80. [DOI] [PubMed] [Google Scholar]
- 48. Grossmann WM, Koeberle B. The dose‐response relationship of acetazolamide on the cerebral blood flow in normal subjects. Cerebrovasc Dis. 2000;10(1):65‐69. [DOI] [PubMed] [Google Scholar]
- 49. Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Veluw SJ, Hou SS, Calvo‐Rodriguez M, et al. Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron. 2020;105(3):549‐561.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta‐analysis. Neurobiol Aging. 2003;24(1):1‐23. [DOI] [PubMed] [Google Scholar]
- 52. Conti E, Galimberti G, Tremolizzo L, et al. Cholinesterase inhibitor use is associated with increased plasma levels of anti‐Aβ 1‐42 antibodies in Alzheimer's disease patients. Neurosci Lett. 2010;486(3):193‐196. [DOI] [PubMed] [Google Scholar]
- 53. Proulx ST. Cerebrospinal fluid outflow: a review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell Mol Life Sci. 2021;78(6):2429‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8(1):1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahn JH, Cho H, Kim JH, et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. 2019;572:62‐66. [DOI] [PubMed] [Google Scholar]
- 56. Decker Y, Kramer J, Xin L, et al. Magnetic resonance imaging of cerebrospinal fluid outflow after low‐rate lateral ventricle infusion in mice. JCI Insight. 2022;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Antila S, Karaman S, Nurmi H, et al. Development and plasticity of meningeal lymphatic vessels. J Exp Med. 2017;214(12):3645‐3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Erlich SS, McComb JG, Hyman S, Weiss MH. Ultrastructural morphology of the olfactory pathway for cerebrospinal fluid drainage in the rabbit. J Neurosurg. 1986;64(3):466‐473. [DOI] [PubMed] [Google Scholar]
- 61. Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol. 1993;19(6):480‐488. [DOI] [PubMed] [Google Scholar]
- 62. Norwood JN, Zhang Q, Card D, Craine A, Ryan TM, Drew PJ. Anatomical basis and physiological role of cerebrospinal fluid transport through the murine cribriform plate. eLife. 2019;8:1‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jacob L, de Brito Neto J, Lenck S, et al. Conserved meningeal lymphatic drainage circuits in mice and humans. J Exp Med. 2022;219(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jackson RT, Tigges J, Arnold W. Subarachnoid space of the CNS, nasal mucosa, and lymphatic system. Arch Otolaryngol. 1979;105(4):180‐184. [DOI] [PubMed] [Google Scholar]
- 65. Bradbury MW, Westrop RJ. Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J Physiol. 1983;339:519‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non‐human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Spera I, Cousin N, Ries M, et al. Open pathways for cerebrospinal fluid outflow at the cribriform plate along the olfactory nerves. EBioMedicine. 2023;91:104558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016;132(3):317‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. De Leon MJ, Li Y, Okamura N, et al. Cerebrospinal fluid clearance in Alzheimer disease measured with dynamic PET. J Nucl Med. 2017;58(9):1471‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mehta NHLK, Wang X, Spector E, et al. Reduced Nasal Turbinate [11C]‐Butanol Clearance in Amyloid Positive Elderly Subjects. Paper presented at: AD/PD 2023; 2023; Gothenburg, Sweden. [Google Scholar]
- 71. Ethell DW. Disruption of cerebrospinal fluid flow through the olfactory system may contribute to Alzheimer's disease pathogenesis. J Alzheimer's Dis: JAD. 2014;41(4):1021‐1030. [DOI] [PubMed] [Google Scholar]
- 72. Zaragoza R, Miulli D, Kashyap S, et al. Impairment of CSF egress through the cribriform plate plays an apical role in Alzheimer's disease etiology. medRxiv. 2021. 2021.10.04.21264049. [Google Scholar]
- 73. Rezek DL. Olfactory deficits as a neurologic sign in dementia of the Alzheimer type. Arch Neurol. 1987;44(10):1030‐1032. [DOI] [PubMed] [Google Scholar]
- 74. Michael CC, Carol AM. Reconnecting eye to brain. J Neurosci. 2016;36(42):10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Erskine L, Herrera E. Connecting the retina to the brain. ASN Neurol. 2014;6(6):1759091414562107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Patton N, Aslam T, MacGillivray T, Pattie A, Deary IJ, Dhillon B. Retinal vascular image analysis as a potential screening tool for cerebrovascular disease: a rationale based on homology between cerebral and retinal microvasculatures. J Anat. 2005;206(4):319‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shi H, Koronyo Y, Rentsendorj A, et al. Retinal vasculopathy in Alzheimer's disease. Front Neurosci. 2021;15:731614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mirzaei N, Shi H, Oviatt M, et al. Alzheimer's retinopathy: seeing disease in the eyes. Front Neurosci. 2020;14:921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Doustar J, Torbati T, Black KL. Optical coherence tomography in Alzheimer's disease and other neurodegenerative diseases. Front Neurol. 2017;8:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hart NJ, Koronyo Y, Black KL, Koronyo‐Hamaoui M. Ocular indicators of Alzheimer's: exploring disease in the retina. Acta Neuropathol. 2016;132(6):767‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Snyder PJ, Alber J, Alt C, et al. Retinal imaging in Alzheimer's and neurodegenerative diseases. Alzheimers Dement. 2021;17(1):103‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic‐nerve degeneration in Alzheimer's disease. N Engl J Med. 1986;315(8):485‐487. [DOI] [PubMed] [Google Scholar]
- 83. Koronyo‐Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54:S204‐S217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof‐of‐concept imaging trial in Alzheimer's disease. JCI Insight. 2017;2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koronyo Y, Rentsendorj A, Mirzaei N, et al. Retinal pathological features and proteome signatures of Alzheimer's disease. Acta Neuropathol. 2023;145(4):409‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schultz N, Byman E, the Netherlands Brain B, Wennström M. Levels of retinal amyloid‐β correlate with levels of retinal IAPP and hippocampal amyloid‐β in neuropathologically evaluated individuals. J Alzheimer's Dis. 2020;73:1201‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. La Morgia C, Ross‐Cisneros FN, Koronyo Y, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. 2016;79(1):90‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lee S, Jiang K, McIlmoyle B, et al. Amyloid beta immunoreactivity in the retinal ganglion cell layer of the Alzheimer's eye. Front Neurosci. 2020:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Grimaldi A, Pediconi N, Oieni F, et al. Neuroinflammatory processes, A1 astrocyte activation and protein aggregation in the retina of Alzheimer's disease patients, possible biomarkers for early diagnosis. Front Neurosci. 2019;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu QA, Boerkoel P, Hirsch‐Reinshagen V, et al. Müller cell degeneration and microglial dysfunction in the Alzheimer's retina. Acta Neuropathol Commun. 2022;10(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hart de Ruyter FJ, Morrema THJ, Den Haan J, et al. Phosphorylated tau in the retina correlates with tau pathology in the brain in Alzheimer's disease and primary tauopathies. Acta Neuropathol. 2023;145(2):197‐218. [DOI] [PubMed] [Google Scholar]
- 92. Schön C, Hoffmann NA, Ochs SM, et al. Long‐term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS One. 2013;7(12):e53547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Asanad S, Ross‐Cisneros FN, Nassisi M, Barron E, Karanjia R, Sadun AA. The retina in Alzheimer's disease: histomorphometric analysis of an ophthalmologic biomarker. Invest Ophthalmol Vis Sci. 2019;60(5):1491‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cao KJ, Kim JH, Kroeger H, et al. ARCAM‐1 facilitates fluorescence detection of amyloid‐containing deposits in the retina. Transl Vis Sci Technol. 2021;10(7):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Den Haan J, Morrema THJ, Verbraak FD, et al. Amyloid‐beta and phosphorylated tau in post‐mortem Alzheimer's disease retinas. Acta Neuropathol Commun. 2018;6(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Du X, Koronyo Y, Mirzaei N, et al. Label‐free hyperspectral imaging and deep‐learning prediction of retinal amyloid β‐protein and phosphorylated tau. PNAS Nexus. 2022;1(4):pgac164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nilson AN, English KC, Gerson JE, et al. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J Alzheimer's Dis. 2017;55:1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer's disease. Brain Res. 1989;501(2):364‐372. [DOI] [PubMed] [Google Scholar]
- 99. Blanks JC, Torigoe Y, Hinton DR. Blanks RHI. Retinal pathology in Alzheimer's disease. I. Ganglion cell loss in foveal/parafoveal retina. Neurobiol Aging. 1996;17(3):377‐384. [DOI] [PubMed] [Google Scholar]
- 100. Qiu Y, Jin T, Mason E, Campbell MCW. Predicting thioflavin fluorescence of retinal amyloid deposits associated with Alzheimer's disease from their polarimetric properties. Transl Vis Sci Technol. 2020;9(2):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shi H, Koronyo Y, Rentsendorj A, et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer's disease retina. Acta Neuropathol. 2020;139(5):813‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shi H, Koronyo Y, Fuchs D‐T, et al. Retinal arterial Aβ(40) deposition is linked with tight junction loss and cerebral amyloid angiopathy in MCI and AD patients. Alzheimer Dementia. 2023. n/a(n/a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ning A, Cui J, To E, Ashe KH, Matsubara J. Amyloid‐beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci. 2008;49(11):5136‐5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S. Beta‐amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci. 2009;50(2):793‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Habiba U, Descallar J, Kreilaus F, et al. Detection of retinal and blood Aβ oligomers with nanobodies. Alzheimers Dement. 2021;13(1):e12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Doustar J, Rentsendorj A, Torbati T, et al. Parallels between retinal and brain pathology and response to immunotherapy in old, late‐stage Alzheimer's disease mouse models. Aging Cell. 2020;19(11):e13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vit JP, Fuchs DT, Angel A, et al. Color and contrast vision in mouse models of aging and Alzheimer's disease using a novel visual‐stimuli four‐arm maze. Sci Rep. 2021;11(1):1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tsai Y, Lu B, Ljubimov AV, et al. Ocular changes in TgF344‐AD rat model of Alzheimer's disease. Invest Ophthalmol Vis Sci. 2014;55(1):523‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhang J, Gao F, Ma Y, Xue T, Shen Y. Identification of early‐onset photoreceptor degeneration in transgenic mouse models of Alzheimer's disease. iScience. 2021;24(11):103327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Shi H, Yin Z, Koronyo Y, et al. Regulating microglial miR‐155 transcriptional phenotype alleviates Alzheimer's‐induced retinal vasculopathy by limiting Clec7a/Galectin‐3(+) neurodegenerative microglia. Acta Neuropathol Commun. 2022;10(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Grimaldi A, Brighi C, Peruzzi G, et al. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer's disease in the 3xTg‐AD mouse model. Cell Death Dis. 2018;9(6):685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer's disease. Neuroreport. 2011;22(12):623‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stiebing C, Jahn IJ, Schmitt M, et al. Biochemical characterization of mouse retina of an Alzheimer's disease model by Raman Spectroscopy. ACS Chem Neurosci. 2020;11(20):3301‐3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Georgevsky D, Retsas S, Raoufi N, Shimoni O, Golzan SM. A longitudinal assessment of retinal function and structure in the APP/PS1 transgenic mouse model of Alzheimer's disease. Transl Neurodegener. 2019;8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mirzaei M, Pushpitha K, Deng L, et al. Upregulation of proteolytic pathways and altered protein biosynthesis underlie retinal pathology in a mouse model of Alzheimer's disease. Mol Neurobiol. 2019;56(9):6017‐6034. [DOI] [PubMed] [Google Scholar]
- 116. More SS, Beach JM, Vince R. Early detection of amyloidopathy in Alzheimer's mice by hyperspectral endoscopy. Invest Ophthalmol Vis Sci. 2016;57(7):3231‐3238. [DOI] [PubMed] [Google Scholar]
- 117. Gupta VK, Chitranshi N, Gupta VB, et al. Amyloid β accumulation and inner retinal degenerative changes in Alzheimer's disease transgenic mouse. Neurosci Lett. 2016;623:52‐56. [DOI] [PubMed] [Google Scholar]
- 118. Mei X, Yang M, Zhu L, et al. Retinal levels of amyloid beta correlate with cerebral levels of amyloid beta in young APPswe/PS1dE9 transgenic mice before onset of Alzheimer's disease. Behav Neurol. 2020;2020:1574816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Habiba U, Merlin S, Lim JKH, et al. Age‐Specific retinal and cerebral immunodetection of amyloid‐β plaques and oligomers in a rodent model of Alzheimer's disease. J Alzheimer's Dis : JAD. 2020;76(3):1135‐1150. [DOI] [PubMed] [Google Scholar]
- 120. Ferreira H, Serranho P, Guimarães P, et al. Stage‐independent biomarkers for Alzheimer's disease from the living retina: an animal study. Sci Rep. 2022;12(1):13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ngolab J, Donohue M, Belsha A, et al. Feasibility study for detection of retinal amyloid in clinical trials: the anti‐amyloid treatment in asymptomatic Alzheimer's disease (A4) trial. Alzheimers Dement. 2021;13(1):e12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tadokoro K, Yamashita T, Kimura S, et al. Retinal amyloid imaging for screening Alzheimer's disease. J Alzheimer's Dis: JAD. 2021;83(2):927‐934. [DOI] [PubMed] [Google Scholar]
- 123. Dumitrascu OM, Lyden PD, Torbati T, et al. Sectoral segmentation of retinal amyloid imaging in subjects with cognitive decline. Alzheimers Dement (Amst). 2020;12(1):e12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Dumitrascu OM, Rosenberry R, Sherman DS, et al. Retinal venular tortuosity jointly with retinal amyloid burden correlates with verbal memory loss: a pilot study. Cells. 2021;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hadoux X, Hui F, Lim JKH, et al. Non‐invasive in vivo hyperspectral imaging of the retina for potential biomarker use in Alzheimer's disease. Nat Commun. 2019;10(1):4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. More SS, Beach JM, McClelland C, Mokhtarzadeh A, Vince R. In vivo assessment of retinal biomarkers by hyperspectral imaging: early detection of Alzheimer's disease. ACS Chem Neurosci. 2019;10(11):4492‐4501. [DOI] [PubMed] [Google Scholar]
- 127. Lemmens S, Van Craenendonck T, Van Eijgen J, et al. Combination of snapshot hyperspectral retinal imaging and optical coherence tomography to identify Alzheimer's disease patients. Alzheimers Res Ther. 2020;12(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sharafi SM, Sylvestre JP, Chevrefils C, et al. Vascular retinal biomarkers improves the detection of the likely cerebral amyloid status from hyperspectral retinal images. Alzheimers Dement (N Y). 2019;5:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shi H, Koronyo Y, Fuchs DT, et al. Retinal capillary degeneration and blood‐retinal barrier disruption in murine models of Alzheimer's disease. Acta Neuropathol Commun. 2020;8(1):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dumitrascu OM, Koronyo‐Hamaoui M. Retinal vessel changes in cerebrovascular disease. Curr Opin Neurol. 2020;33(1):87‐92. [DOI] [PubMed] [Google Scholar]
- 131. Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL. Retinal abnormalities in early Alzheimer's disease. Invest Ophthalmol Vis Sci. 2007;48(5):2285‐2289. [DOI] [PubMed] [Google Scholar]
- 132. Chua J, Hu Q, Ke M, et al. Retinal microvasculature dysfunction is associated with Alzheimer's disease and mild cognitive impairment. Alzheimers Res Ther. 2020;12(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chiara C, Gilda C, Daniela M, et al. A two‐year longitudinal study of retinal vascular impairment in patients with amnestic mild cognitive impairment. Front Aging Neurosci. 2022;14:993621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Xie J, Yi Q, Wu Y, et al. Deep segmentation of OCTA for evaluation and association of changes of retinal microvasculature with Alzheimer's disease and mild cognitive impairment. Br J Ophthalmol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yeh TC, Kuo CT, Chou YB. Retinal microvascular changes in mild cognitive impairment and Alzheimer's disease: a systematic review, meta‐analysis, and meta‐regression. Front Aging Neurosci. 2022;14:860759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Costanzo E, Lengyel I, Parravano M, et al. Ocular biomarkers for Alzheimer disease dementia: an umbrella review of systematic reviews and meta‐analyses. JAMA Ophthalmol. 2023;141(1):84‐91. [DOI] [PubMed] [Google Scholar]
- 137. Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof‐of‐concept imaging trial in Alzheimer's disease. JCI Insight. 2017;2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Carrano A, Hoozemans JJ, Van Der Vies SM, Van Horssen J, De Vries HE, Rozemuller AJ. Neuroinflammation and blood‐brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis. 2012;10(1‐4):329‐331. [DOI] [PubMed] [Google Scholar]
- 139. Carrano A, Hoozemans JJM, Van Der Vies SM, Rozemuller AJM, Van Horssen J, De Vries HE. Amyloid beta induces oxidative stress‐mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signaling. 2011;15(5):1167‐1178. [DOI] [PubMed] [Google Scholar]
- 140. Park SW, Kim JH, Mook‐Jung I, Kim KW, Park WJ, Park KH. Intracellular amyloid beta alters the tight junction of retinal pigment epithelium in 5XFAD mice. Neurobiol Aging. 2014;35(9):2013‐2020. [DOI] [PubMed] [Google Scholar]
- 141. Morin PJ, Abraham CR, Amaratunga A, et al. Amyloid precursor protein is synthesized by retinal ganglion cells, rapidly transported to the optic nerve plasma membrane and nerve terminals, and metabolized. J Neurochem. 1993;61(2):464‐473. [DOI] [PubMed] [Google Scholar]
- 142. Campbell M, Humphries P. The blood‐retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol. 2012;763:70‐84. [PubMed] [Google Scholar]
- 143. Zenaro E, Piacentino G, Constantin G. The blood‐brain barrier in Alzheimer's disease. Neurobiol Dis. 2017;107:41‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of blood‐brain barrier in Alzheimer's disease. J Alzheimer's Dis: JAD. 2018;63(4):1223‐1234. [DOI] [PubMed] [Google Scholar]
- 145. Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood‐brain barrier transport of circulating Alzheimer's amyloid beta. Biochem Biophys Res Commun. 1993;197(3):1034‐1040. [DOI] [PubMed] [Google Scholar]
- 146. Do TM, Dodacki A, Alata W, et al. Age‐dependent regulation of the blood‐brain barrier influx/efflux equilibrium of amyloid‐beta peptide in a mouse model of Alzheimer's disease (3xTg‐AD). J Alzheimer's Dis: JAD. 2015;49(2):287‐300. [DOI] [PubMed] [Google Scholar]
- 147. Banks WA, Robinson SM, Verma S, Morley JE. Efflux of human and mouse amyloid beta proteins 1‐40 and 1‐42 from brain: impairment in a mouse model of Alzheimer's disease. Neuroscience. 2003;121(2):487‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid‐beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295(5563):2264‐2267. [DOI] [PubMed] [Google Scholar]
- 149. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhao Z, Sagare AP, Ma Q, et al. Central role for PICALM in amyloid‐beta blood‐brain barrier transcytosis and clearance. Nat Neurosci. 2015;18(7):978‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang X, Lou N, Eberhardt A, et al. An ocular glymphatic clearance system removes beta‐amyloid from the rodent eye. Sci Transl Med. 2020;12(536). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wostyn P, De Groot V, Van Dam D, Audenaert K, Killer HE, De Deyn PP. The glymphatic hypothesis of glaucoma: a unifying concept incorporating vascular, biomechanical, and biochemical aspects of the disease. Biomed Res Int. 2017;2017:5123148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Silva I, Silva J, Ferreira R, Trigo D. Glymphatic system, AQP4, and their implications in Alzheimer's disease. Neurol Res Pract. 2021;3(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wostyn P, Van Dam D, Audenaert K, Killer HE, De Deyn PP, De Groot V. A new glaucoma hypothesis: a role of glymphatic system dysfunction. Fluids Barriers CNS. 2015;12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hughes TM, Kuller LH, Barinas‐Mitchell EJ, et al. Pulse wave velocity is associated with beta‐amyloid deposition in the brains of very elderly adults. Neurology. 2013;81(19):1711‐1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Shi H, Koronyo Y, Rentsendorj A, et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer's disease retina. Acta Neuropathol. 2020;139(5):813‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Deal JA, Sharrett AR, Rawlings AM, et al. Retinal signs and 20‐year cognitive decline in the Atherosclerosis risk in communities study. Neurology. 2018;90(13):e1158‐e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Baker ML, Marino Larsen EK, Kuller LH, et al. Retinal microvascular signs, cognitive function, and dementia in older persons: the cardiovascular health study. Stroke. 2007;38(7):2041‐2047. [DOI] [PubMed] [Google Scholar]
- 159. Cabrera DeBuc D, Somfai GM, Arthur E, Kostic M, Oropesa S, Mendoza Santiesteban C. Investigating multimodal diagnostic eye biomarkers of cognitive impairment by measuring vascular and neurogenic changes in the retina. Front Physiol. 2018;9:1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ong SS, Proia AD, Whitson HE, Farsiu S, Doraiswamy PM, Lad EM. Ocular amyloid imaging at the crossroad of Alzheimer's disease and age‐related macular degeneration: implications for diagnosis and therapy. J Neurol. 2019;266(7):1566‐1577. [DOI] [PubMed] [Google Scholar]
- 161. La Mantia A, Kurt RA, Mejor S, et al. Comparing fundus fluorescein angiography and swept‐source optical coherence tomography angiography in the evaluation of diabetic macular perfusion. Retina. 2019;39(5):926‐937. [DOI] [PubMed] [Google Scholar]
- 162. Memon AS, Memon NA, Mahar PS. Role of optical coherence tomography angiography to differentiate intraretinal microvascular abnormalities and retinal neovascularization in diabetic retinopathy. Pak J Med Sci. 2022;38(1):57‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Mochi T, Anegondi N, Girish M, Jayadev C, Sinha Roy A. Quantitative comparison between Optical coherence tomography angiography and fundus fluorescein angiography images: effect of vessel enhancement. Ophthalmic Surg Lasers Imaging Retina. 2018;49(11):e175‐e181. [DOI] [PubMed] [Google Scholar]
- 164. Marmor MF, Ravin JG. Fluorescein angiography: insight and serendipity a half century ago. Arch Ophthalmol. 2011;129(7):943‐948. [DOI] [PubMed] [Google Scholar]
- 165. Cunha‐Vaz J, Santos T, Alves D, et al. Agreement between OCT leakage and fluorescein angiography to identify sites of alteration of the blood‐retinal barrier in diabetes. Ophthalmol Retina. 2017;1(5):395‐403. [DOI] [PubMed] [Google Scholar]
- 166. Schallek J, Geng Y, Nguyen H, Williams DR. Morphology and topography of retinal pericytes in the living mouse retina using in vivo adaptive optics imaging and ex vivo characterization. Invest Ophthalmol Vis Sci. 2013;54(13):8237‐8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Virchow R. Ueber die Erweiterung kleinerer Gefäfse. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1851;3(3):427‐462. [Google Scholar]
- 168. Weed LH. Studies on Cerebro‐Spinal Fluid. No. III : the pathways of escape from the subarachnoid spaces with particular reference to the Arachnoid Villi. J Med Res. 1914;31(1):51‐91. [PMC free article] [PubMed] [Google Scholar]
- 169. Woollam DH, Millen JW. The perivascular spaces of the mammalian central nervous system and their relation to the perineuronal and subarachnoid spaces. J Anat 1955;89(2):193‐200. [PMC free article] [PubMed] [Google Scholar]
- 170. Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow‐Robin) spaces in the human cerebrum. J Anat. 1990;170:111‐123. [PMC free article] [PubMed] [Google Scholar]
- 171. Carare R, Bernardes‐Silva M, Newman T, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34(2):131‐144. [DOI] [PubMed] [Google Scholar]
- 172. Morris AWJ, Sharp MM, Albargothy NJ, et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131(5):725‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Criswell TP, Sharp MM, Dobson H, et al. The structure of the perivascular compartment in the old canine brain: a case study. Clin Sci (London). 2017;131(22):2737‐2744. [DOI] [PubMed] [Google Scholar]
- 174. Hutchings M, Weller RO. Anatomical relationships of the pia mater to cerebral blood vessels in man. J Neurosurg. 1986;65(3):316‐325. [DOI] [PubMed] [Google Scholar]
- 175. Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. Brain MR: pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow‐Robin spaces. AJR Am J Roentgenol. 1988;151(3):551‐558. [DOI] [PubMed] [Google Scholar]
- 176. Maclullich AM, Wardlaw JM, Ferguson KJ, Starr JM, Seckl JR, Deary IJ. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry. 2004;75(11):1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Paradise MB, Beaudoin MS, Dawes L, et al. Development and validation of a rating scale for perivascular spaces on 3T MRI. J Neurol Sci. 2020;409:116621. [DOI] [PubMed] [Google Scholar]
- 178. Potter GM, Chappell FM, Morris Z, Wardlaw JM. Cerebral perivascular spaces visible on magnetic resonance imaging: development of a qualitative rating scale and its observer reliability. Cerebrovasc Dis. 2015;39(3‐4):224‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12(8):822‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ding J, Sigurðsson S, Jónsson PV, et al. Large perivascular spaces visible on magnetic resonance imaging, cerebral small vessel disease progression, and risk of dementia: the age, gene/environment susceptibility‐Reykjavik Study. JAMA Neurol. 2017;74(9):1105‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke. 2010;41(3):450‐454. [DOI] [PubMed] [Google Scholar]
- 182. Heier LA, Bauer CJ, Schwartz L, Zimmerman RD, Morgello S, Deck MD. Large Virchow‐Robin spaces: MR‐clinical correlation. AJNR Am J Neuroradiol. 1989;10(5):929‐936. [PMC free article] [PubMed] [Google Scholar]
- 183. Rouhl RP, Van Oostenbrugge RJ, Knottnerus IL, Staals JE, Lodder J. Virchow‐Robin spaces relate to cerebral small vessel disease severity. J Neurol. 2008;255(5):692‐696. [DOI] [PubMed] [Google Scholar]
- 184. Brown R, Benveniste H, Black SE, et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018;114(11):1462‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Charidimou A, Meegahage R, Fox Z, et al. Enlarged perivascular spaces as a marker of underlying arteriopathy in intracerebral haemorrhage: a multicentre MRI cohort study. J Neurol Neurosurg Psychiatry. 2013;84(6):624‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Charidimou A, Boulouis G, Pasi M, et al. MRI‐visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2017;88(12):1157‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Banerjee G, Kim HJ, Fox Z, et al. MRI‐visible perivascular space location is associated with Alzheimer's disease independently of amyloid burden. Brain. 2017;140(4):1107‐1116. [DOI] [PubMed] [Google Scholar]
- 188. Hurford R, Charidimou A, Fox Z, Cipolotti L, Jager R, Werring DJ. MRI‐visible perivascular spaces: relationship to cognition and small vessel disease MRI markers in ischaemic stroke and TIA. J Neurol Neurosurg Psychiatry. 2014;85(5):522‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Cavallari M, Egorova S, Healy BC, et al. Evaluating the association between enlarged perivascular spaces and disease worsening in multiple sclerosis. J Neuroimaging. 2018;28(3):273‐277. [DOI] [PubMed] [Google Scholar]
- 190. Feldman RE, Rutland JW, Fields MC, et al. Quantification of perivascular spaces at 7T: a potential MRI biomarker for epilepsy. Seizure. 2018;54:11‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Groeschel S, Brockmann K, Hanefeld F. Virchow‐Robin spaces on magnetic resonance images of children with adrenoleukodystrophy. Eur J Paediatr Neurol. 2007;11(3):142‐145. [DOI] [PubMed] [Google Scholar]
- 192. Smeijer D, Ikram MK, Hilal S. Enlarged perivascular spaces and dementia: a systematic review. J Alzheimer's Dis. 2019;72(1):247‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Spalice A, Guido CA, Nicita F, Biasi CD, Zicari AM, Giannini L. Dilated Virchow‐Robin spaces in children with seizures. A possible correlation? Med Hypotheses. 2020;136:109481. [DOI] [PubMed] [Google Scholar]
- 194. Jost G, Frenzel T, Lohrke J, Lenhard DC, Naganawa S, Pietsch H. Penetration and distribution of gadolinium‐based contrast agents into the cerebrospinal fluid in healthy rats: a potential pathway of entry into the brain tissue. Eur Radiol. 2017;27(7):2877‐2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Deike‐Hofmann K, Reuter J, Haase R, et al. Glymphatic pathway of Gadolinium‐Based contrast agents through the brain: overlooked and misinterpreted. Invest Radiol. 2019;54(4):229‐237. [DOI] [PubMed] [Google Scholar]
- 196. Naganawa S, Suzuki K, Yamazaki M, Sakurai Y. Serial scans in healthy volunteers following intravenous administration of gadoteridol: time course of contrast enhancement in various cranial fluid spaces. Magn Reson Med Sci. 2014;13(1):7‐13. [DOI] [PubMed] [Google Scholar]
- 197. Naganawa S, Ito R, Kawamura M, Taoka T, Yoshida T, Sone M. Peripheral retinal leakage after intravenous administration of a Gadolinium‐based contrast agent: age dependence, temporal and inferior predominance and potential implications for eye homeostasis. Magn Reson Med Sci. 2023;22(1):45‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Benveniste H, Lee H, Ozturk B, et al. Glymphatic cerebrospinal fluid and solute transport quantified by MRI and PET imaging. Neuroscience. 2021;474:63‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ringstad G, Valnes LM, Dale AM, et al. Brain‐wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight. 2018;3(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Naganawa S, Nakane T, Kawai H, Taoka T. Gd‐based contrast enhancement of the perivascular spaces in the basal ganglia. Magn Reson Med Sci. 2017;16(1):61‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Naganawa S, Nakane T, Kawai H, Taoka T. Differences in signal intensity and enhancement on MR images of the perivascular spaces in the basal ganglia versus those in white matter. Magn Reson Med Sci. 2018;17(4):301‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. MacGregor Sharp M, Saito S, Keable A, et al. Demonstrating a reduced capacity for removal of fluid from cerebral white matter and hypoxia in areas of white matter hyperintensity associated with age and dementia. Acta Neuropathol Commun. 2020;8(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Naganawa S. The technical and clinical features of 3D‐FLAIR in neuroimaging. Magn Reson Med Sci. 2015;14(2):93‐106. [DOI] [PubMed] [Google Scholar]
- 204. Rasschaert M, Weller RO, Schroeder JA, Brochhausen C, Idée JM. Retention of gadolinium in brain parenchyma: pathways for speciation, access, and distribution. A critical review. J Magn Reson Imaging: JMRI. 2020;52(5):1293‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information