

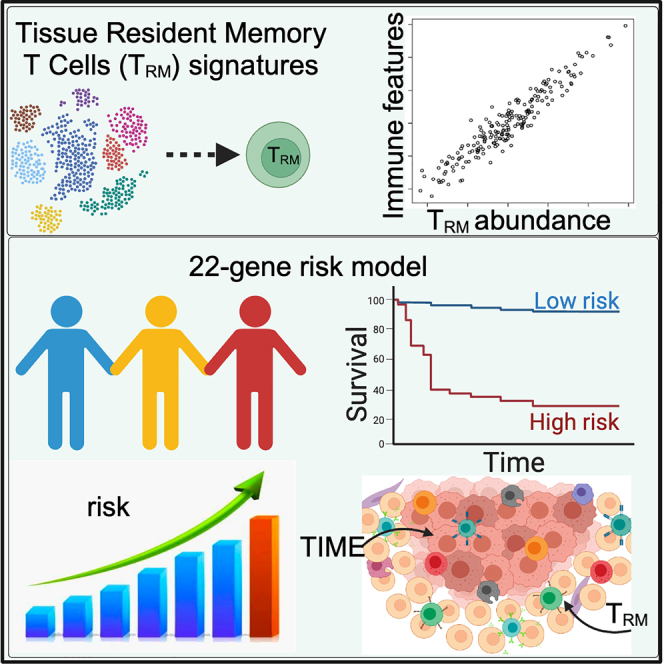
Summary
Tissue-resident memory T cells (TRM) are a specialized T cell population residing in peripheral tissues. The presence and potential impact of TRM in the tumor immune microenvironment (TIME) remain to be elucidated. Here, we systematically investigated the relationship between TRM and melanoma TIME based on multiple clinical single-cell RNA-seq datasets and developed signatures indicative of TRM infiltration. TRM infiltration is associated with longer overall survival and abundance of T cells, NK cells, M1 macrophages, and memory B cells in the TIME. A 22-gene TRM–derived risk score was further developed to effectively classify patients into low- and high-risk categories, distinguishing overall survival and immune activation, particularly in T cell-mediated responses. Altogether, our analysis suggests that TRM abundance is associated with melanoma TIME activation and patient survival, and the TRM-based machine learning model can potentially predict prognosis in melanoma patients.
Subject areas: Microenvironment, Immunology, Cancer
Graphical abstract
Highlights
-
•
A signature to evaluate tissue-resident memory T cells (TRM) abundance in melanoma
-
•
TRM influence the tumor immune microenvironment (TIME)
-
•
TRM are associated with TILs, NK cells, and M1 macrophages in TIME
-
•
A TRM-derived 22-gene risk score associated with melanoma patient prognosis
Microenvironment; Immunology; Cancer
Introduction
Tissue-resident memory T cells (TRM) are a specialized population of T cells that reside in peripheral tissues, such as the skin, gut, and lungs.1,2 TRM are present in many cancer types,3,4,5,6,7,8,9,10 including melanoma, the most malignant skin cancer that develops from the pigment-producing cells known as melanocytes.11 TRM possess unique characteristics that set them apart from circulating T cells, including the ability to persist long-term in tissues without recirculating.12 Recent studies suggest that TRM contribute to cancer immunosurveillance13,14,15,16,17,18,19,20,21,22 and may play a role in immune response in melanoma.22,23,24,25 TRM cells are strategically positioned at the interface between the tumor and its surrounding tissue, enabling interaction with both tumor cells and other immune cells within the tumor immune microenvironment (TIME).12,26,27 The presence and functional properties of TRM cells within the tumor microenvironment have sparked significant interest in harnessing their potential for cancer immunotherapy.20 Evaluating the TRM cell abundance and understanding the role of TRM that regulate the development and maintenance of the immune cells within the TME is also crucial for developing effective immunotherapeutic interventions. The gene signature derived from single-cell RNA sequencing (scRNA-seq) captures a more comprehensive range of genetic information, providing a more nuanced understanding of TRM cell characteristics and behavior. This holistic view allows for a more accurate evaluation of TRM cell abundance and a more robust prognostic assessment for melanoma patients, compared to the limited insight provided by the expression of just one or two TRM marker genes.9,28 However, the presence and potential impact of TRM in the TIME remain to be elucidated.24,25 Reports were mixed regarding whether the TRM promotes better outcomes in melanoma patients.3,8,11 A systematic study examining all available patient scRNA-seq and bulk RNA-seq data to comprehensively assess the impact of TRM on the remaining immune cells in the tumor milieu and to extract TRM-related molecular signatures that can provide prognostic value in clinical settings have been lacking.
In this study, we aimed to bridge this gap and further enhance the impact of TRM by exhaustively analyzing all available single-cell and bulk RNA-seq datasets to extract a minimal but comprehensive TRM-related TRM signatures representative of TRM impact in tumor milieu and providing prognostic value of patient risk and survival. We validated the melanoma TRM signatures obtained from scRNA-seq of melanoma patients, confirming their representation of TRM cell abundance in these patients. We then investigated the relationship between TRM, TIME, and patients’ prognosis. As the role of TRM cells in cancer immunity is complex and context-dependent, we systematically evaluated TRM and its TIME in available melanoma single-cell and bulk RNA-seq datasets, discovering that TRM presence and signatures are consistently associated with various immune cell populations in TIME and with patient prognosis. Immune-related genes (IRGs) associated with TRM characterized immune features predictive of overall survival. A 22-gene risk score derived from TRM signatures across independent datasets stratified low- and high-risk melanoma patients. Furthermore, a 22-gene TRM-derived risk score was developed to effectively classify patients into low- and high-risk categories, distinguishing overall survival and investigating the potential mechanisms underlying the role of TRM in the prognosis of melanoma patients. The workflow of the present study is illustrated in Figure 1.
Figure 1.

The workflow of the entire study
Eleven distinct melanoma TRM signatures reflective of TRM infiltration have been extracted utilizing multiple independent single-cell RNA-seq data from human melanoma samples. The role of TRM infiltration in the TIME and the prognosis of the melanoma patients were systematically investigated. A LASSO Cox regression prognostic model for survival was developed and tested against the TCGA dataset and independently validated across multiple melanoma cohorts. The effects of the TRM infiltration on melanoma patient TIME were also investigated. Figure created with BioRender.com.
Results
TRM signatures in melanoma
Two independent human melanoma single-cell RNA-seq cohorts29,30 were used to generate TRM gene expression signatures and infer TRM abundance in melanoma patients. We isolated all the TRM clusters designated by the study authors and derived 11 TRM cluster-specific maker gene sets, as shown in Table S1. For each TRM cluster, we generated a gene expression signature (see STAR Methods) and compared the relationship between our TRM signatures and the proportion of TRM in melanoma patients.
The 11 selected TRM signatures are positively correlated with TRM proportion in melanoma, which was demonstrated by building a simulated bulk RNA-seq cohort and calculating the TRM cells’ infiltration using the selected 11 TRM signatures (Figure 2A). The high correlation between the TRM signatures and the proportion of TRM indicates that these signatures represent TRM abundance in melanoma, and TRM signature scores are associated with high TRM abundance in melanoma.
Figure 2.

The TRM signatures can represent the abundance of the TRM cells in melanoma
(A) The correlation of the TRM signatures and the proportion of the TRM cells in the single cell RNA-seq cohort.29
(B) The heatmap of the TRM signatures infiltration score in the TCGA-SKCM cohort.
(C) Survival result of the high TRM cells infiltration group and low TRM cells infiltration group. HR = hazard ratio. P = Log rank p value.
We then clustered TCGA-SKCM patients based on their TRM infiltration profile and observed two groups of patients who had either high (red) or low (blue) TRM signature scores (Figure 2B). The survival results showed that the high TRM signature group had a better prognosis than the low TRM signature group (Figure 2C).
TRM are associated with other immune cells in TIME and patient survival
We next assessed the correlation of the TRM signatures with other immune cells in the TIME. TRM were positively correlated with CD8+ and CD4 T cells, NK cells (especially the activated NK cells), monocyte-derived macrophages, and memory B cells according to multiple independent methods that evaluate immune cell infiltration in TIME, such as BASE31 and xCell32 (Figures 3A and S1A).
The majority of TRM marker genes, immune checkpoint genes, and immune-related genes are associated with the TRM signatures, suggesting the role of TRM in modulating TIME (Figure S2). The 11 TRM signatures were strongly associated with overall survival in melanoma patients with hazard ratios less than 1.033 (Figures 3B, 3C, and S1B). TRM marker genes are highly expressed in TRM and were positively associated with patient survival (Figure S2D). The inferred TRM cell abundance was correlated with TRM marker and immune-related genes (Figure S2B), including immune checkpoint genes, such as PD1(PDCD1), PDL1(CD274), CTLA4, LAG3, TIGIT, HAVCR2(TIM3), BTLA, CD40, TNFRSF4(OX40), TNFRSF18(GITR), and so on (Figure S2C). Hence, higher TRM infiltration inferred from the signatures was associated with longer overall survival.
Figure 3.

TRM cell abundance is positively associated with melanoma prognosis
The PC1 score can represent the 11 TRM signatures and the TRM cell proportion in melanoma.
(A) The correlation of TRM signatures with immune infiltration in TCGA-SKCM.
(B) Forest plot showing almost all the 11 TRM signatures were significantly associated with good prognosis.
(C) Survival analysis for TRM signatures.
(D) The correlation of the TRM signatures with each other.
(E) Principal Component Analysis (PCA) on the expression of the 11 TRM signatures in TCGA-SKCM patients.
(F) The correlation of PC1 and the TRM cell abundance.
(G) Kaplan-Meier plot showing the association between overall survival and Principal Component 1 (PC1) in TCGA-SKCM.
(H) Forest plot depicting hazard ratios of univariate Cox regression models evaluating the association between overall survival and several clinical variables.
The TRM signatures from the different datasets and clusters were highly correlated, suggesting high overlap in the signals captured by these different signatures (Figure 3D), and these results were supported in another independent dataset (GSE65904). These TRM signatures were highly correlated with each other and positive for patient prognosis. TRM were also positively correlated with CD8+ and CD4 T cells, NK cells, and memory B cells. These results solidify the reliability and persuasiveness of this study (Figure S3). Then, we utilized dimensionality reduction (PCA) to unify these signatures to generate a single signature that represents all TRM signatures. The first principal component (PC1) was highly correlated with all TRM signatures and captured 82.86% of the variation among patients; the TCGA-SKCM patients can be divided into high PC1 (red) and low PC1(blue) groups, by using the median value of PC1 as the cutoff34 (Figure 3E). The PC1 was highly correlated with the TRM cell abundance in melanoma (Figure 3F). The patients with high PC1 scores archived significantly longer survival (Figure 3G). Notably, the PC1 score was highly associated with survival when evaluated in a multivariate model with several clinical variables (Figure 3H).
TRM abundance (PC1) is associated with immune cells, immune checkpoint genes, and immune regulatory pathways
The melanoma TIME is usually infiltrated by different kinds of immune cells.35,36 We investigated the relationship between the TRM cells and the melanoma TIME. The results suggest that TRM cell abundance in melanoma is associated with the expression of immune checkpoint genes (Figure 4A) and TRM main marker genes expression (Figure 4B), such as CD69 and ITGAE (Figure 4C). TRM abundance is also correlated with immune regulatory pathways and immune response, such as the tumor-infiltrating lymphocytes (TIL), leukocyte, stromal cells, lymphocyte infiltration, T cell receptor (TCR) richness and Shannon, and IFN-γ response (Figures 4D–4F). Moreover, the tumors with high TRM cell abundance may correspond to a high mutation burden and rich neoantigens (Figure 4G), which suggests that these tumors could be more sensitive to immune checkpoint inhibiter (ICI) therapies.37
Figure 4.

TRM cell abundance is associated with the expression of immune checkpoint genes and immune regulatory pathways
(A) The Spearman correlation coefficient (SCC) between PC1 and the expression of several immune checkpoint genes.
(B) SCC between PC1 and marker genes expressed in TRM.
(C) SCC between PC1 and CD69 and ITGAE(CD103).
(D) SCC between PC1 and immune infiltration score.
(E) SCC between PC1 and lymphocyte infiltration.
(F) SCC between PC1 and TCR richness.
(G) SCC between PC1 and mutations.
TRM are correlated with CD8+ T cells, Th1 cells, and patients survival time, as shown in the Figure S4A. TRM cells were also correlated with Th1 cells, lymphocytes, M1 macrophages, and activated NK cells (Figure S4B). At the same time, we noticed that TRM were negatively correlated with mast cells, M0 and M2 macrophages (Figure S4C).
A 22-gene risk score for improving prognostic prediction in melanoma
As shown previously, TRM cell infiltration is associated with survival independently of clinical variables (Figure 3H). However, the inference of TRM abundance throughout our studies has been based on many genes contributing to the final infiltration score. Aiming to reduce the number of genes into a simpler signature that can potentially be used to risk-stratify melanoma patients in the clinic, we used the Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression for feature (gene) selection and ended up with a 22-gene risk score (STAR Methods, Figure 5A). All of the genes were significantly correlated with patient survival (Figure 5B). Many of these TRM signature genes are related to tumor progression or immune pathways, such as the BCAS4,38 CAMK4,39 GIMAP2,40,41 IFITM,42 KPNA2,43 MKI67,44 NCCRP1,45 NCL,46 PRKAR2B,47 TTC39C.48
Figure 5.

Stratified survival analysis of the 22-gene risk score model
Kaplan-Meier survival analysis for the patients in independent datasets by the 22-gene risk score model.
(A) Development of a TRM risk score for GLIOMA by LASSO Cox regression analysis.
(B) Hazard ratios of univariate Cox regression models evaluating the association between overall survival and the 22 genes.
(C) Patients in the TCGA-SKCM, GSE65904, and GSE8401 cohorts.
(D) Forest plot of hazard ratios derived from multivariable Cox regression analysis including the listed variables. The y axis is the risk group (High vs. Low risk), age (>50 vs. ≤ 50), gender (Male vs. Female), tumor stages (High vs. Low stage), and TNM cancer staging (High vs. Low stages of T, N, and M, respectively) of melanoma patients. For the TNM cancer staging system, TNM stands for Tumor, Nodes, and Metastasis. T is assigned based on the extent of involvement at the primary tumor site, N reflects the extent of involvement in regional lymph nodes, and M indicates for distant spread.
(E) Time-dependent of the TCGA-SKCM.
The expression of these genes was weighted and summed to generate a final risk score, . The refers to the weight of each gene in the model. A higher value of means that this gene is more important to the patients’ prognosis, and the weight is higher in this model. represents the expression value of the gene in a patient.34,49,50,51,52 This signature was significantly associated with melanoma survival in the TCGA training dataset, and in the independent datasets, such as the GSE65904 and GSE8401, higher risk scores were associated with shorter overall survival (Figures 5C and S5). Notably, the risk score was highly associated with survival when evaluated in a multivariate model with several clinical variables. This TRM-derived risk score seemed to perform better than other main clinical features, such as age, gender, tumor stages, and TNM cancer staging (T, N, and M, respectively), in melanoma patients (Figure 5D). For the TNM cancer staging system, TNM stands for Tumor, Nodes, and Metastasis. T is assigned based on the extent of involvement at the primary tumor site, N reflects the extent of involvement in regional lymph nodes, and M indicates distant spread. These results suggest that the LASSO Cox regression model is effective for the prognostic prediction of melanoma patients, with the risk model outperforming other clinical variables. The study aimed to generate a clinically useful tool by significantly reducing the number of genes in a signature that can be used to risk-stratify melanoma patients.
Prognostic significance of risk model in melanoma patients based on clinicopathological characteristics
We compared the survival differences between the high- and low-risk patients in main clinical features, such as gender, age, and stages, respectively. The stratification analysis demonstrated again that a patient with a high-risk score has a significantly poor prognosis, as shown in Figure 6. The results showed that low-risk patients consistently achieved better prognoses in male and female (Figures 6A and 6B), young and old populations (Figures 6C and 6D), tumor stages (Figures 6E and 6F), and TNM cancer staging (T, N, and M, Figures 6G‒6L, respectively). Taken together, the 22-gene risk score demonstrated a significant independent prognostic risk factor for patients with melanoma.
Figure 6.

Stratified survival analysis of the 22-gene risk score model in clinicopathological factors
(A) The risk model in male.
(B) The risk model in female.
(C) The risk model in old (age >50).
(D) The risk model in young (age ≤50).
(E) The risk model in low tumor stage patients.
(F) The risk model in high tumor stage patients. For the TNM cancer staging system, TNM stands for Tumor, Nodes, and Metastasis. T is assigned based on the extent of involvement at the primary tumor site, N reflects the extent of involvement in regional lymph nodes, and M indicates for distant spread.
(G) The risk model in low T stage patients.
(H) The risk model in high T stage patients.
(I) The risk model in low N stage patients.
(J) The risk model in high N stage patients.
(K) The risk model in low M stage patients.
(L) The risk model in high M stage patients.
Low-risk patients with significantly up-regulated memory T cell and immune-related pathways in melanoma
GSEA analysis revealed that many T cells and immune response-related pathways are significantly up-regulated in low-risk patients (Figure 7). For example, T cell differentiation, co-stimulation, proliferation, and activation-related pathways were significantly up-regulated in low-risk patients. Especially Th1, Th2, Th17, antigen, T cell receptor signaling, and immune-check point (PD1/PD-L1) related pathways are also activated in the low-risk patients (Figure 7A). Memory T cell differentiation, T cell selection, T cell co-stimulation, T cell migration, T cell regulation, T cell activation, and memory T cell-related immune response pathways were also significantly changed in melanoma patients (Figure 7). Low-risk patients had high TRM cell infiltration (Figure 7B). These pathways related to memory T cells and immune response were also upregulated in patients with high TRM cell abundance compared to those with low TRM cell abundance (Figure S6). T cells mediated immune response, especially TRM-related pathways, were up-regulated in the low-risk patients. Malignant skin cells, especially melanocyte differentiation, proliferation, and migration, were also downregulated with increasing TRM abundance (Figure 7C). These findings suggested that melanoma patients with low-risk scores (higher TRM infiltration) had more immune activation, especially in the T cell-mediated response.
Figure 7.

The Gene Set Enrichment Analysis (GSEA) analysis
(A) Low-risk patients with significant up-regulated memory T cell and immune-related pathways in the melanoma.
(B) The relationship between the PC1 and risk score.
(C‒F) GSEA indicated the tendency of individual pathways to be up- or down-regulated in response to high PC1 compared to low PC1 patients or low risk compared to high-risk patients. T cell-mediated immunity pathway is up-regulated (C), memory T cell differentiation pathway is up-regulated (D), and skin epidermis development and melanocyte differentiation-related pathways are down-regulated (E and F, respectively).
Discussion
The melanoma TIME is infiltrated by various immune cells.35,36 TRM are a specialized population of T cells that reside in peripheral tissues, especially, the skin.1,2 From scRNA-seq datasets of human melanoma, we constructed TRM signatures accurately representing TRM cell abundance in melanoma patients. A high TRM signature score was found to be associated with a better prognosis (Figure 2). TRM signatures were correlated with CD8+ and CD4+ T cells, NK cells, monocytes, memory B cells, immune checkpoint genes, and immune-related genes. These analyses suggest that the presence of TRM may indicate a more active melanoma TIME, which leads to better patient outcomes.
TRM signatures derived from different datasets were highly correlated with one another, suggesting high overlap in the signals captured by these different signatures. PC1 generated by the PCA analyses captures overall TRM infiltration. The PC1 score was highly associated with patients’ survival and better than other clinical variables, when being evaluated in a multivariate model (Figure 3), indicating its potential as a prognostic marker for melanoma patients. The findings significantly highlight the role of TRM cells in melanoma prognosis, and the potential of TRM signatures as prognostic markers. TRM signatures can potentially indicate which patients may benefit more from immunotherapies.
A higher TRM abundance seems to indicate a more active TIME suggested by the correlation of PC1 across different signatures with T cell activation, TIL regulation, leukocyte, stromal cells, lymphocyte infiltration, and immune response-related pathways. Based on the PC1 signature, we developed a high-precision 22-gene risk score that can stratify high and low-risk patients irrespective of the predictive ability of clinical variables. This risk score was validated in independent datasets and was negatively associated with prognosis. Many of these TRM signature genes play important roles in the T cell activating, antigen-presenting, tumor progression, and immune response, such as the BCAS4, CAMK4, GIMAP2, IFITM1, KPNA2, MKI67, NCCRP1, NCL, PRKAR2B, and TTC39C.38,39,40,41,42,43,44,45,46,47,48 Low-risk patients could with high TRM cell infiltration, and our results also showed that the T cell (such as the memory T cells, Th1 cells, Th2 cells, and Th17 cells), immune checkpoint, and other immune regulation-related pathways were significantly up-regulated in the low-risk or high TRM infiltration patients, which could be further investigated to understand the interplay between TRM and the TIME.
Limitations of the study
Although our study confirmed an association between TRM cell infiltration and melanoma prognosis, the immune inference methods we used cannot distinguish causal mechanisms behind this association. Since the abundance of TRM cells are also associated with the presence of other immune cell types such as T cells and macrophages, the observed association may result from collective effects in the TIME.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| RNA-seq data and clinical information for TCGA-SKCM | Broad Institute | TCGA (https://gdac.broadinstitute.org/) |
| TCGA mutation annotation format (MAF) files | Genomic Data Commons | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Bulk RNA-seq and related clinical information of 215 melanoma patients validation data | Cabrita et al.53 | GEO: GSE65904 |
| Bulk RNA-seq and related clinical information of 50 melanoma patients validation data | Xu et al.54 | GEO: GSE8401 |
| Immune-related genes (IRGs) | Charoentong et al.55 Bindea et al.56 Xu et al.57 |
Table S6 of Charoentong et al.55Table S2 of Bindea et al.56Table S1A of Xu et al.57 |
| Software and algorithms | ||
| BASE | Chen et al.31 | |
| R (v4.2.0) | R CRAN | https://cran.r-project.org/ |
| glmnet R package (v4.1-2) | Friedman et al.58 | https://cran.r-project.org/web/packages/glmnet/index.html |
| tidyr R package (v1.1.3) | Wickham59 | https://cran.r-project.org/web/packages/tidyr/index.html |
| dplyr R package (v1.0.7) | Wickham et al.60 | https://cran.r-project.org/web/packages/dplyr/index.html |
| data.table R package (v1.14.0) | Dowle et al.61 | https://cran.r-project.org/web/packages/data.table/index.html |
| survival R package (v3.2-11) | Therneau et al.62 | https://cran.r-project.org/web/packages/survival/index.html |
| survivalROC R package (v1.0.3) | Heagerty et al.63 | https://cran.r-project.org/web/packages/survivalROC/index.html |
| xCELL | Aran et al.32 | https://github.com/dviraran/xCell |
| fgsea R package (v1.26.0) | Korotkevich et al.64 | https://cran.r-project.org/web/packages/fgsea/index.html |
| prcomp R package (v3.6.2) | Venables et al.65 | https://cran.r-project.org/web/packages/prcomp/index.html |
| factoextra R package (v1.0.7) | Kassambara et al.66 | https://github.com/kassambara/factoextra |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xiling Shen (xiling.shen@terasaki.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data available in this study is publicly available. The RNA-seq data and clinical information for TCGA-SKCM were obtained from TCGA on FireBrowse (gdac.broadinstitute.org/). TCGA mutation annotation format (MAF) files for gene mutation analyses were obtained from https://gdc.cancer.gov/about-data/publications/pancanatlas. Accession numbers for these datasets are listed in the key resources table. All codes in this study and any additional information required in this paper is available from the lead contact upon request.
Method details
Data utilization
Level 3 TCGA RNA-seq data and clinical information for skin cutaneous melanoma (SKCM) were obtained from TCGA on FireBrowse (gdac.broadinstitute.org/). TCGA MAF files for gene mutation analyses were obtained from https://gdc.cancer.gov/about-data/publications/pancanatlas. All genes in which non-silent mutations occurred were considered to be mutated. Total mutation burden (TMB) was represented as the sum of all non-silent mutations in a given TCGA sample. The copy number variation (CNV) data were also downloaded from Firehose, which provided DNA segments that deviated from normal copy numbers (copy number = 2) in each tumor sample. The CNV burden of a sample was calculated as the total size (in bp) of genomic regions covered by those segments. Macrophage regulation scores, leukocyte and lymphocyte infiltration scores, and IFNγ response and TGFβ response scores for TCGA-SKCM samples were downloaded as a supplementary file (Table S2) from prior work.67 The gene expression of 215 melanoma patients and related clinical information data were collected from the GSE65904 of the open Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65904). The gene expression of 50 melanoma patients and related clinical information data were collected from the GSE8401 of the open Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE8401).
Curation of immune-related genes (IRGs)
Immune-related genes (IRGs) were obtained from the Supplementary Table 6 of Charoentong et al.,55 Table S2 of Bindea et al.,56 and Table S1A of Xu et al.57 All genes from immune cells were collected (marker genes attributed to cancer cells were excluded) and combined into a single list of 831 IRGs genes.
Immune cell inference
Immune infiltration scores of six immune cells were calculated using Binding Association with Sorted Expression (BASE),31 a rank-based gene set enrichment method. Previous publications have detailed and validated immune cell infiltration using this method.31,34,68,69,70,71 BASE uses immune cell-specific weight profiles and patient gene expression data to infer immune cell infiltration for each patient and immune cell type. Briefly, BASE orders genes for a patient's gene expression profile from high to low expression and then uses weights from each immune cell weight profile to weigh the patient's gene expression values. BASE calculates two running sums, one representing the cumulative distribution of the patient's weighted gene expression values (foreground function) and another representing the cumulative distribution of the patient's complementary weighted (1-weight) gene expression values (background function). In the presence of a high amount of infiltrate from a specific immune cell type, the foreground function increases quickly, as the highly expressed genes in a patient’s profile tend to be the ones with high immune cell weights, while the background function increases slowly. The maximal absolute difference between the foreground and background functions represents the immune infiltration level and, after a normalization procedure, results in the final immune infiltration score. Full details on the calculation and validation of the immune infiltration scores can be found in.68,69 Similarly, BASE was used to calculate single cell-based TRM scores using TRM signatures (see next section).
Generation of TRM signatures
Melanoma single cell RNA-seq datasets from human melanoma were obtained from previous publications.29,30 Cluster annotations were also obtained from these publications. For each melanoma cluster, a list of marker genes was provided by identifying genes that are over-expressed in the corresponding cluster than all the other clusters. These cluster-specific marker gene sets were used as TRM signatures. In total, 11 human TRM signatures were defined, including 8 CD8+ sources, 1 CD4+ source, 1 lamina propria (LP) source, and 1 generalized TRM signature. Given a melanoma gene expression dataset, the BASE algorithm was used to calculate sample-specific TRM scores for each signature. The TRM signatures were represented as gene sets without assigning weights to the member genes. In this case, the BASE algorithm degenerated into a method like the single-sample GSEA analysis.72 A high TRM score indicates that the corresponding TRM cells are strongly infiltrated in the tumor.
Lasso Cox-regression
The TCGA SKCM dataset was randomly divided into a training and testing set with a 1:1 ratio. The training set was analyzed to identify potential prognostic genes, and both the testing set and the entire set were used for validation. First, univariate Cox-proportional hazards regression analysis was used to evaluate the association between the expression of the 11 TRM signatures genes, TRM marker genes, IRGs and their according overall survival. Genes with a P value of < 0.05 based on the log-rank test were selected as candidate genes. Second, Lasso Cox-regression analysis from the glmnet R package (v4.1-2)58 was employed to screen these genes most associated with overall survival in a multivariate model, which resulted in 22 genes (BCAS4, CAMK4, CCT6B, CD33, CTBS, GIMAP2, HAPLN3, HCP5, HSPA2, HSPA7, HSPB6, IFITM1, KPNA2, MKI67, NCCRP1, NCL, NOTCH3, PRG2, PRKAR2B, SLC25A12, TMBIM6, and TTC39C). These 22 genes composed the final risk score, which is described as follows:
The refers is the weight of each gene in the model and represents the expression value of the gene in a patient.34,49,50,51,52 The expression of genes with positive value worsens patient prognosis whereas negative values enhances patient prognosis and survival. The values for all 22 genes are shown in the Table S3.
Survival analysis
For univariate and multivariate survival analyses, Cox proportional hazards models were calculated using the “coxph” function from the R “survival” package (v3.2-11).62 Survival curves were visualized using Kaplan-Meier curves using the “survfit” function from the R “survival” package. Median immune cell infiltration scores were used to stratify patients into “high” and “low” groups for univariate analyses. For multivariate analyses, an infiltration score of 0 was used as a separator to stratify patients into “high” and “low” groups. Differences in survival distributions in each Kaplan-Meier plot were calculated using a log-rank test using the “survdiff” function from the R “survival” package.
Enrichment pathway analysis
The fgsea R package (v1.26.0),64 was used to perform the gene set enrichment analysis (GSEA) with hallmark pathways from the Molecular Signatures Database (MSigDB)73 to investigate which hallmark pathways were significantly (adjust P value < 0.05).
Quantification and statistical analysis
The major analysis and package development were based on R (v4.2.0)64 and R libraries (tidyr R package (v1.1.3),59 dplyr R package (v1.0.7),60 data.table R package (v1.14.0),61 and so on). The Spearman correlation coefficient (SCC) was reported for all correlation analyses as the assumptions underlying the Pearson correlation (i.e., normal distribution, homoscedasticity, or linearity) were not met. SCC was calculated using the R function cor, and significance was assessed using the cor.test. Principal component analysis (PCA) was performed using the prcomp R function by the prcomp R package (v3.6.2).65 Principal component coordinates for each sample were extracted using the factoextra R package (v1.0.7).66 Principal component 1 (PC1) was used to represent TRM infiltration. The sensitivity and specificity of the diagnostic and prognostic prediction models were analyzed by the ROC curve and quantified based on the area under the ROC curve (AUC), which were calculated by survivalROC R package (v1.0.3).63 All statistical tests were two-sided; P values < 0.05 were considered statistically significant.
Acknowledgments
This work is supported by the National Institutes of Health, USA (NIH) R01 DK119795 and R35 GM122465. This work is was also funded by the Cancer Prevention Research Institute of Texas (CPRIT) (RR180061). We would like to give special thanks to Qiang Huang and other members of the Xiling Shen and Chao Cheng labs for their valuable discussions and critical feedback. We especially thank Xiuying Li, Chenyang Li, and Xinlei Gao for the valuable suggestions. We would like to thank BioRender.com for providing valuable materials in Figure 1.
Author contributions
C.C., C.M.J., and X.L.S. conceived the project. C.M.J., C.-C.C., J.R.L., A.S., and X.G. prepared and analyzed the results. C.M.J. and C.C. evaluated the conclusions and wrote the manuscript. C.M.J., C.-C.C., X.G., V.J., C.C., and X.L.S. reviewed and revised the content. All authors read and approved the final manuscript.
Declaration of interests
Author C.-C.C. was employed by the company Biomap, Inc. The remaining authors declare that the research was conducted without commercial or financial relationships that could be construed as a potential conflict of interest.
Published: February 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109277.
Contributor Information
Chongming Jiang, Email: chongming.jiang@terasaki.org.
Chao Cheng, Email: chao.cheng@bcm.edu.
Xiling Shen, Email: xiling.shen@terasaki.org.
Supplemental information
References
- 1.Szabo P.A., Miron M., Farber D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4:eaas9673. doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shin H., Iwasaki A. Tissue-resident memory T cells. Immunol. Rev. 2013;255:165–181. doi: 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganesan A.-P., Clarke J., Wood O., Garrido-Martin E.M., Chee S.J., Mellows T., Samaniego-Castruita D., Singh D., Seumois G., Alzetani A., et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017;18:940–950. doi: 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koh J., Kim S., Kim M.Y., Go H., Jeon Y.K., Chung D.H. Prognostic implications of intratumoral CD103+ tumor-infiltrating lymphocytes in pulmonary squamous cell carcinoma. Oncotarget. 2017;8:13762–13769. doi: 10.18632/oncotarget.14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 6.Komdeur F.L., Wouters M.C.A., Workel H.H., Tijans A.M., Terwindt A.L.J., Brunekreeft K.L., Plat A., Klip H.G., Eggink F.A., Leffers N., et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget. 2016;7:75130–75144. doi: 10.18632/oncotarget.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webb J.R., Milne K., Nelson B.H. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol. Res. 2015;3:926–935. doi: 10.1158/2326-6066.CIR-14-0239. [DOI] [PubMed] [Google Scholar]
- 8.Lohneis P., Sinn M., Bischoff S., Jühling A., Pelzer U., Wislocka L., Bahra M., Sinn B.V., Denkert C., Oettle H., et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur. J. Cancer. 2017;83:290–301. doi: 10.1016/j.ejca.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Savas P., Virassamy B., Ye C., Salim A., Mintoff C.P., Caramia F., Salgado R., Byrne D.J., Teo Z.L., Dushyanthen S., et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z.Q., Milne K., Derocher H., Webb J.R., Nelson B.H., Watson P.H. CD103 and intratumoral immune response in breast cancer. Clin. Cancer Res. 2016;22:6290–6297. doi: 10.1158/1078-0432.CCR-16-0732. [DOI] [PubMed] [Google Scholar]
- 11.Djenidi F., Adam J., Goubar A., Durgeau A., Meurice G., de Montpréville V., Validire P., Besse B., Mami-Chouaib F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015;194:3475–3486. doi: 10.4049/jimmunol.1402711. [DOI] [PubMed] [Google Scholar]
- 12.Crowl J.T., Heeg M., Ferry A., Milner J.J., Omilusik K.D., Toma C., He Z., Chang J.T., Goldrath A.W. Tissue-resident memory CD8+ T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat. Immunol. 2022;23:1121–1131. doi: 10.1038/s41590-022-01229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mami-Chouaib F., Blanc C., Corgnac S., Hans S., Malenica I., Granier C., Tihy I., Tartour E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer. 2018;6:87. doi: 10.1186/s40425-018-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menares E., Gálvez-Cancino F., Cáceres-Morgado P., Ghorani E., López E., Díaz X., Saavedra-Almarza J., Figueroa D.A., Roa E., Quezada S.A., Lladser A. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-12319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdeljaoued S., Arfa S., Kroemer M., Ben Khelil M., Vienot A., Heyd B., Loyon R., Doussot A., Borg C. Tissue-resident memory T cells in gastrointestinal cancer immunology and immunotherapy: Ready for prime time? J. Immunother. Cancer. 2022;10:e003472. doi: 10.1136/jitc-2021-003472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Granier C., Blanc C., Karaki S., Tran T., Roussel H., Tartour E. Tissue-resident memory T cells play a key role in the efficacy of cancer vaccines. OncoImmunology. 2017;6 doi: 10.1080/2162402X.2017.1358841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosato P.C., Beura L.K., Masopust D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017;22:44–50. doi: 10.1016/j.coviro.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yenyuwadee S., Sanchez-Trincado Lopez J.L., Shah R., Rosato P.C., Boussiotis V.A. The evolving role of tissue-resident memory T cells in infections and cancer. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abo5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christian L.S., Wang L., Lim B., Deng D., Wu H., Wang X.-F., Li Q.J. Resident memory T cells in tumor-distant tissues fortify against metastasis formation. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okła K., Farber D.L., Zou W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021;218:e20201605. doi: 10.1084/jem.20201605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craig D.J., Creeden J.F., Einloth K.R., Gillman C.E., Stanbery L., Hamouda D., Edelman G., Dworkin L., Nemunaitis J.J. Resident Memory T Cells and Their Effect on Cancer. Vaccines. 2020;8:562. doi: 10.3390/vaccines8040562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumauthioz N., Labiano S., Romero P. Tumor resident memory T cells: New players in immune surveillance and therapy. Front. Immunol. 2018;9:2076. doi: 10.3389/fimmu.2018.02076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pizzolla A., Keam S.P., Vergara I.A., Caramia F., Thio N., Wang M., Kocovski N., Tantalo D., Jabbari J., Au-Yeung G., et al. Tissue-resident memory T cells from a metastatic vaginal melanoma patient are tumor-responsive T cells and increase after anti-PD-1 treatment. J. Immunother. cancer. 2022;10:e004574. doi: 10.1136/jitc-2022-004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S.L., Buzzai A., Rautela J., Hor J.L., Hochheiser K., Effern M., McBain N., Wagner T., Edwards J., McConville R., et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature. 2019;565:366–371. doi: 10.1038/s41586-018-0812-9. [DOI] [PubMed] [Google Scholar]
- 25.Ganesan A.P., Ottensmeier C.H.H. Melanoma-reactive T cells take up residence. Nat. Cancer. 2021;2:253–255. doi: 10.1038/s43018-021-00189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams J.B., Kupper T.S. Resident Memory T Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020;1273:39–68. doi: 10.1007/978-3-030-49270-0_3. [DOI] [PubMed] [Google Scholar]
- 27.Lange J., Rivera-Ballesteros O., Buggert M. Human mucosal tissue-resident memory T cells in health and disease. Mucosal Immunol. 2022;15:389–397. doi: 10.1038/s41385-021-00467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrne A., Savas P., Sant S., Li R., Virassamy B., Luen S.J., Beavis P.A., Mackay L.K., Neeson P.J., Loi S. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nat. Rev. Clin. Oncol. 2020;17:341–348. doi: 10.1038/s41571-020-0333-y. [DOI] [PubMed] [Google Scholar]
- 29.Luoma A.M., Suo S., Williams H.L., Sharova T., Sullivan K., Manos M., Bowling P., Hodi F.S., Rahma O., Sullivan R.J., et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell. 2020;182:655–671.e22. doi: 10.1016/j.cell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J., Zhao Y., Shirai K., Molodtsov A., Kolling F.W., Fisher J.L., Zhang P., Yan S., Searles T.G., Bader J.M., et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat. Cancer. 2021;2:300–311. doi: 10.1038/s43018-021-00180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng C., Yan X., Sun F., Li L.M. Inferring activity changes of transcription factors by binding association with sorted expression profiles. BMC Bioinf. 2007;8:452. doi: 10.1186/1471-2105-8-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aran D., Hu Z., Butte A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spruance S.L., Reid J.E., Grace M., Samore M. Hazard ratio in clinical trials. Antimicrob. Agents Chemother. 2004;48:2787–2792. doi: 10.1128/AAC.48.8.2787-2792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaafsma E., Jiang C., Nguyen T., Zhu K., Cheng C. Microglia-Based Gene Expression Signature Highly Associated with Prognosis in Low-Grade Glioma. Cancers. 2022;14 doi: 10.3390/cancers14194802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giraldo N.A., Sanchez-Salas R., Peske J.D., Vano Y., Becht E., Petitprez F., Validire P., Ingels A., Cathelineau X., Fridman W.H., Sautès-Fridman C. The clinical role of the TME in solid cancer. Br. J. Cancer. 2019;120:45–53. doi: 10.1038/s41416-018-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu D., Yang X., Wu X. Tumor Immune Microenvironment Characterization Identifies Prognosis and Immunotherapy-Related Gene Signatures in Melanoma. Front. Immunol. 2021;12:663495. doi: 10.3389/fimmu.2021.663495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jardim D.L., Goodman A., de Melo Gagliato D., Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39:154–173. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabaie H., Talebi M., Gharesouarn J., Asadi M.R., Jalaiei A., Arsang-Jang S., Hussen B.M., Taheri M., Jalili Khoshnoud R., Rezazadeh M. Identification and Analysis of BCAS4/hsa-miR-185-5p/SHISA7 Competing Endogenous RNA Axis in Late-Onset Alzheimer’s Disease Using Bioinformatic and Experimental Approaches. Front. Aging Neurosci. 2022;14 doi: 10.3389/fnagi.2022.812169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koga T., Kawakami A. The role of CaMK4 in immune responses. Mod. Rheumatol. 2018;28:211–214. doi: 10.1080/14397595.2017.1413964. [DOI] [PubMed] [Google Scholar]
- 40.Limoges M.A., Cloutier M., Nandi M., Ilangumaran S., Ramanathan S. The GIMAP Family Proteins: An Incomplete Puzzle. Front. Immunol. 2021;12:679739. doi: 10.3389/fimmu.2021.679739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komatsu M., Saito K., Miyamoto I., Koike K., Iyoda M., Nakashima D., Kasamatsu A., Shiiba M., Tanzawa H., Uzawa K. Aberrant GIMAP2 expression affects oral squamous cell carcinoma progression by promoting cell cycle and inhibiting apoptosis. Oncol. Lett. 2022;23:49. doi: 10.3892/ol.2021.13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yánez D.C., Ross S., Crompton T. The IFITM protein family in adaptive immunity. Immunology. 2020;159:365–372. doi: 10.1111/imm.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X., Zhang J., Gao F., Fan S., Dai L., Zhang J. KPNA2-Associated Immune Analyses Highlight the Dysregulation and Prognostic Effects of GRB2, NRAS, and Their RNA-Binding Proteins in Hepatocellular Carcinoma. Front. Genet. 2020;11 doi: 10.3389/fgene.2020.593273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu S.Y., Liao P., Yan L.Y., Zhao Q.Y., Xie Z.Y., Dong J., Sun H.T. Correlation of MKI67 with prognosis, immune infiltration, and T cell exhaustion in hepatocellular carcinoma. BMC Gastroenterol. 2021;21:416. doi: 10.1186/s12876-021-01984-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miwa T., Kanda M., Koike M., Iwata N., Tanaka H., Umeda S., Tanaka C., Kobayashi D., Hayashi M., Yamada S., et al. Identification of NCCRP1 as an epigenetically regulated tumor suppressor and biomarker for malignant phenotypes of squamous cell carcinoma of the esophagus. Oncol. Lett. 2017;14:4822–4828. doi: 10.3892/ol.2017.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ying J., Pan R., Tang Z., Zhu J., Ren P., Lou Y., Zhang E., Huang D., Hu P., Li D., et al. Downregulation of NCL attenuates tumor formation and growth in HeLa cells by targeting the PI3K/AKT pathway. Cancer Med. 2022;11:1454–1464. doi: 10.1002/cam4.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia L., Sun J., Xie S., Chi C., Zhu Y., Pan J., Dong B., Huang Y., Xia W., Sha J., Xue W. PRKAR2B-HIF-1α loop promotes aerobic glycolysis and tumour growth in prostate cancer. Cell Prolif. 2020;53:1–13. doi: 10.1111/cpr.12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rong H., Peng J., Ma K., Zhu J., He J.T. Ttc39c is a potential target for the treatment of lung cancer. BMC Pulm. Med. 2022;22:391. doi: 10.1186/s12890-022-02173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q., Qiao W., Zhang H., Liu B., Li J., Zang C., Mei T., Zheng J., Zhang Y. Nomogram established on account of Lasso-Cox regression for predicting recurrence in patients with early-stage hepatocellular carcinoma. Front. Immunol. 2022;13:1019638. doi: 10.3389/fimmu.2022.1019638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W., Liu W. Integration of gene interaction information into a reweighted Lasso-Cox model for accurate survival prediction. Bioinformatics. 2021;36:5405–5414. doi: 10.1093/bioinformatics/btaa1046. [DOI] [PubMed] [Google Scholar]
- 51.Tang Z., Shen Y., Zhang X., Yi N. The spike-and-slab lasso Cox model for survival prediction and associated genes detection. Bioinformatics. 2017;33:2799–2807. doi: 10.1093/bioinformatics/btx300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLernon D.J., Giardiello D., Van Calster B., Wynants L., van Geloven N., van Smeden M., Therneau T., Steyerberg E.W., topic groups 6 and 8 of the STRATOS Initiative Assessing Performance and Clinical Usefulness in Prediction Models With Survival Outcomes: Practical Guidance for Cox Proportional Hazards Models. Ann. Intern. Med. 2023;176:105–114. doi: 10.7326/M22-0844. [DOI] [PubMed] [Google Scholar]
- 53.Cabrita R., Lauss M., Sanna A., Donia M., Skaarup Larsen M., Mitra S., Johansson I., Phung B., Harbst K., Vallon-Christersson J., et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 54.Xu L., Shen S.S., Hoshida Y., Subramanian A., Ross K., Brunet J.-P., Wagner S.N., Ramaswamy S., Mesirov J.P., Hynes R.O. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol. Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charoentong P., Finotello F., Charoentong P., Angelova M., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 56.Bindea G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., Angell H., Fredriksen T., Lafontaine L., Berger A., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 57.Xu L., Deng C., Pang B., Zhang X., Liu W., Liao G., Yuan H., Cheng P., Li F., Long Z., et al. Tip: A web server for resolving tumor immunophenotype profiling. Cancer Res. 2018;78:6575–6580. doi: 10.1158/0008-5472.CAN-18-0689. [DOI] [PubMed] [Google Scholar]
- 58.Friedman J.D., Reece G.R., Eldor L. The Utility of the Posterior Thigh Flap for Complex Pelvic and Perineal Reconstruction. Plast. Reconstr. Surg. 2010;126:146–155. doi: 10.1097/PRS.0b013e3181da8769. [DOI] [PubMed] [Google Scholar]
- 59.Wickham H., Vaughan D., Girlich M. tidyr: Tidy Messy Data. 2024. https://CRAN.R-project.org/package=tidyr
- 60.Wickham H., Romain F., Lionel H. 2021. Kirill Mu€ller (2020) Dplyr: A Grammar of Data Manipulation. R Packag. version 0.8. 4. [Google Scholar]
- 61.Dowle M., Srinivasan A., Gorecki J., Chirico M., Stetsenko P., Short T., Lianoglou S., Antonyan E., Bonsch M., Parsonage H., et al. Vol. 596. 2019. (Package ‘data. table.’ Ext. ‘data. Fram.). [Google Scholar]
- 62.Therneau T.M., Grambsch P.M. Modeling Survival Data: Extending the Cox Model. Statistics for Biology and Health; 2000. The Cox Model; pp. 39–77. [DOI] [Google Scholar]
- 63.Heagerty P.J., Saha-Chaudhuri P., Saha-Chaudhuri M.P. San Fr. GitHub; 2013. Package ‘survivalROC’. [Google Scholar]
- 64.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 65.Venables W.N., Ripley B.D. Modern Applied Statistics with S-PLUS. Statistics and Computing. Springer; 1997. Tree-based Methods; pp. 413–430. [DOI] [Google Scholar]
- 66.Kassambara A. 2017. Practical Guide To Principal Component Methods in R: PCA, M(CA), FAMD, MFA, HCPC, factoextra (STHDA) [Google Scholar]
- 67.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.-H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The Immune Landscape of Cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varn F.S., Tafe L.J., Amos C.I., Cheng C. Computational immune profiling in lung adenocarcinoma reveals reproducible prognostic associations with implications for immunotherapy. OncoImmunology. 2018;7:e1431084. doi: 10.1080/2162402X.2018.1431084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varn F.S., Wang Y., Mullins D.W., Fiering S., Cheng C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. 2017;77:1271–1282. doi: 10.1158/0008-5472.CAN-16-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng C., Nguyen T.T., Tang M., Wang X., Jiang C., Liu Y., Gorlov I., Gorlova O., Iafrate J., Lanuti M., et al. Immune infiltration in tumor and adjacent non-neoplastic regions co-determines patient clinical outcomes in early-stage lung cancer. J. Thorac. Oncol. 2023;18:1184–1198. doi: 10.1016/j.jtho.2023.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schaafsma E., Jiang C., Cheng C. B cell infiltration is highly associated with prognosis and an immune-infiltrated tumor microenvironment in neuroblastoma. J. Cancer Metastasis Treat. 2021;7 doi: 10.20517/2394-4722.2021.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis : A knowledge-based approach for interpreting genome-wide. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data available in this study is publicly available. The RNA-seq data and clinical information for TCGA-SKCM were obtained from TCGA on FireBrowse (gdac.broadinstitute.org/). TCGA mutation annotation format (MAF) files for gene mutation analyses were obtained from https://gdc.cancer.gov/about-data/publications/pancanatlas. Accession numbers for these datasets are listed in the key resources table. All codes in this study and any additional information required in this paper is available from the lead contact upon request.