

Summary
Here, we present a protocol to deliver nanoliter volumes of Toll-like receptor (TLR) agonist onto a culture of nuclear factor κB (NF-κB) reporter macrophages using fluidic force microscopy and a micron-scale probe. We describe steps for quantifying the dose of agonist by modeling their diffusion with experimental inputs. We then detail procedures for quantifying and categorizing macrophage responses to individual and varied doses and combining agonist concentration and macrophage response to analyze the NF-κB response to localized TLR stimulation.
For complete details on the use and execution of this protocol, please refer to Mulder et al. (2024).1
Subject areas: Biophysics, Atomic Force Microscopy, AFM, Cell Biology, Cell culture, Single Cell, Cell-based Assays, Immunology, Microscopy
Graphical abstract
Highlights
-
•
Stimulate single cells or clusters of cells live in culture via fluidic force microscopy
-
•
Quantify single-cell agonist dose with mathematical model of agonist diffusion
-
•
Combine agonist gradient, culture density, and single-cell responses for analysis
-
•
Apply bootstrap statistics to calculate error, significance, and effect size
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol to deliver nanoliter volumes of Toll-like receptor (TLR) agonist onto a culture of nuclear factor κB (NF-κB) reporter macrophages using fluidic force microscopy and a micron-scale probe. We describe steps for quantifying the dose of agonist by modeling their diffusion with experimental inputs. We then detail procedures for quantifying and categorizing macrophage responses to individual and varied doses and combining agonist concentration and macrophage response to analyze the NF-κB response to localized TLR stimulation.
Before you begin
Culture NF-κB reporter RAW Macrophages2 for at least 2 passages.
Culture NF-κB reporter RAW macrophages
Timing: 4–6 days
-
1.
Thaw 1 vial of NF-κB reporter macrophages and plate entire contents (5–10 million cells) in T75 Flask in 15–20 mL Dulbecco’s Modified Eagle Medium (DMEM) with 20% Fetal Bovine Serum (FBS).
-
2.
Grow 2 days at 37°C and 5% CO2.
-
3.
Passage 1 million cells to a new T75 culture flask with DMEM + 10% FBS.
-
4.
Grow 2–4 days at 37°C and 5% CO2 until ready to passage again.
-
5.Plate cells:
-
a.500,000 cells in 2 mL media in 50 mm microscope dish for 16–24 h incubation.
-
b.200,000 cells in 2 mL media in 50 mm microscope dish for 40–48 h incubation.
-
c.Shake dish gently up and down and side to side to distribute cells evenly.
-
a.
-
6.
Incubate cells at 37°C and 5% CO2 for 16–24 h or 40–48 h depending on plating density. Final culture density should be ∼ 500 cells/mm2, evenly distributed with few clumps.
CRITICAL: Macrophages should be used between passages 2 and 8 and within 30 days for most consistent results. Do not start experiment if you see visible cell debris, cell blebbing, or elevated resting NF-κB activation.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| R848 (resiquimod) | InvivoGen | tlrl-r848 |
| CO2 independent medium | Fisher Scientific | 18045088 |
| DMEM (Dulbecco’s modified Eagle’s medium) | Thermo Fisher Scientific | 11995073 |
| FBS (fetal bovine serum) | Thermo Fisher Scientific | 26140079 |
| L-glutamine (200 mM solution) | Thermo Fisher Scientific | 25030081 |
| NovoRinse | Agilent | 872B603 |
| NovoClean | Agilent | 872B602 |
| Hoechst 33342, trihydrochloride, trihydrate | Fisher Scientific | H3570 |
| Alexa Fluor 488 | Thermo Fisher Scientific | 33077A |
| Deposited data | ||
| Data referenced in this paper | This paper | Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2 |
| Experimental models: Cell lines | ||
| Murine: RAW264.7 macrophages, GFP NF-kB reporter | Iain Fraser Lab Sung et al. 2014 |
RRID: CVCL_0493, ATCC TIB71 |
| Software and algorithms | ||
| Fiji | Schindelin et al. 2012 | RRID: SCR_002285 https://fiji.sc |
| CellProfiler | McQuin et al. 2018 | RRID: SCR_007358 https://cellprofiler.org |
| Cytosurge FluidFM software | Cytosurge | https://documentation.cytosurge.com/cora/ |
| FluidFM C3000 controller software | Nanosurf | https://www.nanosurf.com/en/software/c3000 |
| ZEISS ZEN Pro microscope software | ZEISS | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| RStudio | RStudio Team 2020 | RRID: SCR_000432 http://www.rstudio.com |
| GraphPad Prism | GraphPad Software | RRID: SCR_002798 http://graphpad.com |
| Mathematica | Wolfram Research | RRID: SCR_014448 https://www.wolfram.com/mathematica/ |
| Code referenced in this paper | This paper | Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2 |
| Other | ||
| FlexAFM | Nanosurf | https://www.nanosurf.com/en/products/flexafm |
| FluidFM micropipette probes 2 N/m stiffness, 2 micron aperture, FlexAFM | Cytosurge | www.cytosurge.com/shop/product/fluidfm-micropipette-208#attr=187,172,52 |
| Axio Observer 7 inverted optical microscope with live cell incubation box | ZEISS | 000000-2624-360 |
| ORCA-Flash4.0 V3 xCMOS camera | Hamamatsu | C13440-20CU-KIT |
| Microscopy dishes 50 mm × 7 mm | Ted Pella | 14027-200 |
| Cover glass 24 mm × 50 mm | Fisher Scientific | 22-050-232 |
| Syringe filter, 0.2 μm, PTFE | Corning | 431212 |
Materials and equipment
CO2 Independent medium with L-Glutamine and HI-FBS
| Reagent | Concentration | Amount |
|---|---|---|
| L-glutamine (200 mM) | 2% of medium volume | 0.26 mL |
| HI-FBS | 10% of total volume | 1.5 mL |
| CO2 Independent Medium | N/A | 13.24 mL |
| Total | N/A | 15 mL |
Store at 4°C, discard if not used within two weeks.
Short recipes
-
•
Media supplemented with FBS: DMEM with 10% of FBS [store at 4°C, discard if not used within two weeks]
Critical materials and equipment
-
•
FlexAFM installed on inverted optical microscope with live cell incubation box.
-
•
Micropipette probes compatible with FlexAFM.
-
•
Microscopy dishes that fit on FluidFM stage underneath controller.
-
•
CO2 Independent Medium.
Alternative materials and equipment
-
•
Alternative NF-κB reporter cell line instead of those developed by Sung et al., 2014.
-
•
Alternative culture media instead of DMEM.
-
•
Alternative nuclear stain such as DAPI instead of Hoechst.
-
•
Alternative cleaning solutions for the probe: any ethanol-based solution can substitute for NovoRinse, and a dilute bleach solution can substitute for NovoClean.
-
•
Alternative TLR agonists: any that flow through probe channel.
-
•
Alternative fluorophore for modeling agonist: any with molecular weight the same order of magnitude as TLR agonist detectible with your microscope.
-
•
Alternative to CellProfiler pipelines: write an ImageJ macro for segmentation and quantification.
-
•
Alternative to Mathematica, R, and GraphPad Prism: Any coding language or software for data processing and plotting.
Step-by-step method details
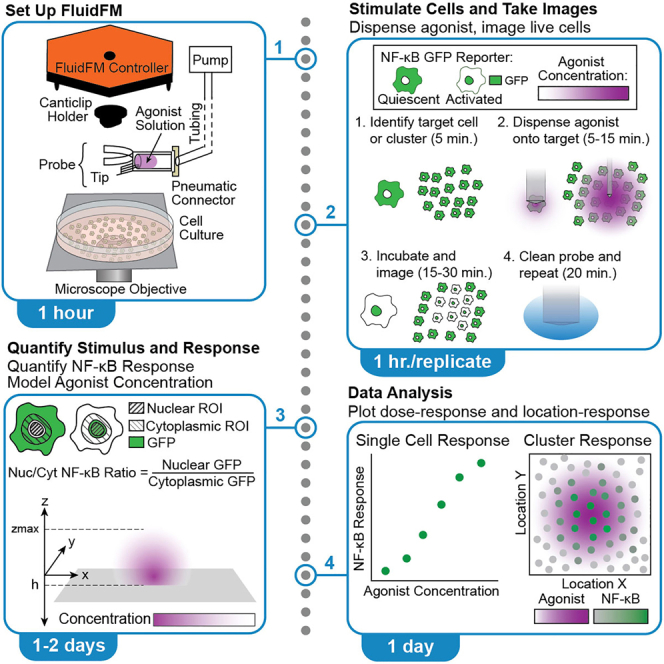
Use Fluidic Force Microscopy3,4,5 to dispense TLR agonist onto macrophages in culture (Figure 1A). Stimulate single macrophages or clusters of macrophages (Figure 1B). Quantify the agonist gradient and single cell NF-κB responses and analyze the results.
Figure 1.

Set up FluidFM for controlled dispensing of agonist onto live macrophage culture
(A) FluidFM set up for macrophage stimulation. Probe with agonist in reservoir attached to underside of controller, lowered into macrophage culture sitting on microscope stage. Green indicates reporter GFP fluorescence, purple indicates agonist concentration.
(B) Diagram of single cell and cluster stimulation modes.
(C) Validation of RAW macrophage NF-κB activity in CO2 Independent Medium compared with normal culture conditions in the incubator and commonly used buffer HEPES.
(D) Probe on canticlip holder, showing location of tip, channel, and reservoir.
(E) Holder clips onto magnetic stand, and probe clips onto holder, pneumatic connector attaches to back of the reservoir.
(F) Controller on stage with power cable, laser cable, and pneumatic nozzle connected. Top view shows location of screws for aligning laser.
(G) Probe attached to underside of controller.
(H) How to tilt up controller to swap samples underneath.
Set up FluidFM and prepare cell culture
Load and prepare the FluidFM probe, prepare the cell dish.
-
1.Warm up microscope and open software.
-
a.Turn on microscope and FluidFM and open their software: Zeiss Zen Pro, Nanosurf C3000 Controller, and Cytosurge User Interface.
-
b.Warm up microscope incubation box to 37°C.
-
a.
-
2.Prepare the cell culture:
-
a.Prepare 15 mL CO2 independent medium with L-Glutamine and HIFBS (Materials and Equipment). Warm to 37°C. (CO2 independent medium allows the RAW macrophages to retain NF-κB activity at atmospheric CO2 levels, Figure 1C).
-
b.Dilute Hoechst 33342 nuclear stain (10 mg/mL) 1:1000 in PBS (10 μg/mL).
-
c.Remove growth media from 50 mm culture dishes plated as described above.
-
d.Replace with 2 mL CO2 independent medium with HIFBS.
-
e.Add Hoechst solution 1:100 (final concentration 100 ng/mL).
-
f.Incubate at 37°C and atmospheric CO2 for 1 h. Protect from light.CRITICAL: you must allow the cells to equilibrate to their new media at 37°C before stimulating them, minimum 1 hr., but no more than 3 hours.
 Pause point: You can pause here while the cells incubate or continue with setup.
Pause point: You can pause here while the cells incubate or continue with setup.
-
a.
-
3.Prepare agonist solution:
-
a.Dilute TLR agonist in sterile filtered, endotoxin-free water to desired concentration. For R848, use concentrations between 100 ng/mL and 2 μg/mL.
-
a.
Optional: To reduce chance of clogging, sterile filter solution with a 0.22 μm membrane.
Note: Due to diffusion, the final concentration of agonist “seen” by each cell will be less than that loaded into the probe and will depend on cell locations and dispensing conditions. The section “diffusion modeling to determine concentration gradient of the agonist” describes how to compute actual agonist concentrations in the dish.
-
4.Load agonist into probe:
-
a.Following manufacturer instructions, load 10 μL of agonist solution into a clean micropipette probe (2 μm opening, 2 N/m stiffness) (Figure 1D). If you encounter issues, see troubleshooting tips.CRITICAL: Take care not to touch anything near probe tip!
-
b.Attach pneumatic tubing to back of probe (Figure 1E). If you encounter issues, see troubleshooting tips.Note: Take care not to press on tubing where it joins the connector – this weak point splits easily!
-
a.
-
5.Set up FluidFM for dispensing:
-
a.Set FluidFM controller on microscope stage. Attach pneumatic nozzle to side with provided screwdriver and plug in laser cable and power cable (Figure 1F).
-
b.Pick up controller and hold upside-down. Clip magnetic canticlip holder with probe onto underside of controller and connect the pneumatic connector to the nozzle (Figure 1G).
-
c.Using the Cytosurge software, align the optical feedback laser in air. If encountering issues aligning the laser, check Cytosurge Support or troubleshooting tips.
-
d.Using the Cytosurge software, apply overpressure to fill the probe from the reservoir. You should see liquid dispense from the tip within a few minutes. If encountering issues filling the probe, see Cytosurge Support or troubleshooting tips.
-
e.Place a 500 μL drop of endotoxin-free water on coverslip beneath the controller (Figure 1H), submerge the probe, and align the laser again.
-
a.
Note: risk of breaking probe with pipette tip, be careful to leave extra room between the probe and the coverslip.
Note: make sure the water drop makes good contact with the probe to avoid bubbles.
Note: If having trouble aligning laser, see troubleshooting tips.
-
6.Rinse probe in water twice, 5 min each, to remove any agonist from the outer surface:
-
a.Remove the previous coverslip and water drop. Place a new coverslip on the stage.
-
b.Pipette two drops of endotoxin-free water approximately ∼250 μL each on either end of the coverslip. Position one in the center underneath the probe.
-
c.Lower the controller so the probe is submerged in the first drop. Let sit 5 min.
-
d.Lift up the controller and move the coverslip so the second drop is centered.
-
e.Lower the probe into the second drop. Let sit 5 min.
-
a.
-
7.Place in cell dish:
-
a.Remove the coverslip and water.
-
b.Place the cell dish on the stage underneath the controller with the lid removed.
-
c.Replace the controller on the stage to submerge the probe in the media.
-
d.Check the laser is still aligned.
-
e.Proceed immediately to the next section.
-
a.
Stimulate and image target area in culture
Stimulate an area in the culture by dispensing agonist with the FluidFM. Image with fluorescence microscopy to monitor cell response. Clean probe and repeat. (Figure 2A).
-
8.
Locate a quiescent cluster of macrophages: look at the GFP fluorescence to identify an area of the dish with healthy, unactivated cells and an even density, not too sparse or overgrown.
-
9.Approach and position probe (Figure 2B):
-
a.Use FluidFM controller at 200 mV setpoint to approach probe to an empty spot in your target area. Troubleshooting: Approach Failed.
-
b.Retract probe to dispensing height using the “safety retraction” feature in the Cytosurge software. Use 4 μm for single cell targeting, and 20 μm for cluster targeting; different combinations of dispensing height and rate will stimulate different sized areas of the culture (Figure 1B). You can change the retraction height in the AFM settings menu of the Cytosurge software.
-
c.Position probe over target cell or spot using the knobs on the stage.
-
a.
-
10.Image cells before stimulation:
-
a.Use the DAPI, GFP, and Bright-field or DIC channels.
-
a.
Note: If the GFP signal is too low, you may need to average together successive acquisitions to improve image quality.
Note: Since the Hoechst nuclear stain excitation wavelength is in the ultraviolet range, minimize excitation intensity and exposure time to prevent macrophage activation.
Note: If encountering issues with nuclear image quality, see troubleshooting tips.
-
11.Dispense the agonist:
-
a.Apply overpressure to dispense agonist for the desired stimulation time. Use 4 mbar overpressure for 4 μm height single cell targeting, and 400 mbar pressure for 20 μm height cluster targeting. I used stimulation times of 5 and 15 min.
-
b.At end of stimulation time, change pressure setpoint back to zero and retract probe 100 μm. Leave controller in place until imaging is completed.
-
a.
-
12.Image cells to monitor the NF-κB response:
-
a.Image cells in 5- or 15-min intervals for up to 40 min, as needed, to monitor NF-κB response. Save a set of Hoechst, GFP, and Bright-field/DIC images at each time point.
-
a.
Note: The reporter macrophages should reach maximum response by 30–40 minutes, but they can respond in as little as 10–15 minutes.
-
13.Clean probe for next use (Figure 2C):
-
a.When imaging is completed, remove cell dish from underneath controller, place aside in a safe place, and replace lid.
-
b.Put a coverslip on the microscope stage and put a drop of NovoRinse solution in the center.Alternative: use another ethanol-based cleaning solution.
-
c.Replace controller on stage so probe is submerged. Soak 10 min.
-
d.Swap out the NovoRinse slide for a clean slide and place two drops of water on it, as described in Step 6.
-
e.Soak the probe in each drop of water for 5 min.CRITICAL: multiple rinses are required to remove all of the NovoRinse from the probe, which will irritate the macrophages if not completely rinsed off.Pause point: you can leave the probe soaking in water for longer if needed.
-
f.Remove the water slide and replace the cell dish back underneath the controller as described in Step 7.
-
g.Repeat Steps 8–13 to take a second dataset. On last dataset, skip step 13 and proceed to cleanup (below) instead.
-
a.
Figure 2.

Stimulation of single cells or clusters in culture with FluidFM
(A) Workflow overview for single run. Green indicates reporter GFP fluorescence, purple indicates agonist concentration.
(B) How to approach target cell or spot. 1: approach probe to empty spot, 2: retract to dispensing height (4 μm for single cell targeting, 20 μm for cluster targeting), 3: move probe over target cell. Image is example of resting culture from raw data in Mulder et al. 2024,1 GFP: NF-κB reporter, 10 μm scale.
(C) FluidFM set up for cleaning probe. Drop of NovoRinse solution or water on coverslip on stage, probe lowered into drop to soak.
Clean up and save images
Clean, empty, and store probe for future use. Export data for processing and analysis. [Figure 6: Empty Probe and Soak for Reuse]
-
14.Clean probe:
-
a.Soak the probe in a drop of NovoClean solution for 10 min (as described in Step 13).Alternative: use a dilute bleach solution. Be careful, though, as too much exposure will corrode the metal contact at the base of the canticlip holder.
-
b.Rinse 5 min with water.
-
a.
-
15.Empty and store probe (Figure 3).
-
a.Remove probe from FluidFM controller and replace on pedestal.
-
b.Remove tubing and empty reservoir with pipette.
-
c.Rinse out reservoir with 10 μL sterile water.
-
d.Replace tubing. Detach nozzle from FluidFM controller and reattach to tubing to reconnect probe to pneumatic system (Figure 3A).
-
e.Locate the syringe port in the pneumatic line. Attach an empty 30 mL syringe to the port. Turn the valve to connect syringe to the probe (Figure 3A).
-
f.Apply negative pressure with the syringe to vacuum residual solution from the probe channel into the reservoir. Check channel is empty (Figure 3B).Alternative: you can apply positive syringe pressure to empty the probe as well. It will take longer and has a risk of clogging, but it has lower risk of introducing bubbles into the channel.Note: If reservoir is not empty before applying negative pressure, media can get into the pneumatic tubing. For cleaning instructions, see troubleshooting tips.
-
g.Release the syringe pressure. Disconnect tubing from nozzle and probe and store.
-
h.Empty and rinse probe reservoir again.
-
i.Remove probe from canticlip holder and place back in box. Add 1 mL sterile water to box to soak probe (Figure 3C).
-
j.Tape box closed and store at 20°C–25°C.
-
a.
-
16.Save Images:
-
a.In Zen Pro software, decrease GFP channel max brightness to brighten image; use the same value for all GFP images. Adjust DAPI channel min and max to give best contrast for nuclear stain. Do this for all images and save.
-
b.Export images of resting and activated macrophages as TIFF files. Export channels separately.
-
a.
-
17.Store FluidFM and shut down instruments/computers:
-
a.Detach laser cable and pneumatic tubing from controller. You may also unplug the power cable.Note: Be careful detaching the laser cable, pneumatic tubing, and power cable! Bending the cables near their attachment points can lead to bent pins, frayed wires, or split tubing.
-
b.Store the FluidFM controller on the microscope stage or in its box.
-
c.Shut down the instruments and software.
-
a.
Figure 6.

Quantify fluorophore brightness to convert intensity to concentration
(A) Diagram of setup for imaging reference slides: FluidFM controller with canticlip holder attached but no probe.
(B) Two reference slides, with and without coverslip spacer to create a wider, thinner drop (left) or narrower, taller drop (right). Labeled with drop concentration, volume, and area. Area border drawn and measured in ImageJ. Drop volume and area used to calculate drop thickness.
(C and D) Results of imaging calibration slides with 40x (C) and 10x (D) objective. Plots of drop concentration ∗ thickness vs. modal intensity. Linear fit in GraphPad Prism to determine conversion factor needed to fit the model.
Figure 3.

Empty probe and soak for reuse
(A) Setup for emptying probe channel after removing remaining agonist from reservoir. A syringe is connected directly to the probe instead of the pump.
(B) A probe with an empty channel.
(C) Fill well in box to soak probe for storage.
Negative controls
Run the above protocol with dispensing endotoxin-free water. If there is still NF-κB activation, see troubleshooting tips.
Segment images and quantify NF-κB translocation with CellProfiler
Note: CellProfiler6 pipeline files available on Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2.
Pipeline for 100x images
Note: Scale for 100x images is 15.3 px/micron. Adjust pixel values in pipeline as needed for images of a different resolution.
-
18.
Load GFP and HOECHST images.
-
19.
Extract metadata from file names (dataset, channel, time point).
-
20.
In NamesAndTypes match up the pairs of channel images and put them in order for processing.
-
21.
Group the datasets to be processed separately (so the cell tracking only applies within each dataset).
-
22.
Run “rescale intensity” to brighten the nuclear images (to improve segmentation): set values of 0.6 and above to 1.0.
-
23.
Smooth the nuclei with a 30 px median filter.
-
24.
Run Identify Primary Objects on the smoothed nuclei. Object diameters 80–250 px, use global thresholding, Otsu method, two classes, correction factor 0.9 (adjust as needed).
-
25.
Run Track Objects. Tracking method LAP, search radius 3 SD, search radius limit 100–2000 px, no second phase. Save “tracked cells” image.
-
26.
Expand nuclei objects by 100 px and save as “cell regions” objects.
-
27.
Expand the nuclei objects by 10 px and save as “outer ring”. This will form the outer boundary of an ROI to capture the bit of the cytoplasm closest to the nucleus.
-
28.
Shrink the nuclei by 16 px to capture a nuclear ROI and correct for oversized nuclei from the brightening in Step 22 and save as “shrunken nuclei.”
-
29.
Run Identify Tertiary Objects and subtract the nuclei objects from the outer ring objects and save as “cytoplasm” (captures the ring of cytoplasm right outside the nucleus).
Note: In wide-field images the difference in thickness between cytoplasm and nucleus parts of the cell cause the nucleus to seem brighter and the cytoplasm dimmer and this throws off the results. As in Ding et al. 1998,7 sampling the portion of cytoplasm nearest to the nucleus, where the thicknesses are more similar, is a better measure of nuclear vs cytoplasmic GFP fluorescence.
-
30.
Create a masked image where the “cell regions” objects are masked.
-
31.
Measure the intensity of the masked image to get a measure of background intensity.
-
32.
Use Image Math to subtract the mean intensity of the masked image from the GFP image, save as “NF-κB background subtracted.”
-
33.
Measure the GFP intensity in the shrunken nuclei objects.
-
34.
Measure the GFP intensity in the cytoplasm objects.
-
35.
Use Calculate Math to divide the mean intensity of the shrunken nucleus by the mean intensity of the cytoplasm for each object.
-
36.
Make an Overlay Outlines image showing the nucleus, cytoplasm, and cell regions.
-
37.
Use Display Data on Image to add the cell numbers from “track objects” and the nuc/cyt ratio from “calculate math” to write these values by each cell on the outlines overlay image.
-
38.
Save the annotated images (to check quality of cell tracking after).
-
39.Export the data to spreadsheet (manually select which values to export):
-
a.Any metadata extracted in step 19.
-
b.Object tracking labels from step 25.
-
c.Object location X and Y from step 25.
-
d.Nuc/Cyt ratio values from step 35.
-
a.
Note: the file exported by CellProfiler will include by default some columns you do not need, duplicate columns, and two header rows. Clean up each dataset in Excel to remove duplicates and unnecessary columns and consolidate header rows to one row. This will make processing in R much easier. Save as CSV.
Pipeline for 10x images
Note: Scale for 10x images is 1.53 px/micron. Adjust pixel values in pipeline as needed for images of a different resolution.
-
40.
Pipeline for 10x images is the same as for 100x images, except divide all pixel distance values by 10 to account for decrease in scale.
-
41.
For datasets without a time series, omit grouping (step 21) and cell tracking (step 25). Cell numbering will instead come from the “identify primary objects” module. Make sure to adjust the data export settings accordingly.
-
42.
For FluidFM Cluster Stimulation data taken with 10x objective, omit steps 30–32 (the background subtraction). There is significant reflection from the underside of the FluidFM controller in these images, and it artificially brightens the background. In this case, background subtraction makes the data quality worse.
Take calibration data to determine activation cutoff
Take images of resting and activated macrophage populations under the same imaging conditions as in the FluidFM experiments. Quantify Nuclear/Cytoplasmic NF-κB ratio for each population and determine activation cutoff value (Figure 4).
-
43.Prepare cells:
-
a.Grow reporter macrophages in 50 mm microscope dish as for FluidFM experiment (Figure 4A).Pause point: The macrophages will need to grow for 1–2 days before continuing.
-
b.Warm up microscope incubation box to 37°C.
-
c.Change cell dish media to 2 mL CO2 Independent Medium with HIFBS (materials and equipment). Add Hoechst 33342 stain (working concentration 100 ng/mL).
-
d.Incubate microscope dishes in microscope incubation box for 1 h. Protect from light.Pause point: The macrophages can incubate for up to 3 hours before continuing.
-
a.
-
44.Prepare agonist and FluidFM:
-
a.Prepare >20 μL of agonist solution at 100x activating concentration in water or PBS.
-
b.Put the FluidFM controller on the microscope stage with the canticlip holder attached, but no probe. You don’t need to attach any cables, the FluidFM just needs to be in the light-path as it would be in an experiment (Figure 4B).
-
c.Repeat Step 42 with both the 100x objective and 10x objective to obtain calibration data for both single-cell targeting experiments, and cluster targeting experiments, respectively.
-
a.
-
45.Stimulate and Image:
-
a.Place the cell dish underneath the controller.
-
b.Image several spots in the dish before stimulation: Hoechst and GFP (Figure 4B).
-
c.Remove cell dish, mix in 20 μL of the 100x agonist solution (Figure 4C).
-
d.Incubate cell dish at 37°C for 15–30 min, until all cells are brightly activated.
-
e.Image several spots in the dish: Hoechst and GFP (Figure 4C).
-
a.
-
46.Data Processing.
-
a.Export images for quantification and analysis (Figure 4D) as described in Clean Up and Save Images.Pause point: Image quantification and analysis can be done at any time.
-
b.Quantify images as described Steps 18–39 (Figure 4E).
-
c.The data table should have the following columns: “Label” which is the number assigned to each nuclear region by CellProfiler, “Group” which was extracted from image filenames and is either “resting” or “activated”, and “Nuc_cyt_ratio” which is the ratio of GFP mean intensity in the nuclear region to the cytoplasmic region calculated by CellProfiler.
-
a.
-
47.Find activation cutoff in R (Figure 4F).
-
a.Setup and data import.knitr::opts_chunk$set(echo = TRUE)library(tidyverse)setwd(file location)data <-read_csv(file name)#filter out the cells that don't have a labeldata <- drop_na(data, Label)
-
b.Find the cutoff value for the top 5% of the resting cells.n_rest <- nrow(filter(data, Group == "resting"))#extract the top 5% of the resting population at the final timepointtop5rest <- data %>%filter(Group == "resting") %>%slice_max(order_by = Nuc_cyt_ratio, n=ceiling(n_rest∗0.05))#get the cutoff value: the mean of the last two in the top 5% groupcutoff <-round(mean(top5rest[nrow(top5rest),"Nuc_cyt_ratio"], top5rest[nrow(top5rest)-1,"Nuc_cyt_ratio"]),3)
-
c.Calculate the power of the cutoff.n_act <- nrow(filter(data, Group == "activated"))power <- nrow(filter(data, Group == "activated" & Nuc_cyt_ratio >= cutoff))/n_act
-
a.
Figure 4.

Activation cutoff distinguishes between activated and unactivated macrophages
(A) Dish of resting macrophages before stimulation.
(B) Set up for imaging resting macrophages. Include controller and canticlip holder so imaging conditions are the same as for a FluidFM experiment.
(C) Treat macrophages with agonist at a highly activating concentration and image again.
(D) Two sets of images, resting cells before stimulation, and activated cells after stimulation.
(E) Quantify activation of each cell. Nuclear boundaries are drawn from the nuclear stain, then a nuclear ROI is inside that boundary (right diagonal stripes), and the cytoplasmic ROI is a ring just outside the nuclear boundary (left diagonal stripes). This method is most reliable for wide-field images. Measure mean GFP intensity (green) in the two ROIs for each cell. The Nuc/Cyt NF-κB ratio is the ratio of these two measurements. A higher value indicates higher NF-κB activation.
(F) Histogram of the resting and activated macrophage populations. The activation cutoff is set just below the top 5% of the resting population (blue). The power of the cutoff is the percentage of activated cells (pink) above the cutoff. It’s important to make sure there is good contrast between these populations before continuing with quantifying your experiment data, so you can accurately distinguish between activated and unactivated cells. The quantitative cutoff is used to compute the fraction activated metric used in most of the data analysis.
Diffusion modeling to determine concentration gradient of the agonist
Dispense a small molecule fluorophore of similar molecular weight to the TLR agonist using the FluidFM in order to fit a model of point-source diffusion and determine the concentration of the agonist in the dish for a given set of dispensing parameters (flow rate and height) (Figures 5, 6, and 7). I used Alexa Fluor 488 (AF488) to model the agonist Resiquimod (R848); this method can be adapted to other small molecule agonists.
Figure 5.

Visualization of FluidFM dispensing with Alexa Fluor 488
(A) Diagram showing FluidFM setup: coverslip with water drop on microscope stage, FluidFM probe filled with fluorophore solution and attached to pneumatic connector and canticlip holder, which is in turn attached to the underside of the FluidFM controller. The controller will rest on the stage with the probe submerged in the water drop.
(B) Time-course images of Alexa Fluor 488 fluorescence (green) dispensed at 4 mbar (top row) and 400 mbar (bottom row) overpressure. Three frames are shown at the start of dispensing in 30 s intervals, then the frame right before the pressure was stopped, and then three frames after the pressure was stopped. Top row: 100 μm scale bar, 40x objective, bottom row: 300 μm scale bar, 10x objective.
(C) Example of measuring intensity vs. distance as described in step 3 below. Green fluorescence from AF488. Intensity measured along the white lines from the maxima. Scale bar 50 μm.
(D) Example of data obtained by the method in (C). Error bars are standard deviation (SD) of 3 replicates.
Figure 7.

Fit dispensing data to analytical models to quantify diffusion of dispensed agonist
(A) Growth and decay of fluorophore peak intensity (normalized to equilibrium value) during and after dispensing. End of dispensing marked by dashed line. Time constants with uncertainty from exponential fit in GraphPad Prism. Error bars are SD or 3 or more replicates.
(B) Comparison of fluorophore dispensing at different molecular weights. Small molecule fluorophores (< 1 kg/mol) behave similarly, larger molecules do not. Error bars SD or 3 or more replicates.
(C) Diagram of the point source model of diffusion in 3D, and the 2D images taken with the microscope. By integrating the 3D model in z, you get a 2D radially symmetric distribution that can be fit to the analytical model.
(D) Measurement of the distance between the probe and the canticlip holder window, used to set a limit on the parameter “zmax”. Ruler at left of image shows mm scale.
(E) Best fits of the model to dispensing data for 4 mbar and 400 mbar dispensing. Error bars SD or 3 or more replicates.
(F) Diagram of how the 3D concentration gradient for the agonist can be reconstructed from the 2D fit of the fluorophore diffusion in (E).
Set up FluidFM to dispense fluorophore solution
-
48.
Make AF488 solution in water (100 μg/mL for dispensing at 4–40 mbar overpressure, 10 μg/mL for 40–400 mbar, and 1 μg/mL for 400–1000 mbar) and pass through 0.22 μm filter.
-
49.
Fill micropipette probe (2 μm opening, 2 N/m stiffness) reservoir with 10 μL AF488 solution.
-
50.
Open Zeiss Zen software, Nanosurf C3000 FluidFM software, and Cytosurge software.
-
51.Mount probe on FluidFM controller and position controller on microscope stage as in Step 5 (Figure 5A).
-
a.Attach pneumatic tubing to back of probe.
-
b.Position controller on stage and connect power cable, pneumatic nozzle, and laser cable.
-
c.Lift up probe and clip on to controller, attach pneumatic tubing to nozzle. Place coverslip on stage to protect objective. Place controller gently back down, check to make sure probe does not crash into slide.
-
d.Align FluidFM laser as in Step 5.
-
e.Fill probe as in Step 5.
-
f.Check fluorescence with microscope software.
-
g.Place 500 μL drop of water on coverslip beneath probe.
-
h.Align FluidFM laser in water as in Step 5.
-
i.Approach probe to surface using software, then retract to dispensing height; take data at both 4 μm height and 20 μm height to model single-cell targeting and cluster targeting, respectively (Step 9).
-
j.Center probe in field of view of microscope.
-
a.
Dispense AF488 and image
-
52.Set up time-series imaging on microscope:
-
a.Check image brightness during dispensing and adjust excitation laser intensity and/or camera exposure time so that both the brightest and the dimmest images show up well. Avoid maxing out the detector with the brightest image.
-
b.Choose objective to capture dispensed fluorophore “cloud” in field of view – I used the 40x for single-cell targeting, and the 10x for cluster targeting.
-
c.Set up time series to image in 30 s intervals for 20 min.
-
a.
-
53.
Start dispensing by applying overpressure: 4 mbar for dispensing at 4 μm height, and 400 mbar for dispensing at 20 μm height, again to model single cell and cluster targeting. Immediately start imaging. Turn off dispensing just before the 15-min image and remove probe by swiping it out of the field of view with the control knobs on the side of the stage. Let imaging run until completed (Figure 5B).
-
54.
Export images as TIFF files for analysis.
-
55.Clean up:
-
a.Remove probe from controller and empty reservoir.
-
b.Store in box with 1 mL water to soak. Tape closed and store at 20°C–25°C.
-
c.Place canticlip holder back on controller and controller back on stage for the next step.
-
a.
Reference slides
-
56.
Prepare a series of slides with 1 μL drop of AF488 solution between two coverslips: prepare a range of AF488 concentrations and make 2 slides per concentration, one with the drop flat between two coverslips (tape together), and one with square coverslip spacers on each end to hold the long coverslips a small distance apart.
-
57.
Image the center of each drop with the same imaging settings used to image the dispensed AF488, including keeping the FluidFM controller and canticlip holder in the light path (Figure 6A).
-
58.
Take a photo of the calibration slides to measure the drop area (Figure 6B).
-
59.
Export images as TIFF files for analysis.
Use ImageJ to extract intensity profiles for each run
Note: ImageJ8 Macro included in supplemental information and available at Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2
-
60.
Open the last image taken before end of dispensing for the run.
-
61.
Find the maximum location with “find maxima”.
-
62.
Define a function that takes in endpoint coordinates and draws a line, calls “getProfile”, and saves the plot profile results by calling “updateResults”. Takes in start and end coordinates of a line (x1,y1,x2,y2) in px, number (n) for each line, and image scale (s) in px/micron.
function radProfile(x1,y1,x2,y2,n,s){
l=sqrt((x2-x1)∗(x2-x1)+(y2-y1)∗(y2-y1))/s;
makeLine(x1,y1,x2,y2);
profile = getProfile();
for (i=0; i<profile.length; i++){
setResult("Position "+n, i, l∗i/profile.length);
updateResults;
setResult("Intensity "+n, i, profile[i]);
updateResults;
}
run("Draw","slice");
}
-
63.
Run the function for five lines from the maximum location to each of the left, right, and bottom edges of the image and to the corners, avoiding the probe.
-
64.
Save the image with the lines on it and the results window (Figure 6C).
-
65.
In Excel, average replicates to get an average and SD for concentration vs. distance from the dispensing point for each set of dispensing conditions (AF488 concentration, probe height, and applied pressure) (Figure 1D). Save as CSV file. (Figure 6D).
Use ImageJ to process the AF488 dispensing time series images
Note: ImageJ Macro included in supplemental information and available at Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2
-
66.
Open all images in a run and loop over them in batch mode.
-
67.
Set global variables: dispensing time and maxima locations (for images with and without cantilever).
-
68.
Subtract image of cantilever before dispensing from images containing the probe.
-
69.
Run the moment calculator to get the variance of the image in x and y.
-
70.
Draw an ROI around each maxima that is 14% of the standard deviation in width (corresponding to 99% of peak intensity if the cloud was Gaussian).
-
71.
Measure the peak intensity inside the ROI.
-
72.
Save the images with the ROI on them and data.
Process reference slides and compute conversion factor
-
73.
Measure the size of each drop with ImageJ by tracing with the outline and taking the area measurement for that ROI (Figure 6B). Use the coverslip dimensions to set the scale.
-
74.
Measure the modal intensity of each drop’s fluorescence image.
-
75.
In Excel, compile a table of reference slide concentration, area, and modal intensity. Use the known volume and measured area to compute drop thickness.
-
76.
Plot thickness∗concentration against modal intensity. Do a linear fit. The slope is the conversion factor from concentration∗thickness to intensity (Figures 6C and 6D).
Fit analytical model to fluorophore dispensing data
-
77.Peak Intensity vs. Time data (Figure 7A).
-
a.Fit exponential growth (to plateau) and decay (to zero) to the time series curves, broken up into before and after end of dispensing.
-
b.The time constants describe how quickly the fluorophore cloud reaches an equilibrium concentration.
-
a.
-
78.
Choice of fluorophore and molecular weight: comparison of peak intensity at different dispensing rates for different fluorophores (normalized to fluorophore concentration and brightness) show that small molecule fluorophores behave similarly, while larger molecules (I tested 10 kg/mol) do not (Figure 8B). However, the similarity between AF488 (838 g/mol) and AF350 (349 g/mol) lends support to using AF488 to model R848 (314 g/mol). Larger molecular weight agonists may not match the model as well.
-
79.Analytical model derivation:
-
a.Definitions:
-
i.C0 is the concentration loaded into the probe.
-
ii.V is the dispensing rate in volume/time.
-
iii.D is the diffusion coefficient of the dispensed molecule.
-
iv.k is the conversion factor from integrated concentration to brightness.
-
v.r is distance from the dispensing point in 3D space.
-
vi.ρ is distance from the dispensing point location in the XY plane.
-
i.
-
b.Differential equation for steady state diffusion from a constant source:
-
c.A point source at dispensing at a constant rate of mass per time:
-
d.Differential equation for the point source
-
e.Solution to the differential equation
-
f.Add parameter R to prevent C from going to infinity at r = 0
-
g.Apply the boundary condition C = C0 at r = 0 to solve for R
-
h.Final expression for C(r):
-
i.Convert to intensity (Figure 8C): Integral does not converge; can only solve computationally. In order to solve the integral, limits of integration were chosen that match the physical dimensions of the apparatus: the dispensing point was 4 μm above the surface of the coverslip, and the distance from the dispensing point to the top of the water drop (where it contacted the FluidFM instrument) was measured to be 2 mm (Figure 7D). So, the intensity was predicted computationally as follows: Where h is the height of the probe above the surface of the slide (4 μm in this case) and zmax is a fit parameter describing the height of the dispensed “cloud” of AF488. The value of zmax is constrained by the height of the water drop, and assuming spherical symmetry should be about the same as the radius of the cloud in the x-y plane. The best value fit for zmax (900 μm for 4 mbar dispensing of AF488, Figure 7E) is within those parameters.
-
a.
Figure 8.

Quantify macrophage clustering by counting number of individual cell neighbors
Image is raw data from Mulder et al. 2024,1 GFP: NF-κB. Black is FluidFM probe shadow. Yellow numbers are the number of neighbors for each cell computed by the algorithm. Scale bar 50 μm.
The point source prediction for C®, including an overall fit parameter A, is:
-
80.
Import intensity vs. distance information for each dispensing condition, save as a table with distance, intensity value, and error in 3 columns.
Note: the following code is for Mathematica.9 Full code included in supplemental, and at Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2.
data = Import[file, "CSV"];
distance = data[[All, 1]];
data4mbar = data[[All, 2]];
error4mbar = data[[All, 3]];
Data4mbar = Transpose[{distance[[2 ;;]],Table[data4mbar[[i]] ± error4mbar[[i]], {i, 2, Length[data4mbar]}]}];
-
81.
Define a function for concentration vs. distance, following the theoretical model, then define a function for intensity vs. distance, using the conversion factor you got from the calibration slides.
concentration[r_, d_, c0_, p_, a_] := a∗c0/(1+(4∗Pi∗d∗r/(160∗p)));
k = 0.88;
intensity[rho_, d_, c0_, p_, a_, zmin_, zmax_] := NIntegrate[k∗concentration[Sqrt[rhoˆ2+zˆ2], d, c0, p, a], {z, zmin, zmax}];
-
82.
Use the SEGWE (Stokes–Einstein Gierer-Wirtz Estimation) calculator developed by Evans et al.10 to estimate the diffusion coefficients for AF488 in 25°C water, and your agonist in 37°C water.
-
83.Use a chi-square fit to fit the theoretical expression to the dispensing data (Figure 7E).
-
a.The parameters zmax and A were fit by minimizing chi-squared for the difference between the dispensing data between 0 and 100 μm distance from the dispensing point and the predicted value for those same points. (In the single cell targeting experiments most cells are within 100 μm of the dispensing point. Including larger distances weighted the distribution too heavily towards the tail, rather than the center where accuracy is most important in order to compute the concentration at the target cell location).
-
b.Define a function to compute the chi-sq distance between two lists:chisq[exp_, pred_] := Module[ {},Sum[With[{i = i},If[exp[[i]]≠0,(exp[[i]] - pred[[i]])ˆ2 / exp[[i]],0]],{i, Length[exp]}]];
-
c.Range of zmax values: 400–1000 μm, step size 100 μm Range of A values: 0.4 to 0.7, step size 0.05.alist = Table[i, {i, 0.4, 0.7, 0.05}];zmaxlist = Table[i, {i, 400, 1000, 100}];
-
d.Generate predicted curve for each pair of A and zmax values forNote: cut off data at r = 235 μm (index 1450) because data is unreliable past this point.predictedData = ParallelTable[intensity[r, 356, 100, 4, a, 0, zmax] +intensity[r, 356, 100, 4, a, 0, 4],{zmax, zmaxlist},{a, alist},{r, distance[[2 ;; 1450]]}];
-
e.Compute chi-square distance between AF488 dispensing data and the predicted curves.chisqvals = ParallelTable[chisq[data4mbar[[2 ;; 1450]], predictedData[[i]][[j]]],{i, Length[zmaxlist]},{j, Length[alist]}];
-
f.Find pair of parameters that have the smallest chi-square value.Position[chisqvals, Min[chisqvals]]Output = [n1, n2]zmaxlist[[n1]]alist[[n2]]
-
g.Best values for 4 mbar dispensing at 4 μm height: A = 0.55, zmax = 900 μm Best values for 400 mbar dispensing at 20 μm height: A = 1.1, zmax = 700 μm.
-
a.
-
84.
Once you have the fit parameter, use the model to plot concentration vs. distance from the dispensing point at the dish surface for your agonist (Figure 7F).
Note: Change from the spherical coordinate rho to the polar coordinate r so that we can later match concentration values to cell location coordinates in the plane of the dish.
conc[rho_, d_, c0_, p_, h_, a_] := concentration[Sqrt[rhoˆ2 + hˆ2], d, c0, p, a];
Plot[{conc[r, 731, 1, 400, 20, 1.1]},{r, 1, 750}, PlotRange → {0, 0.3}]
Determine the “dilution factor”
-
85.
Use the concentration model to determine the ratio of concentration at the center of the dish vs. concentration loaded into the probe, called the “dilution factor.”
-
86.Use the dilution factor to calculate what concentration of agonist to use in order to stimulate the cells at a desired concentration.
-
a.For cluster targeting (400 mbar dispensing at 20 μm height), after fitting, the dilution factor for R848 is 0.28. Use this number to estimate R848 concentration for the FluidFM cluster targeting experiments.conc[0, 731, 1, 400, 20, 1.1] = 0.28
-
b.For single cell targeting (4 mbar dispensing at 4 μm height), the distance between the probe opening and the target cell surface varied due to variation in cell height. This introduced additional uncertainty into the calculation of concentration at the target cell surface. We calculated R848 concentrations from the best fit 4 mbar model for distances 0–5 μm from the dispensing point (probe opening).Table[N[concentration[r, 731, 1, 4, 0.55]], {r, 0, 5, 1}]In the single cell targeting condition, the target cell surface is estimated to be 1–4 μm from the probe opening. The relative concentration of agonist for those distances is shown in the table in Figure 3 (highlighted portion). Accounting for this uncertainty, the dilution factor (the relative concentration of agonist at the target cell position compared to the concentration loaded into the probe, C0) was estimated to be 0.02 +/- 0.01. Use this number to estimate R848 concentration for the FluidFM single cell targeting experiments.Pause point: Data analysis can be done later.
-
a.
Data analysis in R
Note: Code files for R11 and example data (sampled from Mulder et al. 20241) included in supplemental and on Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2.
Single-cell targeting datasets
Full code in supplemental “R_Analysis_fluidfm_single_cell_targeting.”
-
87.Setup:
-
a.Set up r-markdown document.
-
b.Set up libraries: tidyverse.
-
c.Import cell data and metadata.
-
a.
-
88.Data wrangling (cell data and metadata):
-
a.Make unique run identifier column from metadata.
-
b.Compute Cmax from C0 using the dilution factor from the diffusion model.
-
c.Compute distance from target cell (15.3 px/micron scale) for each cell.
-
d.Pivot wider: make data tables with one row per run, with initial and final N/C values and initial position values and target position.
-
e.Compute the ratio of final and initial time point Nuc/Cyt NF-κB values (“rNC”).
-
a.
-
89.
Define bootstrap function for calculating uncertainty in fraction activated:
#input list of activation data (binary, cutoff already applied)
bootstrap <- function(data)
{
#initialize an array to hold the fraction activated values
a <- as.numeric(rep(NA,5000))
n <- length(data) #size of each sample
#sample with replacement and get 5000 fraction activated values
for(i in 1:5000){
s <- sample(data, size = n, replace = T) #sample the data
a[i] <- sum(s,na.rm = T)/sum(!is.na(s)) #compute fraction activated
}
sd(a) #compute the variance in the fraction activated values
}
-
90.Bin by distance from target cell to compute fraction activated:
-
a.Create “binned_distance” column to group cells by their distance from the target cell.Note: Bin widths defined asymmetrically to highlight areas of greater change in fraction activated while still containing enough data points for accurate statistics.
-
b.Create binary “activated” columns using the cutoffs determined from calibration data.
-
c.Summarize data and compute fraction activated per stimulation condition.
-
d.Plot fraction activated vs. distance from target cell.
-
a.
-
91.Target cell dose-response curve:
-
a.Filter target cells from the data.
-
b.Summarize replicates and calculate fraction activated with uncertainty from bootstrap algorithm.
-
c.Compute uncertainty in Cmax from the uncertainty in Step 83.
-
d.Plot dose-response curve for target cells.
-
a.
Cluster targeting datasets
Full code in supplemental “R_Analysis_fluidfm_cluster_targeting”.
-
92.Setup:
-
a.Set up r-markdown document.
-
b.Set up libraries: tidyverse, patchwork, and ggforce.
-
c.Import cell data, cell metadata, and agonist concentration data.
-
a.
-
93.Data wrangling:
-
a.Calculate distance from dispensing point at x = 1025 px, y = 1025 px, z = 20 μm (for 10x images, the conversion factor is 1.53 px/micron).
-
b.Join metadata to data.
-
c.Add a column for binary activation (apply cutoff from calibration data).
-
d.Use pivot_wider to match up cells across time points by label.
-
e.Compute FC in Nuc/Cyt ratio from initial to final time point.
-
a.
-
94.Count number of nearest neighbors:
-
a.Define a function to count the number of nearest neighbors by counting nuclei within 18 μm of target cell nucleus.
-
b.Apply the function, add to data table.
-
c.Check accuracy of counting by printing one example plot and overlay on image (Figure 8).
-
a.
-
95.Local vs. culture density comparison:
-
a.Filter cells in the center activation zone so as not to weight the data with the outlying unstimulated cells. Distance cutoff depends on Cmax, determined to be 30 and 100 μm from center for 90 nM and 900 nM, respectively.
-
b.Determine culture density for each run.
-
c.Plot, group by number of neighbors and culture density.
-
a.
-
96.Bin data in xy grid to calculate fraction activated, join concentration information:
-
a.Join concentration information to cell data.
-
b.Create an xy grid of bins, join relative concentration info.
-
c.Sort cells into bins by location.
-
d.Group and summarize, join concentration information to cell information.
-
a.
-
97.
Plot binned data with concentration information in an xy grid (filter by stimulation concentration and time).
Statistical analysis
-
98.
Permutation analysis for comparing small group to large group: Takes in: combined activation data for two groups you want to compare (binary), size of smaller group for sampling, and experimental fraction activated of small group.
Returns: p-value for comparison between small and large group.
perm <- function(data, n, exp_a) {
a <- as.numeric(rep(NA,5000))
for(i in 1:5000){
s <- sample(data, size = n, replace = T)
a[i] <- sum(s,na.rm = T)/sum(!is.na(s))
}
pval = sum(a >= exp_a)/5000
return(pval)
}
-
99.
Permutation ANOVA code: Takes in: 2-column data-frame with group names in column 1 and continuous quantitative values in column 2. Returns: p-value for comparison between all groups, experimental F-statistic, list of F-statistics from sampled data.
permutation_anova <- function(dat){
colnames(dat) <- c("g", "d")
F_exp <- summary(aov(d∼g, data=dat))[[1]][1,4]
groups <- unique(dat$g)
F_vals <- vector(mode = "numeric")
for (i in 1:5000) {
sampled_data <- data.frame(g = dat$g, d = as.numeric(NA))
n = nrow(sampled_data)
sampled_data$d <- sample(dat$d, size=n, replace=T)
F_vals[i] <- summary(aov(d∼g, data=sampled_data))[[1]][1,4]
}
p_val <- sum(F_vals >= F_exp)/5000
return(list(p_val, F_exp, F_vals))
}
-
100.
Effect size code: Takes in: 2 column data frame with group names in column 1 and binary activation values in column 2, plus the names of two groups to compare change in fraction activated between. Returns: difference in fraction activated between the groups, uncertainty in effect size (from bootstrap method), and the fraction activated for each group.
effect_size3 <- function(dat, g1, g2) {
#rename columns of dataframe to d and g
colnames(dat) <- c("g", "d")
a <- as.numeric(rep(NA,5000))
group1 <- filter(dat, g==g1)
group2 <- filter(dat, g==g2)
n1 <- nrow(group1)
n2 <- nrow(group2)
a1 <- sum(group1$d,na.rm = T)/sum(!is.na(group1$d))
a2 <- sum(group2$d,na.rm = T)/sum(!is.na(group2$d))
d_exp <- a2 - a1
for (i in 1:5000) {
sample1 <- unlist(sample(group1$d, size=n1, replace=T))
sample2 <- unlist(sample(group2$d, size=n2, replace=T))
s1 <- sum(sample1,na.rm = T)/sum(!is.na(sample1))
s2 <- sum(sample2,na.rm = T)/sum(!is.na(sample2))
a[i] <- s2 - s1
}
SE <- sd(a, na.rm = T)
return(list(d_exp, SE, a1, a2))
}
-
101.
Example: filter data you want to compare, run the permutation ANOVA for all groups, use the Wilcoxon test (non-parametric pairwise comparisons) to determine pairwise significance, and run the effect size code. “df” is the data frame, “density” is the categorical variable for grouping, and “rNC” is the result to be compared.
dat1 = data.frame(
g = filter(fg_nn_center, C0_μm == 0.318)$density,
d = filter(fg_nn_center, C0_μm == 0.318)$rNC
)
permutation_anova(dat1)[[1]] #result: 0/5000
wilcox.test(filter(fg_nn_center, C0_μm == 0.318 & density == "sparse")$rNC,
filter(fg_nn_center, C0_μm == 0.318 & density == "dense")$rNC)
effect_size(dat1, "sparse", "dense")
Interpretation of example data
Single-cell stimulation data:
| Column name | Data type | Interpretation |
|---|---|---|
| Single-cell example data | ||
| Date | Character | Date of experiment |
| Dish | Numeric | Dish number of experiment |
| Spot | Numeric | Spot in dish of experiment |
| Timepoint | Numeric | Imaging timepoint (“before start of dispensing” noted as -1, at start of dispensing noted as 0, later time points given in minutes. |
| Math_nuc_cyt_ratio | Numeric | Nuclear/Cytoplasmic NF-κB Ratio, quantitative measure of activation |
| Location_Center_X | Numeric | Location of nuclear ROI center (in pixels), horizontal, from left |
| Location_Center_Y | Numeric | Location of nuclear ROI center (in pixels), vertical, from top |
| TrackObjects_Label | Numeric | Numbering of cell objects by CellProfiler TrackObjects module |
| Single-cell example data: metadata | ||
| Date | Character | Date of experiment |
| Dish | Numeric | Dish number of experiment |
| Spot | Numeric | Spot in dish of experiment |
| Timepoint | Numeric | Imaging timepoint (“before start of dispensing” noted as -1, at start of dispensing noted as 0, later time points given in minutes. |
| Target_Cell | Numeric | The TrackObjects_Label belonging to the target cell stimulated in that dataset. |
| Pressure.mbar | Numeric | Overpressure applied to dispense from FluidFM |
| C0.uM. | Numeric | Concentration of agonist loaded into the probe (μM) |
| Dispensing_Time.min. | Numeric | Time during which dispensing overpressure was applied (stimulation time) in minutes. |
| Condition | Character | Concentration of agonist at the target cell location, with uncertainty, and simulation time. |
Cluster Stimulation data:
| Column name | Data type | Interpretation |
|---|---|---|
| Cluster example data | ||
| Date | Character | Date of experiment |
| Dish | Numeric | Dish number of experiment |
| Spot | Numeric | Spot in dish of experiment |
| Timepoint | Numeric | Imaging timepoint (“before start of dispensing” noted as -1, at start of dispensing noted as 0, later time points given in minutes. |
| Math_nuc_cyt_ratio | Numeric | Nuclear/Cytoplasmic NF-κB Ratio, quantitative measure of activation |
| Location_Center_X | Numeric | Location of nuclear ROI center (in pixels), horizontal, from left |
| Location_Center_Y | Numeric | Location of nuclear ROI center (in pixels), vertical, from top |
| TrackObjects_Label | Numeric | Numbering of cell objects by CellProfiler TrackObjects module |
| Cluster example data: metadata | ||
| Date | Character | Date of experiment |
| Dish | Numeric | Dish number of experiment |
| Spot | Numeric | Spot in dish of experiment |
| Timepoint | Numeric | Imaging timepoint (“before start of dispensing” noted as -1, at start of dispensing noted as 0, later time points given in minutes. |
| Pressure_mbar | Numeric | Overpressure applied to dispense from FluidFM |
| C0_ug.mL | Numeric | Concentration of agonist loaded into the probe (μg/mL) |
| Stim_Time_min | Numeric | Time during which dispensing overpressure was applied (stimulation time) in minutes. |
| Cluster example data: R848 diffusion model | ||
| distance | Numeric | Distance from center of dish (below dispensing point) in μm |
| relative_concentration | Numeric | Concentration relative to C0 (concentration of agonist in probe) |
Expected outcomes
You should now have microscopy images showing single-cell NF-κB translocation over time, plus nuclear images for segmentation and quantification, and bright-field images showing probe position. The targeted cell(s) should have increased GFP fluorescence in the nucleus, while untargeted surrounding cells should remain quiescent, with GFP fluorescence in the cytoplasm (Figures 9A and 9B). The quantified NF-κB response is combined with the agonist diffusion model to analyze the single macrophage response to localized TLR stimulation in an otherwise unstimulated culture (Figure 9C).
Figure 9.

Expected outcome: target cell or cluster exhibits higher nuclear NF-κB after stimulation, analysis determines single and cluster activation thresholds
(A-B) Single cell (A) or cluster (B) stimulation before, during, and after dispensing agonist. Target cell/cluster becomes activated, neighboring cells do not respond (or respond less strongly). Green indicates reporter GFP fluorescence, purple indicates agonist concentration.
(C) Quantified individual cell responses and agonist diffusion data are combined during data analysis to compare stimulus and response on the single cell level. Results can be visualized as a plot of activation vs. location in target area, or as activation vs. agonist concentration at each location.
Criteria for data exclusion
-
•
Errors in cell tracking prevented the same cell from being identified in the initial and final time point images. Data point is excluded.
-
•
Cell divided, died, or moved out of view. Dataset is excluded.
-
•
Negative controls failed: Target cell Nuclear/Cytoplasmic NF-κB value for the negative controls are above the activation threshold. Exclude all datasets taken using that cell dish and/or probe and start again with fresh materials.
Limitations
Probe clogging: any molecule you may want to dispense must be able to flow through the probe without clogging the channel. Anything sticky or prone to aggregation may clog the probe. Likewise, whichever solvent you dissolve your molecule in must not clog or damage the probe. Always test dispensing before planning an experiment.
Passive diffusion: even when no overpressure is applied, agonist will diffuse slowly from the probe opening into the dish and may activate the target cell if close enough. Use low concentrations of the agonist to minimize any activation from passive diffusion.
Dish size: the FluidFM stage and controller dimensions limit what kinds of dishes and slides can be used with the microscope. Dishes and slides must be more than 40 mm wide and less than 9 mm high if the controller is on the stage. The stage is also not compatible with standard 96-well plates, so the microscope will not be able to accommodate those when the FluidFM is not in use.
Low-throughput: this protocol recommends performing single cell experiments one at a time, limiting the total number of experiments that can be completed in a day to less than 10. It might be possible to adapt the workflow to do a few runs in parallel, but it is still a low-throughput approach.
Diffusion model: the diffusion model used to determine agonist concentration has limited precision from the goodness of fit to the fluorophore dispensing data, and uncertainty in target cell height (and thus distance from dispensing point at low dispensing height).
Approach force: the FluidFM controller uses a force setpoint to determine when it has touched the dish surface or a cell. If the setpoint is too high, the probe will squish the cell when it makes contact, before it stops the approach. If the setpoint is too low, movement of the media and any stray vibrations will trigger the approach stop and it will be difficult to reach the dish surface. This is why I have used an approach of finding the dish surface, instead of the cell surface, to avoid disturbing the target cell by making forceful contact.
Troubleshooting
Problem 1
Cannot get probe on/off canticlip holder: newer probes can be difficult to get on and off the canticlip holder without breaking the tip. You may accidentally brush a fingertip or glove tip against the probe tip while trying to get it into place, or the canticlip holder may unclip from the magnetic stand and cause you to drop the probe. You may encounter this issue in Step 4 or Step 51.
Potential solution
To avoid breaking the tip, make sure your gloves fit snugly and be cautious to keep your fingertips away from the probe tip at all times. To put the probe on the holder: hold the top and bottom of the probe reservoir with one hand, position it above the holder, and with the other gently ease the legs of the probe down onto the holder with your fingernails (Figure 10). To get the probe off the holder: with one thumbnail hold the canticlip holder on the stand (use just the fingernail to keep the rest of your hand away from the probe tip) and with your other hand grasp the back of the probe and rock back and forth to loosen the legs until you can pull the probe off vertically.
Figure 10.

How to hold probe when attaching/detaching from holder
Grasp top and bottom of probe reservoir with index finger and thumb of one hand and use thumbnail of other hand to secure holder to magnetic stand as you remove the probe.
Problem 2
Difficulty attaching/detaching pneumatic tubing to/from probe. Especially when tubing is new, it can be difficult to correctly attach the pneumatic connector to the probe. Additionally, there is a high risk of splitting the tubing and/or breaking the probe tip when attaching or detaching the connector. You may encounter this issue in Step 4 or Step 51.
Potential solution
Best way to attach tubing (Figure 11).
-
•
Brace the probe: with the fingertips of your non-dominant hand, hold the arms of the probe onto the holder. Take care to keep away from probe tip! This will prevent the probe from coming off the holder, or the holder coming off the stand, when you twist the connector on/off.
-
•
With your dominant hand, press the connector vertically against the reservoir with the O-ring centered on the opening. The tubing should be facing away from you, if the probe tip is pointing to the left, and towards you if the probe tip is pointing to the right.
-
•
Firmly twist the connector clockwise 90 degrees, using your forefinger and thumb to press against the corners of the connector (NOT the bottom where the tubing attaches!) until it clicks into place horizontally. You will have to use a lot of force here, especially if the O-ring is new.
-
•
Check the connector is level and centered on the reservoir opening. If it is not, take off and try again.
Figure 11.

How to attach/detach pneumatic tubing
Secure probe arms and press on corners of tubing attachment to attach/detach tubing without splitting fragile tubing attachment point.
Best way to detach tubing (Figure 11).
-
•
Brace the probe arms as described above.
-
•
Twist the connector counterclockwise 90 degrees, again pressing against the corners and NOT the bottom of the connector, until it detaches.
Problem 3
Cannot find laser and/or probe. After mounting probe on controller, cannot see probe tip or laser light in camera field of view. You may encounter this issue in Step 5 or Step 51.
Potential solution
Potential causes: the controller is out of alignment, the probe is far out of focus, the probe is far off-center, the laser is far off-center, the laser is not on or plugged in, the laser light is being scattered by something, or the position knobs are far off center. If the laser or the probe are far off-center when you attempt to focus the microscope, the range of motion allowed by the stage control screws or laser control screws will not allow you to find them.
If you cannot find the probe:
-
•
Check that the controller is firmly seated in the divots in the stage beneath each leg.
-
•
Check that the probe is clipped onto the holder and the holder to the controller as far down as possible, centered, and level.
-
•
Try using a lower-magnification objective to locate the probe and center it, before switching to a higher magnification objective that has a smaller field of view.
-
•
Try moving the stage to find the edge of the chip where the tip is attached, then move along the edge until you see the tip come into view, then focus and center the tip (Figure 12).
Figure 12.

How to find probe went off-center
Move probe with stage control knobs until you find the edge of the chip (1), then move along the edge until you see the tip (2), and then focus and center the tip (3).
If you cannot find the laser:
-
•
Try looking for the lens flare/reflection of the laser light to give some direction (Figure 13).
-
•
Check that the laser cable is fully plugged in and the "laser off" box is unchecked in the Nanosurf software (see user manual).
-
•
Try moving the stage and laser to their limits to search the space (Figure 4).
-
•
Check that the probe is centered, because focusing on an off-center probe can lead to the laser being out of view.
-
•
Try taking the probe off the controller and replacing it, and repositioning everything.
-
•
Try moving the probe to home position, where the laser should be in view.
Figure 13.

How to find laser went off-center using reflections
Reflections from the laser point back towards its position.
Problem 4
Cannot align laser in air. When attempting to align the laser in air, cannot get a strong enough signal to use the optical force feedback system. You may encounter this issue in Step 5 or Step 48.
Potential solution
-
•
Check that the laser is centered on the probe tip. Sometimes a strong reflection can be mistaken for the laser if it is far off-center (Figure 14A).
-
•
Try moving the detector screws so that the detector indicator is far off-center - this can help in picking up a weak signal. In the example, “poor” or “bad” alignment is better than a false “good” that really indicates a lack of signal (Figure 14B).
-
•
Try also moving the laser further up the probe length, and it sometimes gives a stronger signal than at the very tip (Figure 14C).
-
•
Clean the probe of any gunk that may be scattering the laser light. Soak in an ethanol or bleach-based solution, followed by water (Figure 2C).
-
•
For a stronger cleaning, use oxygen plasma. Make sure the probe is dry, and place in a small Pyrex dish. Use a plasma etch instrument to apply oxygen plasma, 20 cc/min, at 200 W for 3–5 min. Do not use longer times as it will damage the glue that holds the tip onto the probe.
-
•
Clean the canticlip holder window and apply an anti-fogging spray (see key resources table). Clean the window surface with a drop of EtOH and a strip of lens paper folded into a small “brush”. Then spray the anti-fog solution onto the window once. Clean off the excess with lens paper. Make sure there are not bubbles or streaks on the window. (Figure 14D).
-
•
If none of this helps, switch to a new probe.
Figure 14.

Improving laser alignment in air
(A) Laser reflections can be mistaken for the laser, check you have found the brightest point.
(B) A false “good” will display on the detector position when there is no signal – moving the detector off-center may improve signal.
(C) Moving the laser further up the tip may improve signal.
(D) Apply anti-fog spray to the probe holder inner window using a folded lens tissue “brush” to improve signal.
Problem 5
Probe will not fill or dispense. Solution loaded into reservoir will not fill probe channel or dispense out of tip. You may encounter this issue in Step 5 or Step 51.
Potential solution
There are two possible explanations for a failure to dispense; either there is a clog preventing the probe from filling, or there is a pressure leak somewhere in the pneumatic system.
-
•
To test for a leak, apply manual pressure with a 10 mL syringe of air applied to the port in the pressure line (Figure 3A). If you are able to push the plunger all the way down, there is a leak.
-
•
You can also visually inspect the probe channel (Figure 1D) while applying syringe pressure (Figure 3A) to see if you can see liquid move from the reservoir into the channel. If there is no motion, there is likely a pressure leak. If the channel fills partway and stops, there is likely a clog in the probe tip.
To fix a leak: The most common place for a leak to occur is in the pneumatic connector, right behind the attachment to the probe (Figure 15). If the tubing is split, replace with a new pneumatic connector, or patch with a small amount of epoxy.
-
•
Sometimes the probe will leak if the chip has come unglued from the plastic part of the probe. If this is the case, replace the probe.
Figure 15.

Pneumatic connector split point
The fragile tubing often splits at the attachment point, causing a leak.
To clear a clog.
-
•
Try soaking the tip for 10 min in a drop of water, ethanol-based solution, or dilute bleach solution followed by water, as described previously (Figure 2C).
-
•
You can also try and clear a clog with increased pressure. If the maximum pump pressure is insufficient, try using a 10 mL syringe of air, attached at the in-line port (Figure 3A). You should feel resistance as you compress the syringe, and it should stop around the 2 mL mark. You can use a syringe pump to hold the plunger down.
-
•
If the above does not work, clean the probe with oxygen plasma as described in the “cannot align laser in air” section or replace the probe.
Problem 6
Cannot align laser in liquid. When attempting to align the laser in water/media, cannot get a strong enough signal. You may encounter this issue in Step 5 or Step 51.
Potential solution
Potential causes:
-
•
Air pockets between the canticlip window and the probe (Figure 16A). Especially if the probe is dry before lowering into liquid, this is likely to occur. The probe might be blurry even when in focus, or you might see a shadow cast by an air bubble.
-
•
Condensation on the inside of the canticlip holder window (Figure 16B).
-
•
Dirty probe or damaged reflective coating.
Figure 16.

Improving laser alignment in liquid
(A) Air bubbles can get stuck between the probe and the holder window and scatter the laser.
(B) Condensation can form on the inner surface of the holder window and scatter the laser.
(C) The water drop needs to make good contact between the slide and the probe to avoid scattering from an air-water interface.
(D) Water on the window method to dislodge air bubbles and make good liquid contact: pipette a small drop of water onto the holder window.
Solutions:
-
•
Try and dislodge any air bubbles by lifting up and replacing the controller to remove/replace the probe in the water drop.
-
•
Add more water to the drop underneath the probe to ensure it is fully submerged (Figure 16C).
-
•
Hold the controller upside down and VERY CAREFULLY pipette 40 μL of water onto the window to wet it before replacing the controller on the stage (Figure 16D).
-
•
Reapply anti-fogging spray to the inside of the window as described in the “cannot align laser in air” section.
-
•
Clean probe as described in “cannot align laser in air” section.
-
•
If nothing works, replace probe.
Problem 7
Probe automated approach failed. When trying to approach the probe to the dish surface using the controller automated approach, you may get the error message “approach failed, check laser alignment,” or the approach may stop with no error message before you have reached the surface. You may encounter this issue in Step 9 or Step 51.
Potential solution
Potential causes:
-
•
Laser is off.
-
•
Laser lost alignment.
-
•
Automatic stop was triggered by vibrations in the room or movement of media in the dish.
Solutions:
-
•
Check that the laser is on and still aligned. Sometimes a laser that was badly aligned when the probe was submerged in water will lose signal when the probe is placed in the media dish.
-
•
Improve the signal quality by following the suggestions in the “cannot align laser” troubleshooting sections.
-
•
Try increasing the force set-point (in the AFM settings menu) to avoid triggering the automatic stop from stray vibrations.
-
•
Sometimes you will get the "approach failed" error when you contact the surface, because the deflection of the probe moved the laser beyond the limits of the detector. That’s okay because you’ve still approached the surface successfully.
Problem 8
Negative control activating. The macrophages are activating even when you are not dispensing agonist. You may encounter this issue in Step 8 and Negative controls.
Potential solution
If your negative controls are causing activation of the macrophages, review your probe cleaning protocols. Any stray ethanol or bleach solution on the probe will irritate the cells. You could increase the number or duration of water rinses. Also make sure the water drop makes complete contact with the probe and holder. Keep your rinsing water as sterile as possible.
If still experiencing issues, try dispensing at a greater height or lower pressure, so the dispensed water does not displace the media but rather diffuses into it. Do not dispense when probe is in contact with cell, as the cell may start up-taking the dispensed fluid directly.
Check on your incubation conditions for the cells. Is the pH of their media correct? Are they in the correct temperature range? A temperature a little too high (even just 38°C) will cause irritation, and too low will slow down NF-κB responses. For pH, use of CO2 Independent Medium is best, as other buffer methods such as HEPES do not work well with this protocol. If the HIFBS in your media is too old, that may cause problems with cell responses. Check on the exposure time used in imaging your nuclear stain – the UV wavelength used to excite DAPI and Hoechst stains will also irritate the cells if the exposure time is too long.
Problem 9
Poor quality nuclear stain, probe reflection. If the nuclear stain is dim, the FluidFM probe may cause a reflection in the Hoechst channel images that will interfere with segmentation later (Figure 17). It is essential for data quality to be able to draw accurate boundaries around each nucleus, so the quality of the nuclear image is important. You may encounter this issue in Step 12.
Figure 17.

Poor nuclear staining interferes with segmentation during image processing
Example of poor nuclear stain quality image, showing reflection from the probe surface. Blue is Hoechst 33342 nuclear stain, scale bar 25 μm.
Potential solution
-
•
Use fresh stain and media to maximize cell health and stain uptake.
-
•
Add more stain and/or increase staining time.
-
•
Average multiple frames together, adjust the image threshold, or increase exposure time.
-
•
Background subtraction or flatfield correction in ImageJ.
-
•
Use ImageJ to select and delete areas with the probe reflection by hand.
Problem 10
Media in tubing. Sometimes when using negative pump pressure, media from the cell dish can get into the pneumatic tubing. You may encounter this issue in Step 15.
Potential solution
To clean the pneumatic tubing, detach the lengths of tubing from the pump and trap and hang them vertically. Use a syringe to push water through the tubing. Then push through air until no more liquid comes out the bottom. Hang to air dry overnight, then reconnect.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aaron Esser-Kahn (aesserkahn@uchicago.edu).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Elizabeth J Mulder (ejmulder@uchicago.edu).
Materials availability
This study did not generate new unique materials.
Data and code availability
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2.
Acknowledgments
This work was supported in part by the National Institute of Allergy and Infectious Diseases (75N93019C00041 and 5U01AI124286-06), Defense Threat Reduction Agency (HDTRA11810052), National Science Foundation (OMA-2121044), The Pew Charitable Trusts: Pew Scholars, and the Camille Dreyfus Teacher-Scholar Award. This work made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center, supported by the National Science Foundation under award number DMR-2011854. NF-κB reporter cells were a generous gift from the Fraser Lab. Thanks to Dr. Kenneth Mulder for his advice on statistical analysis. Thanks to Nina Coyle and Aurora Ireland for their advice on the diffusion model derivations. Thanks to Dr. Justin Jureller and Dr. Edward Nelson for assistance with FluidFM setup and troubleshooting. Thanks to Dr. Jamie Scott and Dr. Matthew Curtis for assistance with microscopy setup and troubleshooting.
Author contributions
Conceptualization, B.M., E.M., R.S., and A.E.-K.; methodology, E.M., B.M., J.D., and R.S.; investigation, E.M. and J.D.; validation, formal analysis, data curation, writing – original draft, and visualization, E.M.; writing – review and editing, E.M., B.M., and A.E.-K.; supervision, project administration, and funding acquisition, A.E.-K.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Elizabeth J. Mulder, Email: ejmulder@uchicago.edu.
Aaron P. Esser-Kahn, Email: aesserkahn@uchicago.edu.
References
- 1.Mulder E.J., Moser B.A., Delgado J., Steinhardt R.C., Esser-Kahn A.P. Evidence of Collective Influence in Innate Sensing using Fluidic Force Microscopy. Front. Immunol. 2024;15:1664–3224. doi: 10.3389/fimmu.2024.1340384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li N., Sun J., Benet Z.L., Wang Z., Al-Khodor S., John S.P., Lin B., Sung M.-H., Fraser I.D.C. Development of a cell system for siRNA screening of pathogen responses in human and mouse macrophages. Sci. Rep. 2015;5:9559. doi: 10.1038/srep09559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meister A., Gabi M., Behr P., Studer P., Vörös J., Niedermann P., Bitterli J., Polesel-Maris J., Liley M., Heinzelmann H., Zambelli T. FluidFM: Combining Atomic Force Microscopy and Nanofluidics in a Universal Liquid Delivery System for Single Cell Applications and Beyond. Nano Lett. 2009;9:2501–2507. doi: 10.1021/nl901384x. [DOI] [PubMed] [Google Scholar]
- 4.Guillaume-Gentil O., Potthoff E., Ossola D., Franz C.M., Zambelli T., Vorholt J.A. Force-controlled manipulation of single cells: from AFM to FluidFM. Trends Biotechnol. 2014;32:381–388. doi: 10.1016/j.tibtech.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Aebersold M.J., Dermutz H., Demkó L., Cogollo J.F.S., Lin S.-C., Burchert C., Schneider M., Ling D., Forró C., Han H., et al. Local Chemical Stimulation of Neurons with the Fluidic Force Microscope (FluidFM) ChemPhysChem. 2018;19:1234–1244. doi: 10.1002/cphc.201700780. [DOI] [PubMed] [Google Scholar]
- 6.McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding G.J., Fischer P.A., Boltz R.C., Schmidt J.A., Colaianne J.J., Gough A., Rubin R.A., Miller D.K. Characterization and Quantitation of NF-κB Nuclear Translocation Induced by Interleukin-1 and Tumor Necrosis Factor-α DEVELOPMENT AND USE OF A HIGH CAPACITY FLUORESCENCE CYTOMETRIC SYSTEM. J. Biol. Chem. 1998;273:28897–28905. doi: 10.1074/jbc.273.44.28897. [DOI] [PubMed] [Google Scholar]
- 8.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfram Research, Inc., Mathematica, Version 12.0, Champaign, IL (2022). https://www.wolfram.com/mathematica.
- 10.Evans R., Dal Poggetto G., Nilsson M., Morris G.A. Improving the Interpretation of Small Molecule Diffusion Coefficients. Anal. Chem. 2018;90:3987–3994. doi: 10.1021/acs.analchem.7b05032. [DOI] [PubMed] [Google Scholar]
- 11.RStudio Team (2020). RStudio: Integrated Development for R. RStudio, PBC, Boston, MA URL http://www.rstudio.com/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/kgtt279fp7.2.