

Key Points
-
•
We identified a novel miRNA signature in patients with BCP-ALL at high risk of relapse, which is independent of known genetic subtypes.
-
•
This miRNA signature is often newly acquired at relapse and is associated with the upregulation of MYC target genes.
Visual Abstract
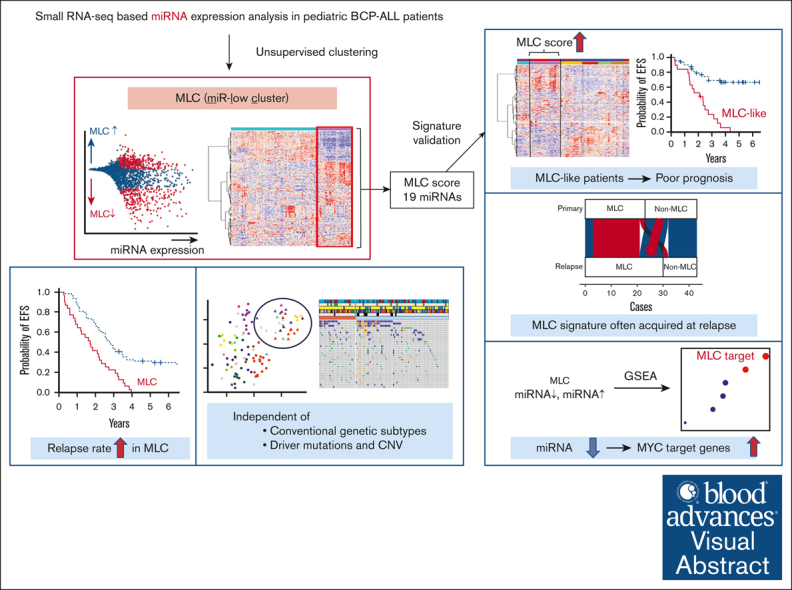
Abstract
Aberrant micro-RNA (miRNA) expression profiles have been associated with disease progression and clinical outcome in pediatric cancers. However, few studies have analyzed genome-wide dysregulation of miRNAs and messenger RNAs (mRNAs) in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL). To identify novel prognostic factors, we comprehensively investigated miRNA and mRNA sequencing (miRNA-seq and mRNA-seq) data in pediatric BCP-ALL samples with poor outcome. We analyzed 180 patients, including 43 matched pairs at diagnosis and relapse. Consensus clustering of miRNA expression data revealed a distinct profile characterized by mainly downregulation of miRNAs (referred to as an miR-low cluster [MLC]). The MLC profile was not associated with any known genetic subgroups. Intriguingly, patients classified as MLC had significantly shorter event-free survival (median 21 vs 33 months; log-rank P = 3 ×10−5). Furthermore, this poor prognosis was retained even in hyperdiploid ALL. This poor prognostic MLC profiling was confirmed in the validation cohort. Notably, non-MLC profiling at diagnosis (n = 9 of 23; Fisher exact test, P = .039) often changed into MLC profiling at relapse for the same patient. Integrated analysis of miRNA-seq and mRNA-seq data revealed that the transcriptional profile of MLC was characterized by enrichment of MYC target and oxidative phosphorylation genes, reduced intron retention, and low expression of DICER1. Thus, our miRNA-mRNA integration approach yielded a truly unbiased molecular stratification of pediatric BCP-ALL cases based on a novel prognostic miRNA signature, which may lead to better clinical outcomes.
Introduction
Recent genomic and transcriptomic analyses of pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL) have revealed novel genetic subgroups.1 These novel genetic subgroups and their driver mutations affect prognosis; therefore, treatment stratification based on risk classification has been proposed to improve treatment outcomes.2,3 However, ∼20% of pediatric patients with BCP-ALL relapse, and the prognosis remains poor; indeed, <50% of the patients who relapse survive long term,4 whereas, approximately half of the patients who relapse have a favorable prognosis; these include patients with hyperdiploidy and ETV6::RUNX1.5 Although minimal residual disease (MRD), a marker of response to therapy, is the strongest prognostic factor,6 many patients who achieve undetectable MRD experience relapse; therefore, current prognostic factors are not thought to be sufficiently stratified.
Micro RNAs (miRNAs) regulate posttranscriptional gene expression by functioning as messenger RNA (mRNA) translation repressors or degraders.7 Aberrant expression of miRNAs by tumor cells is a novel biomarker or therapeutic target for various malignancies.8 Recent studies have revealed characteristic expression patterns according to genetic subgroups of pediatric BCP-ALL as well as their prognostic significance.9,10 In addition, novel prognostic factors identified by integrated analysis, including miRNAs and mRNAs, have been reported in several types of malignant tumors11; however, few studies have conducted an integrated analysis focusing on miRNA profiles in a large cohort of pediatric BCP-ALL.
In this study, we aimed to clarify the expression profile of miRNAs in pediatric BCP-ALL by performing an integrative analysis of miRNA sequencing (miRNA-seq) data. We then used this information to investigate their clinical significance and identify prognostic factors on which we could base a more refined risk stratification approach.
Methods
Patients
The TARGET ALL-P2 cohorts have been published previously, and data were used with permission from the database of Genotypes and Phenotypes (accession number phs000218).1 The study included 111 primary samples of BCP-ALL (of which 35 patients had paired sample data at relapse) from the TARGET cohort, a high-risk cohort with clinical information, for whom both miRNA-seq and mRNA sequencing (mRNA-seq) data were available (supplemental Tables 1 and 2). Of these 111 cases, 105 had single nucleotide polymorphism (SNP) array data (Affymetrix SNP Array 6.0), and 53 had methylation data (HpaII tiny fragment enrichment by ligation-mediated PCR [HELP]) Assay with Roche NimbleGen). The samples sequenced in this study were derived from 69 primary samples (including 8 paired samples at relapse) provided by multiple institutions in Japan (supplemental Tables 1 and 3). All samples were obtained with written consent from the patient or their parent/guardian under the protocols approved by the institutional review board of each institution. All studies were conducted in accordance with the international ethical guidelines for biomedical research involving human subjects. The chemotherapy regimens used in the TARGET cohort were AALL023212 and AALL0331,13 and in the Japanese cohort, were based on JACLS14 or TCCSG15 protocols.
miRNA-seq
miRNA-seq was performed in the Japanese cohort (validation cohort). Total RNA was extracted using the miRNeasy Kit (Qiagen). Libraries for miRNA-seq were generated using the NEXTflex Small RNA Sequencing kit version 3 (PerkinElmer) and sequenced using the Illumina NextSeq 550 using a 75 bp single-end read protocol. miRNA-seq analysis was performed using the nf-core/smrnaseq pipeline version 1.0.0.16 Briefly, all steps consisted of the removal of 3′ adapter sequences by TrimGalore version 0.6.5, mapping to mature and hairpin miRNAs using miRbase version 22.1, and mapping to the GRCh37 human reference genome using Bowtie version 1.3.0. After alignment and trimming, sorted binary alignment map (BAM) files were used for further analysis using edgeR version 3.32 for reads per million normalization, and mirtop version 0.4.23 for raw counts. Normalized count data obtained from the regularized logarithm function of the R package DESeq2 version 1.26.0 were used for clustering analysis. Cluster stability was determined by consensus clustering with 1000 iterations using the R package ConsensusClusterPlus version 1.50.0. The function removeBatchEffect from the R package limma package version 3.42.2 was used to remove batch effects for merged miRNA expression during unsupervised clustering analysis. Differential expression analysis was performed using the DESeq2. After the rlog gene-expression level was calculated using DESeq2, Rtsne in the R package was used to map the samples to a 2-dimensional t-distributed stochastic neighbor embedding (tSNE) plot of the top 500 most variable miRNAs (on the basis of median absolute deviation), and the tSNE perplexity parameter was set to 30.
mRNA-seq
RNA was isolated from fresh/frozen samples using the NucleoSpin Triprep (Macherey-Nagel). Sequencing libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs). The library was sequenced using an Illumina HiSeq X platform using a 150-bp paired-end read protocol. mRNA-seq was performed in the Japanese cohort. Sequence alignment and read counting were conducted using the Genomon version 2.6.2 pipeline (http://genomon.readthedocs.io/ja/latest/index.html). Read counting was also normalized to obtain fragments per kilobase million values. Fusion transcripts were detected by the CICERO software (supplemental Table 4).17 To identify BCR::ABL1–like and ETV6::RUNX1 ALL, 198 genes were identified using the recognition of outliers by sampling ends (ROSE) method,18 including the top 25 genes in clusters 1 to 8 in the TARGET cohort (supplemental Figure 1). Ward hierarchical clustering method was then used for clustering analysis of the ROSE gene set. Samples in cluster 5 had a signature similar to that of BCR::ABL1 ALL and were, therefore, labeled BCR::ABL1–like, whereas samples in cluster 8 had a signature similar to that of ETV6::RUNX1 ALL and were labeled ETV6::RUNX1–like.
We used mRNA-seq data to examine driver mutations in variant calls, following the Genome Analysis Toolkit (GATK) best practices for calling variants (https://gatk.broadinstitute.org/hc/). Variants were called by the GATK HaplotypeCaller version 4.1 according to the GATK best practices for variant calling in mRNA-seq, as implemented in the CRG-CNAG/CalliNGS-NF pipeline (https://github.com/CRG-CNAG/CalliNGS-NF). Overall, 110 known or putative driver genes and target genes of activation-induced cytidine deaminase in BCP-ALL were filtered (supplemental Table 5).3 The filtered variants were then annotated against the COSMIC database version 92 (a cancer mutation catalog) from the GRCh37 assembly to retain those variants previously identified as cancer mutations.19
Additional details for “Methods” are provided in the supplemental Data.
Results
Consensus clustering of miRNAs reveals a novel high-risk subgroup with a distinct miRNA profile
To characterize the miRNA profile of patients with BCP-ALL having poor outcome, we focused on the TARGET ALL-P2 cohort, which mostly consisted of cases with early bone marrow relapse. We included 111 cases for which miRNA-seq, mRNA-seq data, and clinical information were all available and used them as the discovery cohort (supplemental Tables 1 and 2). First, we performed unsupervised consensus clustering of miRNA expression data obtained from primary samples. The analysis revealed a strong cluster (n = 31) that was not associated with well-known genetic subtypes (cluster 2 in Figure 1A-B; supplemental Figure 2A-B). This cluster included diverse genetic subtypes, such as hyperdiploidy (n = 7) and ETV6::RUNX1 (n = 4) fusions. Differential expression analysis of miRNAs with high average expression across the whole population composing this subgroup demonstrated that they were mainly downregulated (Figure 1C-D; supplemental Table 6). Hereafter, we refer to this subgroup with a distinct miRNA expression profile as the miR-low cluster (MLC). Notable differentially expressed miRNAs in the MLC included tumor suppressive (eg, miR-26b,20 miR-101,21 miR-29, miR-142,22 miR-146b,23 and miR-32624) and oncogenic (eg, miR-130b25 and miR-92a26) miRNAs linked to leukemia. Consecutive 2-step unsupervised consensus clustering of the MLC and non-MLC group identified 4 clusters respectively, which generally clustered together with well-known subtypes (supplemental Figure 2C), further highlighting the distinct features of the aberrant miRNA expression profile identified in the MLC.
Figure 1.

miRNA expression clusters in 111 BCP-ALL cases based on unsupervised consensus clustering and clinical impact of the MLC miRNA signature. (A) Heat map of miRNA expression by 111 primary samples from the TARGET cohort, along with the clinical information for each case. Two stable clusters were identified by consensus clustering of the 111 samples using 500 miRNAs (the distance method: Pearson) (supplemental Figure 2A). (B) Gene expression profiling of the 111 primary samples is shown as a 2-dimensional tSNE plot. Each dot represents 1 sample. The top 1000 most variable mRNA genes (based on the median absolute deviation) (top) and the top 500 most variable miRNAs (bottom) were selected and processed by the tSNE algorithm, with a perplexity score of 30. Genetic BCP-ALL subtypes are highlighted in different colors. Circles and triangles indicate cluster 1 and cluster 2, respectively. (C) MA plot for differential expression analysis between cluster 1 and cluster 2 generated by DESeq2: for each miRNA, the log2 (fold change) (log2[cluster 2/cluster 1]) is plotted (A, y-axis) against the log2 (average normalized expression) of the gene in the 2 clusters (M, x-axis). Significantly differentially expressed genes with an adjusted P value < .05 are shown in red. (D) Volcano plot showing differences in miRNAs expressed in cluster 2 samples (compared with cluster 1 samples) among the highly expressed miRNAs with a median of ≥25 reads per million. Significantly differentially expressed miRNAs showing a log2-fold change (cluster 2/cluster 1) > 1 or < −1, and an adjusted P value < .05, are shown in red. (E) Kaplan-Meier survival curves of EFS and OS for 111 BCP-ALL cases and 23 hyperdiploid BCP-ALL cases with and without the MLC signature. P values were calculated by the log-rank test. (F) Forest plot showing multivariate Cox regression analysis of the effect of different parameters on EFS. Squares represent the hazard ratio, and horizontal lines represent the CI. WBC at Dx, white blood cell count at diagnosis.
Next, we assessed the impact of the miRNA signature of the MLC on the clinical outcome of pediatric patients with BCP-ALL. In the TARGET ALL cohort, 83 patients experienced relapse. We found that the MLC group had significantly shorter event-free survival (EFS) than the non-MLC group (median, 21 vs 33 months, respectively; 95% confidence interval [CI], 11.1-26.8; log-rank P = 3 × 10−5; Figure 1E). Although not significant, the MLC group also showed a trend toward shorter overall survival (OS) (median, 29 vs 64 months, respectively; 95% CI, 21.2-84.9; log-rank P = .09). In addition, when the analysis was restricted to patients with hyperdiploidy (n = 23), the MLC group (7 of 23 patients) had significantly shorter EFS (median, 28 months; 95% CI, 3.3-39.1; log-rank P = 5 × 10−4). Although the difference in OS was not statistically significant, there was a trend toward shorter OS in the MLC group of those with hyperdiploidy (log-rank P = .15). Multivariable Cox regression analysis of MLC status and established prognostic factors, including end-induction MRD assessed by multicolor flow cytometry,27,28 showed that MLC status was an independent predictor of relapse (hazard ratio, 2.59; 95% CI, 1.56-4.3; P < .001; Figure 1F). This was also true in the analysis in which each genetic subtype was included as a covariate (supplemental Figure 3). Taken together, these results suggest that the miRNA signature of MLC could be a novel prognostic factor that predicts relapse better than current prognostic factors, such as MRD.
Validation of the poor prognostic MLC profile in the validation cohort
To validate the poor prognostic miRNA profile of MLC, we evaluated the miRNA profile in the Japanese BCP-ALL cohort. We collected primary samples of 69 patients, 34 of whom relapsed (validation cohort; supplemental Tables 1 and 3). Two-step consecutive unsupervised consensus clustering (supplemental Figure 4A-B) identified a cluster (n = 19) that was independent of known genetic subtypes (cluster 2 in supplemental Figure 4C), as observed for the MLC in the TARGET cohort. To identify patients similar to the MLC in this small Japanese cohort, we applied a statistical regression algorithm based on the least absolute shrinkage selection operator.19,29 This analysis identified a 19-miRNA signature characterizing the MLC (Figure 2A). Next, the weighted sum of the expression levels of these 19 miRNAs was calculated for each patient in the validation cohort and was expressed as the MLC score. The group identified by the 2-step consecutive unsupervised clustering method described earlier (supplemental Figure 4C) had MLC scores almost exclusively in the highest tertile within the validation cohort; therefore, we referred to this group as patients with MLC-like characteristics (Figure 2B). Compared with the non–MLC-like group, patients with MLC-like characteristicswere more likely to relapse (Figure 2C) and had a significantly shorter EFS (median, 25 months; 95% CI, 16.1-33.0; log-rank P = 1 × 10−6) and OS (median, 42.6 months; 95% CI, 22.3-78.4; log-rank P = 8 × 10−4; Figure 2D). Furthermore, evaluation of the area under the curve of the time-dependent receiver-operating characteristic curve, which measures the prognostic ability of the MLC score and miRNAs to predict EFS and OS at 2 years, showed that the MLC score was one of the best predictors of poor prognosis compared with each single miRNA in both the TARGET and Japanese cohorts (Figure 2E; supplemental Tables 7 and 8). In particular, among the elements of the MLC score, high expression of miR-92b-3p and miR-1304-3p were good predictors of poor prognosis (supplemental Figure 5A-B). Taken together, these data confirm the use of the MLC profile for predicting poor prognosis in the validation cohort.
Figure 2.

Validation of the MLC signature in the Japanese cohort. (A) Strategy used to identify and test the MLC score for the 19-miRNA signature. (B) Heat map of miRNA expression by 69 primary samples from the Japanese cohort, along with clinical information for each case. Six clusters were identified by consensus clustering of the 69 samples using 500 miRNAs (the distance method, Pearson). Cases with an MLC score in the third quartile or higher were defined as having a high MLC score. Samples in Cluster 2 had a high MLC score and were labeled as MLC-like (n = 19). (C) The distribution of MLC scores and EFS in the Japanese cohort. (D) Kaplan-Meier survival curves of EFS and OS for BCP-ALL cases with and without an MLC-like signature. P values are based on the log-rank test. (E) Time-dependent receiver-operating characteristic curve analysis and area under the curve (AUC) of the predictive power of MLC score, miR-92b-3p, and miR-1304-3p expression on 2-year EFS and OS in the TARGET and Japanese cohort. FP, false-positive rate; RPM, reads per million; TP, true-positive rate.
MLC status is not associated with previously reported genetic alterations
To investigate the relationship between MLC status and previously reported genetic alterations in BCP-ALL, we examined driver mutations in short variants and copy number alterations from the discovery cohort (supplemental Tables 9 and 10). In general, the distribution of these genetic alterations did not differ significantly according to MLC status, although alterations in IKZF1 or TP53 and rearrangement in ZNF384 were significantly mutually exclusive with MLC status (Figure 3A-B; supplemental Figure 6A). Variant call and copy number analysis were also performed using whole-exome sequencing from the validation cohort (supplemental Figure 6B-C; supplemental Tables 11 and 12), but we found no abnormalities characterizing MLC-like cases. Analysis of focal copy number (supplemental Figure 7) and DNA methylation data (supplemental Figure 8) from the TARGET ALL cohort did not identify characteristic abnormalities for MLC. In addition, the poor risk chromosomal profile for hyperdiploidy30 was observed in only 1 case in the MLC (supplemental Table 2). Thus, the MLC miRNA profile does not seem to be associated with any of the well-known high-risk genetic subgroups, driver sequence mutations, copy number alterations, such as IKZF1 or IKZF1plus, or aberrant DNA methylation, making it a strong candidate for novel risk stratification.
Figure 3.

Gene mutations in the MLC BCP-ALL cases. (A) Recurrent driver mutations identified by variant calls from RNA sequencing and copy number variations from the single nucleotide polymorphism array, along with clinical information, for 111 BCP-ALL primary samples from the TARGET cohort. Cases are grouped as MLC or non-MLC according to miRNA expression clusters. (B) Gene mutations and MLC signature pairs showing co-occurrence or exclusivity in the 111 BCP-ALL primary samples from the TARGET cohort are illustrated as a triangular matrix. Green indicates a tendency toward co-occurrence, whereas brown indicates exclusivity. The point indicates P < .1, and the asterisk indicates P < .05. WBC, white blood cell.
Non-MLC samples frequently acquired MLC-like miRNA signature at relapse
Next, we analyzed paired samples (obtained at diagnosis and relapse) to determine whether the miRNA profile changes at the point of relapse. Unsupervised clustering of 43 paired primary and relapse samples identified 2 clusters (Figure 4A). Cluster 1 contained all 20 primary samples classified as MLC at diagnosis; this was considered to be the group with the MLC profile. Eighteen of these were also categorized into the same cluster at relapse (Figure 4B; supplemental Table 13). By contrast, 9 of 23 patients classified as having a non-MLC profile at diagnosis showed a change in their miRNA profile from cluster 2 at diagnosis to cluster 1 at relapse (Fisher exact test, P = .039). This suggests that patients with non-MLC can acquire an MLC profile at relapse, which is consistent with the fact that MLC scores were significantly higher at relapse than at diagnosis in these 9 patients (Wilcoxon signed-rank test, P = .021; Figure 4C). Furthermore, evaluation of the MLC scores for the paired primary and relapse samples from the 23 non-MLC cases showed that the MLC score increased significantly at relapse (Wilcoxon signed-rank test, P = .014; Figure 4D). These results imply that an MLC profile may confer resistance to treatment and contribute to disease progression and relapse.
Figure 4.

The MLC signature of relapsed BCP-ALL. (A) Heat map showing miRNA expression by 86 paired primary and relapse samples from the TARGET cohort (35 cases) and the Japanese cohort (8 cases), along with clinical information for each case. Two clusters were identified by consensus clustering of the 86 samples using 400 miRNAs (the distance method, Pearson). Primary samples with an MLC or MLC-like signature are denoted as MLC at diagnosis. (B) An alluvial plot illustrates the transition of the miRNA cluster from the primary to relapse samples from 43 cases; red indicates primary samples classified as MLC (20 cases), and blue indicates those classified as non-MLC (23 cases). (C-D) Box plots comparing the MLC scores between paired primary and relapse samples (C) in the 9 cases that acquired MLC-like miRNA profiles at relapse, and (D) in the 23 non-MLC cases at diagnosis. P values are calculated using the Wilcoxon signed-rank test.
The MLC is characterized by enrichment of MYC target and oxidative phosphorylation genes, and by reduced IR
To identify genes targeted by deregulated miRNAs in the MLC, we performed an integrated analysis of miRNAs and mRNAs in the TARGET cohort (Figure 5A; supplemental Table 14).11 Gene set enrichment analysis of the upregulated genes in the 2331 pairs showing negative correlation between miRNAs and mRNAs revealed significant enrichment of MYC proto-oncogene, bHLH transcription factor (MYC) target and oxidative phosphorylation genes (Figure 5B) as well as genes related to RNA splicing (Figure 5C). Furthermore, gene set enrichment analysis of genes upregulated in the paired primary and relapse samples from the 9 patients who acquired an MLC profile at relapse also showed significant enrichment of MYC target genes and genes related to oxidative phosphorylation (Figure 5D-E). Collectively, these results suggest that these pathways contribute to the aggressive phenotype of the MLC.
Figure 5.


Integrative miRNA-mRNA analysis of BCP-ALL. (A) Workflow for the integrative miRNA-mRNA analysis of 111 BCP-ALL cases from the TARGET ALL cohort. (B-C) Dot plot showing the top enriched (B) Hallmark gene sets, and (C) Gene Ontology terms ranked according to gene ratio based on upregulated genes in sample pairs showing decreased miRNA expression and increased mRNA expression in the 111 BCP-ALL primary samples from the TARGET cohort. The adjusted P value is represented by the color scale, and the size of the dot reflects the number of genes assigned to the gene sets. Over-representation analysis was performed based on a 1-sided Fisher exact test conducted using the R package clusterProfiler. (D) Workflow for gene set enrichment analysis of genes significantly upregulated in relapse samples compared with primary samples from the 9 cases that acquired an MLC profile at relapse. (E) Dot plot showing the top enriched Hallmark gene sets, ranked according to the gene ratio of the upregulated genes shown in panel D. (F) Type and number of splicing alterations significantly associated with MLC compared with non-MLC cases in the TARGET cohort. The bars on the right denote alternative-splicing events that were increased in MLC cases. The bars on the left denote those that were decreased in MLC cases. (G) Venn diagram of decreased IR in the MLC compared with that in normal pro–B-cell and non-MLC samples, genes showing increased expression in the MLC compared with non-MLC samples, and genes with negatively correlated miRNA-mRNA pairs in MLC samples. (H) miR-30e-5p and MTA1 show inversely correlated expression patterns in 111 cases from the TARGET ALL cohort. Red dots represent MLC cases and blue dots represent non-MLC cases. (I) Heat map and clustering based on gene-set variation analysis enrichment scores for genes upregulated in normal B-precursor cells from 111 cases in the TARGET ALL cohort (left). Gene set enrichment analysis and enrichment plot for MLC and non-MLC cases, showing enrichment of genes upregulated in pre–B cells from MLC cases (right). CLP, common lymphoid progenitor; FDR, false discovery rate; FPKM, fragments per kilobase million; NES, normalized enrichment score; RPM, reads per million.
Given the enrichment of RNA splicing terms within the genes upregulated in the MLC, we next focused on splicing alterations in the MLC. We identified that almost all of the alternative-splicing events in MLC cases were reductions in intron retention (IR) (Figure 5F; supplemental Table 15), which was also observed when compared with normal pro–B cells, and in 9 relapsed cases that acquired the MLC-like profile at relapse (supplemental Figure 9A-B). Genes showing decreased IR and increased expression because of a reduction in miRNA expression may be crucial for the MLC; therefore, we looked for genes that met all of the following criteria: (1) genes for which expression increased in MLC compared with that in non-MLC samples (supplemental Table 16); (2) genes for which an miRNA reduction in the MLC may be associated with an increase in mRNA expression (compared with that in non-MLCs); and (3) genes with decreased IR (compared with that in non-MLCs and normal pro–B cells). This resulted in identification of the MTA1 gene, an RNA-binding protein gene that posttranscriptionally activates MYC31 (Figure 5G). Notably, miR-30e-5p, a previously identified MTA1 suppressor,32 showed a significant inverse correlation with MTA1 expression in the TARGET cohort (Figure 5H) and may play a role in increasing MYC activity in the MLC. Next, we performed gene-set variation analysis.33 Based on the B-lineage cluster-specific gene set,34 the gene-expression pattern of MLC leukemic blasts resembled that of pre–B cells; this was not the case for non-MLC patterns (Figure 5I). A previous study shows that MYC-driven transcriptional programs contribute to pre–B-cell transformation and that the pre–B-cell receptor signaling expression pattern could serve as a prognostic marker for high-risk BCP-ALL.34,35 Similarly for MLC, it is suggested that increased pre–B-cell signaling upon activation of the MYC pathway (caused by miRNA aberrations) may be associated with a poor prognosis.
Downregulation of DICER1 may dysregulate miRNA expression in the MLC
Finally, to identify the mechanism underlying the downregulation in key miRNA expression in the MLC, we explored abnormalities in the miRNA biogenesis pathway (supplemental Figure 10A). We found that expression of DICER1, also a crucial factor for B-cell development,36 differed significantly between MLC, non-MLC, and normal B-progenitor cells (Kruskal-Wallis test, P = 5.0 × 10−6); DICER1 was downregulated significantly in the MLC compared with the non-MLC samples (Wilcoxon rank-sum test, Bonferroni-corrected, P = 2.0 × 10−5; Figure 6A), which was also the case in the patients with MLC-like characteristics of the Japanese cohort (Wilcoxon rank-sum test, P = .0153; Figure 6B). In addition, DICER1 was downregulated significantly at relapse, compared with that in the paired primary sample, in all 9 patients who acquired an MLC profile at relapse (Wilcoxon signed-rank test, P = 7.81 × 10−3; Figure 6C). Furthermore, DICER1 expression was significantly lower in patients at relapse than at diagnosis in the other B-ALL cohort37 (supplemental Figure 10B), and patients with low expression of DICER1 were associated with poor prognosis, although there was no significant difference between the intermediate and high expression groups in the TARGET cohort, probably because of the high proportion of relapsed cases (supplemental Figure 10C). Indeed, sequencing of mature miRNAs revealed a marked reduction in the 5p- to 3p-mature miRNA ratio in MLC compared with non-MLC samples (Wilcoxon signed-rank test, P = 6.59 × 10−15; Figure 6D), which is consistent with the finding that DICER1 loss-of-function mutations lead to depletion of 5p miRNAs in other cancers.38 In vitro experiments showed that although DICER1 knockdown of REH cell lines had no effect on cell growth, it did confer resistance to cytarabine, for which oxidative phosphorylation has been reported to be involved in resistance,39 suggesting that DICER1 is crucial for chemoresistance of BCP-ALL (Figure 6E-F). Taken together, these observations suggest that the downregulation of DICER1 is a potential mechanism underlying the miRNA dysregulation that characterizes the MLC. Although reduced expression of DICER1 because of DNA methylation or copy number alterations promotes tumorigenesis in other tumors,40 these abnormalities were not observed in MLC (supplemental Figure 10D).
Figure 6.

DICER1 expression in BCP-ALL. (A) Box plots comparing DICER1 expression (expressed as FPKM) by normal pro–B cells (n = 4), pre–B cells (n = 4), immature B cells (n = 4), non-MLC cases (n = 80), and MLC cases (n = 31) from the TARGET ALL cohort. The P value was calculated using the Wilcoxon rank-sum test, adjusted using Bonferroni-correction. (B) Box plots comparing DICER1 expression (FPKM) in non-MLC-like cases (n = 50) and MLC-like cases (n = 19) from the Japanese cohort. The P value was calculated using the Wilcoxon rank-sum test. (C) Box plots comparing DICER1 expression (FPKM) in paired primary and relapse samples from 9 cases that acquired an MLC signature at relapse. The P value was calculated using the Wilcoxon signed-rank test. (D) Box plots showing quantification of miRNA processing, based on the median 5p to (5p + 3p) miRNA ratio in non-MLC cases (n = 80) and MLC cases (n = 31) from the TARGET ALL cohort. The P value was calculated using the Wilcoxon rank-sum test. (E) Immunoblotting to detect DICER1 and GAPDH expression by REH cells transduced with control (shLuc) or DICER1 short hairpin RNAs (shDICER1). Cells were treated for 72 hours with 3 μM doxycycline (DOX) or an equivalent amount of dimethyl sulfoxide (DMSO). (F) Box plots comparing the viability of REH cells transduced with control or shDICER1 after exposure to cytarabine. REH cells were transduced with shLuc or shDICER1 and then cultured in the presence of 3 μM doxycycline, or an equivalent amount of DMSO, for 72 hours. After the indicated concentrations of cytarabine were added to the culture medium, cell viability (%) relative to that of control wells containing culture medium and the equivalent amount of vehicle was measured after 48 hours in a water-soluble tetrazolium 8 (WST-8) assay using Cell Counting Kit-8 (n = 6). P values were calculated using the Wilcoxon signed-rank test. ∗P < .05, ∗∗P < .01. FPKM, fragments per kilobase million; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
This study describes a novel RNA-seq–based miRNA signature that identifies pediatric patients with BCP-ALL at high risk of relapse and can lead to optimized treatment stratification as a novel risk factor. Unlike mRNA expression profiles that are dependent on existing subtypes,41 the miRNA profile of the MLC was independent of existing subtypes, driver mutations, and methylation changes, meaning that it cannot be identified in the absence of miRNA-seq data. Several studies report prognostic prediction based on miRNA signatures in pediatric ALL.42,43 However, these studies were limited to analysis of specific miRNAs by quantitative polymerase chain reaction or microarrays, and did not analyze their association with comprehensive genetic alterations in patients with BCP-ALL. Previous studies of miRNA expression in cases of pediatric ALL are inconsistent with respect to methodology and study design, which reduces reproducibility and prevents adequate meta-analysis; therefore, no definitive prognostic miRNAs have been identified.10 Among miRNA profiling methods, miRNA-seq is the most accurate, sensitive, and specific, and it can quantify expression over a wide dynamic range.44 Thus, comprehensive integrative analysis using miRNA-seq allowed us to identify an aberrant miRNA signature in the MLC.
Predicting relapse is particularly important for standard-risk BCP-ALL. Recently, several novel prognostic factors for ALL have been reported. However, we found that IKZF1plus45 and a focal 22q11.22 deletion combined with IKZF1 alterations,46 both of which are poor prognostic factors, were not associated with the MLC. Other recently identified poor prognostic factors, including the poor risk chromosomal profile for hyperdiploidy30 and the prognostic epigenetic regulator mutations1 (ie, TBL1XR1 and SETD2), were not associated with the MLC. The miRNA profile observed in the MLC could be a predictor of relapse even in hyperdiploid ALL; indeed, it may be a better predictor of relapse than MRD. More precise identification of patients at high risk for relapse at the time of diagnosis based on miRNA expression would improve the prognosis of pediatric BCP-ALL through risk-stratified treatment, including induction of immunotherapy.47 Because evaluation of the miRNA transcriptome is still difficult in clinical practice, it was suggested that high expression of miRNA-92b-3p and miRNA-1304-3p could alternatively predict MLC profile and prognosis, suggesting the potential use of their individual assessment, but further validation is needed.
Our integrative miRNA-mRNA analysis showed that a distinctive reduction in miRNA expression in the MLC may lead to increased expression of MYC target genes and genes related to oxidative phosphorylation, which are associated with a poor prognosis and cytarabine resistance in BCP-ALL.48 Given the challenges associated with direct targeting of MYC, exploring the therapeutic potential of drugs targeting the oxidative phosphorylation pathway, such as venetoclax,39 warrants further investigation. Elucidating the functional basis for the association between the MLC miRNA profile and adverse outcomes and the founding genetic lesion underlying DICER1 downregulation is beyond the scope of this study, but further research is warranted.
The MLC was characterized by reduced IR, accompanied by increased expression of RNA processing genes. Aberrant alternative splicing (AS) is relevant to BCP-ALL.49 In addition, a recent study of BCP-ALL found that recurrent alterations in the RNA machinery genes1 as well as RNA splicing alterations50 have prognostic implications. IR is a type of AS event that occurs when an intron is retained in the final mature mRNA transcript. Many malignancies are characterized by increased IR; however, we found (unexpectedly) that the MLC had lower levels of IR than the non-MLC and normal B-cell precursors. Decreased IR has been reported in SF3B1-mutated myelodysplastic syndrome51 and breast cancer52, and an inverse relationship between IR and cell proliferation has been observed during B-cell development.53 Reduced IR in the MLC is presumably due to altered expression of spliceosome components through dysregulated miRNAs or to increased MYC activity.54 Because mRNA transcripts containing introns are degraded by nonsense-mediated decay of host genes, reduced IR is associated with the upregulation of gene products. In the MLC, reduced IR of the MTA1 gene, an activator of MYC, has been implicated in disease pathogenesis. Overexpression of MTA1 results in spontaneous development of B-cell lymphoma, and PAX5 has been identified as a potential target of MTA1,55 which could be associated with increased pre-BCR signaling. Pre-BCR signaling may be a useful therapeutic target because a subset of BCP-ALL depends on it for survival,56 and Bruton tyrosine kinase inhibitors, such as ibrutinib are effective.57
This study has several limitations. First, we did not perform functional analyses of the aberrant miRNAs in MLC. The phenotypic changes conferred by these miRNA alterations in BCP-ALL cells in vitro and in vivo need to be investigated. Second, because this study was a retrospective cohort study with a relatively small sample size and various treatment contexts, caution should be exercised when interpreting the results. Future validation is needed to determine whether the results can be replicated in the modern era when immunotherapy is available for relapsed cases. Because only limited miRNA-seq data for pediatric BCP-ALL are still available, further data accumulation in large prospective cohorts will be required to optimize prognostic models for clinical application. Recently, whole-genome sequencing identified new genetic abnormalities in BCP-ALL,58 and future analyses combining transcriptome and whole-genome sequencing are needed to obtain a complete overview of the genomics of miRNAs in BCP-ALL.
In conclusion, our miRNA-mRNA integration analysis comprehensively identified the miRNA profiles associated with pediatric BCP-ALL and clearly identified the novel prognostic miRNA signature in a group of patients at high risk of relapse. Risk stratification based on miRNA expression profiles is expected to improve the treatment outcomes for pediatric patients with BCP-ALL.
Conflict-of-interest disclosure: The authors declare no conflicting financial interests.
Acknowledgments
The authors thank the patients and their families for their cooperation. The authors also thank K. Kodama for technical assistance.
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers JP17H04224, JP18K19467, JP20H00528, and JP21K19405 (J.T.); the Project for Cancer Research and Therapeutic Evolution (grant no. JP19cm0106509h9904), the Project for Promotion of Cancer Research and Therapeutic Evolution (grant no. JP22cm0106xxxh000x and 23ama221505h0002), the Practical Research for Innovative Cancer Control (grant no. JP19ck0106468h0001) from the Japan Agency for Medical Research and Development, the Princess Takamatsu Cancer Research Fund (all to J.T.); the Takeda Hosho Grants for Research Manitoba (J.T.); and Ishizue from Kyoto University Research Administration (J.T.).
Authorship
Contribution: H.K., H.U., H.H., and J.T. designed the study; M.H., D.H., and T. Imamura provided specimens and collected clinical data; H.K. and K.T. prepared the samples; H.K., N.K., Y.N., and S.O. performed sequencing; H.K., H.U., and T. Isobe. performed bioinformatics analysis; S.S., I.K., and K.U. participated in helpful discussions and commented on the research direction; H.K., H.H., and J.T. wrote the manuscript; and all authors reviewed the manuscript during preparation.
Footnotes
The miRNA-seq, mRNA-seq, and whole-exome sequencing data in the Japanese cohort that support the findings of this study are deposited in the Japanese genotype-phenotype archive (accession number JSUB000865).
Publicly available RNA sequencing data for normal B-cell progenitors were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE115656).
Other data are available on request from the corresponding authors, Hidefumi Hiramatsu (hiramatu@kuhp.kyoto-u.ac.jp) and Junko Takita (jtakita@kuhp.kyoto-u.ac.jp).
The full-text version of this article contains a data supplement.
Contributor Information
Hidefumi Hiramatsu, Email: hiramatu@kuhp.kyoto-u.ac.jp.
Junko Takita, Email: jtakita@kuhp.kyoto-u.ac.jp.
Supplementary Material
References
- 1.Brady SW, Roberts KG, Gu Z, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54(9):1376–1389. doi: 10.1038/s41588-022-01159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeha S, Choi J, Roberts KG, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease–directed therapy. Blood Cancer Discov. 2021;2(4):326–337. doi: 10.1158/2643-3230.BCD-20-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueno H, Yoshida K, Shiozawa Y, et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020;4(20):5165–5173. doi: 10.1182/bloodadvances.2019001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teachey DT, Pui CH. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20(3):e142–e154. doi: 10.1016/S1470-2045(19)30031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irving JAE, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128(7):911–922. doi: 10.1182/blood-2016-03-704973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teachey DT, Hunger SP. Predicting relapse risk in childhood acute lymphoblastic leukaemia. Br J Haematol. 2013;162(5):606–620. doi: 10.1111/bjh.12442. [DOI] [PubMed] [Google Scholar]
- 7.Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13(4):271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 8.Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther. 2016;1 doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziętara KJ, Lejman J, Wojciechowska K, Lejman M. The importance of selected dysregulated microRNAs in diagnosis and prognosis of childhood B-cell precursor acute lymphoblastic leukemia. Cancers (Basel) 2023;15(2):428. doi: 10.3390/cancers15020428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kyriakidis I, Kyriakidis K, Tsezou A. MicroRNAs and the diagnosis of childhood acute lymphoblastic leukemia: systematic review, meta-analysis and re-analysis with novel small RNA-seq tools. Cancers (Basel) 2022;14(16):3976. doi: 10.3390/cancers14163976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim EL, Trinh DL, Ries RE, et al. MicroRNA expression-based model indicates event-free survival in pediatric acute myeloid leukemia. J Clin Oncol. 2017;35(35):3964–3977. doi: 10.1200/JCO.2017.74.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen EC, Devidas M, Chen S, et al. Dexamethasone and high-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: a report from Children’s Oncology Group Study AALL0232. J Clin Oncol. 2016;34(20):2380–2388. doi: 10.1200/JCO.2015.62.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maloney KW, Devidas M, Wang C, et al. Outcome in children with standard-risk B-cell acute lymphoblastic leukemia: results of Children’s Oncology Group trial AALL0331. J Clin Oncol. 2020;38(6):602–612. doi: 10.1200/JCO.19.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa D, Imamura T, Yumura-Yagi K, et al. Japan Association of Childhood Leukemia Study Group JACLS Risk-adjusted therapy for pediatric non-T cell ALL improves outcomes for standard risk patients: results of JACLS ALL-02. Blood Cancer J. 2020;10(2):23. doi: 10.1038/s41408-020-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi H, Kajiwara R, Kato M, et al. Treatment outcome of children with acute lymphoblastic leukemia: the Tokyo Children’s Cancer Study Group (TCCSG) study L04-16. Int J Hematol. 2018;108(1):98–108. doi: 10.1007/s12185-018-2440-4. [DOI] [PubMed] [Google Scholar]
- 16.Ewels PA, Peltzer A, Fillinger S, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38(3):276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 17.Tian L, Li Y, Edmonson MN, et al. CICERO: a versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol. 2020;21(1):126. doi: 10.1186/s13059-020-02043-x. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng SWK, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016. 2016;540(7633):433–437. doi: 10.1038/nature20598. 540:433. [DOI] [PubMed] [Google Scholar]
- 20.Yuan T, Yang Y, Chen J, et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: a novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia. 2017;31(11):2355–2364. doi: 10.1038/leu.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian L, Zhang W, Lei B, et al. MicroRNA-101 regulates T-cell acute lymphoblastic leukemia progression and chemotherapeutic sensitivity by targeting Notch1. Oncol Rep. 2016;36(5):2511–2516. doi: 10.3892/or.2016.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X-S, Gong J-N, Yu J, et al. MicroRNA-29a and microRNA-142-3p are regulators of myeloid differentiation and acute myeloid leukemia. Blood. 2012;119(21):4992–5004. doi: 10.1182/blood-2011-10-385716. [DOI] [PubMed] [Google Scholar]
- 23.Mitsumura T, Ito Y, Chiba T, et al. Ablation of miR-146b in mice causes hematopoietic malignancy. Blood Adv. 2018;2(23):3483–3491. doi: 10.1182/bloodadvances.2018017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghodousi ES, Aberuyi N, Rahgozar S. Simultaneous changes in expression levels of BAALC and miR-326: a novel prognostic biomarker for childhood ALL. Jpn J Clin Oncol. 2020;50(6):671–678. doi: 10.1093/jjco/hyaa025. [DOI] [PubMed] [Google Scholar]
- 25.Malouf C, Antunes ETB, O’Dwyer M, et al. miR-130b and miR-128a are essential lineage-specific codrivers of t(4;11) MLL-AF4 acute leukemia. Blood. 2021;138(21):2066–2092. doi: 10.1182/blood.2020006610. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Vecchiarelli-Federico LM, Li YJ, et al. The miR-17-92 cluster expands multipotent hematopoietic progenitors whereas imbalanced expression of its individual oncogenic miRNAs promotes leukemia in mice. Blood. 2012;119(19):4486–4498. doi: 10.1182/blood-2011-09-378687. [DOI] [PubMed] [Google Scholar]
- 27.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111(12):5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children’s Oncology Group study AALL0232. Blood. 2015;126(8):964–971. doi: 10.1182/blood-2015-03-633685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
- 30.Enshaei A, Vora A, Harrison CJ, Moppett J, Moorman AV. Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol. 2021;8(11):e828–e839. doi: 10.1016/S2352-3026(21)00304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y-T, Liu C-J, Kao J-H, et al. Metastatic tumor antigen 1 contributes to hepatocarcinogenesis posttranscriptionally through RNA-binding function. Hepatology. 2023;77(2):379–394. doi: 10.1002/hep.32356. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Tang J, Yang H, et al. MTA1 modulated by miR-30e contributes to epithelial-to-mesenchymal transition in hepatocellular carcinoma through an ErbB2-dependent pathway. Oncogene. 2017;36(28):3976–3985. doi: 10.1038/onc.2016.491. [DOI] [PubMed] [Google Scholar]
- 33.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen D, Zheng J, Gerasimcik N, et al. The expression pattern of the pre-B cell receptor components correlates with cellular stage and clinical outcome in acute lymphoblastic leukemia. PLoS One. 2016;11(9) doi: 10.1371/journal.pone.0162638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee RD, Munro SA, Knutson TP, LaRue RS, Heltemes-Harris LM, Farrar MA. Single-cell analysis identifies dynamic gene expression networks that govern B cell development and transformation. Nat Commun. 2021;12(1):6843. doi: 10.1038/s41467-021-27232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koralov SB, Muljo SA, Galler GR, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132(5):860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 37.McLeod C, Gout AM, Zhou X, et al. St. Jude Cloud: a pediatric cancer genomic data-sharing ecosystem. Cancer Discov. 2021;11(5):1082–1099. doi: 10.1158/2159-8290.CD-20-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vedanayagam J, Chatila WK, Aksoy BA, et al. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat Commun. 2019;10(1):3682. doi: 10.1038/s41467-019-11610-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen C, Hao X, Lai X, et al. Oxidative phosphorylation enhances the leukemogenic capacity and resistance to chemotherapy of B cell acute lymphoblastic leukemia. Sci Adv. 2021;7(11) doi: 10.1126/sciadv.abd6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14(10):662–672. doi: 10.1038/nrc3802. [DOI] [PubMed] [Google Scholar]
- 41.Li JF, Dai YT, Lilljebjörn H, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. 2018;115(50):E11711–E11720. doi: 10.1073/pnas.1814397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang Y-H, Jou S-T, Yen C-T, et al. A microRNA signature for clinical outcomes of pediatric ALL patients treated with TPOG protocols. Am J Cancer Res. 2022;12(10):4764–4774. [PMC free article] [PubMed] [Google Scholar]
- 43.Rashed WM, Hamza MM, Matboli M, Salem SI. MicroRNA as a prognostic biomarker for survival in childhood acute lymphoblastic leukemia: a systematic review. Cancer Metastasis Rev. 2019;38(4):771–782. doi: 10.1007/s10555-019-09826-0. [DOI] [PubMed] [Google Scholar]
- 44.Godoy PM, Barczak AJ, DeHoff P, et al. Comparison of reproducibility, accuracy, sensitivity, and specificity of miRNA quantification platforms. Cell Rep. 2019;29(12):4212–4222.e5. doi: 10.1016/j.celrep.2019.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanulla M, Dagdan E, Zaliova M, et al. TRANSCALL Consortium. International BFM Study Group IKZF1 plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric b-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240–1249. doi: 10.1200/JCO.2017.74.3617. [DOI] [PubMed] [Google Scholar]
- 46.Mangum DS, Meyer JA, Mason CC, et al. Association of combined focal 22q11.22 deletion and IKZF1 alterations with outcomes in childhood acute lymphoblastic leukemia. JAMA Oncol. 2021;7(10):1521–1528. doi: 10.1001/jamaoncol.2021.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Locatelli F, Zugmaier G, Rizzari C, et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):843–854. doi: 10.1001/jama.2021.0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmadi SE, Rahimi S, Zarandi B, Chegeni R, Safa M. MYC: a multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J Hematol Oncol. 2021;14(1):121. doi: 10.1186/s13045-021-01111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Black KL, Naqvi AS, Asnani M, et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018;46(21):11357–11369. doi: 10.1093/nar/gky946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Closa A, Reixachs-Solé M, Fuentes-Fayos AC, et al. A convergent malignant phenotype in B-cell acute lymphoblastic leukemia involving the splicing factor SRRM1. NAR Cancer. 2022;4(4):zcac041. doi: 10.1093/narcan/zcac041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shiozawa Y, Malcovati L, Gallì A, et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun. 2018;9(1):3649. doi: 10.1038/s41467-018-06063-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shah JS, Milevskiy MJG, Petrova V, et al. Towards resolution of the intron retention paradox in breast cancer. Breast Cancer Res. 2022;24(1):100. doi: 10.1186/s13058-022-01593-1. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ullrich S, Guigó R. Dynamic changes in intron retention are tightly associated with regulation of splicing factors and proliferative activity during B-cell development. Nucleic Acids Res. 2020;48(3):1327–1340. doi: 10.1093/nar/gkz1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu TYT, Simon LM, Neill NJ, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525(7569):384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balasenthil S, Gururaj AE, Talukder AH, et al. Identification of Pax5 as a target of MTA1 in B-cell lymphomas. Cancer Res. 2007;67(15):7132–7138. doi: 10.1158/0008-5472.CAN-07-0750. [DOI] [PubMed] [Google Scholar]
- 56.Geng H, Hurtz C, Lenz KB, et al. Self-enforcing feedback activation between BCL6 and Pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell. 2015;27(3):409–425. doi: 10.1016/j.ccell.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim E, Hurtz C, Koehrer S, et al. Ibrutinib inhibits pre-BCR+ B-cell acute lymphoblastic leukemia progression by targeting BTK and BLK. Blood. 2017;129(9):1155–1165. doi: 10.1182/blood-2016-06-722900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryan SL, Peden JF, Kingsbury Z, et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia. 2023;37(3):518–528. doi: 10.1038/s41375-022-01806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.