

Abstract
Background
T cells in people with human immunodeficiency virus (HIV) demonstrate an exhausted phenotype, and HIV-specific CD4+ T cells expressing programmed cell death 1 (PD-1) are enriched for latent HIV, making antibody to PD-1 a potential strategy to target the latent reservoir.
Methods
This was a phase 1/2, randomized (4:1), double-blind, placebo-controlled study in adults with suppressed HIV on antiretroviral therapy with CD4+ counts ≥350 cells/μL who received 2 infusions of cemiplimab versus placebo. The primary outcome was safety, defined as any grade 3 or higher adverse event (AE) or any immune-related AE (irAE). Changes in HIV-1–specific polyfunctional CD4+ and CD8+ T-cell responses were evaluated.
Results
Five men were enrolled (median CD4+ count, 911 cells/μL; median age, 51 years); 2 received 1 dose of cemiplimab, 2 received 2 doses, and 1 received placebo. One participant had a probable irAE (thyroiditis, grade 2); another had a possible irAE (hepatitis, grade 3), both after a single low-dose (0.3 mg/kg) infusion. The Safety Monitoring Committee recommended no further enrollment or infusions. All 4 cemiplimab recipients were followed for 48 weeks. No other cemiplimab-related serious AEs, irAEs, or grade 3 or higher AEs occurred. One 2-dose recipient of cemiplimab had a 6.2-fold increase in polyfunctional, Gag-specific CD8+ T-cell frequency with supportive increases in plasma HIV RNA and decreases in total HIV DNA.
Conclusions
One of 4 participants exhibited increased HIV-1-specific T-cell responses and transiently increased HIV-1 expression following 2 cemiplimab infusions. The occurrence of irAEs after a single, low dose may limit translating the promising therapeutic results of cemiplimab for cancer to immunotherapeutic and latency reversal strategies for HIV.
Clinical Trials Registration. NCT03787095.
Keywords: anti-PD-1 inhibitor, HIV, HIV cure, HIV latency, immune checkpoint inhibitors
One of 4 participants administered anti-PD-1 antibody exhibited increased HIV-1–specific T-cell responses and transiently increased HIV-1 expression. Significant immune-related adverse events after a single, low dose of anti-PD-1 antibody may limit translating the benefits of anti-PD-1 therapies from cancer to HIV.
Immune checkpoint therapy (ICT) targeting programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 [CTLA-4]) has revolutionized immunotherapy for cancer [1] and is standard of care for several malignancies. Cancer and chronic viral infections both increase PD-1 expression on activated effector T cells, leading to an exhausted T-cell phenotype with ineffective immune responses to tumor and viral antigens. With chronic viral infections including human immunodeficiency virus type 1 (HIV-1), persistent antigenic stimulation causes T-cell expression of inhibitory co-receptors (eg, PD-1) associated with downregulation of immune responses [2–4]. The resulting T-cell “exhaustion” starts with loss of proliferative potential, cytotoxic responses, and polyfunctionality, followed by defects in cytokine production such as interferon gamma (IFN-γ) [5–7].
Inhibitory co-receptor expression on CD4+ and/or CD8+ T cells is associated with disease progression in untreated people with HIV (PWH) [8–10]. PD-1 expression on HIV-1–specific CD8+ and CD4+ T cells decreases with antiretroviral therapy (ART) [8, 11], but in some studies remained elevated compared to uninfected persons [8, 12], including on CD4+ effector memory T cells in PWH starting ART early [13]. High levels of PD-1 expression on CD4+ T cells correlated inversely with HIV-specific responses and directly with HIV DNA levels in PWH on ART [14]. Moreover, receptor density of PD-1 ligand (PD-L1) on T cells remains elevated despite ART suppression [12, 15, 16]. Data suggest that CD4+ T cells expressing PD-1 are enriched for latent HIV, making anti-PD-1 a promising strategy to target the latent reservoir and reverse immune exhaustion [17–21]. Furthermore, the inability of CD8+ T cells from PWH to kill infected, resting CD4+ T cells after viral reactivation suggests diminished CTL function despite ART suppression [22].
In a macaque simian immunodeficiency virus (SIV) model, multiple doses of anti-PD-1 increased SIV-specific CD8+ T cells and polyfunctional response, significantly reduced plasma SIV RNA, and prolonged survival without ART [23]. In ART-suppressed PWH, a single, low dose (0.3 mg/kg) of anti-PD-L1 (BMS-936559) appeared to increase HIV-1–specific CD8+ T-cell responses and frequencies of polyfunctional HIV-1 Gag-specific cells in 2 of 6 participants; findings correlated with pretreatment ex vivo CD8+ T-cell proliferative responses to Gag following PD-L1 exposure [24].
Cemiplimab (REGN2810), a high-affinity, hinge-stabilized, IgG4P human antibody to PD-1 receptor (PDCD1, CD279), blocks PD-1/PD L1–mediated T-cell inhibition and is US Food and Drug Administration approved at a fixed 350-mg dose every 3 weeks for advanced cutaneous squamous cell, basal cell, and non-small-cell lung carcinoma treatment. A5370 was designed to assess the safety of cemiplimab and its impact on anti-HIV immune function in PWH on ART. Study enrollment stopped prematurely due to suspected immune-related AEs (irAEs) in 2 participants, probably and possibly related to study treatment previously described [25]. Herein, we report on the pharmacokinetics/pharmacodynamics (PK/PD) for cemiplimab and receptor occupancy (RO), as well as immunologic and virologic outcomes in the context of safety events following low-dose cemiplimab 0.3 mg/kg infusions.
METHODS
Study Design
ACTG protocol A5370 (NCT03787095) was a phase 1/2, randomized, double-blind, placebo-controlled study of 2 infusions of cemiplimab (REGN2810) at weeks 0 and 6 with sequential, dose-escalating cohorts of 0.3, 1, 3 and 10 mg/kg. In cohort 1, 5 participants were centrally randomized 4:1 by permuted block method to receive cemiplimab or placebo (normal saline). Eligible participants were healthy PWH aged 18–70 years who were on ART for ≥24 months with CD4+ T-cell counts ≥350 cells/µL and plasma HIV-1 RNA <50 copies/mL. Exclusion criteria comprised active hepatitis B or C, latent tuberculosis, type 1 or 2 diabetes, or autoimmune disorder including but not limited to hypothyroidism, hyperthyroidism, or adrenal insufficiency. The primary outcome was any grade 3 or higher AE, or grade 1 or higher irAE, at least possibly related to study treatment, assessed by the blinded core study team through week 48. Fasting glucose, thyroid-stimulating hormone (TSH), free T4, morning cortisol, follicle-stimulating hormone, and free testosterone were tested at screening, preentry, and weeks 4, 6, 12, 16, 20, 24, 28, 36, and 48. Samples were cryopreserved for future assays including antithyroperoxidase antibody.
Patient Consent Statement
Local institutional review boards approved the study. All study participants provided written informed consent and completed an assessment of understanding [25].
HIV-Specific Immune Responses
Cryopreserved peripheral blood mononuclear cells (PBMCs) obtained before and at multiple timepoints postinfusion were analyzed for HIV-1–specific polyfunctional responses. In brief, cells were incubated with HIV-1 peptide pools (Gag, Env, and Pol consensus B pool, 15mers overlapping with 11aa; National Institutes of Health [NIH] Reagent Program) or phorbol myristate acetate/ionomycin in the presence of brefeldin A, monensin, CD28/49 (all BD Biosciences) and CD107a (Biolegend) at 37°C for 6 hours followed by Live/Dead staining (Amcyan; Invitrogen) and surface staining with anti-CD3, anti-CD4, and anti-CD8 antibodies (all BD Biosciences). Cells were permeabilized using 1X BD Perm/Wash buffer (BD Biosciences), incubated with anti–IFN-γ, anti–interleukin 2, and anti–tumor necrosis factor alpha (TNF-α) antibodies (all BD Biosciences), fixed and analyzed using BD LSRFortessa within 24 hours. Flow cytometry data were analyzed using FlowJo v10.8 software. Polyfunctional response was defined as expressing 2 or more effector cytokines following HIV peptide stimulation.
Checkpoint Inhibitor Expression and T-Cell Activation
To evaluate frequencies of T cells expressing 1 or more checkpoint receptors and levels of T-cell activation (defined as percentage of T cells co-expressing HLA-DR and CD38) and cell cycling (Ki-67), cryopreserved PBMCs obtained at multiple timepoints underwent staining with antibodies for CD3, CD4, CD8, HLA-DR, CD38, Ki-67, PD-1, PD-L1, TIM3 (all BD Biosciences), CTLA4, LAG3, TIGIT (all Biolegend), and Live/Dead (Invitrogen). Cells were analyzed using BD LSRFortessa and FlowJo v10.8.
Ex Vivo Evaluation of Proliferative Response With Cemiplimab
Proliferative responses following stimulation with HIV peptides were evaluated in the presence or absence of cemiplimab, as previously described [24]. In brief, cryopreserved cells obtained preinfusion were thawed then labeled with CellTrace Violet for 20 minutes at 37°C, then washed and incubated with Gag, Env, and Pol consensus B peptide pools (2.5 μg/mL; NIH AIDS Reagent Program) with or without cemiplimab (10 μg/mL) for 6 days at 37°C, washed, then incubated with Live-Dead solution. Cells were washed again and stained with anti-CD3, CD4, and CD8 antibodies, and analyzed using BD LSRFortessa and FlowJo v10.8.
PK Analysis and Receptor Occupancy
Serum samples were collected for PK analysis at preentry, preinfusion at weeks 0 and 6, and weeks 1, 2, 7, 8, 10, 12, 16, 20, 24, 28, 36, and 48. Samples were analyzed for functional cemiplimab concentrations using a validated enzyme immunoassay, utilizing rHu PD-1 extracellular domain as the capture reagent detected using a biotinylated anti-human IgG4 monoclonal antibody with lower limit of quantification (LLOQ) 0.078 mg/L [26]. Peripheral blood cemiplimab PD-1 RO was measured using flow cytometry detection of cell surface bound drug. In brief, PBMC samples were incubated with fluorescently labeled CD4, CD8, and anti-human IgG4 antibody for detection of T-cell surface bound cemiplimab, directly ex vivo, or following incubation with saturating concentration of cemiplimab, for detection of total PD-1 receptor levels. The PD-1 RO was determined based on the ratio of human IgG4-mediated detection of bound and total PD-1 receptor in peripheral T cells. Exploratory time-dependent PK/PD relationships (Hysteresis Plots) of cemiplimab RO to CD3+ T cells as a function of cemiplimab concentrations in serum after cemiplimab 0.3 mg/kg intravenous (IV) single dose were conducted.
Residual Viremia, Total HIV-1 DNA, and Cell-Associated RNA
Intracellular DNA and RNA were isolated from cryopreserved peripheral PBMCs using the AllPrep DNA/RNA Mini Kit (Qiagen). Unspliced, cell-associated RNA (CA-RNA) and total HIV DNA (CA-DNA) levels were quantified using real-time polymerase chain reaction (rtPCR) with primers/probes targeting conserved regions of HIV LTR/Gag as previously described [27, 28]. Cell numbers were quantified by rtPCR measurement of CCR5 copy numbers. Cellular integrity for RNA analysis was assessed by measurement of total extracted RNA and evaluation of the IPO8 housekeeping gene [29]. Total, intact, 3′ defective, and 5′ defective proviral reservoirs were quantified in CD4+ cells using the intact proviral reservoir assay performed at AccelevirDx as described [30]. Plasma HIV residual viremia was measured using the validated ultrasensitive integrase single-copy assay [31]. Plasma from participants was spiked with an internal replication-competent avian sarcoma leukosis virus long terminal repeat with a Splice acceptor virion standard as a control for RNA extraction efficiency [32]. rtPCR reactions were performed with a Roche LightCycler 480 system using primers and probes specific to conserved regionS of the HIV integrase gene [31].
Statistical Considerations
With the reduced sample size following early closure, descriptive and graphical approaches were used for data analyses.
RESULTS
Twenty-seven individuals were screened; 22 were ineligible, most commonly due to laboratory results outside of eligibility cutoffs for hemoglobin A1C, morning cortisol, and hemoglobin. Five participants enrolled across 3 sites; 4 received cemiplimab and 1 received placebo. All participants were male with a median age of 51 years (range, 27–57 years), with 4 White and 1 multiple race, 2 of whom were of Hispanic ethnicity. The pretreatment median CD4+ T-cell count was 911 cells/μL (range, 348–1957 cells/μL).
Study enrollment stopped prematurely due to 2 suspected irAEs in 2 different participants, probably and possibly related to study treatment as described below and previously [25]. Two participants completed both infusions of 0.3 mg/kg of cemiplimab at weeks 0 and 6, and 2 participants received 1 dose of cemiplimab 0.3 mg/kg due to irAEs. One participant received 1 placebo dose and withdrew at week 7 given study cessation. All participants who received cemiplimab were followed until week 48.
Cemiplimab infusions were well-tolerated in 2 participants who received 2 infusions with no treatment-related AEs. Transient, grade 1 increases in aspartate aminotransferase (AST)/alanine aminotransferase (ALT) occurred in 1 of these participants (weeks 0, 6, 10, 12, 20, 24, 28, 36, but normal at weeks 2, 4, 8, 16, 48) and were assessed as unrelated to study treatment. Grade 2 AST/ALT elevations occurred at week 36 in the other participant following resumption of heavy alcohol use. The placebo recipient had grade 1 elevated AST/ALT assessed as not related to blinded study treatment.
Two participants experienced a probable or possible irAE following a single 0.3 mg/kg cemiplimab infusion [26]. One participant developed grade 2 thyroiditis at week 4 (TSH 0.02 µg/mL, free T4 2.73 ng/dL) that resolved by week 16. Retrospective testing revealed a positive, pretreatment antithyroglobulin antibody level (7.00 IU/mL) that increased markedly postinfusion (Figure 1A) and elevated TPO antibodies (28.0 IU/mL, week 16) persisting through week 48 (Figure 1B). Another participant, with undisclosed, chronic alcohol use, developed grade 3 AST/ALT elevation (Figure 1C and 1D) at week 2 (after consuming 6 beers, 2 whiskey drinks, and acetaminophen 500 mg the evening before laboratory testing), assessed as drug-induced liver injury and/or possibly immune-related due to cemiplimab (no biopsy performed) [25].
Figure 1.

Antithyroglobulin antibodies (A) and thyroperoxidase antibodies (B) over time for each participant. Participant (Pt) #1 and Pt #2 received 2 doses of cemiplimab. Pt #3 and #4 received a single dose of cemiplimab due to suspected adverse events. Pt #5 received 1 placebo dose. Pt #3 (shown in blue, thyroiditis immune-related adverse event [AE]) had a detectable pretreatment antithyroglobulin antibody level and thyroperoxidase antibody level with an increase in both measures after cemiplimab infusion. Alanine aminotransferase (ALT; C) and aspartate aminotransferase (AST; D) over time for each participant. Pt #4 (shown in green, liver injury AE) experienced increased AST and ALT at week 2. Elevated AST and ALT (grade 1–2) recurred for this participant at weeks 20, 29, and 48, attributed to ongoing alcohol use.
Serum cemiplimab concentrations after one 0.3 mg/kg IV dose reached a maximum concentration (Cmax) of 5.04–16.7 mg/L and were detectable at week 6 (>0.5 mg/L in 3 participants, 0.089 mg/L in 1 participant). In 2 participants who received only a single dose of cemiplimab, detectable drug concentrations persisted until week 10 (LLOQ 0.078 mg/mL). Notably, cemiplimab RO on CD3+ T cells remained high at low cemiplimab concentrations in the 2 participants who received a single cemiplimab 0.3 mg/kg infusion (Figure 2). Two participants who received a second dose on week 6 showed a second Cmax of 2.38 to 8.43 mg/L, with concentrations becoming undetectable at weeks 12 and 16, respectively.
Figure 2.
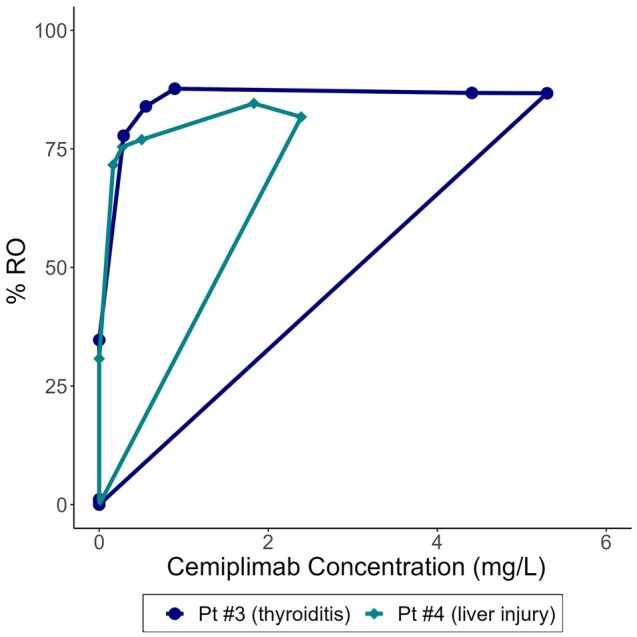
Hysteresis plot of cemiplimab receptor occupancy (RO) on CD3+ T cells relative to cemiplimab concentrations in serum in people with human immunodeficiency virus after a single intravenous (IV) dose of cemiplimab 0.3 mg/kg IV. Cemiplimab RO on CD3+ T cells at low cemiplimab concentrations are shown for the 2 participants (Pt #3 and #4) who received only a single cemiplimab infusion to demonstrate persistent receptor occupancy at the lowest cemiplimab concentration levels.
PD-1 expression was observed at baseline in all participants and decreased following the initial cemiplimab infusion, rising again around week 12 (Figure 3). For all 4 treated participants, PD-1 RO increased from baseline values of <2%, remained >70% for 4–8 weeks after the last dose, then returned to baseline levels (Figure 4). Extended PD-1 RO was observed in participants who received 2 infusions and corresponded with decreased PD-1 expression from weeks 2 through 12.
Figure 3.
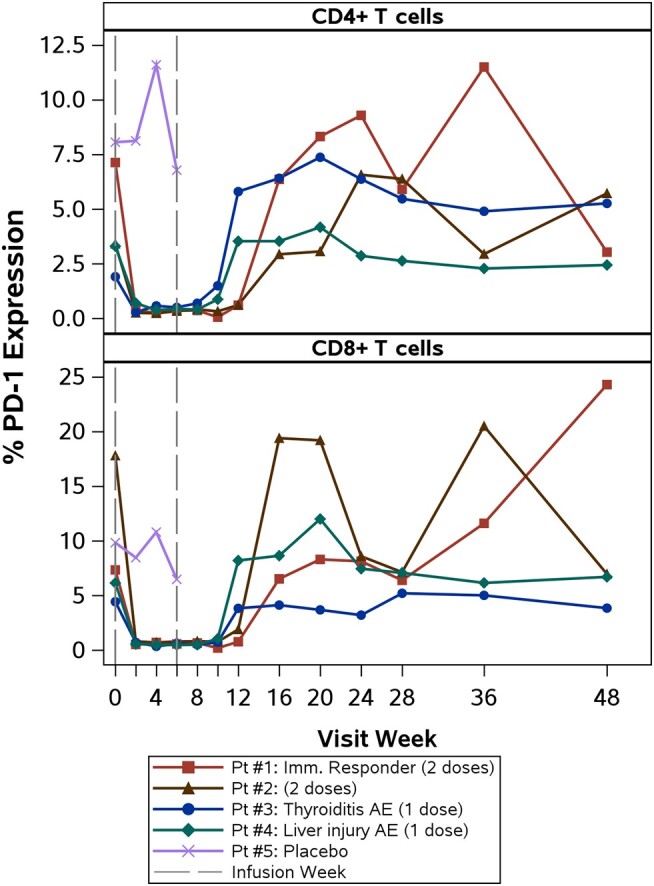
Programmed cell death 1 (PD-1) expression on CD4+ and CD8+ T cells over time for each participant. All participants had decreased frequency of PD-1+ T cells after the first cemiplimab infusion. Participant (Pt) #1 (shown in red, immune responder) had the highest pretreatment PD-1 expression percentage on CD4+ T cells. Pt #3 and #4 received a single dose of cemiplimab; Pts #1 and #2 received 2 doses of cemiplimab. Abbreviations: AE, adverse event; PD-1, programmed cell death 1.
Figure 4.
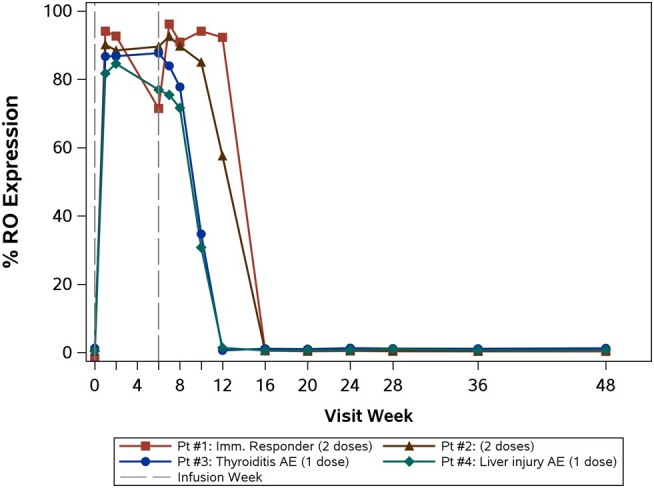
Programmed cell death 1 receptor occupancy expression over time for each participant. Pt #1 and #2, who received 2 doses of cemiplimab, returned to baseline levels at week 16 versus at week 12 following a single cemiplimab infusion in Pt #3 and #4. Abbreviations: AE, adverse event; RO, receptor occupancy.
Among the 4 treated participants, the frequency of Gag-specific, polyfunctional CD8+ and CD4+ T cells (mean of weeks 2–12) increased from baseline by a median of 2.2-fold and 1.8-fold, respectively. This median increase in polyfunctional T cells was driven largely by 1 participant deemed an immunologic responder (Figure 5A), who received 2 cemiplimab infusions with 6.2-fold and 3.4-fold increases in Gag-specific polyfunctional CD8+ and CD4+ T-cell response, respectively. Increases in the frequencies of Env- and Pol-specific polyfunctional T cells were also observed (Figure 5B and 5C), but were apparent at later timepoints with median increases at weeks 2–12 of 1.3-fold and 1.3-fold for Pol responses for CD8+ and CD4+ T cells, respectively, and with corresponding Env response median increases of 1.1-fold and 1.0-fold.
Figure 5.

Gag-stimulated (A), Env-stimulated (B), and Pol-stimulated (C) polyfunctional CD4+ and CD8+ T-cell responses over time for each participant. Pt #1 (immune responder) had increased gag-, env-, and pol-stimulated polyfunctional CD4+ and CD8+ T-cell responses following the second cemiplimab infusion. Pt #4 demonstrated increases in human immunodeficiency virus–specific polyfunctional responses coinciding with symptoms consistent with an upper respiratory illness at week 19. Pt #3 and #4 received a single dose of cemiplimab; Pt #1 and #2 received 2 doses of cemiplimab. Abbreviation: AE, adverse event.
The apparent immune responder had the highest pretreatment frequency of PD-1–positive CD4+ T cells (Figure 3); his pretreatment intracellular cytokine panel was otherwise unremarkable (Table 1). Notably, pretreatment expression of multiple checkpoint receptors was lower in the other participant who received 2 cemiplimab infusions without an immune response. No apparent associations were observed between pretreatment frequencies of CD4+ and CD8+ T cells expressing PD-L1 and HIV-1–specific polyfunctional responses following cemiplimab infusion. Similarly, we did not observe associations between pretreatment frequencies of T cells expressing multiple checkpoint receptors and HIV-1–specific immune responses.
Table 1.
Baseline Clinical and Immunologic Parameters for Treated Participants
| Parameter | Participant 1 | Participant 2 | Participant 3 | Participant 4 | Mean |
|---|---|---|---|---|---|
| Immunologic Responder | Thyroiditis AE | Liver Injury AE | |||
| Race | White | White | White | White | |
| Ethnicity | Not Hispanic/Latino | Not Hispanic/Latino | Hispanic/Latino | Not Hispanic/Latino | … |
| Age, y | 27 | 51 | 50 | 57 | 46 |
| CD4+ T-cell count, cells/μL | 528 | 1391 | 1957 | 911 | 1197 |
| CD8+ T-cell count, cells/μL | 1508 | 1524 | 2220 | 867 | 1530 |
| HIV-1 RNA, copies/mL | <40 | <40 | <40 | <40 | … |
| Inhibitory receptor expression | |||||
| % PD-1+ on CD4+ T cells | 7.1 | 3.4 | 1.9 | 3.3 | 3.9 |
| % 2 or more inhibitory receptors on CD4+ T cells | 2.5 | 1.4 | 1.5 | 2.1 | 1.9 |
| % PD-1+ on CD8+ T cells | 7.3 | 17.8 | 4.4 | 6.1 | 8.9 |
| % 2 or more inhibitory receptors on CD8+ T cells | 6.5 | 11.9 | 3.1 | 4.8 | 6.6 |
| Polyfunctional response | |||||
| % response on CD4+ T cells, Gag stimulation | 0.3 | 0.3 | 0.2 | <0.1 | 0.2 |
| % response on CD4+ T cells, Env stimulation | 8.6 | 2.6 | 0.6 | 1.0 | 3.2 |
| % response on CD4+ T cells, Pol stimulation | 2.2 | 3.6 | 0.4 | 1.7 | 2.0 |
| % response on CD8+ T cells, Gag stimulation | 0.2 | 0.7 | <0.1 | <0.1 | 0.2 |
| % response on CD8+ T cells, Env stimulation | 6.5 | 3.1 | 0.5 | 0.3 | 2.6 |
| % response on CD8+ T cells, Pol stimulation | 1.7 | 4.6 | 0.2 | 0.4 | 1.7 |
Abbreviations: AE, adverse event; HIV-1, human immunodeficiency virus type 1; PD-1, programmed cell death 1.
Both participants who received 2 infusions demonstrated baseline HIV-1 RNA levels <40 copies/mL by standard assays. One participant with pretreatment HIV-1 RNA <20 copies/mL and no apparent immune response had an HIV-1 RNA of 44 copies/mL at week 6 before the second infusion. The potential immune responder demonstrated HIV-1 RNA levels <20 copies/mL (target not detected) and 23 copies/mL pretreatment, then 76 copies/mL, 71 copies/mL, and 57 copies/mL at weeks 6 (before the second dose), 7 and 12, respectively. No participants experienced virologic failure (2 consecutive HIV-1 RNA levels ≥200 copies/mL), and all participants had HIV-1 RNA below LLOQ at end of the study. No consistent trends in CA-RNA or total CA-DNA following 1 or 2 cemiplimab doses were detected. However, the immune responder showed a decrease in proviral HIV-1 DNA by 2 separate assays (Figure 6A and 6C) and a transient increase in CA-RNA (Figure 6B). Levels of intact proviral DNA (IPD) (Figure 6D) are difficult to interpret in this participant due to low levels detected and the limited pretreatment CD4+ T-cell equivalents tested; baseline IPD was undetectable (<20/106 CD4+ T cells) based on 48 000 CD4+ T-cell equivalents assayed. At week 2, IPD was measurable (estimated 25/106 CD4+ T cells based on 69 000 CD4+ T cells), while at weeks 6–24 IPD was undetectable (with CD4+ T-cell equivalents >200 000). At week 48, IPD was 85/106 CD4+ T cells.
Figure 6.

Total cell-associated DNA (A), cell-associated RNA (B), total proviral human immunodeficiency virus type 1 (HIV-1) DNA (C), and intact proviral HIV-1 DNA (D) over time for each participant. Pt #1 (immune responder) had decreased total cell-associated DNA, proviral HIV-1 DNA, and cell-associated RNA. For the proviral HIV-1 DNA results, this participant had a limited number of CD4+ T-cell equivalents available at baseline and week 2, as well as an undetectable proviral HIV-1 DNA result at week 12 (represented by an open square in the figure). This participant also had undetectable intact proviral HIV-1 DNA at multiple time points. Abbreviations: AE, adverse event; n.d., not detected.
Prior to treatment, an increase in proliferative response to HIV-1 Gag peptides following ex vivo exposure to cemiplimab was not observed in CD8+ T cells from the potential immune responder, contrasting with a prior correlation between ex vivo proliferative response and Gag-specific CD8+ T-cell responses following anti-PD-L1 antibody in PWH [24].
DISCUSSION
Motivated by expanding ICT success in reversing immune dysregulation for some cancers [33], we evaluated an anti-PD-1 antibody (cemiplimab) in PWH as an immune-enhancing strategy to target the HIV latent reservoir. Unfortunately, the study closed after suspected irAEs following a single infusion in the lowest dose cohort. Although irAEs due to ICT for cancer therapy are well-described [34], it was unknown whether healthy PWH would experience irAEs with comparable frequency or severity as cancer patients, many of whom would previously received chemotherapy or radiation. Studies of ICT in PWH and cancer demonstrated similar safety profiles and frequency of irAEs compared to ICT in participants without HIV [35, 36].
Despite the low initial dose (<10% of approved dose) in our study, at least 1 irAE was probably related to study treatment. Thyroid dysfunction is a commonly reported irAE, with frequencies ranging from 5% to 50% with ICT. In 1 study, 6 of 30 (20%) PWH receiving pembrolizumab for cancer developed hypothyroidism, but none had elevated antibodies to thyroglobulin or TPO at baseline [35]. In our study, timing of thyroiditis (elevated free T4/suppressed TSH), the concurrent rise in anti-thyroglobulin antibody following cemiplimab administration, and normalization of TSH/free T4 by week 24 without thyroid hormone replacement strongly support a causal relationship. Immune-related thyroiditis may progress to permanent hypothyroidism, but resolution without treatment has occurred in cancer patients receiving ICT [37, 38]. Although autoimmune disease–related antibody screening has not predicted many types of irAEs in cancer patients, preexisting thyroid autoimmunity predicting thyroid dysfunction may be an exception [38] and warrant anti-thyroglobulin antibody screening in future HIV remission studies of ICT. For the participant with grade 3 transaminitis, recurrent AST/ALT elevations through week 48 with ongoing alcohol use suggest that these irAE-suspected elevations may not have reflected immune-mediated hepatitis due to cemiplimab.
There are other notable findings from our study. Substantial biologic effect was not anticipated with the lowest dose (0.3 mg/kg); however, this dose clearly had immune-mediating effects as demonstrated by the irAE (thyroiditis). PK and PD for cemiplimab were rigorously characterized for the first time, PK/PD relationships for RO at low concentrations were explored, and PD-1 RO >70% for 4–8 weeks following low-dose cemiplimab correlated with undetectable PD-1 expression. While the small N limits interpretation, 1 participant had a convincing HIV-specific immune response with a substantial increase in polyfunctional Gag-, Pol-, and Env-specific responses for both CD4+ and CD8+ T cells. Notably, this participant had the highest baseline frequency of CD4+ T cells expressing PD-1 and >70% RO through 8 weeks postinfusion [39]. Pretreatment, low-level plasma viremia in this participant appeared to increase following both cemiplimab infusions, and HIV DNA levels declined. These findings support the conclusion that cemiplimab stimulated both reservoir disruption and HIV-specific immune enhancement, or, alternatively, could have resulted from an increase in circulating HIV-1 antigen and not immune enhancement.
The major limitation of this study was early closure and inability to complete dose escalation. The small N and single placebo recipient are major limitations to drawing conclusions about HIV-specific activity. Detailed immunologic and virologic analyses provide biologically consistent results from 1 participant presumed to respond to cemiplimab. Importantly, in cancer treatment, substantial clinical responses have occurred only in a subset of patients, and prescreening for PD-L1 on tumor cells improves response rates [40]. The fact that the potential immune responder in this small cohort had the highest expression of PD1 on CD4+ T cells at entry is consistent with previous findings.
Eight of 10 ART-treated rhesus macaques receiving anti-PD-1 and anti–interleukin 10 antibodies maintained viral suppression for 14 weeks following ART interruption with 4-log10 lower viral loads versus pre-ART viral loads and significantly lower than controls; no significant AEs occurred [41]. While animal data and ICT success in cancer are promising, early study closure due to suspected irAEs highlights challenges in translating cancer immunotherapies to HIV remission strategies. Although limited, ICT for PWH with cancer has demonstrated an acceptable safety profile with the anticipated benefit of enhanced short-term survival, justifying the risk of irAEs. In cancer therapy, immune-related thyroiditis progressing to hypothyroidism is not considered a reason to discontinue ICT. Lower ICT doses (<0.3 mg/kg) as an HIV remission strategy have been considered, although risk of irAEs in cancer patients has not correlated with dose or number of doses and may limit this approach to mitigating risk. However, high PD-1 RO, which persisted for 4–8 weeks following a low cemiplimab dose in our study, suggests an immunologic effect and potential role for low-dose immune checkpoint therapy in combination with other interventions to target the HIV reservoir. The initial 0.3 mg/kg cemiplimab dose was selected based on prior investigations of dosing in cancer patients and as a starting point in PWH. Dose-response observations that enhanced HIV-1–specific immunity and RO data were considered for dose selection [1, 2]. Cemiplimab linear PK informed that 0.3 mg/kg would exhibit serum concentrations above reported preclinical half-maximal inhibitory concentration values (0.085–5.61 nM) for binding to human PD-1 [42].
In summary, although only 4 participants received study treatment, there is evidence for both immunologic and virologic responses in 1 participant and a probable irAE in another, at the lowest anti-PD1 antibody dose studied. These results call for caution when considering ICT as an immune-enhancing tool in healthy PWH but raise the possibility of a more targeted or combination treatment approach to improve the risk:benefit ratio.
Contributor Information
Cynthia L Gay, Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
Ronald J Bosch, Department of Biostatistics, Center for Biostatistics and AIDS Research, Harvard T. H. Chan School of Public Health, Boston, Massachusetts, USA.
Ashley McKhann, Department of Biostatistics, Center for Biostatistics and AIDS Research, Harvard T. H. Chan School of Public Health, Boston, Massachusetts, USA.
Raymond Cha, Center for Integrated Global Biomedical Sciences, University at Buffalo, Buffalo, New York, USA.
Gene D Morse, Center for Integrated Global Biomedical Sciences, University at Buffalo, Buffalo, New York, USA.
Chanelle L Wimbish, Department of Clinical Research, Social and Scientific Systems, Inc, a DLH Company, Silver Spring, Maryland, USA.
Danielle M Campbell, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California, USA.
Kendall F Moseley, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
Steven Hendrickx, Department of Medicine, University of California, San Diego, San Diego, California, USA.
Michael Messer, Department of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, USA.
Constance A Benson, Department of Medicine, University of California, San Diego, San Diego, California, USA.
Edgar T Overton, Department of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, USA; North America Medical Affairs, ViiV Healthcare, Durham, North Carolina, USA.
Anne Paccaly, Departments of Clinical Sciences, Translational Medicine and Precision Medicine, Regeneron Pharmaceuticals, Inc, Tarrytown, New York, USA.
Vladimir Jankovic, Departments of Clinical Sciences, Translational Medicine and Precision Medicine, Regeneron Pharmaceuticals, Inc, Tarrytown, New York, USA.
Elizabeth Miller, Departments of Clinical Sciences, Translational Medicine and Precision Medicine, Regeneron Pharmaceuticals, Inc, Tarrytown, New York, USA.
Randall Tressler, HIV Research Branch, Division of AIDS, National Institute of AIDS, National Institutes of Health, Rockville, Maryland, USA.
Jonathan Z Li, Division of Infectious Diseases, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA.
Daniel R Kuritzkes, Division of Infectious Diseases, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA.
Bernard J C Macatangay, Division of Infectious Diseases, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
Joseph J Eron, Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
W David Hardy, Division of Infectious Diseases, Keck School of Medicine, University of Southern California, Los Angeles, California, USA.
for the A5370 Team:
Amanda Tipton, Susan Pedersen, Bernadette Jarocki, Scott Anderson, Lynette Purdue, Kyle Whitson, Sara Zabih, Cheryl Jennings, Pamela Lankford-Turner, Patrick Mehta, and Thomas Uldrick
Notes
Acknowledgments. We sincerely thank all of the participants who participated in this study. Additional ACTG A5370 Team Members are as follows: Amanda Tipton and Susan Pedersen (field representatives), Bernadette Jarocki, BS, and Scott Anderson (data managers), Lynette Purdue, PharmD, BCPS (Division of AIDS pharmacist), Kyle Whitson, (laboratory data manager), Sara Zabih (laboratory specialist), Cheryl Jennings and Pamela Lankford-Turner (laboratory technologists), Patrick Mehta (Pitt Immunology Specialty Laboratory), and Thomas Uldrick, MD, MS (Regeneron).
Author contributions. Study design was done by C. L. G., W. D. H., J. J. E., C. L. W., R. J. B., G. D. M., B. J. M., D. R. K., and R. T. Data generation/assay performance was done by C. L. G., W. D. H., J. J. E., R. J. B., A. M., B. J. M., R. C., D. R. K., M. M., S. H., C. B., and E. T. O. Drafting or editing of the manuscript was done by C. L. G., W. D. H., J. J. E., R. J. B., A. M., R. C., D. M. C., K. M., B. J. M., G. D. M., R. T., D. R. K., E. M., C. B., and E. T. O.
Disclaimer. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Financial support. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (award numbers UM1 AI068634, UM1 AI068636, UM1 AI106701, UM1 AI69452, UM1 AI069412, UM1 AI069432, UM1 AI069465, and UM1 AI069423); the University of North Carolina at Chapel Hill Center for AIDS Research (grant number P30 AI50410); and the National Center for Advancing Translational Sciences (cooperative agreement UL1TR001111). Cemiplimab was provided by Regeneron Pharmaceuticals.
References
- 1. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013; 342:1432–3. [DOI] [PubMed] [Google Scholar]
- 2. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 2009; 106:8623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 2006; 12:1198–202. [DOI] [PubMed] [Google Scholar]
- 5. Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492–9. [DOI] [PubMed] [Google Scholar]
- 6. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 2003; 77:4911–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology 2010; 129:474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006; 443:350–4. [DOI] [PubMed] [Google Scholar]
- 9. Kaufmann DE, Kavanagh DG, Pereyra F, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol 2007; 8:1246–54. [DOI] [PubMed] [Google Scholar]
- 10. Boasso A, Hardy AW, Landay AL, et al. PDL-1 upregulation on monocytes and T cells by HIV via type I interferon: restricted expression of type I interferon receptor by CCR5-expressing leukocytes. Clin Immunol 2008; 129:132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Souza M, Fontenot AP, Mack DG, et al. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol 2007; 179:1979–87. [DOI] [PubMed] [Google Scholar]
- 12. Sachdeva M, Fischl MA, Pahwa R, Sachdeva N, Pahwa S. Immune exhaustion occurs concomitantly with immune activation and decrease in regulatory T cells in viremic chronically HIV-1-infected patients. J Acquir Immune Defic Syndr 2010; 54:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cockerham LR, Jain V, Sinclair E, et al. Programmed death-1 expression on CD4(+) and CD8(+) T cells in treated and untreated HIV disease. AIDS 2014; 28:1749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macatangay BJC, Gandhi RT, Jones RB, et al. T cells with high PD-1 expression are associated with lower HIV-specific immune responses despite long-term antiretroviral therapy. AIDS 2020; 34:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fenwick C, Joo V, Jacquier P, et al. T-cell exhaustion in HIV infection. Immunol Rev 2019; 292:149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosignoli G, Cranage A, Burton C, et al. Expression of PD-L1, a marker of disease status, is not reduced by HAART in aviraemic patients. AIDS 2007; 21:1379–81. [DOI] [PubMed] [Google Scholar]
- 17. Khoury G, Fromentin R, Solomon A, et al. Human immunodeficiency virus persistence and T-cell activation in blood, rectal, and lymph node tissue in human immunodeficiency virus–infected individuals receiving suppressive antiretroviral therapy. J Infect Dis 2017; 215:911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fromentin R, Bakeman W, Lawani MB, et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog 2016; 12:e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lau JSY, McMahon JH, Gubser C, et al. The impact of immune checkpoint therapy on the latent reservoir in HIV-infected individuals with cancer on antiretroviral therapy. AIDS 2021; 35:1631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rasmussen TA, Rajdev L, Rhodes A, et al. Impact of anti-PD-1 and anti-CTLA-4 on the human immunodeficiency virus (HIV) reservoir in people living with HIV with cancer on antiretroviral therapy: the AIDS Malignancy Consortium 095 study. Clin Infect Dis 2021; 73:e1973–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uldrick TS, Adams SV, Fromentin R, et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci Transl Med 2022; 14:eabl3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shan L, Deng K, Shroff NS, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012; 36:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009; 458:206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gay CL, Bosch RJ, Ritz J, et al. Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis 2017; 215:1725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gay CL, Bosch RJ, McKahnn A, et al. Suspected immune-related adverse events with an anti-PD-1 inhibitor in otherwise healthy people with HIV. J Acquir Immune Defic Syndr 2021; 87:e234–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dengler AF, Weiss R, Truong T, et al. Bioanalytical challenges due to prior checkpoint inhibitor exposure: interference and mitigation in drug concentration and immunogenicity assays. AAPS J 2021; 23:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malnati MS, Scarlatti G, Gatto F, et al. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat Protoc 2008; 3:1240–8. [DOI] [PubMed] [Google Scholar]
- 28. Li JZ, Etemad B, Ahmed H, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2016; 30:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ledderose C, Heyn J, Limbeck E, Kreth S. Selection of reliable reference genes for quantitative real-time PCR in human T cells and neutrophils. BMC Res Notes 2011; 4:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bruner KM, Wang Z, Simonetti FR, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019; 566:120–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cillo AR, Vagratian D, Bedison MA, et al. Improved single-copy assays for quantification of persistent HIV-1 viremia in patients on suppressive antiretroviral therapy. J Clin Microbiol 2014; 52:3944–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palmer S, Wiegand AP, Maldarelli F, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 2003; 41:4531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma P, Siddiqui BA, Anandhan S, et al. The next decade of immune checkpoint therapy. Cancer Discov 2021; 11:838–57. [DOI] [PubMed] [Google Scholar]
- 34. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One 2016; 11:e0160221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uldrick TS, Goncalves PH, Abdul-Hay M, et al. Assessment of the safety of pembrolizumab in patients with HIV and advanced cancer—a phase 1 study. JAMA Oncol 2019; 5:1332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El Zarif T, Nassar AH, Adib E, et al. Safety and activity of immune checkpoint inhibitors in people living with HIV and cancer: a real-world report from the cancer therapy using checkpoint inhibitors in people living with HIV–international (CATCH-IT) consortium. J Clin Oncol 2023; 41:3712–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iyer PC, Cabanillas ME, Waguespack SG. Immune-related thyroiditis with immune checkpoint inhibitors. Thyroid 2018; 28:1243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lurain K, Ramaswami R, Yarchoan R, Uldrick TS. Anti-PD-1 and anti-PD-L1 monoclonal antibodies in people living with HIV and cancer. Curr HIV/AIDS Rep 2020; 17:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuazo M, Arasanz H, Bocanegra A, et al. Systemic CD4 immunity as a key contributor to PD-L1/PD-1 blockade immunotherapy efficacy. Front Immunol 2020; 11:586907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity 2018; 48:434–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strongin Z, Ribeiro SP, Hoang TN, et al. Dual IL-10 and PD-1 blockade in SIVmac239 infected macaques promotes sustained virologic control in absence of ART. J Int AIDS Soc 2021; 24:68–70. [Google Scholar]
- 42. US Food and Drug Administration Drug Evaluation and Research . BLA 761097. NDA/BLA multidisciplinary review and evaluation. 2018. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761097Orig1s000MultidisciplineR.pdf.