

Abstract
INTRODUCTION
We studied how biomarkers of reactive astrogliosis mediate the pathogenic cascade in the earliest Alzheimer's disease (AD) stages.
METHODS
We performed path analysis on data from 384 cognitively unimpaired individuals from the ALzheimer and FAmilies (ALFA)+ study using structural equation modeling to quantify the relationships between biomarkers of reactive astrogliosis and the AD pathological cascade.
RESULTS
Cerebrospinal fluid (CSF) amyloid beta (Aβ)42/40 was associated with Aβ aggregation on positron emission tomography (PET) and with CSF p‐tau181, which was in turn directly associated with CSF neurofilament light (NfL). Plasma glial fibrillary acidic protein (GFAP) mediated the relationship between CSF Aβ42/40 and Aβ‐PET, and CSF YKL‐40 partly explained the association between Aβ‐PET, p‐tau181, and NfL.
DISCUSSION
Our results suggest that reactive astrogliosis, as indicated by different fluid biomarkers, influences the pathogenic cascade during the preclinical stage of AD. While plasma GFAP mediates the early association between soluble and insoluble Aβ, CSF YKL‐40 mediates the latter association between Aβ and downstream Aβ‐induced tau pathology and tau‐induced neuronal injury.
Highlights
Lower CSF Aβ42/40 was directly linked to higher plasma GFAP concentrations.
Plasma GFAP partially explained the relationship between soluble Aβ and insoluble Aβ.
CSF YKL‐40 mediated Aβ‐induced tau phosphorylation and tau‐induced neuronal injury.
Keywords: AD cascade, astrogliosis, biomarkers, chitinase‐3‐like protein 1 (YKL‐40), glial fibrillary acidic protein (GFAP), preclinical Alzheimer's disease, structural equation modeling
1. BACKGROUND
There is increased recognition that glial cells play an active role in the pathogenesis of Alzheimer's disease (AD). 1 , 2 Astrocytes are important regulators of the brain's inflammatory response to injury and have been shown to become activated in reaction to the deposition of misfolded protein aggregates. 3 Moreover, several studies have demonstrated that reactive astrocytes surround amyloid beta (Aβ) plaques and tau deposits early in AD 4 , 5 , 6 and shown a strong correlation between astrocyte reactivity and increased accumulation of AD pathology. 7 , 8 , 9 These activated astrocytes in turn release pro‐inflammatory molecules such as cytokines and chemokines, which may contribute to neurotoxic effects and exacerbate the progression of AD. 4 , 10 , 11 However, the specific impact of reactive astrogliosis on key pathological events early in the AD continuum remains uncertain. A deeper understanding of how central disease mechanisms are mediated by activated astrocytes may provide us with insight into pathogenic mechanisms underlying AD.
Two robust fluid biomarkers for measuring astrocyte reactivity in vivo are glial fibrillary acidic protein (GFAP) and chitinase‐3‐like protein 1 (YKL‐40), 12 both of which have consistently been found to be elevated in the dementia phase of AD. 13 , 14 , 15 Recent work has suggested that changes in astrocytes arise very early in the course of AD, prior to frank neurodegeneration and cognitive impairment, demonstrating an upregulation of GFAP and YKL‐40 levels in Aβ‐positive cognitively unimpaired (CU) individuals. 16 , 17 , 18 , 19 , 20 , 21 , 22 In particular, plasma GFAP, rather than GFAP in cerebrospinal fluid (CSF), has demonstrated superior performance in detecting Aβ‐positive CU individuals. 17 , 23 , 24 Moreover, some studies have hypothesized that astrogliosis may even precede the formation of amyloid plaques. 25 , 26 , 27
This Aβ‐induced astroglial response could in turn impact downstream pathological events, including further aggregation of Aβ, tau pathology, neuronal damage, and cognitive decline. 4 , 28 , 29 , 30 However, the impact of reactive astrocytes on disease progression has been shown to be very heterogeneous, and reactive astrocytes may respond differently depending on disease stage, specific pathology, biomarker, brain region, and genetic background. 31 , 32 , 33 , 34 Therefore, we aimed to study how two astrocyte biomarkers (plasma GFAP and CSF YKL‐40), probably reflecting different astrocyte phenotypes, mediate the early pathogenic cascade in the preclinical stages of AD. Through structural equation modeling (SEM), we aimed to analyze the relationships among multiple pathological hallmarks of AD, including biomarkers of amyloid pathology (CSF Aβ42/40 and Aβ‐positron emission tomography [PET]), tau pathology (CSF p‐tau181), neuronal damage (CSF NfL), and cognitive performance simultaneously, and test whether and how these relationships are affected by reactive astrocytes. This may help us untangle the complex interplay among pathological changes occurring in the earliest stages of AD.
2. METHODS
2.1. Study participants
Participants were selected from the ALFA+ study, a longitudinal research cohort of CU individuals aged 45 to 74, enriched for a family history of AD or Apolipoprotein E (APOE) ε4 carriership. All participants scored above pre‐established cut‐off values on the following neuropsychological tests: Mini‐Mental State Examination (≥26), Memory Impairment Screen (≥6), Time Orientation Subtest of the Barcelona Test II (≥68), verbal semantic fluency (naming animals ≥12), and a Clinical Dementia Rating (CDR) of 0. A more detailed description of the study protocol can be found in Molinuevo et al. (2016). 35 The study was approved by an independent ethics committee ‘Parc de Salut Mar’, Barcelona, and is registered at Clinicaltrials.gov (Identifier: NCT02485730).
2.2. Fluid biomarker sampling and analysis
CSF samples were obtained by lumbar puncture following a standardized protocol 36 and then collected in 15‐mL polypropylene tubes (Sarstedt catalogue no. 62.554), aliquoted into 0.5‐mL polypropylene tubes (Sarstedt catalogue no. 72.730.005), and frozen at −80°C within 2 h after lumbar puncture. Blood samples were collected using a 20‐ or 21‐g needle gauge into a 10‐mL EDTA tube (BD Hemogard, 10 mL, K2EDTA, catalogue no. 367525). 37 Tubes were gently inverted five to 10 times and centrifuged at 2000 × g for 10 min at 4°C. The supernatant was aliquoted in volumes of 0.5 mL into sterile poly(propylene) tubes (Sarstedt Screw Cap Micro Tube, 0.5 mL, PP, ref. no. 72.730.105) and immediately frozen at −80°C. The samples were processed at room temperature. The time between collection and freezing of both CSF and plasma samples was < 30 min.
CSF p‐tau181 was measured using the electrochemiluminescence Elecsys immunoassay on a fully automated cobas e601 module (both Roche Diagnostics International Ltd., Rotkreuz, Switzerland). CSF Aβ40, Aβ42, NfL, and YKL‐40 were measured using the Roche NeuroToolKit immunoassays (Roche Diagnostics International Ltd.) on a cobas e411 or e601 analyzer. Plasma GFAP was quantified on the Simoa HD‐X (Quanterix, Billerica, MA, USA) using the commercial single‐plex assay. All CSF and plasma measurements were performed at the Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden. A‐T+ individuals, as determined by a CSF Aβ42/40 ratio > 0.071 (A−) and CSF p‐tau181 > 24 pg/mL (T+), were removed from further analysis as they were considered to reflect non‐AD pathological changes. 22
2.3. Image acquisition and processing
Amyloid PET scans were acquired on a Siemens Biograph mCT scanner, following a cranial computed tomography (CT) scan for attenuation correction. Four frames (4 × 5 min) were collected 90 to 110 min after the injection of 185 MBq [18F]flutemetamol. 38 An OSEM3D algorithm with eight iterations and 21 subsets was used to reconstruct the images with a point spread function and time‐of‐flight corrections into a 1.02 × 1.02 × 2.03‐mm matrix. The averaged PET images were co‐registered to the corresponding T1‐weighted (T1w) magnetic resonance imaging (MRI) images. Three‐dimensional (3D) high‐resolution T1w turbo field echo (TFE) images (voxel size 1 mm3 isotropic, TR/TE/TI: 6.16/2.33/450 ms, flip angle = 12°) were obtained using a 3T scanner (Ingenia CX, Philips Healthcare, Best, The Netherlands). The T1‐weighted images and co‐registered PET images were then warped to Montreal Neurological Institute (MNI) space with SPM12. The standardized uptake value ratio (SUVR) was calculated in MNI space using the standard target region (https://www.gaain.org/centiloid‐project) with the whole cerebellum as a reference region. We then transformed the SUVR values into the centiloid (CL) scale using a previously calibrated conversion equation. 39 , 40 Aβ‐PET was available for a subset of participants (n = 195).
RESEARCH IN CONTEXT
Systematic review: We reviewed the literature using PubMed and previously published reviews. Recent publications investigating astrocyte biomarkers and describing the biological mechanisms underlying AD are cited throughout the manuscript.
Interpretation: Our results indicate that astrocyte reactivity, as measured by increased plasma GFAP and CSF YKL‐40 concentrations, is associated with the build‐up of Aβ plaques and downstream neurodegenerative events in the earliest stages of the AD continuum.
Future directions: Longitudinal studies across the full spectrum of AD are needed to increase our understanding of how the influence of astrogliosis on the progression of AD may change over time and differ by disease stage.
2.4. APOE genotyping
Total DNA was obtained from the blood cellular fraction by proteinase K digestion followed by alcohol precipitation. APOE genotype was obtained from the allelic combination of the rs429358 and rs7412 variants. All participants were classified as APOE ε4 carriers or APOE ε4 non‐carriers.
2.5. Neuropsychological evaluation
In this study, a modified version of the Preclinical Alzheimer Cognitive Composite (PACC) score was used, 41 which consisted of the Free and Cued Selective Reminding Test (total immediate recall), 42 the Logical Memory test of the Wechsler Memory Scale (total delayed recall), 43 the WAIS‐IV Coding subtest, 44 and semantic fluency (animals within 1 min). 45 All raw test scores were standardized into z‐scores using the mean and standard deviation (SD) from CU A‐T‐ participants as a reference and then averaged into a composite score.
2.6. Statistical analysis
Non‐normally distributed CSF and plasma biomarkers were log10‐transformed. Biomarker values three times outside the interquartile range below Q1 or above Q3 were considered outliers and were removed from further analysis.
To investigate the association between astrocyte phenotypes and pathological hallmarks of AD, we applied linear regression analysis using astrocyte biomarkers as predictors and individual core AD and neurodegeneration biomarkers as outcomes while adjusting for age, sex, and APOE ε4 carriership. Additionally, raw associations between all model parameters were examined by performing a cross‐correlation using Pearson's r.
To model a potential mediating role of reactive astrogliosis (ie, elevations in plasma GFAP and CSF YKL‐40) on the association between biomarkers in the AD cascade (ie, changes in amyloid, tau, and neuronal injury biomarkers), we built a path model. Our model was hypothesis‐based with reference to the current literature and followed the amyloid neuropathological cascade pathway. 29 , 46 , 47 , 48 To quantify complex multivariate relationships in our AD cascade model simultaneously, we used SEM. In contrast to standard regression modeling in which all variable coefficients are calculated separately, each structural equation coefficient is computed while considering the direct and indirect effects between all biomarkers. Therefore, SEM makes it possible to test more complicated mediation models in a single analysis. Our model is hierarchical in nature and structured in seven levels: (I) covariates age, sex, and APOE ε4 carriership; (II) CSF Aβ42/40 ratio as an initial pathological trigger; (III) astrocytic biomarkers, which may serve as potential mediators along all associations in this cascade; (IV) Aβ‐PET global CL level; (V) CSF p‐tau181 level; (VI) NfL concentration in CSF; and finally (VII) cognitive performance as measured by the PACC. In SEM, a variable can appear as a predictor in several equations as well as the outcome in others. The direct effect of a predictor variable on a higher‐level outcome can be interpreted as the net effect of a predictor when adjusting for the other predictors (ie, variables of preceding levels) in the equation, and is visualized by an arrow in the path model. The indirect effect is the effect mediated by the reactive astrocyte variables.
All variables in the model were standardized (z‐score), so that 1 SD change in the independent variable predicts 1 SD change in the dependent variable (while holding the other variables in the model constant) and to allow for direct comparisons of beta estimates. The model parameters (effects) were computed by maximum likelihood estimation. Effects were considered significant at p < 0.05 false discovery rate (FDR) corrected. The 95% confidence interval (CI) of the parameters was estimated using Monte Carlo bootstrapping (1000 iterations). Model fit was evaluated by a comparative fit index ≥ 0.90, which indicates that the model accounts for most of the variance in the data and by a root mean square error of approximation statistic < 0.05, indicating low residual values not accounted for by the model. 49 All statistical analyses were carried out using R version 4.2.2, with the lavaan package for SEM analysis. 50
3. RESULTS
The sample characteristics are summarized in Table 1. There were a total of 384 CU participants, 61% of whom were female, and 54% were APOE ε4 carriers; the mean age was 61 years old. The average Mini‐Mental State Examination (MMSE) score was 29.2 (range 27.0 to 30.0), and centiloid values ranged from −21.15 to 63.10, with a mean value of 4.39.
TABLE 1.
Participant characteristics.
| Total (N = 384) | |
|---|---|
| Age (y) | 61.1 [49.6 to 73.4] |
| Sex, F | 235 (61.2%) |
| Education (y) | 13.5 (3.5) |
| MMSE | 29.2 [27.0 to30.0] |
| PACC (z‐score) | 0.00 [−2.01 to 1.40] |
| APOE ε4 carrier | 208 (54.2%) |
| Aβ‐PET (CL)* | 4.39 [−21.15 to 63.10] |
| Plasma GFAP (pg/mL) | 94.8 (44.4) |
| CSF Aβ42/40 | 0.07 (0.02) |
| CSF YKL‐40 (pg/mL) | 145.3 (51.7) |
| CSF p‐tau181 (pg/mL) | 307.7 (141.6) |
| CSF NfL (pg/mL) | 80.7 (25.7) |
Note: Data is presented as mean (SD), mean [range], or N (%); * n = 195.
Abbreviations: Aβ, β‐amyloid; APOE, apolipoprotein E; CL, centiloid; CSF, cerebrospinal fluid; F, female; GFAP, glial fibrillary acidic protein; MMSE, Mini‐Mental State Examination; NfL, neurofilament light; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; p‐tau, phosphorylated tau; y, years; YKL‐40, chitinase‐3‐like protein 1.
Linear regression analysis of the relationship of astrocyte biomarkers with core AD and neurodegeneration biomarkers (Figure 1; Table 2) revealed that higher CSF YKL‐40 was associated with higher Aβ centiloid values, higher CSF p‐tau181, and higher CSF NfL. Higher CSF YK‐L40 was also weakly associated with a lower Aβ42/40 ratio and worse cognitive performance. Higher plasma GFAP was associated with lower CSF Aβ42/40, higher Aβ‐PET load, and higher CSF p‐tau181 and showed a weaker association with higher CSF NfL. In addition, plasma GFAP showed no significant association with cognitive performance or with CSF YKL‐40 after adjusting for age, sex, and APOE ε4 carriership. Additional analysis using a subset of participants (n = 195) with all biomarker measurements available showed similar results (Figure S1).
FIGURE 1.
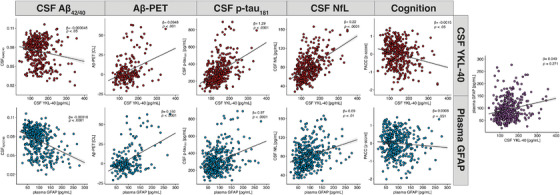
Scatterplots showing the relation between astrocyte biomarkers (ie, YKL‐40 and GFAP) with biomarkers of the AD cascade (ie, Aβ42/40, Aβ‐PET, p‐tau181, NfL, PACC). All models included age, sex, and APOE ε4 allele status. Abbreviations: Aβ, β‐amyloid; CL, centiloid; CSF, cerebrospinal fluid; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; p‐tau, phosphorylated tau; YKL‐40, chitinase‐3‐like protein 1.
TABLE 2.
Linear regression analysis: Individual associations between CSF YKL‐40 and plasma GFAP with AD cascade biomarkers.
| CSF YKL‐40 | Plasma GFAP | |
|---|---|---|
| CSF Aβ42/40 | −0.00005 (0.00002)* | −0.00016 (0.00002)**** |
| Aβ‐PET (CL) | 0.09 (0.02)*** | 0.15 (0.02)**** |
| CSF p‐tau181 | 1.29 (0.14)**** | 0.99 (0.17)**** |
| CSF NfL | 0.22 (0.02)**** | 0.09 (0.03)** |
| PACC | −0.0015 (0.0007)* | 0.0008 (0.0008) |
Note: Data are represented as beta (standard error); betas are unstandardized. All models included age, sex, APOE ε4 allele status.
Abbreviations: Aβ, β‐amyloid; CL, centiloid; CSF, cerebrospinal fluid; GFAP, glial fibrillary filament protein; NFL, neurofilament light; PACC, Preclinical Alzheimer Cognitive Composite; p‐tau, phosphorylated tau; YKL‐40, Chitinase 3‐like 1.
* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
3.1. Structural equation model
The results of our path model are shown in Figure 2, which displays all significant direct associations between the biomarkers. All possible (ie, significant and non‐significant) associations and their corresponding estimates with 95% CI are shown in Table 3. We observed that older age (β = −0.24; 95% CI = −0.32 to −0.15) and particularly APOE ε4 carriership (β = −0.74; 95% CI = −0.92 to −0.56) showed a direct significant association with lower CSF Aβ42/40 ratio. Moreover, older age (β = 0.36; 95% CI = 0.27 to 0.45) and early Aβ pathology, as indicated by lower CSF Aβ42/40 ratio (β = −0.33; 95% CI = −0.43 to −0.23), were directly related to increased plasma GFAP. Older age (β = 0.37; 95% CI = 0.27 to 0.47), higher Aβ load on PET (β = 0.21; 95% CI = 0.10 to 0.33), higher CSF p‐tau181 (β = 0.31; 95% CI = 0.22 to 0.39), and increased CSF NfL (β = 0.18; 95% CI = 0.07 to 0.27) were directly associated with higher CSF YKL‐40. There was a strong direct effect of lower CSF Aβ42/40 ratio (β = −0.54; 95% CI = −0.68 to −0.40) on elevated Aβ‐PET and, to a lesser extent, from plasma GFAP (β = 0.17; 95% CI = 0.03 to 0.31) and from CSF YKL‐40 (β = 0.19; 95% CI = 0.06 to 0.33) on Aβ‐PET. Furthermore, a direct association was observed between CSF p‐tau181 and CSF Aβ42/40 (β = −0.26; 95% CI = −0.40 to −0.13), Aβ‐PET load (β = 0.31; 95% CI = 0.13 to 0.48), and CSF YKL‐40 (β = 0.40; 95% CI = 0.30 to 0.50). In addition, older age (β = 0.20; 95% CI = 0.11 to 0.30), male sex (β = 0.57; 95% CI = 0.41 to 0.74), higher CSF p‐tau181 (β = 0.18; 95% CI = 0.06 to 0.31), and higher CSF YKL‐40 (β = 0.31; 95% CI = 0.20 to 0.41) were directly associated with increased CSF NfL. Finally, older age (β = −0.19; 95% CI = −0.28 to −0.11) was directly associated with worse cognitive performance, as measured by the PACC score. Note that in SEM, all tested associations are corrected for parameters of preceding levels. A descriptive cross‐correlation matrix across all model parameters separately is provided in Supplementary Figure 2.
FIGURE 2.
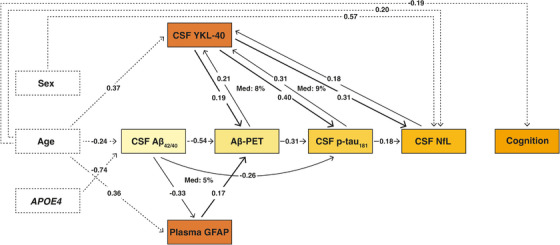
Path analysis showing the impact of CSF YKL‐40 and plasma GFAP on Amyloid‐Tau‐Neurodegeneration and cognition. Cognition was measured by the Preclinical Alzheimer Cognitive Composite (PACC) score. Arrows show the direct effects of significant associations at p < .05 FDR‐corrected between all biomarker relationships (z‐score) from the structural equation model. The beta estimates represent the unique contribution of a specific variable to the change in a dependent variable after controlling for the effects of all variables of preceding levels in the model.
TABLE 3.
Structural equation model coefficients of Figure 2 displaying all direct and indirect effects of the path model.
| Biomarker | β (95% CI) | p value |
|---|---|---|
| Cognition (PACC) | ||
| NfL | 0.014 (−0.074 to 0.114) | 0.974 |
| p‐tau181 | −0.015 (−0.118 to 0.091) | 0.974 |
| Aβ‐PET | 0.027 (−0.120 to 0.155) | 0.974 |
| YKL‐40 | −0.078 (−0.173 to 0.018) | 0.358 |
| GFAP | 0.031 (−0.045 to 0.113) | 0.974 |
| Aβ42/40 | 0.025 (−0.080 to 0.123) | 0.974 |
| Sex, male | 0.100 (−0.049 to 0.243) | 0.511 |
| Age | −0.192 (−0.280 to −0.109) | <0.001* |
| APOE ε4 carrier | 0.098 (−0.04 to 0.235) | 0.471 |
| NfL | ||
| p‐tau181 | 0.182 (0.057 to 0.305) | 0.015* |
| Mediated by YKL‐40 | 0.094 (0.055 to 0.138) | <0.001* |
| Aβ‐PET | 0.076 (−0.065 to 0.225) | 0.505 |
| YKL‐40 | 0.306 (0.196 to 0.406) | <0.001* |
| GFAP | 0.072 (−0.009 to 0.154) | 0.225 |
| Aβ42/40 | 0.027 (−0.090 to 0.133) | 0.796 |
| Sex, male | 0.570 (0.410 to 0.739) | <0.001* |
| Age | 0.202 (0.107 to 0.304) | <0.001* |
| APOE ε4 carrier | 0.009 (−0.150 to 0.176) | 0.939 |
| p‐tau181 | ||
| Aβ‐PET | 0.305 (0.133 to 0.481) | 0.002* |
| Mediated by YKL‐40 | 0.082 (0.041 to 0.139) | 0.002* |
| YKL‐40 | 0.398 (0.302 to 0.497) | <0.001* |
| GFAP | 0.059 (−0.036 to 0.150) | 0.294 |
| Aβ42/40 | −0.261 (−0.404 to −0.131) | <0.001* |
| Sex, male | 0.055 (−0.117 to 0.208) | 0.564 |
| Age | −0.061 (−0.154 to 0.030) | 0.276 |
| APOE ε4 carrier | 0.002 (−0.171 to 0.170) | 0.999 |
| Aβ‐PET | ||
| YKL‐40 | 0.194 (0.059 to 0.328) | 0.008* |
| GFAP | 0.170 (0.034 to 0.312) | 0.030* |
| Aβ42/40 | −0.541 (−0.677 to −0.402) | <0.001* |
| Mediated by GFAP | −0.055 (−0.106 to −0.009) | 0.043* |
| Sex, male | −0.061 (−0.296 to 0.171) | 0.635 |
| Age | 0.010 (−0.119 to 0.129) | 0.896 |
| APOE ε4 carrier | −0.104 (−0.328 to 0.130) | 0.430 |
| YKL‐40 | ||
| NfL | 0.177 (0.067 to 0.274) | 0.004* |
| p‐tau181 | 0.309 (0.220 to 0.392) | <0.001* |
| Aβ‐PET | 0.206 (0.103 to 0.325) | 0.001* |
| Aβ42/40 | −0.085 (−0.196 to 0.033) | 0.193 |
| Sex, male | −0.188 (−0.396 to 0.036) | 0.113 |
| Age | 0.370 (0.269 to 0.466) | <0.001* |
| APOE ε4 carrier | −0.112 (−0.306 to 0.106) | 0.368 |
| GFAP | ||
| NfL | 0.008 (−0.03 to 0.115) | 0.965 |
| p‐tau181 | 0.014 (−0.065 to 0.089) | 0.823 |
| Aβ‐PET | 0.107 (−0.001 to 0.225) | 0.124 |
| Aβ42/40 | −0.327 (−0.429 to −0.228) | <0.001* |
| Sex, male | −0.084 (−0.266 to 0.090) | 0.430 |
| Age | 0.361 (0.274 to 0.446) | <0.001* |
| APOE ε4 carrier | −0.167 (−0.367 to 0.030) | 0.142 |
| Aβ42/40 | ||
| Sex, male | 0.091 (−0.102 to 0.271) | 0.430 |
| Age | −0.235 (−0.315 to −0.152) | <0.001* |
| APOE ε4 carrier | −0.743 (−0.922 to −0.562) | <0.001* |
Note. Structural equation model showing standardized coefficients with bootstrapped 95% confidence intervals. Model shown in Figure 2. N = 384 (Aβ‐PET n = 195). P values adjusted for multiple comparisons using false discovery rate were considered significant at p < 0.05.
Abbreviations: Aβ, β‐amyloid; APOE, apolipoprotein E; CI, confidence interval; GFAP, glial fibrillary acidic protein; NfL, neurofilament light; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; p‐tau, phosphorylated tau; YKL‐40, chitinase‐3‐like protein 1.
3.2. Mediation effects
We observed that part of the relationship between CSF Aβ42/40 and Aβ‐PET load could be explained by plasma GFAP (Figure 2; Proportion mediated 5%; β = −0.06; 95% CI = −0.11 to −0.01). Furthermore, CSF YKL‐40 mediated the association of Aβ‐PET with CSF p‐tau181 (Proportion mediated 8%; β = 0.08; 95% CI = 0.04 to 0.14), as well as the association of CSF p‐tau181 with CSF NfL (Proportion mediated 9%; β = 0.09; 95% CI = 0.06 to 0.14). Since no direct association between CSF Aβ42/40 and CSF YKL‐40 was observed, no CSF Aβ42/40 induced mediation by CSF YKL‐40 on other biomarkers in the cascade was present. Similarly, plasma GFAP showed no direct relationship with CSF p‐tau181, and thus no mediation by GFAP on the effect of CSF Aβ42/40 on CSF p‐tau181 was established.
4. DISCUSSION
Our structural equation model revealed that plasma GFAP and CSF YKL‐40 are important mediators of key events in the AD cascade and strongly contribute to the progression of AD at an early stage of the disease. We observed that the earliest abnormalities in CSF Aβ42/40 triggered an upregulation of GFAP in the blood. The association between CSF Aβ42/40 and Aβ‐PET was partially explained by this increase in plasma GFAP. This suggests that astroglia, particularly those that release GFAP, may have a role in the early balance between soluble and insoluble Aβ aggregates. Furthermore, we observed that the release of YKL‐40 into the CSF occurred slightly later in the pathological cascade and was linked with Aβ‐induced tau phosphorylation and tau‐induced axonal damage. These results further support the evidence that reactive astrogliosis is an early event in AD and a significant component of the pathological cascade driving neurodegeneration.
Our results are in agreement with increasing evidence that suggests that astrocyte changes occur very early in the course of AD. Furthermore, they indicate that distinct stages of the early pathological cascade in preclinical AD are associated with GFAP and YKL‐40 upregulation, which show differential responses to pathological stimuli. This is in line with previous studies showing distinct astrocyte biomarker signatures in response to Aβ and tau pathology, 31 as well as across disease progression. 51
A strong relationship between Aβ aggregation and GFAP, in blood as well as in CSF, has been frequently demonstrated. 23 , 51 , 52 We add to these findings by showing for the first time, to our knowledge, that changes in plasma GFAP are partly responsible for the relationship between soluble Aβ and increased fibrillar Aβ deposition on PET. Previous studies demonstrated that a rise in Aβ oligomers was highly associated with astrocyte reactivity. 25 , 26 , 53 Furthermore, it has been shown that astrocytes are involved in the clearance and degradation of Aβ, 54 , 55 with evidence suggesting that astrocytes can internalize Aβ oligomers and protofibrils but may eventually become overwhelmed and fail to clear Aβ effectively. Moreover, when astrocytes break down, they release the Aβ they have accumulated, actively contributing to the overall accumulation of Aβ plaques. 56 , 57 , 58 Taken together, this implies that astrocytes’ protective mechanisms become impaired in AD and rather gain a neurotoxic function instead. 11 , 25
Recently, Bellaver et al. (2023) 9 reported that reactive astrocytes were a prerequisite for the phosphorylation of tau in Aβ‐positive CU patients. Although we used a different study population and approach, that is, binary versus continuous astrocyte measurements, we demonstrated compatible results and provided more granularity on the underlying mechanism. Our results suggest that increased astrocyte reactivity may influence the balance between soluble and insoluble Aβ and that this formation of Aβ plaques in turn triggers tau phosphorylation in preclinical AD.
CSF concentrations of YKL‐40 have been thought to mainly reflect a response to tau pathology rather than Aβ, and strong correlations with tau pathophysiology have also been found in preclinical stages. 19 , 22 , 31 , 59 , 60 A growing body of studies have demonstrated a positive association of CSF YKL‐40 levels with markers of neuronal injury, including cortical atrophy, CSF t‐tau, and NfL in early stages of AD. 16 , 21 , 28 , 61 These findings suggest that CSF YKL‐40 is particularly related to tau pathology and neuronal injury and support the hypothesis that reactive astrocytes actively contribute to the disruption of neuronal functioning. 4 , 62 , 63 , 64 Moreover, some studies, including our CU cohort, have suggested YKL‐40 may be involved in a non‐amyloid‐related pathway, demonstrating elevated CSF levels of YKL‐40 in A+T+ and A−T+ individuals, but not in A+T− individuals. 16 , 22 , 31 However, in the current study we observed that fibrillar deposits of Aβ could trigger the expression of YKL‐40, and associations between YKL‐40 levels and Aβ‐PET were also previously reported. 65 , 66 YKL‐40 immunoreactivity was demonstrated to be independent of tau in a recent post mortem study, 60 suggesting that the astrocytic responses by GFAP and YKL‐40 might be more complex than an amyloid‐tau dichotomy. 67
In line with previous reports, we observed no direct association between APOE ε4 carriership or sex with astrocyte biomarkers. 15 , 18 , 21 However, as reported in previous studies, higher CSF NfL concentrations were observed in men. 68 , 69 Furthermore, aging contributed significantly to both YKL‐40 and GFAP concentrations, as reported previously. 16 , 17 , 18 , 21 Finally, we found that age was the only factor that was directly related to cognitive performance, while several previous studies did found an association between GFAP and YKL‐40, both in blood and CSF, and cognition. 30 , 70 , 71 The cross‐sectional nature of this study, the early stage on the AD continuum as reflected by the very low mean CL values, and the limited variance in test scores of cognitively normal participants may explain this finding. Additionally, it is important to note that in the current structural equation model, the association between astrocytosis and cognition is adjusted for the effect of Aβ, tau pathology, and NfL.
Our findings, together with previous evidence, indicate that astrogliosis contributes to the pathogenesis of AD through multiple routes/pathways, which can be observed at early asymptomatic stages of AD. Astroglial response may occur in different stages of preclinical AD: the aggregation of Aβ, the formation of tau tangles, and neuronal damage. It can be speculated that once astrocyte activation is induced, there is a release of pro‐inflammatory molecules and neuronal dysfunction, which in turn reactivates astrocytes. Eventually, this astroglial response may contribute to neurodegenerative changes independently of Aβ plaque pathology. 4 , 72 , 73 Taken together, these findings suggest that interventions targeting astrocyte dysfunction involved in Aβ clearing in early preclinical stages may ultimately prevent or delay the onset of AD dementia.
There are some limitations to this study. First, the data were collected cross‐sectionally from CU individuals only. This prevented us from being able to make any claims on the causality of the pathological events. Longitudinal studies across the entire AD continuum are needed to provide information on how the interaction between reactive astrocytes and AD pathology markers may change over time, which is important considering the indications that the role of reactive astrocytes in disease progression likely varies across disease stages. 51 , 72 Second, our path model provides a simplified view of the AD pathological cascade and is by no means a saturated depiction of its complex pathophysiology. To illustrate, multiple lines of evidence suggest an important interaction between microglia and astrocytes that likely acts in a coordinated manner to promote the progression of AD. 74 The strengths of this study include the well‐characterized cohort and the fact that the SEM approach allowed us to examine all direct and indirect effects on each variable in a single model, rather than studying all relationships separately. This is essential in a multifactorial disease such as AD, in which a complex cascade of connected events ultimately contributes to progression.
In conclusion, we provide evidence that the astrocytosis biomarkers plasma GFAP and CSF YKL‐40 increase very early in the AD continuum and mediate several associations between key pathogenic events that occur during this disease stage. These results substantiate the notion that reactive astrocytes in reaction to AD pathology are active players in promoting downstream neurodegenerative events.
CONFLICT OF INTEREST STATEMENT
W.P., M.S., A.B.S., C.M., K.F., A.G.E., N.J.A. have nothing to disclose. J.L.M. is currently a full‑time employee of H. Lundbeck A/S and previously served as a consultant or on advisory boards for the following for‑profit companies or has given lectures in symposia sponsored by the following for‑profit companies: Roche Diagnostics, Genentech, Novartis, Lundbeck, Oryzon, Biogen, Lilly, Janssen, Green Valley, MSD, Eisai, Alector, BioCross, GE Healthcare, and ProMIS Neurosciences. G.K. is a full‑time employee of Roche Diagnostics GmbH. M.C. is a full‑time employee of Roche Diagnostics International Ltd. and an owner of shares in Roche. H.Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures at symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a cofounder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. K.B. has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a cofounder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. M.S.C. has served as a consultant and at advisory boards for Roche Diagnostics International Ltd. and has given lectures at symposia sponsored by Roche Diagnostics, S.L.U. and Roche Farma, S.A. J.D.G. receives research funding from Roche Diagnostics and GE Healthcare and has given lectures at symposia sponsored by Biogen and Philips. G.S.‐B. has served as a consultant for Roche Farma, S.A. O.G.R. receives research funding from F. Hoffmann‐La Roche Ltd. and has given lectures in symposia sponsored by Roche Diagnostics, S.L.U. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants provided written informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This publication is part of the ALFA study (ALzheimer and FAmilies). The authors would like to express their most sincere gratitude to the ALFA project participants and relatives without whom this research would not have been possible. The authors thank Roche Diagnostics International Ltd. for providing the kits to measure CSF biomarkers and GE Healthcare for providing the doses of [18F]flutemetamol PET. The Roche NeuroToolKit is a panel of exploratory prototype assays designed to robustly evaluate biomarkers associated with key pathologic events characteristic of AD and other neurological disorders, used for research purposes only and not approved for clinical use. COBAS and ELECSYS are trademarks of Roche. All other product names and trademarks are the property of their respective owners. Collaborators of the ALFA study are Müge Akinci, Federica Anastasi, Annabella Beteta, Raffaele Cacciaglia, Lidia Canals, Alba Cañas, Carme Deulofeu, Maria Emilio, Irene Cumplido‐Mayoral, Marta del Campo, Carme Deulofeu, Ruth Dominguez, Maria Emilio, Sherezade Fuentes, Marina García, Laura Hernández, Gema Huesa, Jordi Huguet, Laura Iglesias, Esther Jiménez, David López‐Martos, Paula Marne, Tania Menchón, Paula Ortiz‐Romero, Eleni Palpatzis, Albina Polo, Sandra Pradas, Iman Sadeghi, Lluís Solsona, Anna Soteras, Laura Stankeviciute, Núria Tort‐Colet and Marc Vilanova.
The ALFA+ study receives funding from “la Caixa” Foundation (ID 100010434), under agreement LCF/PR/GN17/50300004, and the Alzheimer's Association and an international anonymous charity foundation through the TriBEKa Imaging Platform project (TriBEKa‑17‑519007). Additional support has been received from the Universities and Research Secretariat, Ministry of Business and Knowledge of the Catalan government under grant no. 2017‐SGR‐892. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (2022‐01018 and 2019‐02397), the European Union's Horizon Europe research and innovation program under grant agreement 101053962, Swedish State Support for Clinical Research (ALFGBG‐71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (ADSF‐21‐831376‐C, ADSF‐21‐831381‐C, and ADSF‐21‐831377‐C), the Bluefield Project, the Olav Thon Foundation, the Erling‐Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (FO2022‐0270), the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021‐00694), and the UK Dementia Research Institute at UCL (UKDRI‐1003). KB is supported by the Swedish Research Council (2017‑00915); the Alzheimer Drug Discovery Foundation (ADDF), USA (RDAPB‑201809‑2016615); the Swedish Alzheimer Foundation (AF‑742881); Hjärnfonden, Sweden (FO2017‑0243); the Swedish state under the agreement between the Swedish government and the county councils, the ALF‑agreement (ALFGBG‑715986); the European Union Joint Programme for Neurodegenerative Disorders (JPND2019‑466‑236); the National Institute of Health (NIH), USA (grant 1R01AG068398‑01); and the Alzheimer's Association 2021 Zenith Award (ZEN‑21‑848495). MSC receives funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement 948677), the Instituto de Salud Carlos III (PI19/00155, PI22/00456), and the ERC under the EU's ‘la Caixa’ Foundation (ID 100010434) and from the EU's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant (847648, LCF/BQ/PR21/11840004). JDG is supported by the Spanish Ministry of Science and Innovation (RYC‑2013‑13054). JDG has also received research support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking AMYPAD (grant agreement 115952), EIT Digital (grant 2021), and from Ministerio de Ciencia y Universidades (grant agreement RTI2018‑102261). GS‐B receives funding from the Ministerio de Ciencia e Innovacion, Spanish Research Agency, PID2020‐119556RA‐I00. OGR receives funding from the Alzheimer's Association Research Fellowship Program (2019‐AARF‐644568), from Instituto de Salud Carlos III (PI19/00117), and from the Spanish Ministry of Science, Innovation and Universities (Juan de la Cierva programme IJC2020‐043417‐I).
Pelkmans W, Shekari M, Brugulat‐Serrat A, et al. Astrocyte biomarkers GFAP and YKL‐40 mediate early Alzheimer's disease progression. Alzheimer's Dement. 2024;20:483–493. 10.1002/alz.13450
Contributor Information
Wiesje Pelkmans, Email: wpelkmans@barcelonabeta.org.
Juan Domingo Gispert, Email: jdgispert@barcelonabeta.org.
REFERENCES
- 1. De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164(4):603‐615. doi: 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- 2. Hampel H, Caraci F, Cuello AC, et al. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer's disease. Front Immunol. 2020;11:456. doi: 10.3389/fimmu.2020.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steardo L, Bronzuoli MR, Iacomino A, Esposito G, Steardo L, Scuderi C. Does neuroinflammation turn on the flame in Alzheimer's disease? Focus on astrocytes. Front Neurosci. 2015;9. doi: 10.3389/fnins.2015.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serrano‐Pozo A, Mielke ML, Gómez‐Isla T, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol. 2011;179(3):1373‐1384. doi: 10.1016/j.ajpath.2011.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia. doi: 10.1002/glia.20967. Published online 2010:NA‐NA. [DOI] [PubMed] [Google Scholar]
- 6. Kamphuis W, Mamber C, Moeton M, et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. Ikezu T, ed. PLoS One. 2012;7(8):e42823. doi: 10.1371/journal.pone.0042823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chatterjee P, Doré V, Pedrini S, et al. Plasma glial fibrillary acidic protein is associated with 18f‐smbt‐1 pet: two putative astrocyte reactivity biomarkers for Alzheimer's disease. J Alzheimers Dis. 2023;92(2):615‐628. doi: 10.3233/JAD-220908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salvadó G, Milà‐Alomà M, Shekari M, et al. Cerebral amyloid‐β load is associated with neurodegeneration and gliosis: mediation by p‐tau and interactions with risk factors early in the Alzheimer's continuum. Alzheimers Dement. 2021;17(5):788‐800. doi: 10.1002/alz.12245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bellaver B, Povala G, Ferreira PCL, et al. Astrocyte reactivity influences amyloid‐β effects on tau pathology in preclinical Alzheimer's disease. Nat Med. 2023;29:1775‐1781. doi: 10.1038/s41591-023-02380-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heneka MT, Carson MJ, Khoury JE, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14(4):388‐405. doi: 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanchez‐Mico MV, Jimenez S, Gomez‐Arboledas A, et al. Amyloid‐β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer's disease. Glia. 2021;69(4):997‐1011. doi: 10.1002/glia.23943 [DOI] [PubMed] [Google Scholar]
- 12. Carter SF, Herholz K, Rosa‐Neto P, Pellerin L, Nordberg A, Zimmer ER. Astrocyte biomarkers in Alzheimer's disease. Trends Mol Med. 2019;25(2):77‐95. doi: 10.1016/j.molmed.2018.11.006 [DOI] [PubMed] [Google Scholar]
- 13. Baldacci F, Lista S, Palermo G, Giorgi FS, Vergallo A, Hampel H. The neuroinflammatory biomarker YKL‐40 for neurodegenerative diseases: advances in development. Expert Rev Proteomics. 2019;16(7):593‐600. doi: 10.1080/14789450.2019.1628643 [DOI] [PubMed] [Google Scholar]
- 14. Kester MI, Teunissen CE, Sutphen C, et al. Cerebrospinal fluid VILIP‐1 and YKL‐40, candidate biomarkers to diagnose, predict and monitor Alzheimer's disease in a memory clinic cohort. Alzheimers Res Ther. 2015;7(1):59. doi: 10.1186/s13195-015-0142-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nordengen K, Kirsebom BE, Henjum K, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation. 2019;16(46). doi: 10.1186/s12974-019-1399-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alcolea D, Martínez‐Lage P, Sánchez‐Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 2015;85(7):626‐633. doi: 10.1212/WNL.0000000000001859 [DOI] [PubMed] [Google Scholar]
- 17. Benedet AL, Milà‐Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry . 2021;11(1):27. doi: 10.1038/s41398-020-01137-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Craig‐Schapiro R, Perrin RJ, Roe CM, et al. YKL‐40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry. 2010;68(10):903‐912. doi: 10.1016/j.biopsych.2010.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo Y, Shen XN, Wang HF, et al. The dynamics of plasma biomarkers across the Alzheimer's continuum. Alzheimers Res Ther. 2023;15(1):31. doi: 10.1186/s13195-023-01174-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867‐e877. doi: 10.1212/WNL.0000000000006082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milà‐Alomà M, Salvadó G, Gispert JD, et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimers Dement. 2020;16(10):1358‐1371. doi: 10.1002/alz.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simrén J, Weninger H, Brum WS, et al. Differences between blood and cerebrospinal fluid glial fibrillary Acidic protein levels: the effect of sample stability. Alzheimers Dement. 2022;18:1988‐1992. doi: 10.1002/alz.12806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pereira Diniz L, Tortelli V, Matias I, et al. Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer's disease model. J Neurosci. 2017;37(28):6797‐6809. doi: 10.1523/JNEUROSCI.3351-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodriguez‐Vieitez E, Ni R, Gulyás B, et al. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur J Nucl Med Mol Imaging. 2015;42(7):1119‐1132. doi: 10.1007/s00259-015-3047-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shah D, Gsell W, Wahis J, et al. Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer's disease. Cell Rep. 2022;40(8):111280. doi: 10.1016/j.celrep.2022.111280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gispert JD, Monté GC, Falcon C, et al. CSF YKL‐40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol Aging. 2016;38:47‐55. doi: 10.1016/j.neurobiolaging.2015.10.022 [DOI] [PubMed] [Google Scholar]
- 29. Stocker H, Beyer L, Perna L, et al. Association of plasma biomarkers, p‐tau181, glial fibrillary acidic protein, and neurofilament light, with intermediate and long‐term clinical Alzheimer's disease risk: results from a prospective cohort followed over 17 years. Alzheimers Dement. 2022:1‐11. doi: 10.1002/alz.12614. October 2021. [DOI] [PubMed] [Google Scholar]
- 30. Verberk IMW, Laarhuis MB, van den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic‐based cohort study. Lancet Healthy Longev. 2021;2(2):e87‐e95. doi: 10.1016/S2666-7568(20)30061-1 [DOI] [PubMed] [Google Scholar]
- 31. Ferrari‐Souza JP, Ferreira PCL, Bellaver B, et al. Astrocyte biomarker signatures of amyloid‐β and tau pathologies in Alzheimer's disease. Mol Psychiatry. 2022;10:22269841. doi: 10.1038/s41380-022-01716-2. Published online August. 2022.01.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiwaji Z, Tiwari SS, Avilés‐Reyes RX, et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat Commun. 2022;13(1):135. doi: 10.1038/s41467-021-27702-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matias I, Morgado J, Gomes FCA. Astrocyte Heterogeneity: impact to brain aging and disease. Front Aging Neurosci. 2019;11:59. doi: 10.3389/fnagi.2019.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perez‐Nievas BG, Serrano‐Pozo A. Deciphering the astrocyte reaction in Alzheimer's disease. Front Aging Neurosci. 2018;10:114. doi: 10.3389/fnagi.2018.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Molinuevo JL, Gramunt N, Gispert JD, et al. The ALFA project: a research platform to identify early pathophysiological features of Alzheimer's disease. Alzheimers Dement Transl Res Clin Interv. 2016;2(2):82‐92. doi: 10.1016/j.trci.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Teunissen CE, Tumani H, Engelborghs S, Mollenhauer B. Biobanking of CSF: international standardization to optimize biomarker development. Clin Biochem. 2014;47(4‐5):288‐292. doi: 10.1016/j.clinbiochem.2013.12.024 [DOI] [PubMed] [Google Scholar]
- 37. Suárez‐Calvet M, Karikari TK, Ashton NJ, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer's continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med. 2020;12(12). 10.15252/emmm.202012921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collij LE, Salvadó G, Shekari M, et al. Visual assessment of [18F]flutemetamol PET images can detect early amyloid pathology and grade its extent. Eur J Nucl Med Mol Imaging. 2021;48(7):2169‐2182. doi: 10.1007/s00259-020-05174-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1). doi: 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salvadó G, Molinuevo JL, Brugulat‐Serrat A, et al. Centiloid cut‐off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther. 2019;11(1):27. doi: 10.1186/s13195-019-0478-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite. JAMA Neurol. 2014;71(8):961. doi: 10.1001/jamaneurol.2014.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pena‐Casanova J, Gramunt‐Fombuena N, Quinones‐Ubeda S, et al. Spanish Multicenter Normative Studies (NEURONORMA Project): norms for the Rey‐Osterrieth Complex Figure (Copy and Memory), and Free and Cued Selective Reminding Test. Arch Clin Neuropsychol. 2009;24(4):371‐393. doi: 10.1093/arclin/acp041 [DOI] [PubMed] [Google Scholar]
- 43. Wechsler D, Escala de memoria de Wechsler‐IV. Pearson Educ . Published online 2013.
- 44. Wechsler D, Escala de inteligencia de Wechsler para adultos‐IV (WAIS‐IV). Madr Pearson . Published online 2012.
- 45. Zhao Q, Guo Q, Hong Z. Clustering and switching during a semantic verbal fluency test contribute to differential diagnosis of cognitive impairment. Neurosci Bull. 2013;29(1):75‐82. doi: 10.1007/s12264-013-1301-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bilgel M, Wong DF, Moghekar AR, Ferrucci L, Resnick SM. Causal links among amyloid, tau, and neurodegeneration. Brain Commun. 2022;4(4). doi: 10.1093/braincomms/fcac193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo T, Korman D, Baker SL, Landau SM, Jagust WJ. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer's disease pathophysiology. Biol Psychiatry. 2020(12):1‐9. doi: 10.1016/j.biopsych.2020.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jack CR, Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321(23):2316. doi: 10.1001/jama.2019.7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu L, Bentler PM. Cutoff criteria for fit indexes in covariance structure analysis: conventional criteria versus new alternatives. Struct Equ Model Multidiscip J. 1999;6(1):1‐55. doi: 10.1080/10705519909540118 [DOI] [Google Scholar]
- 50. Rosseel Y. lavaan : an R package for structural equation modeling. J Stat Softw. 2012;48(2). 10.18637/jss.v048.i02 [DOI] [Google Scholar]
- 51. Asken BM, Elahi FM, La Joie R, et al. Plasma glial fibrillary acidic protein levels differ along the spectra of amyloid burden and clinical disease stage. Mielke m, ed. J Alzheimers Dis. 2020;78(1):265‐276. doi: 10.3233/JAD-200755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pontecorvo MJ, Lu M, Burnham SC, et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79(12):1250‐1259. doi: 10.1001/jamaneurol.2022.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Narayan P, Holmström KM, Kim DH, et al. Rare individual amyloid‐β oligomers act on astrocytes to initiate neuronal damage. Biochemistry. 2014;53(15):2442‐2453. doi: 10.1021/bi401606f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gomez‐Arboledas A, Davila JC, Sanchez‐Mejias E, et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia. 2018;66(3):637‐653. doi: 10.1002/glia.23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wyss‐Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid‐β in vitro and in situ. Nat Med. 2003;9(4):453‐457. doi: 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- 56. Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate Ab42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971(2):197‐209. doi: 10.1016/S0006-8993(03)02361-8 [DOI] [PubMed] [Google Scholar]
- 57. Zhao J, O'Connor T, Vasser R. The contribution of activated astrocytes to Ab production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation. 2011;8(150). doi: 10.1186/1742-2094-8-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Söllvander S, Nikitidou E, Brolin R, et al. Accumulation of amyloid‐β by astrocytes result in enlarged endosomes and microvesicle‐induced apoptosis of neurons. Mol Neurodegener. 2016;11(1):38. doi: 10.1186/s13024-016-0098-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Antonell A, Mansilla A, Rami L, et al. Cerebrospinal fluid level of YKL‐40 protein in preclinical and prodromal Alzheimer's disease. J Alzheimers Dis. 2014;42(3):901‐908. doi: 10.3233/JAD-140624 [DOI] [PubMed] [Google Scholar]
- 60. Querol‐Vilaseca M, Colom‐Cadena M, Pegueroles J, et al. YKL‐40 (Chitinase 3‐like I) is expressed in a subset of astrocytes in Alzheimer's disease and other tauopathies. J Neuroinflammation. 2017;14(1):118. doi: 10.1186/s12974-017-0893-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baldacci F, Lista S, Cavedo E, Bonuccelli U, Hampel H. Diagnostic function of the neuroinflammatory biomarker YKL‐40 in Alzheimer's disease and other neurodegenerative diseases. Expert Rev Proteomics. 2017;14(4):285‐299. doi: 10.1080/14789450.2017.1304217 [DOI] [PubMed] [Google Scholar]
- 62. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481‐487. doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mann CN, Devi SS, Kersting CT, et al. Astrocytic α2‐Na+/K+ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model. Sci Transl Med. 2022;14(632):eabm4107. doi: 10.1126/scitranslmed.abm4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guttenplan KA, Weigel MK, Prakash P, et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599(7883):102‐107. doi: 10.1038/s41586-021-03960-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bos I, Vos S, Verhey F, et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer's disease spectrum. Alzheimers Dement. 2019;15(5):644‐654. doi: 10.1016/j.jalz.2019.01.004 [DOI] [PubMed] [Google Scholar]
- 66. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11(12):1‐13. 10.15252/emmm.201911170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Salvadó G, Ossenkoppele R, Ashton NJ, et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol Med. 2023;13. 10.15252/emmm.202217123. Published online March. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bridel C, Van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76(9):1035. doi: 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mielke MM. Consideration of sex differences in the measurement and interpretation of Alzheimer disease‐related biofluid‐based biomarkers. J Appl Lab Med. 2020;5(1):158‐169. doi: 10.1373/jalm.2019.030023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vergallo A, Lista S, Lemercier P, et al. Association of plasma YKL‐40 with brain amyloid‐β levels, memory performance, and sex in subjective memory complainers. Neurobiol Aging. 2020;96:22‐32. doi: 10.1016/j.neurobiolaging.2020.07.009 [DOI] [PubMed] [Google Scholar]
- 71. Sánchez‐Benavides G, Milà‐Alomà M, Suarez‐Calvet M, et al. Higher levels of the astrocytic marker CSF YKL40 are associated with better memory performance only in amyloid‐positive individuals with subjective cognitive decline. Alzheimers Dement. 2021;17(S6). doi: 10.1002/alz.053463 [DOI] [Google Scholar]
- 72. Rodriguez‐Vieitez E, Saint‐Aubert L, Carter SF, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer's disease. Brain. 2016;139(3):922‐936. doi: 10.1093/brain/awv404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang H, Wei W, Zhao M, et al. Interaction between Aβ and tau in the pathogenesis of Alzheimer's disease. Int J Biol Sci. 2021;17(9):2181‐2192. doi: 10.7150/ijbs.57078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jha MK, Jo M, Kim JH, Suk K. Microglia‐astrocyte crosstalk: an intimate molecular conversation. Neuroscientist. 2019;25(3):227‐240. doi: 10.1177/1073858418783959 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information