

Abstract
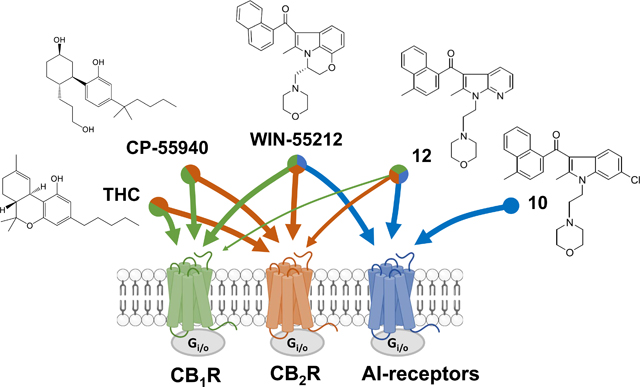
The alkylindole (AI), WIN55212-2, modulates the activity of several proteins, including cannabinoid receptors 1 and 2 (CB1R, CB2R), and at least additional G protein-coupled receptor (GPCR) that remains uncharacterized with respect to its molecular identity and pharmacological profile. Evidence suggests that such AI-sensitive GPCRs are expressed by the human kidney cell line HEK293. We synthesized fourteen novel AI analogues and evaluated their activities at AI-sensitive GPCRs using [35S]GTPγS and [3H]WIN55212-2 binding in HEK293 cell membranes, and performed in silico pharmacophore modeling to identify characteristics that favor binding to AI-sensitive GPCRs versus CB1R/CB2R. Compounds 10 and 12 stimulated [35S]GTPγS binding (EC50s = 3.5 and 1.1 nM, respectively), and this response was pertussis toxin-sensitive, indicating that AI-sensitive GPCRs couple to Gi/o proteins. Five AI analogues reliably distinguished two binding sites that correspond to the high and low affinity state of AI-sensitive GPCRs coupled or not to G proteins. In silico pharmacophore modeling suggest 3 characteristics that favor binding to AI-sensitive GPCRs versus CB1R/CB2R: 1) an s-cis orientation of the two aromatic rings in AI analogues, 2) a narrow dihedral angle between the carbonyl group and the indole ring plane [i.e., O-C(carbonyl)-C3-C2] and 3) the presence of a carbonyl oxygen. The substituted alkylindoles reported here represent novel chemical tools to study AI-sensitive GPCRs.
Keywords: G protein-coupled receptors, Cannabinoids, Alkylindoles
Graphical Abstract
1. Introduction
WIN55212-2 modulates activity of several proteins, including potent activation of CB1R and CB2R, receptors that mediate the bioactivity of Δ9-tetrahydrocannabinol (THC) (Chart 1) [1].
Chart 1.

Chemical structures of the phytocannabinoid Δ9-THC and the bicyclic cannabinoid agonists CP55940 and the alkylindole WIN55212-2.
CB1R couple to Gi/o proteins and are expressed by neurons and many other cell types [2]. CB2R also couple to Gi/o proteins and are principally expressed by hematopoietic cells and various progenitor cells. Numerous AI analogues that selectively activate or antagonize CB1R and CB2R have been developed; however, WIN55212-2 remains the most common of the aminoalkylindole class of agonists used to study the biological functions of cannabinoid receptors [3].
Studies in model systems that lack CB1R and CB2R expression, for example cell lines in culture and CB1R/CB2R-knockout mouse line, indicated that WIN55212-2 modulates the activity of an uncharacterized GPCR [4,5]. WIN55212-2 increases [35S]GTPγS binding in membranes prepared from CB1−/− mouse cerebellum [6]. This GPCR regulates several neuronal functions and behaviors, as shown by WIN55212-2 inhibiting hippocampal neurotransmission in both the hippocampus of CB1R knockout mice and in the nucleus of the solitary tract treated with CB1R and CB2R antagonists [7–9]. WIN55212-2 is anxiogenic in CB1R knockout mice [10]. More recently, our laboratories showed that AI-sensitive receptors inhibit the migration and proliferation of mouse microglia cells in culture without affecting their ability to adopt M1 proinflammatory or M2 anti-inflammatory phenotypes, suggesting that selective activation of AI-sensitive receptors might exhibit anti-inflammatory properties [11]. We also showed that activation of AI-sensitive receptors inhibits the migration of the mouse glioblastoma cell line, DBT, in culture, suggesting that selective activation of AI-sensitive receptors might exhibit anti-tumor properties [11,12]. Of note, the AC-bicyclic analog of Δ9-THC, CP55940, that lacks the dihydropyran ring and exhibits nanomolar potency at CB1R and CB2R, does not compete for this binding site (Chart 1) [13]. Based on this premise, a better understanding of the pharmacological profile of AI-sensitive GPCRs will help unravel its biological role and identity, as well as develop novel therapeutics, including for the treatment of devastating neurological diseases.
In the current study we first tested six commercially available AI analogues for their ability to compete for [3H]WIN55212-2 binding in HEK293 cell membranes, a cell line that lacks CB1R and CB2R and as we show here express AI-sensitive GPCRs. Based on these initial results, we synthesized fourteen new AI analogues to further interrogate the importance of the key moieties responsible for the binding and activation of the AI-sensitive GPCRs compared with CB1 and CB2 receptors. Based on these results, we built an initial pharmacophore model of AI-sensitive GPCRs and identified chemical moieties involved in their selective activation and are different from the chemical moieties involved in CB1R and CB2R activation.
2. Results
2.1. HEK293 cells express AI-sensitive binding sites and lack CB1 and CB2 receptors
We confirmed that [3H]WIN55212-2 binds specifically to a target in HEK293 cell membranes as indicated by saturation of its specific binding, the concentration dependent competition by WIN55212-2 and absence of competition by the enantiomer WIN55212–3 (Fig. 1a–c and Table 1) [14–16]. Significantly, WIN55212-2 produced a concentration-dependent, biphasic competition curve with two high affinity sites (KiHigh = 43 pM and KiLow = 7.2 nM) (Fig. 1c). Two sets of evidence confirmed that HEK293 cells lacked CB1R and CB2R [14–16]. First, using membranes of HEK293 cells heterologous expressing CB1R and CB2R as positive control for [3H]CP55940 specific binding, we found that the cannabinoid agonists, CP55940, the CB1 receptor antagonist SR141617, and the CB2 receptor antagonist SR144528, failed to compete for [3H]CP55940 binding in HEK293 cell membranes (Supplementary Fig. S1). Second, qPCR analysis of HEK293 cells revealed trace amounts of CB1 mRNA (i.e., ~0.1% of the CB1 mRNA levels in U3T3 cells, which endogenously express the CB1 receptor) and absence of CB2 mRNA (Supplementary Table 2). Together, these results confirm and extend studies reporting the absence of functional CB1 and CB2 receptors in HEK293 cells, the likely presence of AI-sensitive GPCRs in this model system, and that AI-sensitive GPCRs do not interact with nanomolar concentrations of Δ9-THC, CP55940, SR141617 and SR145528 [13–15].
Fig. 1. HEK293 cells express AI-sensitive binding sites as determined by specific [3H]WIN55212-2 binding.

HEK293 membranes were incubated with [3H] WIN55212-2 and increasing concentrations of AI analogues, a specific binding determined by filtration assay. a) Diagram depicting [3H]WIN55212-2 binding in HEK293 cell membranes (black arrow) and compounds that compete for this binding and are reported in this figure (gray dotted line arrows). Concentration-dependent competition curves for all other compounds are in Supplementary Fig. 3 b) Saturation of [3H]WIN55212-2 binding in HEK293 membranes (competed by 1 μM WIN55212-2). c) Dose-dependent competition of WIN55212-2 for [3H]WIN55212-2 binding in HEK293 cell membranes. n = 6 P2 fractions prepared from independent cell cultures. Each experiment was performed in triplicate. d-g) Dose-response curves of four AI analogues: d) JWH-450 (R2 = 0.71), e) JWH-042 (R2 = 0.77), 12 (R2 = 0.95) and 13 (R2 = 0.90), that also competed in a biphasic manner for [3H]WIN55212-2 binding in HEK293 cell membranes. Ki values are in Table 1 n = 3–8 fractions prepared from independent cell cultures. Each experiment was performed in triplicate.
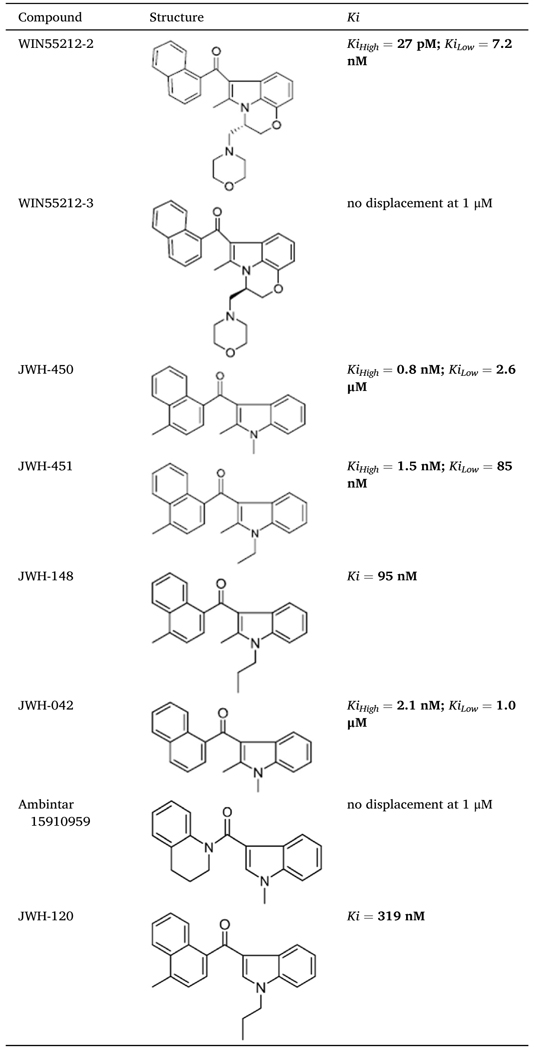
Table 1.
Competition for [3H]WIN 552122-2 binding in HEK293 cell membranes by AI analogues that were either commercially-available or previously synthesized by us. Dose-response curves, 95% confidence intervals and goodness of fit (R2) are provided in Supplementary Figs. 1 and 2.
|
WIN55212-2 modulates the activity of several molecularly-identified GPCRs and ion channels, including GPR55, GPR3, GPR6, GPR12, GPR18, GPR35, GPR40, GPR120, TRPV1, LPA1-LPA5 and 5-HT3 receptors [17]. To determine if any of these targets may account for the [3H]WIN55212-2-binding response measured in HEK293 cell membranes, we tested 23 compounds with known affinities at these identifiable targets. None of the compounds significantly competed for [3H]WIN55212-2 binding, suggesting that the GPCRs and ion channels considered here do not account for the AI binding response measured in HEK293 cell membranes (Supplementary Table 3).
2.2. Initial analysis of the structure-activity-relationship of AI binding sites in HEK293 cells
The six commercially available AI analogues shown in Chart 2 were used as an initial SAR interrogation of three potentially relevant sites along the molecule [18,19]. Specifically, JWH-450, JWH-451 and JWH-148 interrogate the importance of the alkyl chain appended to the indole nitrogen when a 4-methyl is on the naphthoyl moiety. JWH-120 and JWH-148 probe the importance of a C-2 substituent. Comparing JWH-450, JWH-042 and Ambintar 15910959 interrogates the importance of the 4-methyl naphthyl ring, which has been shown to be critical for binding to both CB1 and CB2 receptors [20].
Chart 2.

Six AI analogues interrogate the carbon chain on the indole nitrogen, the C2 substituent on the indole ring and a methyl group on the naphthalene ring.
The concentration dependent competition curves and binding affinities obtained with these AI analogues tested in HEK293 membranes are presented in Table 1, Fig. 1d and e, and Supplementary Fig. S2, and together suggest several chemical characteristics that guided the development of the new AI analogues described below. Specifically, the results obtained with JWH-450, JWH-451 and JWH-148 (which all have a C-4 methyl group on the naphthyl ring) suggested that changes in the N-1 alkyl chain may affect binding affinity. Comparing the affinities of JWH-148 and JWH-120 (Ki = 95 and 319 nM, respectively) suggests that the C-2 substituent on the indole ring also influences binding to AI sites. The presence of a C-4 methyl on the naphthyl ring of JWH-450 fails to alter the affinity for AI sites (compared with the unsubstituted naphthyl in JWH-042). Finally, the lack of binding competition by Ambintar 15910959 suggests that substantial structural modifications of the naphthalene moiety may preclude binding to AI sites.
2.3. Rationale of synthesis of novel AI analogues and their binding affinities at AI-sensitive sites
Based on these results, we developed fourteen novel AI analogues that contain structural modifications in three regions to further interrogate the SAR of compounds interacting with the AI-sensitive sites. Specifically, we considered the prototypical analog, 1,2-dialkyl-3-aroylindole (JWH-450, Chart 3), which represents an elementary chemical scaffold containing an indole ring linked to a naphthalene that can be readily deconstructed into discrete moieties amenable to synthetic manipulation. Note that we kept the methyl-substituted naphthalene constant.
Chart 3.

Three moieties, the phenyl portion of the indole (Aroyl, Ar) and the C-2 and N-1 substituents, were selected for modifications.
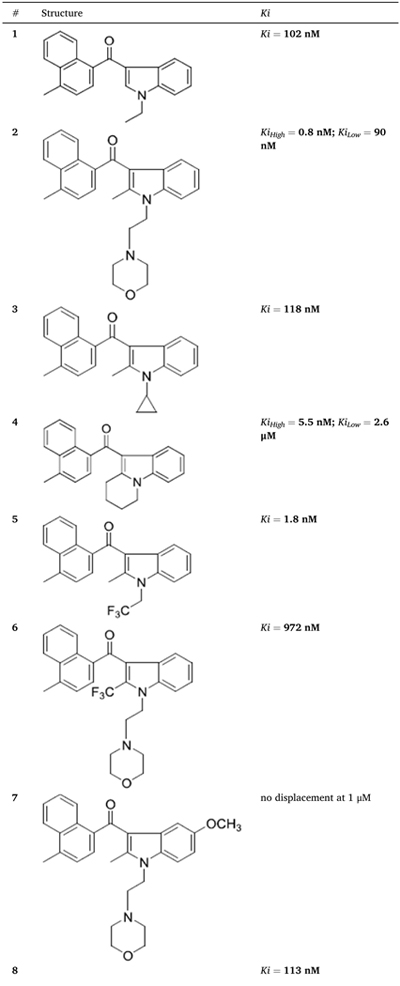
Thus, based on the data reported in Table 1, we sought to interrogate three regions. First, the high degree of sensitivity to the chain length linked to the N-1 and C-2 positions measure in the [3H]WIN55212-2 binding response with JWH-120, JWH-148, JWH-450 and JWH-451 might reflect either intermolecular interactions (ligand-binding site) or intramolecular changes (internal conformational change of the molecule) or some degree of both interactions. To explore how structural modifications of the N-1 and C-2 positions affect AI compound activity, we synthesized analogues 1–6 (Chart 4).
Chart 4.

AI analogues that interrogate the N-1 and C-2 positions.
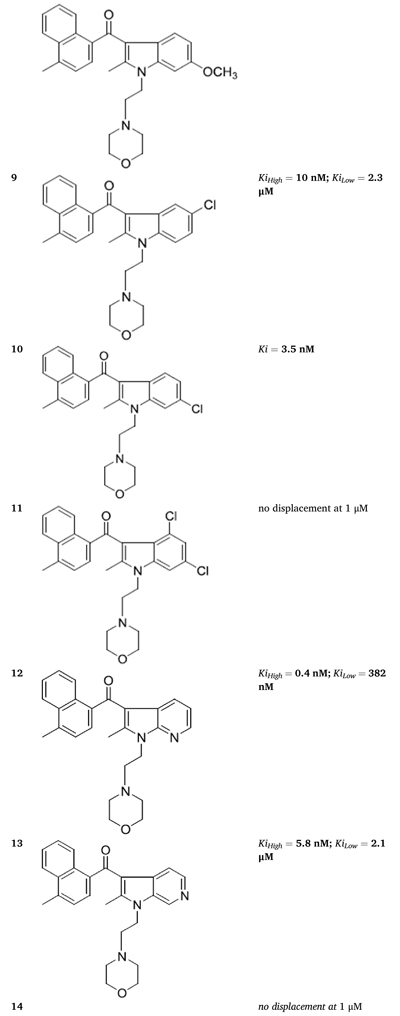
To examine the importance of the aroyl (Ar) moiety, we synthesized analogues 7–13, which primarily alter the electron density of the aromatic ring while also introducing other potential non-covalent contacts with a binding site (Chart 5).
Chart 5.

AI analogues that explore the aroyl (Ar) moiety.
Finally, to validate the importance of the naphthalene moiety, we synthesized compound 14 (Chart 6).
Chart 6.

AI analog that explores the importance of the naphthalene moiety.
Scheme 1 depicts the synthesis routes of the acylindoles 1, 2, 7, 8, 9, 10, 11 and 12, which involve N-alkylation and C-3 Friedel-Crafts acylation [19,21–29].
Scheme 1.

a) KOH, ethyl iodide, and DMSO incubated at room temperature (rt) overnight. b) 4-methyl-1-naphthoyl chloride, EtAlCl2, and CH2Cl2 at rt overnight. c) 2-methanesulfonyl ethylmorpholine, NaH, and DMF at rt overnight. d) 4-methyl-1-naphthoyl chloride, AlCl3, and CH2Cl2 0 °C to rt overnight. e) 2-chloroethylmorpholine HCl, KOH, and DMSO at rt overnight.
For 3, SnCl4 acylation was the preferred chemistry, and the optimal step order was cyclopropanationfollowed by acylation (Scheme 2) [30].
Scheme 2.

a) cyclopropylboronic acid, copper(II) acetate, NaHMDS, DMAP, toluene, and air 95 °C 5 d. b) 4-methyl-1-naphthoyl chloride, SnCl4, nitromethane, and CH2Cl2 0 °C to rt overnight.
The tricyclic compound 4 was prepared as shown in Scheme 3 with assembly of the tetrahydropyridoindole followed by acylation using Scheme 1 reagents and a prolonged reaction time [26,31–33].
Scheme 3.

a) i. n-Bu-Li, CO2(g), and THF −70 °C 30 min ii. t-Bu-Li and THF −70 °C 1 h iii. p-toluenesulfonyl fluoride and THF at −70 °C for 2 h. b) 1,4-dibromobutane, KOH, and DMF at rt for 1.75 h. c) Bu3SnH, AIBN, and toluene reflux for 2 h. d) 4-methyl-1-naphthoyl chloride, EtAlCl2, and CH2Cl2 −70 °C to rt for 5 d.
For analogues 5, 6 and 13, where the synthesis methods outlined in Schemes 1 and 2 were unsuccessful, we mined the literature for alternatives and found that the synthesis of the 2-trifluoromethyl analog 6 required re-ordering of the alkylation/acylation sequence as well as SnCl4 in nitromethane to promote addition of the 4-methylnaphthoyl moiety (Scheme 4) [21,22,24,25,28,29].
Scheme 4.

a) 4-methyl-1-naphthoyl chloride, SnCl4, CH2Cl2, and nitromethane 0 °C to rt overnight. b) 2-chloroethylmorpholine HCl, KOH, and DMSO 80 °C 3 d. c) 4-methyl-1-naphthoyl chloride, EtAlCl2, and CH2Cl2 0 °C to rt 5 d. d) 2,2,2-trifluoroethyltrifluoromethanesulfonate, K2CO3, 18-crown-6, and DMF 80 °C 20 h. e) 4-methyl-1-naphthoyl chloride, SnCl4, CH2Cl2, and nitromethane 0 °C to rt 3 d. f) 2-morpholinoethyl trifluoromethanesulfonate, K2CO3, 18-crown-6, and DMF 80 °C for 6 d.
We used nitromethane as a solvent to maintain the solubility of the indole-Lewis acid complex and facilitate nucleophilic attack of the C-3 carbanion [34]. Accordingly, prolonged reaction times and elevated temperatures were necessary to alkylate the indole nitrogen to obtain modest yields of 6. The same general strategy was used for 5, but both a stronger leaving group in the alkylation and overall more forceful conditions were required in this sequence. To synthesize 28, ethylaluminum dichloride was the preferred Lewis acid, and the reaction was allowed to proceed over the course of several days. As with the alkylation of 27, alkylation of 28 required displacement of a triflate in a polar aprotic solvent at elevated temperatures [35]. In these cases, the reverse sequence of alkylation/acylation produced N-alkylation under milder conditions but was refractory to all acylation methods. Finally, the synthesis of 14 was based on previously reported work (Scheme 5) [24].
Scheme 5.

a) p-anisoyl chloride, EtMgBr, ZnCl2, Et2O and CH2Cl2. b) NaH, EtBr, and DMF.
We tested the fourteen novel AI analogues for their ability to compete for [3H]WIN55212-2 binding in HEK293 cell membranes and found significant differences in their dose-dependent competition response (Table 2, Fig. 1f and g, and Supplementary Fig. 3). Specifically, five AI analogues (2, 4, 9, 12 and 13) competed for [3H]-WIN55212-2 binding in a biphasic manner, six AI analogues (1, 3, 5, 6, 8 and 10) competed for binding in a monophasic manner, and three AI analogues (7, 11 and 14) were inactive.
Table 2.
Novel AI compounds compete for [3H]-WIN55212-2 binding in HEK293 membranes. Competition for [3H]WIN 552122-2 binding in HEK293 cell membranes by novel AI analogues. Dose-response curves, 95% confidence intervals and goodness of fit (R2) are provided in Supplementary Fig. 3.
|
|
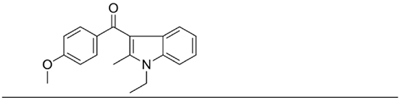
|
3. AI-sensitive receptors couple to Gi proteins
Biphasic displacement of radioligand binding can result from two fundamentally different mechanisms. First, biphasic displacement in binding is a hallmark of GPCRs, as these receptors typically exhibit high affinity binding for an agonist when coupled to a heterotrimeric G protein, and upon activation and dissociation of the G proteins, revert to a low affinity state to facilitate dissociation of the agonist and continuation of the G protein cycle [36]. Alternatively, biphasic displacement might indicate the existence of two protein entities that exhibit different affinities for AI analogues. To differentiate between these two possibilities, we tested whether addition of the non-hydrolyzable GTP analog, GTPγS, to HEK293 membranes affects the amount of [3H]WIN55212-2 binding at the AI sites (Fig. 2a). Specifically, GTPγS incubation increases the dissociation constant of a GPCR by reducing the number of active sites and results in a reduction in the amount of radioligand bound to these receptors at equilibrium. Fig. 2b shows that GTPγS incubation reduces the amount of [3H]WIN55212-2 specific binding in HEK293 cell membranes measured in the presence of JWH-451, suggesting that the distinct AI binding sites likely correspond to different GPCR conformational states. Fig. 2c shows that JWH-451 increases in [35S]GTPγS binding in HEK293 cell membranes, indicating that AI-sensitive receptors couple to Gi/o proteins. This response did not involve CB1R and CB2R as JWH-451 increases in [35S]GTPγS binding in HEK293 cell membranes was insensitive to CB1R and CB2R antagonists, SR141716 and SR144528 (Fig. 2d). Compounds 8, 10 and 12 increases in [35S] GTPγS binding in HEK293 cell membranes (Fig. 2e). The EC50 responses for JWH-451, 10 and 12 were 5, 3.5 and 1.1 nM, respectively, which are within the concentration range of their respective affinities for AIHigh binding sites (KiHigh = 1.5, 3.5 and 0.4 nM reported in Table 2). Significantly, the EC50 for compound 8 was 229 nM, which is also within the concentration range of its monophasic affinity for AI competition (Ki = 113 nM), indicating that the monophasic binding response measured in the presence of this compound likely represents AIHigh sites (AI-sensitive GPCRs in their active state). Together, these results suggest that HEK293 cells express functional AI-sensitive GPCRs that couple to Gi/o proteins and that the biphasic nature of compounds competing for [3H] WIN55212-2 binding in HEK293 membranes reflects AI-sensitive GPCRs that are in an active state (coupled to G proteins) and inactive state (uncoupled to G proteins), respectively.
Fig. 2. AI-sensitive receptors couple to Gi/o proteins.

HEK293 membranes were incubated with [3H]WIN55212-2 and JWH-451, with or without GTPγS, a specific binding determined by filtration assay. a) Diagram depicting [35S]GTPγS binding in HEK293 cell membranes (gray arrow), and alkylindole analogues that increase this binding and are reported in this figure (black arrows). Pertussis toxin (PTX) blocks Gi/o protein function. b) [3H]WIN55212-2 specific binding measured in the presence of JWH-451 is reduced by GTPγS (20 μM), suggesting the coupling to Gi/o proteins. c) JWH-451 increases GTPγS binding in HEK293 cell membranes, and this response is significantly reduced by PTX (100 ng/ml pretreatment of HEK293 cell in culture for 20 h). d) JWH-451 increases [35S]GTPγS binding, and this response is not affected by CB1R antagonist SR141617 (1 μM) and CB2R antagonist SR144528 (1 μM). e) Compounds 8, 10 and 12 increase [35S]GTPγS binding. f) The Z-isomer AI analogues, 31 and 33, stimulate [35S]GTPγS binding in HEK293 cell membranes. Results were obtained using HEK293 cell membranes and are the mean ± SEM of n = 3–4 experiments, each performed in triplicate.
4. Structural features that differentiate binding to AI-sensitive receptors and to CB1R and CB2R
Bell and colleagues were the first to demonstrate the preference of C-2 methyl aminoalkylindoles for the s-cis conformation and to propose that this preference originates from the additional bulk produced by the methyl group at C-2 being better accommodated when the aryl ring is rotated away from C-2 and instead toward C-4 [23]. Evidence obtained with rigid AI analogues that differentiate s-cis and s-trans conformations showed that the s-trans E isomers exhibit higher affinity for the CB1 and CB2 receptors (nanomolar affinity) compared with the s-cis Z-isomers (micromolar affinity) [37]. Fig. 3a illustrates the two minimum energy conformations possible for WIN55212-2 and shows the naphthyl ring oriented over C-4 of the indole ring in the s-cis conformation, whereas the naphthyl ring is oriented over the C-2 methyl group in the s-trans conformation. Consistent with this observation, our conformational analysis indicated that the s-cis conformation for WIN55212-2 is 1.56 kcal/mol lower in energy than the s-trans conformation. Fig. 3b shows the minimum energy conformations for JWH-148 and JWH-120 that only differ in JWH-148’s methyl group at C-2, whereas JWH-120 has a hydrogen atom in that position. Accordingly, the minimum energy conformation for JWH-148 is s-cis (which is 1.01 kcal/mol lower in energy compared with the s-trans), but the minimum energy conformation for JWH-120 is s-trans (which is 1.81 kcal/mol lower in energy compared with s-cis). In Table 1 we report that JWH-148 competes for [3H]WIN55212-2 binding in HEK293 membranes with trending higher affinity compared with JWH-120 (Ki = 95 nM and 319 nM, respectively), suggesting that AI binding sites exhibit a preference for ligands in the s-cis orientation. A preference for s-cis ligands would greatly contrast the preference for ligands in the s-trans orientation exhibited by CB1 and CB2 receptors.
Fig. 3. Illustrations of minimum energy conformers.

Conformational Search were constructed using the Spartan’08 molecular modeling program using the semi-empirical method, AM1, and resultant ab-initio energies used to identify the lowest energy conformation of each compound. a) s-cis and s-trans minimum energy conformers of WIN55,212–2. The s-cis conformation is 1.56 kcal/mol lower in energy compared to the s-trans conformation. b) Global minimum energy conformation for compound JWH-148 (s-cis) and JWH-120 (s-trans). c) Z versus E geometric isomer indenes where the C-2 substituent is a hydrogen. d) Z versus E geometric isomer indenes where the C-2 substituent is a methyl group. e) s-cis and s-trans minimum energy conformers of compound 2. f) s-cis and s-trans minimum energy conformers of compound 4. g-h) 31 and 33 compete and 32 weakly competes for [3H]WIN55212-2 binding in HEK293 membranes, whereas 34 does not compete. Results are the mean ± SEM of 3–6 experiments, each performed in triplicate.
To directly test the importance of the s-cis and s-trans conformation at the AI binding sites, we tested whether (Z)- and (E)-naphthylidene indenes competed for [3H]WIN55212-2 binding in HEK293 membranes. Chart 7 shows the four rigid analogues (31–34) that we selected.
Chart 7.

AI geometric isomers that explore the importance of the s-cis and s-trans conformations.
These four naphthylidene-substituted aminoalkylindenes lack the carbonyl oxygen of our 3-acyl alkylindole series and can be categorized as two pairs of (Z)- and (E)-naphthylidene indenes (C-2 H, 31 and 32; and C-2 CH3, 33 and 34) [37]. The two Z geometric isomers are intended to mimic the s-cis form of the 3-acyl alkylindole, and the two E geometric isomers are intended to mimic the s-trans form (Fig. 3c and d) [37]. When testing these rigid AI analogues against [3H]WIN55212-2 binding in HEK293 cell membranes, we found that Z-isomers exhibited higher affinity (Ki = 437 and 466 nM for 31 and 33) compared with the E-isomers (Ki = 3.4 μM and no significant competition for 32 and 34) (Fig. 3g and h). Furthermore, both 31 and 33 stimulated [35S]GTPγS binding in HEK293 cell membranes (Fig. 2f). Together, these results show that the Z-isomer (s-cis) is likely the form recognized by this AI-sensitive GPCR.
To identify additional chemical features that might differentiate activation of AI-sensitive receptors versus CB1 and CB2 receptors, we tested whether JWH-451, 2, 4, 5 and 12 induced the internalization of HA-tagged human CB1R or human CB2R heterologously-expressed by HEK293 cells in culture as an index of CB1 and CB2 target engagement [38,39]. Fig. 4a shows that 2, 4 and 12 retained significant activity at hCB1R, whereas JWH-451 and 5 were inactive and thus exhibit relative selectivity for AI-sensitive receptors. We also found that these five AI analogues still exhibit significant activity at CB2R (Fig. 4b). Together, these results indicate that there is a degree of relaxed specificity in ligand binding among these three GPCRs, and that further medicinal chemistry efforts will be necessary to improve the selectivity of AI analogues acting at AI-sensitive GPCRs.
Fig. 4. AI analogues activity at CB1R and CB2R.

CB1R and CB2R internalization was measured in HEK293 cells stably expressing either HA-tagged CB1R or HA-tagged CB2R as previously shown [40]. a) Compounds 2, 4 and 12 stimulate significant CB1R internalization and these responses are antagonized by SR141617 (1 μM). JWH-451 and 5 did not induce a significant response. b) JWH-451 and compounds 2, 4, 5, and 12 stimulated significant CB2R internalization and these responses are antagonized by AM630 (1 μM). Compound 12 did not induce a significant response. All AI compounds were tested at 3 μM. Positive control: CP55940 (1 μM). Data are shown as the mean ± SEM of n = 3–5 independent experiments. **P < 0.01 and *P < 0.05 compared to corresponding % vehicle control (ANOVA, one way, Bonferroni’s multiple comparison post-test).
5. Modeling the pharmacophore of AI-sensitive receptors
Eight of the compounds evaluated (WIN55212-2, JWH-450, JWH-451, 2, 4, 9, 12 and 13) exhibited biphasic binding curves in competition with [3H]-WIN55212-2 in HEK293 cell membranes. The SAR analyses presented below assumed these binding sites were two affinity states of a single receptor representing two conformational states that depend on the presence or absence of a G protein and therefore used only the Ki high values for the biphasic compounds. Electrostatic profiling of 2 (Ki = 0.8 nM) and 33 (Ki = 466 nM) provided key information (Fig. 5a). Specifically, while both compounds assume similar s-cis conformations, the highly negative electrostatic potential evoked by the carbonyl of 2 is absent in the Z-indene 33. The resultant decrease in affinity for the AI-sensitive GPCR that the carbonyl oxygen may be important for hydrogen bonding at this target. In contrast, the indenes (E) retained high affinity at CB1R and CB2R despite the loss of the carbonyl oxygen, suggesting that hydrogen bonding of the carbonyl oxygen is not important for aminoalkylindole affinity at CB1R and CB2R. This result clearly distinguishes the AI-sensitive GPCR from CB1 and CB2 receptors [37].
Fig. 5. Global minimum energy conformer molecular electrostatic potential, MEP, density surface maps of AI analogues.

The electrostatic potential density surface was calculated for the global minimum energy conformer of each compound using Spartan’08, the electrostatic potential energy was calculated using the Hartree-Fock method, and the surface was color-coded according to the potential with electron rich regions colored red and electron poor regions colored blue. a) Compound 2, binding with nanomolar affinity to both AI-High and AI-Low, has strong negative potential from the carbonyl oxygen. Compound 33 has mild positive potential at the same position. b) The conformer of compound 13 illustrated is a minimum energy conformer that is 0.05 kcal/mol above its global minimum. The electrostatic scale is provided in units of kJ/mol. Comparison of compounds 12, 10, 13 and 8 with varied MEP at the 6 and 7 positions.
How do aromatic substituents affect the affinity of AI analogues for the AI-sensitive GPCR? To address this question, we analyzed how substituents at C-5 (9, Cl), C-6 of the indole ring (10, Cl), replacements of an indole carbon at C-6 with nitrogen (13) or C-7 with nitrogen (12) might affect their binding affinity to AI sites. The global minimum energy conformer for these compounds was the s-cis conformation, with the s-cis lower by 0.70 kcal/mol for 12, 0.81 kcal/mol for 10, 0.89 kcal/mol for 9, 0.93 kcal/mol for 8 and 1.27 kcal/mol for 13. Fig. 5b illustrates the electrostatic potential density surface maps of the global minimum energy conformer of 12, 10, 13 and 8. The conformer of 12 (KiHigh = 0.4 nM) illustrates a minimum energy conformer that is 0.05 kcal/mol above its global minimum and shows the highly electronegative region at the pyridine nitrogen (red, N-substitution, − 162.8 kJ). Heterocyclic N-substitution at C-7 of the indole system (12) also results in ~1000-fold enhanced activity at AIHigh versus AILow. The chlorine substitution at C-5 in 9 and C-6 in 10 also result in high affinity for AIHigh binding sites (KiHigh = 10 nM and 3.5 nM), and remarkably low affinity for AILow binding sites (KiLow = 2.3 μM and undetectable). The electrostatic potential of the chlorine of 10 in Fig. 5b is negative (yellow, −87.7 kJ), and therefore, the chlorine could serve as a hydrogen bond acceptor. Heterocyclic replacement of C-6 with nitrogen (13) results in an AIHigh affinity of 5.8 nM, and Fig. 5b shows that this nitrogen generates a highly electronegative region (red, −211.4 kJ), which would also be favorable for hydrogen bond acceptance. However, variability inherent to radioligand binding assays prohibits reaching a statistically significant difference between the affinities of compounds 2, 9, 10, 12 and 13. Taken together, the results for 9, 10, 12 and 13 suggest that the existence of a hydrogen bonding residue on the indole ring at the C-5, C-6 or C-7 position does not significantly impact receptor affinity. However, such modifications could be exploited to improve the ADME properties of the lead compound 2. Analysis of the 5 criteria Lipinski’s Rule of these compounds indicated that 9 and 10 exhibit promising ALogP and LogBB values compounds (Supplementary Table S4). Specifically, while the chloro-substituted compounds (9 and 10) yield no overall improvement, the incorporation of nitrogen at C-6 (13) or C-7 (12) leads to LogPs that decrease from 4.77 for 2 to 4.17 (12) and 3.73 (13). Interestingly, methoxy substitution at C-6 in 8 results in a clearly lowered affinity (ki = 113 nM). This loss in affinity is likely due to the increased size of the C-6 substituent (note a significant bulge in 8 in Fig. 5b. The steric restrictions of the binding pocket are again apparent when comparing 7 and 9. Here, the chlorosubstitution at C-5 in 9 is tolerated, but enlargement to a methoxy substituent at C-5 in 7 results in loss of binding.
Compound 6 has poor affinity for the AI-sensitive GPCR (Ki = 972 nM). This lower affinity is likely due to poor adoption of an s-cis conformational state. Fig. 6a presents a comparison of the s-cis conformation adopted by WIN55212-2, 2 and 6 and Fig. 6b illustrates the deviation in the position of the carbonyl group induced by the CF3 group in 6. The carbonyl group in 6 is rotated farther out of the plane of the indole ring [O-C(carbonyl)-C3-C2 = 52°] compared with in 2 [O-C (carbonyl)-C3-C2 = 25 °C] due to the steric influence of the CF3 group (Supplementary Table S5). This effect is very similar to the effect of the 4,6-dichloro analog (11), which has no measurable binding at 1 μM. In 11, the carbonyl group is also out of plane with the indole ring [O-C (carbonyl)-C3-C2 = 52°]. In this analog, it is the chlorine at C-4 that forces this rotation. Together, these results provide a foundation to perform additional medicinal chemistry studies aimed at further exploring the SAR of AI analogues acting on AI-sensitive GPCR(s).
Fig. 6. Steric influence of large C2 position substituents.

Conformations were read into Maestro 9.3, conformers superimposed, single molecular surface generated, a Boolean difference operation, or logical XOR, used to visualize volume occupied and molecular surface maps displayed. a) Comparing the steric influence of the C2 moiety modulating the overall width of each ligand. Vertical bars show the relative position of the naphthyl to the morpholino ring in compounds 2 and 6. WIN55212-2 and compound 2 have a narrow profile compared with compound 6. b) Compound 6 has a wider profile than 2, rotating the naphthyl farther out of the plane of the indole ring, due to the trifluoromethyl steric influence on the carbonyl oxygen.
6. Discussion
We developed novel AI analogues and performed the first SAR analysis of AI-sensitive GPCRs expressed by HEK293 cells. We discovered that these receptors couples to Gi/o proteins and that key moieties are required to activate them and are distinct from moieties required to activate CB1R and CB2R, confirming and extending studies that suggested that AI-sensitive GPCRs that are pharmacologically distinct entities.
There are several fundamental differences between AI analogues that act at AI-sensitive GPCRs versus CB1R and CB2R. The N-morpholinoethyl indoles exhibit affinities ranging from high to low nanomolar in our competition assays, which contrasts with AI analogues binding to CB1R in cerebellum membranes where this N-1 substituent conferred a conspicuous and consistent benefit [21]. AI-sensitive GPCRs recognize the Z-isomer (s-cis), which is consistent with results for JWH-148 versus JWH-120 (Ki = 95 nM and 319 nM, respectively, Supplementary Fig. 2). Because the E-indenes also retained high affinity binding at CB1R and CB2R, these results suggest that the carbonyl oxygen of WIN55212-2 (which is removed in the indenes) is not important for binding to either CB1R or CB2R [37]. The Z-indenes did not retain the higher affinity of JWH-148 for the AIHigh site, suggesting that the carbonyl oxygen in JWH-148 (which is absent in the indenes) may be important for the high affinity binding to this site. Finally, two compounds, JWH-451 and 5, exhibited an initial increase in selectivity due to their reduced interaction with CB1R, indicating that further medical chemistry efforts are needed to increase the selectivity of AI compounds at the AI-sensitive GPCR. Note that based on previous SAR analyses of AI analogues at CB1 receptors, the combined modification of C2 and N moieties likely represents promising strategies to reduce their affinities at CB1R and CB2R [20].
Significant pharmacological and functional differences are likely to exist in AI-sensitive expressed by different cell types and species. Our studies suggest that AI-sensitive receptors couple to Gs in mouse microglia in culture as opposed to Gi/o in the human HEK293 kidney cell lines [11]. While we found that JWH-451, 2, 4, 5 and 12 activate human CB2R as measured by receptor internalization, these compounds do not activate mouse CB2R as measured by receptor internalization (i.e., compounds ST-11, ST-48, ST-23, ST-25 and ST-29 in Ref. [12]). Many fundamental questions about AI-sensitive GPCRs remain unanswered, including the molecular identity, the endogenous ligand(s) that modulate it, and its expression profile and biological functions. The results obtained in our study provide new chemical tools to start answering these questions. In conclusion, the substituted AI analogues developed in this study should help study AI-sensitive GPCRs and guide the development of next generation AI analogues.
Supplementary Material
Acknowledgements
The authors would like to thank Research Triangle Institute International (NC) for providing compounds 31–34. This work was supported by NIH: DA03590 (JWH), DA011322 (KM) DA021696 (KM) DA042157 (ACH and KE), DA021358 (PHR) and DA0144861 (NS). Nephi Stella is employed by Stella Consulting LLC. The terms of this arrangement have been reviewed and approved by the University of Washington in accordance with its policies governing outside work and financial conflicts of interest in research.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115123.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data will be made available on request.
References
- [1].Ward SJ, et al. , Aminoalkylindoles (AAIs): a new route to the cannabinoid receptor? NIDA Res. Monogr 105 (1990) 425–426. [PubMed] [Google Scholar]
- [2].Howlett AC, et al. , International union of pharmacology. XXVII. Classification of cannabinoid receptors, Pharmacol. Rev 54 (2002) 161–202. [DOI] [PubMed] [Google Scholar]
- [3].Wiley JL, Marusich JA, Huffman JW, Moving Around the Molecule: Relationship between Chemical Structure and in Vivo Activity of Synthetic Cannabinoids, Life Sci, 2013. [DOI] [PMC free article] [PubMed]
- [4].Pertwee RG, et al. , International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2), Pharmacol. Rev 62 (4) (2010) 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abood ME, Sorensen SA, Stella N, endoCANNABINOIDS; Actions at Non-CB1/CB2 Cannabinoid Receptors. The Receptors, 24, Elsevier, 2013. [Google Scholar]
- [6].Breivogel CS, et al. , Evidence for a new G protein-coupled cannabinoid receptor in mouse brain, Mol. Pharmacol 60 (2001) 155–163. [PubMed] [Google Scholar]
- [7].Hájos N, Ledent C, Freund TF, Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus, Neuroscience 106 (2001) 1–4. [DOI] [PubMed] [Google Scholar]
- [8].Hájos N, Freund TF, Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers, Neuropharmacology 43 (2002) 503–510. [DOI] [PubMed] [Google Scholar]
- [9].Accorsi-Mendonça D, et al. , Inhibition of spontaneous neurotransmission in the nucleus of solitary tract of the rat by the cannabinoid agonist WIN 55212–2 is not via CB1 or CB2 receptors, Brain Res. 1200 (2008) 1–9. [DOI] [PubMed] [Google Scholar]
- [10].Haller J, et al. , CB1 cannabinoid receptors mediate anxiolytic effects: convergent genetic and pharmacological evidence with CB1-specific agents, Behav. Pharmacol 15 (4) (2004) 299–304. [DOI] [PubMed] [Google Scholar]
- [11].Fung S, et al. , Alkylindole-sensitive receptors modulate microglial cell migration and proliferation, Glia 63 (10) (2015) 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fung S, et al. , Novel indole-based compounds that differentiate alkylindole-sensitive receptors from cannabinoid receptors and microtubules: characterization of their activity on glioma cell migration, Pharmacol. Res 115 (2016) 233–241. [DOI] [PubMed] [Google Scholar]
- [13].Stark S, Pacheco MA, Childers SR, Binding of aminoalkylindoles to noncannabinoid binding sites in NG108–15 cells, Cell. Mol. Neurobiol 17 (5) (1997) 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mukherjee S, et al. , Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors, Eur. J. Pharmacol 505 (1–3) (2004) 1–9. [DOI] [PubMed] [Google Scholar]
- [15].Griffin G, Tao Q, Abood ME, Cloning and pharmacological characterization of the rat CB2 cannabinoid receptor, J. Pharmacol. Exp. Therapeut 292 (2000) 886–894. [PubMed] [Google Scholar]
- [16].Atwood BK, et al. , Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis, BMC Genom. 12 (2011) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kreitzer FR, Stella N, The therapeutic potential of novel cannabinoid receptors, Pharmacol. Ther 122 (2) (2009) 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Poso A, Huffman JW, Targeting the cannabinoid CB2 receptor: modelling and structural determinants of CB2 selective ligands, Br. J. Pharmacol 153 (2) (2008) 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huffman JW, et al. , Structure-activity relationships for 1-alkyl-3-(1-naphthoyl) indoles at the cannabinoid CB(1) and CB(2) receptors: steric and electronic effects of naphthoyl substituents. New highly selective CB(2) receptor agonists, Bioorg. Med. Chem 13 (1) (2005) 89–112. [DOI] [PubMed] [Google Scholar]
- [20].Huffman JW, Padgett LW, Recent developments in the medicinal chemistry of cannabimimetic indoles, pyrroles and indenes, Curr. Med. Chem 12 (12) (2005) 1395–1411. [DOI] [PubMed] [Google Scholar]
- [21].Eissenstat MA, et al. , Aminoalkylindoles: structure-activity relationships of novel cannabinoid mimetics, J. Med. Chem 38 (16) (1995) 3094–3105. [DOI] [PubMed] [Google Scholar]
- [22].Maligres PE, et al. , Practical, highly convergent, asymmetric synthesis of a selective PPAr gamma modulator, Org. Process Res. Dev 13 (2009) 525–534. [Google Scholar]
- [23].Bell MR, et al. , Antinociceptive (aminoalkyl)indoles, J. Med. Chem 34 (3) (1991) 1099–1110. [DOI] [PubMed] [Google Scholar]
- [24].Frost JM, et al. , Indol-3-ylcycloalkyl ketones: effects of N1 substituted indole side chain variations on CB(2) cannabinoid receptor activity, J. Med. Chem 53 (1) (2010) 295–315. [DOI] [PubMed] [Google Scholar]
- [25].Guchhait SK, Kashyap M, Kamble H, ZrCl4-mediated regio- and chemoselective Friedel-Crafts acylation of indole, J. Org. Chem 76 (11) (2011) 4753–4758. [DOI] [PubMed] [Google Scholar]
- [26].Katritzky AR, Akutagawa K, Carbon dioxide: a reagent for the protection of nucleophilic centers and the simultaneous activation of alternative locations to electrophilic attack Part I. A new synthetic method for the 2-substitution of 1-unsubstituted indoles, Tetrahedron Lett. 26 (48) (1985) 5935–5938. [Google Scholar]
- [27].Katritzky AR, et al. , Regiospecific C-acylation of pyrroles and indoles using N-acylbenzotriazoles, J. Org. Chem 68 (14) (2003) 5720–5723. [DOI] [PubMed] [Google Scholar]
- [28].Lézé M-P, et al. , 2- and 3-[(Arul)(azolyl)methyl]indoles as potential non-steroidal aromatase inhibitors, J. Enzym. Inhib. Med. Chem 19 (6) (2004) 549–557. [DOI] [PubMed] [Google Scholar]
- [29].Wynne JH, et al. , 3-Acylindoles via a one-pot, regioselective friedel-crafts reaction, Synthesis 14 (2004) 2227–2282. [Google Scholar]
- [30].Tsuritani T, et al. , N-cyclopropylation of indoles and cyclic amides with copper(II) reagent, Org. Lett 10 (8) (2008) 1653–1655. [DOI] [PubMed] [Google Scholar]
- [31].Caddick S, et al. , Intramolecular radical substitution reactions: a novel approach tofused [ 1,24 indoles, Journal of the Chemical Society of London, Perkin transactions 1 (1996) 675. [Google Scholar]
- [32].Dehaen W, Hassner A, Annulation of heterocycles via IntramolecularNitrile oxide-heterocycle cycloaddition reaction, J. Org. Chem 56 (2) (1991) 896–900. [Google Scholar]
- [33].Tsotinis A, et al. , Synthesis of new tricyclic melatoninergic ligands, Farmaco 56 (9) (2001) 725–729. [DOI] [PubMed] [Google Scholar]
- [34].Ottoni O, et al. , Acylation of indole under Friedel-Crafts conditions-an improved method to obtain 3-acylindoles regioselectively, Org. Lett 3 (7) (2001) 1005–1007. [PubMed] [Google Scholar]
- [35].Menichincheri M, et al. , Cdc7 kinase inhibitors: 5-heteroaryl-3-carboxamido-2-aryl pyrroles as potential antitumor agents. 1. Lead finding, J. Med. Chem 53 (20) (2010) 7296–7315. [DOI] [PubMed] [Google Scholar]
- [36].Mukhopadhyay S, Howlett AC, Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions, Mol. Pharmacol 67 (6) (2005) 2016–2024. [DOI] [PubMed] [Google Scholar]
- [37].Reggio PH, et al. , The bioactive conformation of aminoalkylindoles at the cannabinoid CB1 and CB2 receptors: insights gained from (E)- and (Z)-naphthylidene indenes, J. Med. Chem 41 (26) (1998) 5177–5187. [DOI] [PubMed] [Google Scholar]
- [38].Daigle TL, Kwok ML, Mackie K, Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism, J. Neurochem 106 (1) (2008) 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Atwood BK, et al. , Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands, Mol. Pharmacol 81 (2) (2012) 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wager-Miller J, Mackie K, Quantitation of plasma membrane (G protein-coupled) receptor trafficking in cultured cells, Methods Mol. Biol 1412 (2016) 255–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.