

Abstract
Background
Macroalgae, especially reds (Rhodophyta Division) and browns (Phaeophyta Division), are known for producing various halogenated compounds. Yet, the reasons underlying their production and the fate of these metabolites remain largely unknown. Some theories suggest their potential antimicrobial activity and involvement in interactions between macroalgae and prokaryotes. However, detailed investigations are currently missing on how the genetic information of prokaryotic communities associated with macroalgae may influence the fate of organohalogenated molecules.
Results
To address this challenge, we created a specialized dataset containing 161 enzymes, each with a complete enzyme commission number, known to be involved in halogen metabolism. This dataset served as a reference to annotate the corresponding genes encoded in both the metagenomic contigs and 98 metagenome-assembled genomes (MAGs) obtained from the microbiome of 2 red (Sphaerococcus coronopifolius and Asparagopsis taxiformis) and 1 brown (Halopteris scoparia) macroalgae. We detected many dehalogenation-related genes, particularly those with hydrolytic functions, suggesting their potential involvement in the degradation of a wide spectrum of halocarbons and haloaromatic molecules, including anthropogenic compounds. We uncovered an array of degradative gene functions within MAGs, spanning various bacterial orders such as Rhodobacterales, Rhizobiales, Caulobacterales, Geminicoccales, Sphingomonadales, Granulosicoccales, Microtrichales, and Pseudomonadales. Less abundant than degradative functions, we also uncovered genes associated with the biosynthesis of halogenated antimicrobial compounds and metabolites.
Conclusion
The functional data provided here contribute to understanding the still largely unexplored role of unknown prokaryotes. These findings support the hypothesis that macroalgae function as holobionts, where the metabolism of halogenated compounds might play a role in symbiogenesis and act as a possible defense mechanism against environmental chemical stressors. Furthermore, bacterial groups, previously never connected with organohalogen metabolism, e.g., Caulobacterales, Geminicoccales, Granulosicoccales, and Microtrichales, functionally characterized through MAGs reconstruction, revealed a biotechnologically relevant gene content, useful in synthetic biology, and bioprospecting applications.
Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s40168-023-01740-6.
Keywords: Holobiont, Macroalgae, Sphaerococcus coronopifolius, Asparagopsis taxiformis, Halopteris scoparia, Microbiome, Metagenome-assembled genomes (MAGs), Organohalogens, Halogenation, Dehalogenation
Background
Marine microorganisms are still largely unexplored in many aspects, yet, they represent an extraordinary source of genes, enzymes, and metabolites. These resources have significant potential to encourage the development of novel biotechnological and medical applications, while also being valuable for investigating marine ecosystems and ecological processes [1–6].
Currently, the exploration of metagenome-assembled genomes (MAGs) represents a powerful approach to identify new candidate taxa from uncultivable prokaryotes and clarify their ecological features and potential applications [7–10]. The Tara Oceans Expeditions produced over 2500 bacterial and archaeal MAGs derived from more than 200 marine metagenomic samples. Phylogenomic analysis of these MAGs revealed lineages that had no previously cultured representatives [11]. Nearly 8000 high-quality MAGs of uncultivated bacteria and archaea (UBA) were recovered from over 1500 publicly available metagenomes, noticeably expanding the Earth’s prokaryotic genomic diversity [12].
Among marine multicellular hosts, macroalgae function as a foundation species playing a key role in shaping microbial communities by providing a physical structure and engaging in mutualistic exchanges [13]. Like plants, macroalgae can also be considered holobionts, consisting of the multicellular host and its associated microbial community. In this symbiotic relationship, each partner actively contributes to the maintenance, performance, and resilience of the holobiont [14]. The first studies including prokaryote-macroalga interactions based on MAGs analysis have been published only recently. A comparative genome-centric analysis of reconstructed MAGs from Sargassum spp. macroalgae biofilms revealed that variations in the function of coral reef microbiomes were influenced by seasonality, macroalga biomass abundance, and available nutrients [15]. Another recent study explored the relationship between the kelp Nereocystis luetkeana and its microbiome. Genes involved in moving and assimilating abundant dissolved organic matter produced by kelp were annotated in various microorganisms composing the Nereocystis luetkeana microbiome. This discovery strengthens the concept of the holobiont [16].
Macroalgal–prokaryotic interactions are dependent on the production of different chemical compounds [13]. Notably, halogenated compounds produced by both macroalgae and associated prokaryotes play a significant role in regulating these mutual interactions [17]. Red and brown marine macroalgae are well known to synthesize halogen-containing compounds, which are believed to play a role in regulating biofouling on the algae’s surface, as well as grazing and defending against pathogens [18–22]. Nonetheless, little is known about the metabolic interconnection between the host and microbiota concerning the production and degradation of these compounds. Prokaryotes associated with macroalgae must tolerate the antimicrobial effects of organohalogens synthesized by their host. They achieve this by possessing genes that encode for enzymes capable of managing various halogenated compounds. Conversely, macroalgae-associated microorganisms equipped with biosynthetic functions could enhance their own competitiveness and provide an advantage to the host, shaping the microbial community and controlling the proliferation of harmful taxa. In this contest, MAGs from red and brown marine macroalgae could serve as a valuable genetic resource. They provide new means to explore the complex network of prokaryotic taxa and enzymes involved in biosynthetic and degradative halogenation processes, ultimately aiding in unraveling marine ecological interactions.
Naturally occurring halogenated compounds possess significant medical and industrial value, displaying a wide range of biological activities [23–25]. For instance, the process of biofluorination carried out in engineered Pseudomonas putida is a clear example of synthetic biology [26]. This approach aims to achieve economically feasible and environmentally sustainable production of organohalides [27]. The use of GenoChemetic methods, using genes encoding heterologous halogenases, is a developing approach to generate analogs of natural halogenated products. These derivatives play a crucial role in medicine and agriculture [25]. Genes encoding enzymes involved in dehalogenation pathways are therefore pivotal for developing or improving important biotechnological applications [28–30].
Considering the production of antimicrobial halogenated compounds by macroalgae, our proposal is to investigate the hypothesis that a large proportion of their closely associated bacteria may possess a range of protein-coding genes capable of metabolizing organohalogens. This potential ability might serve as a driving factor in shaping the structure of seaweed holobionts. At the same time, we aim to reveal seaweed microbiomes as a valuable future biotech source. Therefore, we deeply sequenced the epiphytic metagenomes of 2 red (Rhodophyta Division) and 1 brown (Phaeophyta Division) marine macroalgae: Sphaerococcus coronopifolius (Sc), Asparagopsis taxiformis (At), and Halopteris scoparia (Hs). This data enabled the assembly of a total of 98 prokaryotic draft genomes, uncovering novel taxa within each macroalga. Moreover, we detected several genes encoding for enzymes related to halo- and dehalogenation metabolism, annotated using a specialized reference dataset of functions. This exploration revealed both biosynthetic capabilities and a significant biotechnological potential for hydrolytic dehalogenation. To the best of our knowledge, this is the first report describing a genetic resource potentially involved in organohalogen metabolism within microbiomes associated with multicellular eukaryotic organisms generating organohalogens.
Methods
Macroalgae sampling, epiphytic metagenomic DNA extraction, shotgun sequencing, and MAGs detection
Three composite samples of algae (one from each macroalga, consisting of at least three distinct individuals) were collected from the same location, at the harbor bay of Lagosteiros (37° 1′ 9.678′′ N 7° 55′ 49.584′′ W), in southwest Portugal. These macroalgae were sampled from the shallow intertidal area via snorkeling in October 2016. The samples were kept refrigerated for 24 h and transported on ice to the laboratory. Subsequently, they were rinsed 3 times in a sterile Petri dish using artificial seawater filtered through a 0.2 µm filter to remove loosely attached microbes. Using a 50-ml sterile tube, a short centrifugation (500 × g, 2 min per 2 times; Eppendorf 5417R, Hamburg, Germany) was done to remove the remaining seawater from tissue samples. These samples were subsequently stored at −80 °C until DNA extraction.
Metagenomic DNA was extracted from the thawed algal tissue using the kit Quick-DNA™ Fecal/Soil Microbe Midiprep (Zymo Research, Irvine, CA, USA). We followed the recommended guidelines for sample processing (2.5 g per macroalgal composite sample in the present study, a mix including ca. 0.85 g per single individual). However, instead of using a bead beater, we opted to vigorously mix the samples with a vortex (Scientific Industries, Bohemia, NY, USA.) for 10 min at maximum speed. This method was intended to minimize the breakdown of algal tissue and reduce the risk of potential contamination from host genomic DNA. Quantitative PCRs were then performed using serial decimal dilutions of extracted DNA as a template and a specific couple of primers targeting regions of the bacterial 16S rRNA (following the protocol by Mapelli et al. [31]) and seaweed 18S rRNA (following the protocol by Herrero et al. [32]) genes.
One metagenome for each algal species was analyzed for a qualitative comparison among the three microbiomes. No technical and biological replicates were generated.
The DNA libraries for all three metagenomic DNA were prepared following the TruSeq Nano DNA library prep workflow (Illumina, Illumina San Diego, CA, USA), adhering to the manufacturer’s instructions. Subsequently, the three libraries were sequenced on an Illumina NextSeq 500 platform, generating 2 × 75 bp paired-end reads.
Raw reads were quality-trimmed, and the adapters were removed by using trim-galore! v0.6.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The retained reads were assembled in contigs by using metaSPAdes v3.13.1 [33] and binned in MAGs by MetaBAT2 v2.13 [34]. CheckM v1.0.18 [35] was finally applied to evaluate MAGs completeness and contamination. The raw sequencing data and associated metadata have been deposited and can be accessed under the NCBI BioProject PRJNA915238.
Taxonomy and phylogeny
The CheckM ssu_finder script was used to detect prokaryotic SSU rRNA genes within the obtained contigs, and SSU ≥ 300nt were taxonomically annotated in BioMaS [36]. This annotation was done using the release 138 NR 99 of the SILVA databases [37]. Chord diagrams of the taxa shared among the macroalga microbiomes were obtained by using the circlize R library [38].
For Asparagopsis taxiformis, an evaluation of the host genome contamination was performed by mapping trimmed reads on the host genome originating from Guam (JAAEFF000000000.1) through bowtie2 [39]. For the other two macroalgae, no reference genome is available.
The taxonomic classification of all high- and medium-quality MAGs was performed by using KBase implemented GTDB-Tk version 1.7.0 (GTDB R06-RS202 release (https://narrative.kbase.us/#catalog/apps/kb_gtdbtk/run_kb_gtdbtk_classify_wf/release) [9, 40] a software toolkit for assigning objective taxonomic classifications to bacterial and archaeal genomes based on the GTDB [41, 42] constructed from RefSeq and Genbank genomes.
Phylogenetic relatedness among all MAGs was established by SpeciesTreeBuilder v.0.1.3 (https://narrative.kbase.us/#catalog/modules/SpeciesTreeBuilder) in KBase. This process relied on FastTree2 (version 2.1.10) [43] and employed a set of 49 core, universal genes defined by Clusters of Orthologous Groups (COG) gene families. The resulting phylogenetic trees, in Newick format, were then imported into the interactive Tree of Life (iTOL) version 6.6 [44] for visualizing the phylogenetic relationships among the assembled genomes and their associated functional profiles.
Developing BRENDA-EC161 and -EC42 dataset
The enzyme commission serial numbers (EC X.X.X.X) for all enzymes involved in halogen metabolism were retrieved from BRENDA (Braunschweig Enzyme Database, https://www.brenda-enzymes.org) using both the explorer (https://www.brenda-enzymes.org/ecexplorer.php) and full-text search (https://www.brenda-enzymes.org/fulltext.php) functions. The search terms included “halo”, “halides”, “dehalogenase”, and “halogenase”. When necessary, specific source links from KEGG (Kyoto Encyclopedia of Genes and Genome), MetaCyc (Metabolic Pathways From all Domains of Life), and UniProtKB (UniProt Knowledgebase) were used for additional queries. The enzymes that are specifically involved in halo- or dehalogenation reactions and are fully categorized by an EC number are reported in Additional file 2: Table S1.
There are many degradative and biosynthetic reactions involving organohalogens, whose enzymatic components have not been fully characterized, resulting in an incomplete assignment of EC numbers (e.g., brominated pyrroles biosynthesis pathway, https://metacyc.org/META/NEW-IMAGE?type=PATHWAY&object=PWY-7931). Therefore, enzymes with partially assigned EC numbers were not considered because they were not fully characterized.
Metagenomes and MAGs functional annotation
The metagenomic contigs and MAGs were annotated using Prokka v1.14.5 [45], a tool which is available in the DOE Systems Biology Knowledgebase (KBase) platform. KBase is an open-source software and data platform designed for predicting and designing biological functions within microbial communities (https://www.kbase.us/). Default parameters were used in the Prokka annotation process. The resulting feature tables (.tsv format) were then used to extract all annotated EC numbers, from which 161 EC numbers were selected.
For both, single BRENDA target function and clusters, the gene density was estimated as a ratio between the number of annotated genes and the total size (Mbp) of assembled metagenomic contigs.
Results
Macroalga microbiomes
The three macroalga species (Sc, At, and Hs) were sampled from the same Portugal continental coastal area. The primary goal was to minimize contamination originating from the host genome, thus aiming to obtain a metagenome that predominantly contains DNA from epiphytic microbiota. By the quantitative PCRs performed, no algal 18S rRNA amplicons were detected on all the three extracted metagenomic DNA samples up to 10,000-fold DNA dilutions, while 16S rRNA amplicons were observed (data not shown). Consistently, the host At 18S rRNA gene was detected by whole shotgun metagenomic sequencing and annotated (Additional file 1: Table S1) but the DNA genomic contamination of such host was very low (2.93%) (see the “Methods” section), confirming the validity of the extraction method. Unfortunately, the estimation of Sc and Hs genome contamination were not performed due to the unavailability of reference genomes within public genome databases. The extraction of some endophytic microbial genomes is not excluded considering the weak host DNA contamination.
The total size of metagenomic assembled contigs obtained after the assembly of reads were 784.246.579 bp for Sc, 525.823.578 bp for At, and 435.330.334 bp for Hs. For detailed sequencing metadata (e.g., number of reads and contigs), see Additional file 1: Table S2.
Phylogenetic diversity of microbiomes
An initial taxonomic overview of phylogenetic diversity of the microbiomes associated with the three investigated macroalgae, using all contigs encoding SSU rRNA sequences showed a phylogenetic diversity consisting of 10, 11, and 13 phyla in At, Sc, and Hs contigs, respectively (Fig. 1 and Additional file 1: Table S1). The three algae shared 9 phyla.
Fig. 1.
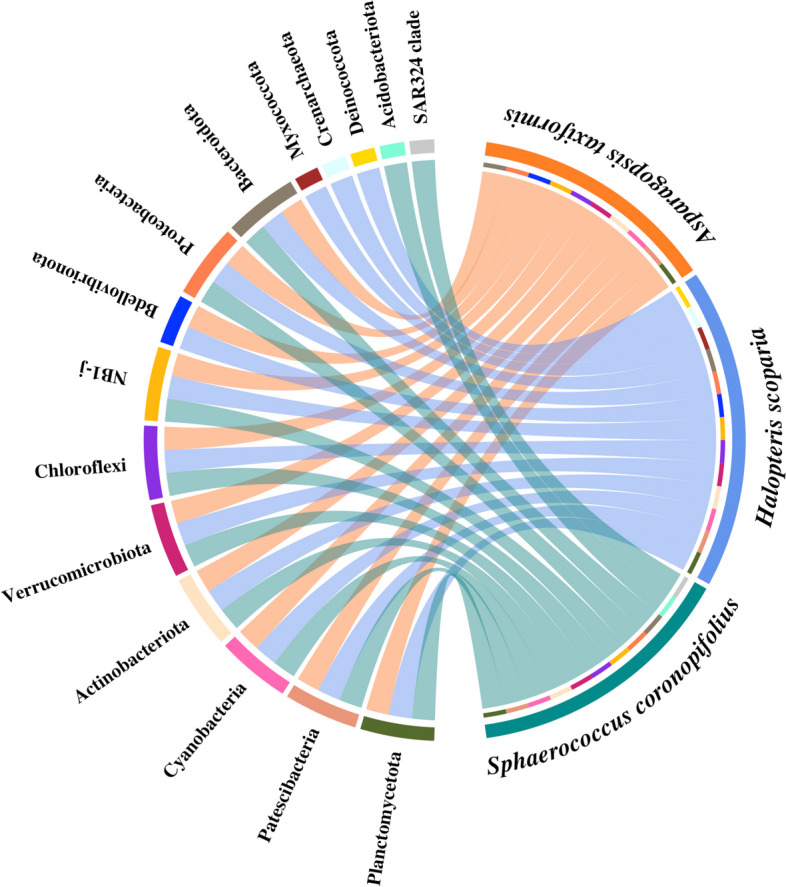
Chord diagram showing the observed prokaryotic phyla based on the taxonomic classification of the SSU rRNA sequences identified in the metagenomic contigs. It illustrates their distribution among the macroalgae-associated microbiomes. Inside the circle: in green the phyla assigned to Sphaerococcus coronopifolius, in blue and in orange those assigned to Halopteris scoparia and Asparagopsis taxiformis, respectively
The SSU rRNAs assigned to Proteobacteria and Bacteroidota phyla were prevalent in all 3 hosts. Exclusive phyla were found in Sc (Acidobacteriota and SAR324 group B) and Hs (Crenarchaeota, Deinococcota, and Myxococcota), while they were absent in At. The phylum Bdellovibrionota was represented in both At and Hs contigs. Only one phylum belonging to Archaea, specifically Hs Crenarchaeota, was detected.
At the order level, the SSU rRNA gene analysis revealed a more evident taxonomic diversity across all 3 microbiomes. In Hs, Sc, and At, contigs of 32, 29, and 28 orders were observed, respectively (Additional file 2: Fig. S1). Hs revealed more exclusive orders (10), followed by Sc (6) and At (6). A further remarkable bacterial diversity was observed at the genus level, as well as a remarkable specificity of taxa represented in the three algae (Additional file 2: Fig. S2).
Taxonomy of MAGs
Of the over 200 MAGs reconstructed (Additional file 1: Table S3), we retained a total of 98 high- (completeness > 90% and contamination < 5%) and medium-quality MAGs (completeness ≥ 50% and contamination < 10%): 49 from Sc (7 high- and 42 medium-quality), 31 from At (10 high- and 21 medium-quality), and 18 from Hs (3 high- and 15 medium-quality) (Additional file 1: Table S4).
The obtained MAGs, classified by using GTDB-Tk (Additional file 1: Table S5), exhibited globally a phylogenetic diversity of 11 phyla (Additional file 2: Fig. S3). Among the 3 MAGs sets, Sc, Hs, and At displayed 10, 6, and 3 phyla, respectively. One medium-quality genome (MAG32) from Hs could not be assigned to any prokaryotic domain (bacteria or archaea). Overall, the phyla assigned to the MAGs were generally consistent with those found in the contigs. However, discrepancies were found, such as the presence of Bdellovibrionota and Myxococcota only in Sc MAGs and absent in the contigs. Conversely, NB1-j, SAR324 clade-Marine group B, and Planctomyceta were exclusively detected in the contigs and not in the MAGs. At the class level, Bacteroidia, Alphaproteobacteria, and Gammaproteobacteria were the most represented, with 27 MAGs (11 Sc, 9 At, 7 Hs), 23 MAGs (11 Sc, 12 At, 0 Hs), and 21 MAGs (8 Sc, 8 At, 5 Hs), respectively (Additional file 2: Fig. S4). However, Alphaproteobacteria MAGs were not detected in the assembly of Hs contigs. At the order level, Flavobacteriales were the most represented with 24 MAGs (10 Sc, 8 At, 6 Hs), followed by Sphingomonadales (3 Sc, 3 At, 0 Hs), Pseudomonadales (3 Sc, 1 At, 2 Hs), Enterobacterales (1 Sc, 4 At, 1 Hs), and Caulobacterales (2 Sc, 4 At, 0 Hs) each accounting for 6 MAGs each (Additional file 2: Fig. S5). The remaining MAGs were distributed among 23 additional orders each containing between one to five MAGs. Flavobacteriales, Pseudomonadales, and Enterobacterales MAGs were observed across all macroalgae.
Out of the 98 MAGs under examination, GTDB-Tk taxonomic placement criteria (considering topology, relative evolutionary divergence, and average nucleotide identity) assigned 90 MAGs into novel ranks: 6 as novel families, 31 as novel genera, and 53 as novel species (Additional file 1: Tables S5 and S6). Seven MAGs were assigned to known species, whereas HsMAG32 could not be classified. Using a phylogenetic tree based on Clusters of Orthologous Groups of proteins (COG) constructed with FastTree2 (Additional file 2: Fig. S6) showed that HsMAG32 represents an independent prokaryotic clade belonging to the Archaea domain related to the candidate genus Nitrosopumilus. Further analysis of the SSU rRNA gene analysis within the Hs metagenome revealed the presence of contigs belonging to the phylum Crenarchaeota (Candidatus_Nitrosopumilus genus) (Additional file 1: Table S1).
Dataset of BRENDA enzymes active in the organohalogen metabolism
To annotate the genetic pool encoding enzymes involved in organohalogen metabolism within At, Sc, and Hs metagenomes and MAGs, a custom dataset composed of 161 BRENDA enzymes, each with a complete EC number, was collected (Additional file 2: Table S1). This dataset served as a reference for the annotation process. For each enzyme, details such as the recommended name, the IUBMB, KEGG, MetaCyc, and UniProtKB annotations related to the catalyzed reactions and target metabolic pathways were provided.
Out of the 161 enzymes (hereinafter called BRENDA-EC161 functions), 42 are directly involved in the genesis or breaking of halide bonds: 16 halogenases and 26 dehalogenases. Hereafter, these 42 enzymes will be referred to as BRENDA-EC42 functions. The remaining 119 enzymes are not directly involved in the genesis or breaking of halogen bonds but play a role in the chemical transformations that precede or follow the halogenation or dehalogenation of substrates.
Biodegradative and biosynthetic potential of BRENDA target genes in the analyzed metagenomes
Using the BRENDRA-EC161 reference collection, a total of 81 functions were annotated across the three metagenomes, with 17 and 64 related to halogenation and dehalogenation pathways, respectively (Additional file 2: Table S2). Most of the functions were shared among the metagenomes (63 out of 81, 77.8%), and only 18 were retrieved in one (Sc or At) or two metagenomes (Sc-At, Sc-Hs, or At-Hs). Genes encoding haloalkane dehalogenases (EC 3.8.1.5) were the most abundant in all metagenomes. The gene density value (expressed as genes/Mb) for this hydrolytic enzyme was 0.41 in Sc, 0.34 in At, and 0.27 in Hs (Fig. 2 and Additional file 1: Table S7A).
Fig. 2.
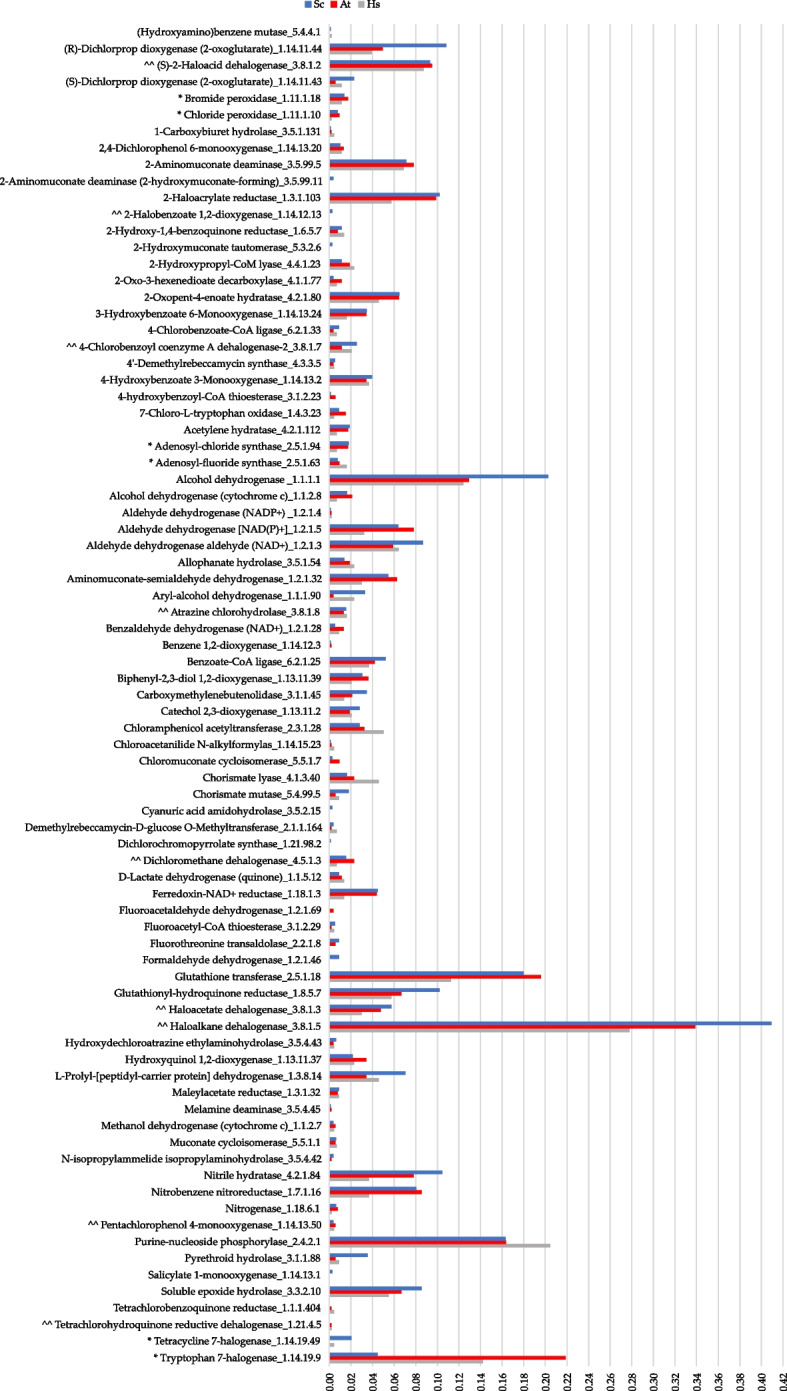
Gene density values of target genes annotated within the Sphaerococcus coronopifolius (Sc, blue bar), Asparagopsis taxiformis (At, red bar), and Halopteris scoparia (Hs, gray bar) sequenced metagenomes. The list indicates the recommended name of the 81 encoded BRENDA enzymes and their associated complete EC number (X.X.X.X). The list shows the enzymes considering enantiomers (R/S), substituent numerical position, and alphabetical order. Annotated BRENDA-EC42 subgroup functions: ^^ dehalogenases, * halogenases, and gene density values (genes/Mbp)
A total of 15 BRENDA-EC42 subgroup functions were annotated of which 12 (80%) were shared among the 3 metagenomes (Fig. 2 and Additional file 2: Table S2). Genes encoding 5 different hydrolytic dehalogenases (EC 3.8.1.X) formed the most conspicuous functional group directly involved in the breaking of carbon-halogen bonds. The tryptophan 7-halogenase (EC 1.14.19.9) gene was the most recurrent in halide bond genesis and was observed in all three metagenomes. The gene density value was particularly high in At (0.22) and Hs (0.14) compared with Sc (0.04) (Fig. 2 and Additional file 1: Table S7).
Based on IUBMB, KEGG, MetaCyc, and UniProtKB annotations (Additional file 2: Table S1), we clustered the 81 BRENDA annotated genes for each macroalga metagenome based on shared categories of degraded molecules or produced metabolites. Subsequently, we calculated the gene density per cluster (expressed as genes/Mbp) (Fig. 3 and Additional file 1: Table S7B). A total of 32 gene clusters were identified in the metagenomes, 23 involved in biodegradation (Fig. 3A), and 9 in biosynthetic processes (Fig. 3B). Thirty-one of the 32 clusters were found to be shared by all 3 metagenomes, indicating that three algal microbiomes shared a similar set of bacterial BRENDA-EC161 functions. The chlortetracycline biosynthetic gene set was not detected in At. Concerning the potential for organohalogens biodegradation, the highest gene densities were observed within the cluster including degradation reactions of chloroalkanes and chloroalkenes (Sc, 1.02; At, 0.83; Hs, 0.69) (cluster 1, Fig. 3A).
Fig. 3.

Gene density values for potential degradative and biosynthetic clusters involved in halogen metabolism depicted for the Sphaerococcus coronopifolius (Sc, blue bar), Asparagopsis taxiformis (At, red bar), and Halopteris scoparia (Hs, gray bar) metagenomes. A Degradative gene clusters (numbered from 1 to 23) along with the chemical types of halogenated molecules degraded. B Biosynthetic gene clusters (numbered from 1 to 9) and chemical types of halogenated metabolites produced. The cluster gene density values are shown as genes/Mbp. The clusters are arranged in descending order of their density values
Cluster 1 revealed a remarkable hydrolytic potential for dehalogenation across all three macroalga metagenomes, especially in genes encoding haloalkane, haloacetate, and (S)-2-haloacid dehalogenases (Additional file 1: Table S7C). Furthermore, a remarkable gene density was also found within the cluster of genes responsible for the activity on chlorocyclohexane, a cyclic aliphatic haloalkane (Fig. 3A).
In addition, several benzene halocompounds appear to be among the main potential targets for degradation. Clusters including genes encoding enzymes responsible for breaking down different haloaromatic compounds, each containing a single benzene ring, were detected. Among them, a high gene density was recorded for the cluster (3) acting on chlorobenzenes. Following this, clusters 5, 6, 8, and 9 were observed to act on chlorophenols, chloronitrobenzenes, fluorobenzoates, and chlorobenzoates, respectively. This trend was notably evident in the two red algae, particularly in Sc. Genes involved in degrading benzene derivates were found to be shared among all 3 metagenomes, including chlorochatecols (cluster 17) and chlorotoluenes (cluster 19).
Gene clusters potentially active in the degradation of other types of halomolecules were also annotated. In particular, genes involved in chloroacrylate degradation (cluster 7; Fig. 3A). Within this cluster, the most represented genes encode for the 2-haloacrylate reductase and the (S)-2-haloacid dehalogenase, which are enzymes responsible for metabolizing chloroacrylate. Gene clusters involved in the degradation of xenobiotics (cluster 4), herbicides (clusters 10, 11, 12, 14, 16, and 23), dioxins (cluster 13), antimicrobials (clusters 15 and 18), pyrethroid pesticides (cluster 21), and chlorosalicylate (cluster 22) were detected in all three metagenomes. Among these, cluster 4, comprising genes encoding for cytochrome P450 family enzymes and glutathione transferases, showed the highest density (Additional file 1: Table S7C).
Considering the 9 biosynthetic clusters, gene density values tended to be lower compared to the degradative ones (Fig. 3). A notable density value was observed for the At cluster involved in biosynthetic reactions for producing the antibiotic rebeccamycin (0.24) (cluster 2). Instead, the cluster associated with Hs chlorination reactions of organic molecules showed a lower density value (0.002) (cluster 9). High-density values were also observed for the Hs and At clusters involved, respectively, in the synthesis of fluoroacetate and fluorothreonine (cluster 1) as well as pyrrolnitrin (cluster 3), both antibiotics.
Other clusters involved in additional antibiotic biosynthesis were also found: pyoluteorin (cluster 5), salinosporamide A (cluster 6), and chlortetracycline (cluster 8). Cluster 5 also included the biosynthesis of brominated pyrroles. Concerning the biosynthesis of other organohalogens, cluster 4 resulted to be potentially active in the genesis of polybrominated phenols, whereas clusters 7 and 9 were associated with the bromination and chlorination of organic molecules, respectively.
Phylogeny of MAGs and taxonomic distribution of BRENDA target functions along the cladogram
The phylogenetic relationships among the 98 MAGs as well as the taxonomic distribution of their annotated BRENDA target functions and related gene density are shown in Fig. 4.
Fig. 4.

Phylogenetic tree showing the relationships between the 98 MAGs and the distribution of BRENDA-EC161 functions density annotated for each MAG, particularly dehalogenases and halogenases (BRENDA-EC42) and hydrolytic and non-hydrolytic dehalogenases gene densities. The 2 phyla including clades enriched in BRENDA-EC161 functions are shown in pink (Actinobacteriota) and light orange (Proteobacteria). MAGs assigned orders are also indicated. (detailed gene density values are in Additional file 1: Table S8). Ref* indicates an unsupervised reference genome of the public KBase genomes database used in phylogenetic tree building. Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis; Hs, Halopteris scoparia
Two phylogenetic clades showed an enrichment of BRENDA-EC161 functions. The first clade was made up by a group of 5 MAGs all belonging to the order Microtrichales (phylum Actinobacteriota). The second clade consisted of nearly half of the total number of MAGs. This large clade comprised several orders all belonging to the phylum Proteobacteria. Within this clade, high-gene density was notable in UBA10353, Granulosicoccales, Pseudomonadales, Enterobacterales, Geminicoccales, Caulobacterales, Rhodobacterales, and Rhizobiales orders. Conversely, a marked lower gene density was observed in the orders Flavobacteriales, Chitinophagales, and Cytophagales of the phylum Bacteroidota. All MAGs contained annotated BRENDA target functions except for ScMAG22 and ScMAG25, which were assigned to the orders Flavobacteriales and BD1-5, respectively (Additional file 1: Table S9).
The largest number of annotated genes (70) was found in the medium-quality AtMAG62, assigned to Granulosicoccales which also revealed the largest number of target functions (30). Additionally, among the high-quality MAGs, the one with a substantial number of annotated target genes was ScMAG11, belonging to Pseudomonadales, with 51 genes (Additional file 1: Table S9).
Focusing on BRENDA-EC42 functions, the genes encoding for putative halogenases revealed a higher density in the orders UBA10353, Enterobacterales, and Caulobacterales compared to other lineages (Fig. 4). In particular, the halogenases stand out in the Caulobacterales. Regarding genes encoding putative hydrolytic dehalogenases, they were broadly distributed along the entire tree, with a particularly notable presence in the Microtrichales clade, the UBA9160 order (phylum Myxococcota), and in certain Proteobacteria clades like UBA10353, Pseudomonadales, Caulobacterales, and Rhizobiales. Genes encoding non-hydrolytic functions were restricted to some lineages, e.g., Granulosicoccales, Enterobacterales, Caulobacterales, and Rhizobiales.
Among the 98 MAGs, the most frequently annotated BRENDA-EC42 function was the haloalkane dehalogenase (Additional file 1: Table S9). Specifically, this function was particularly annotated among the Sc MAGs, with as many as 10 genes observed in the Pseudomonadales MAG11.
Biodegradative and biosynthetic potential of BRENDA-EC161 functions encoded by each MAG and taxonomic affiliations
To establish the types of organo-halogenated molecules potentially metabolized by each MAG (biodegradation or biosynthetic potential), we applied the same approach adopted for the metagenomes. In this process, the annotated target genes of each MAG were clustered based on the common category of degraded molecules or metabolites produced by the BRENDA-EC161 enzymes they encoded. Subsequently, the gene density within the single clusters was calculated for each MAG (Additional file 1: Table S8). Furthermore, the phylogenetic tree involving the 98 MAGs was integrated with the densities of both biodegradative and biosynthetic clusters associated with each MAG. This integration enabled to cross-link these clusters to specific prokaryotic taxa (Fig. 5).
Fig. 5.

Phylogenetic tree showing for each MAG the lowest taxon assigned by GTDB-Tk and the clustered target genes according to the potential type of degraded or synthesized halogenated molecule. From inner to outside: MAGs numbered and macroalga host, colored taxonomic orders, lowest taxon assigned by GTDB-Tk (empty box indicates a novel family), MAG quality (the arrow indicates high-quality MAG), and the bar chart representing cluster density (with synthesized compounds in the inner circle and degraded compounds in the outer circle) for each MAG, aligned accordingly. The names of the compounds are listed in descending order of the detected cluster density values across all MAGs. Ref* indicates an unsupervised reference genome of the public KBase genomes database used in phylogenetic tree building. Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis; Hs, Halopteris scoparia
All 32 types of clusters identified within the metagenomes were also found within the MAGs. Regarding their potential ability to degrade halogenated molecules, MAGs assigned to orders such as Microtrichales, Granulosicoccales, Pseudomonadales, Geminicoccales, Sphingomonadales, Rhodobacterales, and Rhizobiales displayed similar profiles. Generally, these orders turned out to be those with the highest cluster density and a notable number of degradation clusters, reaching up to 18–20 per MAG (e.g., ScMAG6 and ScMAG47).
Genes encoding enzymes involved in aliphatic catabolism (clusters 1 and 2) were found in almost all MAGs with a more marked gene density in Caulobacterales ScMAG17 and Microtrichales ScMAG74.
Clusters associated with the degradation of aromatic compounds were spread along the phylogenetic tree showing specialization for specific halogenated molecules: Microtrichales ScMAG53, Pseudomonadales ScMAG11, Rhizobiales ScMAG47, and UBA10353 ScMAG50 for chlorobenzenes (cluster 3); Rhodobacterales AtMAG35 for chlorophenols (cluster 5); Microtrichales ScMAG53 for chloronitrobenzenes (cluster 6) and Rhizobiales ScMAG109 for fluorobenzoates (cluster 8); Granulosicoccales AtMAG62 and Sphingomonadales ScMAG9 for chlorobenzoates (cluster 9); Enterobacterales AtMAG53 for the chlorocatechols (cluster 17); and the Geminicoccales AtMAG44 for the chlorotoluenes (cluster 19).
Some MAGs contained genes annotated for herbicide degradation. The Microtrichales ScMAG53 and the Pseudomonadales ScMAG11 were characterized by degradation clusters targeting phenoxyalkanoic acid herbicides in both R-enantiomer (cluster 12) and S-enantiomer (cluster 23). These two clusters were detected together within another Microtrichales MAG (ScMAG74). Noteworthy, the S-enantiomer cluster alone was annotated in very few MAGs, almost all belonging to Microtrichales whereas the R-enantiomer cluster was only observed in some MAGs belonging to 9 orders, especially in Microtrichales. The chloroacetanilide cluster (10) was particularly detected in Geminicoccales and Sphyngomonadales MAGs, the bromoxynil cluster (11) within Granulosicoccales and Rhizobiales while the chlorophenoxyacetate cluster (14) in Geminicoccales, Granulosicoccales, and Microtrichales. Lastly, the atrazine cluster (16) was shared among 8 orders. No MAGs containing all the 6 herbicide clusters were found, but Microtrichales ScMAG53 and Pseudomonadales ScMAG11 showed 5 out of 6 clusters.
Degradation of xenobiotics by P450 enzymes (cluster 4) was more common in Granulosicoccales, Geminicoccales, Caulobacterales, Sphingomonadales, Rhodobacterales, and Rhizobiales particularly within the Rhodobacterales AtMAG35. The chloroacrylate cluster (7) was also shared among different orders and the Microtrichales ScMAG74 showed the higher density. The dioxins degradation cluster (13) was mainly observed in Microtrichales MAGs, followed by the Granulosicoccales AtMAG62. As for the fluoroacetate (15), pyrethroid (21), chlorosalicylate (22), and chloramphenicol (18) clusters, the first 3 were observed in Geminicoccales (e.g., ScMAG6) and Rhizobiales (e.g., ScMAG47) whereas the latter was notably detected in Bacteriovoracales (e.g., AtMAG70) and Flavobacteriales (e.g., ScMAG33 and ScMAG38). The dehalogenation cluster responsible for substrates containing fluorine, chlorine, bromine, and iodine in the 4-position (20) was observed only in 8 MAGs and the Bacteriovoracales HsMAG5 and the Rhizobiales ScMAG109 displayed a higher density.
As for the biosynthesis clusters, particularly those related to rebeccamycin (2) and pyrrolnitrin (3) were significantly observed in UBA10353, Pseudomonadales, Caulobacterales, and Enterobacterales MAGs, with a high density within the Caulobacterales (e.g., AtMAG29) (Fig. 5). The cluster related to the chlortetracycline biosynthesis (8) was mainly associated with the Verrucomicrobiales MAGs. In contrast, the fluoroacetate and fluorothreonin cluster (1) showed a large distribution across the tree, with a density range from 0.1 (Granulosicoccales AtMAG62) to 1.7 (Flavobacteriales ScMAG42). Conversely, the salinosporamide A cluster (6) was observed in some orders on the left side of the tree.
The cluster of brominated pyrroles and pyoluteorin (5) was mostly observed in Microtrichales among few others. The polybrominated phenols cluster (4) was recorded in different orders, particularly in all Enterobacterales MAGs. Noteworthy, among the numerous Flavobacteriales this cluster was less represented as compared to the clusters of bromination (7) and chlorination (9) of organic molecules. The same was observed for Caulobacterales MAGs. Clusters 7 and 9 were not widespread across the obtained MAGs. Chlorination was detected in only 4 Microtrichales MAGs and 1 Sphingomonadales MAG (AtMAG30). Interestingly, the Microtrichales ScMAG53 showed all 3 clusters (4, 7, and 9).
Discussion
In red and brown algae, a rapid emission of iodinated halocarbons and other halometabolites has been particularly observed during the induction of oxidative burst [46, 47]. The genetic and biochemical reconstitution of bromoform biosynthesis in At has suggested that bromoform and other halogenated molecules found in marine algae are produced as a part of a cascade involved in the production and manipulation of reactive oxygen species. This cascade converts hydrogen peroxide into defensive halogenated metabolites [48]. Some of these halogenated metabolites have been suggested to possess antibacterial properties against marine prokaryotes, potentially serving as a defense mechanism in certain macroalgal taxa, including the Asparagopsis genus [18, 49–51]. However, this marine macroalgal defense mechanism might reduce the microbial epibiosis, potentially eliminating bacteria crucial for extending the algae’s defense mechanisms. Despite this, there has been no in-depth investigation into the genetic content of prokaryotic fractions within algal microbiomes that might be actively involved in controlling the fate of host-secreted halocompounds. This study presents the first in-depth shotgun-metagenomics analysis on macroalga-associated microbes, focusing on their gene repertoires involved in degrading or transforming halogenated metabolites. The study specifically examines 3 sympatric Atlantic macroalgae: two red, Sphaerococcus coronopifolius (Sc) and Asparagopsis taxiformis (At), and one brown, the Halopteris scoparia (Hs). In our work, we investigated At due to its broad and prolific biosynthesis of halometabolites [51–53], its widespread distribution in warm-temperate, tropical, and subtropical waters [54], and the availability of its genomic sequence [48]. At is also a focus of significant research, particularly in innovative biological feed studies for methane emission reduction in cattle due to its halometabolite content, especially bromoform [55].
To understand whether a similar halo-metabolic landscape is shared in seaweeds, we also collected two other sympatric macroalgae, one red (Sc) and one brown (Hs), inhabiting the same sampling area under similar local conditions of pressure and sharing the same microbial community seeded by seawater.
Sc, At, and Hs are distinct ecological niches, each harboring a unique microbiome
In line with previous studies on macroalgae [56], our survey based on the 16S rRNA gene has revealed a wide range of prokaryotic phyla associated with At, Sc, and Hs macroalgae (Fig. 1). The taxonomic annotation of MAGs has confirmed the presence of several bacterial phyla, including the parasites Patescibacteria and Bdellovibrionota. Additionally, in the Hs microbiome, we identified a unique Archaea phylum, the ammonium oxidizing Crenarchaeota (also known as Thermoproteota), previously found to be associated with some macroalgal hosts [57–59]. This finding suggests a predominant presence of the bacterial fraction in macroalgae-associated communities. Furthermore, as formerly observed in other macroalgae microbiomes [57], the 3 algae shared almost all the observed phyla and classes (e.g., Proteobacteria, Gamma- and Alphaproteobacteria, Bacteroidia, Planctomycetes, Actinobacteria, Verrucomicrobia) while a diversification was noticed at lowest phylogenetic ranks. A low number of shared taxa (17.5%) was observed among the three macroalgae, when only known classified genera were considered (Additional file 2: Fig. S2), suggesting that, despite being exposed to the same seawater community and environmental drivers, they represent distinct ecological niches able to select specific prokaryotic communities. Further, the overlap in taxonomic composition between the red algae (Sc and At) did not show a wider similarity than between the red-brown algae (Sc-Hs and At-Hs).
Only At microbiome has been previously described [60], but our observations did not reveal a complete match (data not shown). Rather, we found that some bacterial orders previously observed separately in the coastal continental and island At samples were both present in our coastal continental At sample. Both coastal and mainland At samples (here and previous work) were nevertheless collected in the same marine area but in different seasons. Overall, these taxonomic data suggest that the shaping of the microbiota in the lower taxonomic levels is not strictly dependent on the taxonomy of the alga, at species or division level.
Macroalgae metagenomes and MAGs reveal a versatile organohalogen degradative and biosynthetic potential involving previously unknown taxa
Despite investigations into the ability of individual marine bacteria or phyla to degrade or synthesize halogenated molecules [61–63], the role of the entire marine microbial community in the management of these compounds remains unknown. Our examination of each macroalga metagenome revealed a consistent potential for organohalogen degradation, surpassing the biosynthetic one. This suggests that the microbes associated with these three algae possess a specialization primarily in degrading halogenated molecules rather than synthesizing them. Interestingly, while the three algae showed a wide diversity in the phylogenetic composition of the associated microbiomes, they basically shared all common categories of degraded molecules or produced metabolites (Fig. 3). A priori one might assume that the assembly of microbial BRENDA-EC161 functions on algae is not based on the selection of specific prokaryotic ranks. The lottery hypothesis stating that bacterial communities assemble based on functional genes rather than species was first introduced through research conducted on the green alga Ulva australis [64]. However, the research performed in our current study, examining BRENDA-EC161 functions within MAGs and their taxonomic affiliations, showed that these functions are primarily clustered within specific bacterial ranks. A novel ecological and genetic finding is the potential involvement of uncultured members from relatively unknown prokaryotic orders, such as Caulobacterales, Granulosicoccales, Geminicoccales, UBA10353, UBA9160, Microtrichales in organohalogen metabolism (Fig. 4). Additionally, newly identified uncultured representatives from more recognized bacterial orders (e.g., Rhizobiales, Rhodobacterales, Sphingomonadales, Enterobacterales, Pseudomonadales) are implicated in this process as well.
A specific richness in BRENDA-EC161 degradative functions was uncovered in the macroalga-associated MAGs assigned to the orders Rhizobiales, Rhodobacterales, Caulobacterales, Sphingomonadales, Geminicoccales, Pseudomonadales, Granulosicoccales, and Microtrichales (Fig. 4). Considering the entire community, from the taxonomic profile based on 16S rRNA gene detected for the Sc, At, and Hs metagenomes, these orders were particularly present in Sc, followed by At and Hs (Additional file 2: Fig. S2). This aspect seems to be in line with the organohalogen degradation cluster density values detected in the metagenomes (Fig. 3A). Basically, an overall trend is observed in the density values, with Sc exhibiting higher densities than At, which in turn are higher than those of Hs. We hypothesize that the higher gene density values of the BRENDA-EC161 degradative clusters observed for the two red algae metagenomes, especially in Sc, might be related to a greater secretion of halometabolites compared to the brown alga. Likely, this could justify the richer microbiome in Sc, followed by At, with bacterial ranks more enriched in BRENDA-EC161 functions that degrade organohalogens. Accordingly, higher levels of halogenated secondary metabolite production in red algae, compared to all macroalgae, have been documented [65, 66].
Regarding the spectrum of organohalogen biodegradative functions, our analysis revealed a preeminent set of genes encoding putative hydrolytic dehalogenases (EC 3.8.1.X) (BRENDA-EC42 subgroup) in both metagenomes (Fig. 2) and MAGs (Fig. 4 and Additional file 1: Table S9). This was particularly evident in clusters associated with degradation reactions of haloalkanes and haloalkenes (Fig. 3A). Notably, among these, the haloalkane dehalogenases (EC 3.8.1.5) displayed the highest density values, following a trend Sc > At > Hs (Fig. 2). The putative hydrolytic dehalogenases encoded by the assembled MAGs are known to convert a wide range of halocarbons, including halogenated alkanes, cycloalkanes, alkenes, ethers, alcohols, ketones, cyclic dienes, alkanoic acids, and acetates [67, 68]. Genes encoding these BRENDA-EC42 functions were particularly abundant in MAGs corresponding with Microtrichales, UBA9160, UBA10353, Pseudomonadales, Caulobacterales, and Rhizobiales (Fig. 4). Several are the macroalga species for which the ability to secrete a multitude of halocarbons was demonstrated, at various latitudes [65, 66, 69]. Specifically, At is a prolific producer of these compounds, especially (mono or poly) iodinated, brominated, and/or chlorinated methanes (particularly bromoform), ethanes, propanes, epoxy-propanes, ethanols, isopropanols, ethylenes, propenes, butenols, acetones, acetates, acetic, and acrylic acids [52, 65, 66]. Notably, At is a haloacrylic acid producer [53], which seems to align with our discovery of a gene cluster involved in chloroacrylate degradation (Fig. 3A). The most annotated genes within this cluster encode for the 2-haloacrylate reductase (EC 1.3.1.103) and the (S)-2-haloacid dehalogenase (EC 3.8.1.2) (Fig. 2). In the soil bacterium Burkholderia sp. WS, these enzymes constitute a probable pathway for metabolizing 2-chloroacrylate into (R)-lactate [70]. The 2-haloacrylate reductase catalyzes the conversion of 2-chloroacrylate into (S)-2-chloropropionate, which is subsequently dehalogenated by (S)-2-haloacid dehalogenase into (R)-lactate [70]. Genes encoding both enzymes were found in several of the assembled MAGs of At and Sc, particularly in those assigned to Granulosicoccales, Geminicoccales, and Rhizobiales (Fig. 5 and Additional file 1: Table S9).
Concerning Sc and Hs, we did not find exhaustive studies reporting the characterization of the secreted organic halogenated fraction, especially halocarbons. Nevertheless, the prevalence of hydrolytic dehalogenases among the annotated BRENDA-EC161 functions in the distinct Sc, At, and Hs metagenomes studied here suggests that hydrolytic dehalogenation could be a common trait among coastal algal microbiomes related to the secretion host halocarbons.
Our analysis also identified gene clusters potentially involved in the breakdown of various organic halogen compounds containing the benzene ring (Fig. 3A). Previous research highlighted the ability of macroalgae to produce haloaryl metabolites, such as halophenols [65, 71]. Despite studies demonstrating the production of phenolic compounds in At extract, the precise chemical structures of these metabolites remain elusive [72]. However, two bromophenols have recently been identified in At [73]. In the present study, genes encoding glutathionyl-hydroquinone reductases (EC 1.8.5.7) (Fig. 2), a recently identified subset of glutathione transferases that catalyze the GSH-dependent reduction of glutathionyl-hydroquinones conjugates to hydroquinones, resulted particularly annotated in the cluster of genes degrading halophenols (Fig. 3A). In bacteria these enzymes also catalyze specific reduction of S-glutathionyl-(chloro)hydroquinone [74–76]. Moreover, we observed gene clusters potentially active on anthropogenic haloaromatic molecules (pollutants, herbicides, and pesticides) revealing a multiform degradative potential of organohalogens. Previous biosorption studies showed the interaction between macroalga biomass and aromatic contaminants like toluene, benzene, and polycyclic molecules [77, 78]. Furthermore, in controlled environments like photobioreactors, there have been recent examples of symbiotic relationships forming between bacteria and unicellular algae. These bacteria possess the ability of controlling the fate of aromatic anthropogenic pollutants [79, 80].
Noteworthy, the analysis of three metagenomes revealed a high-density cluster of genes encoding putative cytochrome P450 enzymes as well as glutathione transferases, able to degrade halogenated molecules. The P450 enzymes’ ability to bind foreign liposoluble halogenated hydrocarbons was among the first biochemical studies of these proteins [81]. These enzymes are present in both prokaryotes and eukaryotes and are known for metabolizing xenobiotics, where hydroxylation converts insoluble hydrocarbons into more soluble for easier elimination [82]. A variety of halogenated xenobiotics (e.g., therapeutic agents and agrochemicals) are bound by the lipophilic P450 active site to be detoxified through a dehalogenation step [83–85]. Prokaryotic glutathione transferases are key enzymes in the cellular detoxification of a broad range of harmful xenobiotics, including aliphatic, aromatic, and heterocyclic molecules with halide groups [86]. These enzymes catalyze glutathione conjugation to electrophilic groups (mainly introduced by P450) of a wide range of hydrophobic toxic compounds, thus promoting their excretion from the cell [87]. Glutathione transferases detoxify several classes of herbicides including triazines, a class of man-made chemicals that includes atrazine, one of the most widely used chlorinated herbicides [86].
Collectively, the genes encoding P450 enzymes, glutathione transferases, hydrolytic dehalogenases, and other brenda-EC161 functions annotated here may be a bacterial integrated system useful in controlling the impact of synthetic, natural, and host-secreted organohalogens on the microbial community. While genes encoding P450 enzymes and glutathione transferases are mostly ubiquitous across the assembled MAGs, they are particularly abundant in those assigned to the Proteobacteria phylum, particularly in Rhizobiales, Rhodobacterales, Sphingomonadales, Geminicoccales, and Granulosicoccales (Fig. 5).
Considering the potential synthesized metabolites by the three algal metagenomes (Fig. 3B), a broader trend was seen for the biosynthesis of antimicrobial compounds (fluoroacetate, fluorothreonine, rebeccamycin, pyoluteorin, pyrrolnitrin, salinosporamide A, chlortetracycline), potentially serving as drivers influencing microbiome assembly. There was a marked heterogeneity in the biosynthetic properties of Sc, At, and Hs metagenomes. Although Sc metagenome encodes a higher number of BRENDA-EC161 functions than At and Hs, its biosynthetic clusters tend to have lower gene density values, particularly with those of rebeccamycin and pyrrolnitrin. This data was confirmed by the biosynthetic gene clustering of MAGs (Fig. 5), where At MAGs (e.g., Enterobacterales, Sphingomonadales, and Caulobacterales) were particularly enriched in genes involved in the biosynthesis of these two antimicrobials. The variability in gene density values within biosynthetic clusters among the three metagenomes suggests that the potential halometabolites production in algal microbiomes likely depends on intrinsic properties of individual holobionts. We hypothesize that in the alga-microbiota symbiotic relationship, the lower ability of the host-microbe to produce halometabolites might correspond to a greater algal ability to synthesize them. However, a heterogeneous bacterial synthesis of halometabolites, in particular antimicrobials (e.g., rebeccamycin and pyrrolnitrin), may contribute along with the algal production of halometabolites to select, assemble, and maintain a distinct algal microbiota characterized by unique species diversity.
Organohalogen metabolism genes: microbiome assembly guided by the holobiont as a defense mechanism against environmental stresses
Investigations into the distribution of specific xenobiotics (pharmaceuticals), herbicides, and pesticides in estuarine and seawater along the Portuguese coast, including sampling sites close to the harbor bay of Lagosteiros, have reported the presence of halogenated chemicals, particularly atrazine and alachlor (a chloroacetanilide) herbicides [88]. Recently, contamination by atrazine and alachlor as well as pesticides such as chlorinated pyrethroids and hexachlorocyclohexane isomers has been detected across seasons in water and sediments sampled from the estuaries of Tagus and Douro Rivers (NE Atlantic Ocean Portuguese coast), while searching for thirty-seven endocrine disruptor compounds [89]. The atrazine and alachlor negative effect on the growth of microalgae in vitro has been reported [90].
We suspect that the identified gene clusters within the here analyzed metagenomes and associated MAGs, potentially involved in the metabolism of anthropogenic organohalogens such as chlorocyclohexane, chlorobenzenes, chlorophenols, xenobiotics, pesticides (e.g., pyrethroids), and herbicides (e.g., chlorocetanilides and atrazine) (Figs. 3A and 5), could represent an adaptative mechanism of the coastal Sc, At, and Hs, intermittently exposed to anthropic perturbations. The bacterial ability to biodegrade halogenated herbicides such as atrazine and chlorocetanilides originating from agricultural runoff into coastal waters may therefore preserve the macroalgal fitness ensuring the survival of the entire holobiont. Our hypothesis is that the macroalgal secretion of halometabolites could be evolutionary linked with the biogenic and abiogenic naturally occurring organohalogen compounds in the marine environment. The secretion drives the selection of a degrading microbial community recruited to control the natural halomolecules with a phytotoxic activity and, in the anthropization era, those of anthropic origin having structural similarity. A costal holobiont active in the metabolism of organohalogen compounds would have formed and evolved over time. The macroalgal holobiont appears to favor bacterial populations encoding enzymes with a broad substrate promiscuity, capable of metabolizing both naturally occurring exudates and structurally similar pollutants, thereby establishing a beneficial cycle for the environment. Marine bacteria with haloalkane dehalogenases showing an exceptionally broad substrate specificity have been previously described [91, 92].
An increased rate of halocarbon secretion by red polar marine macroalgae due to stresses induced by altered environmental conditions has been demonstrated, suggesting that global warming and uncontrolled ocean eutrophication may have a significant role in algal halometabolites secretion [93]. In plants, the so-called “cry-for-help” has been described as a mechanism driven by the modification of root exudate chemistry induced by pathogens and predators, aimed to recruit a beneficial microbiome exerting a plant protection effect. The same mechanism has been recently proposed to occur in plants growing in polychlorobiphenyl contaminated soils, in which the root-exudation-mediated microbial recruitment enriches in the plant rhizosphere a complex metabolic network active in contaminant degradation, in turn allowing the plant to rapidly adapt to the phytotoxic stress conditions [94]. It cannot be excluded that the “cry-for-help” mechanism observed in plants under stress conditions, could also be employed in macroalgae. We hypothesize that organohalogen production in macroalgae, besides defending against pathogens, foulers, and herbivores, might act as an ecological driver for recruiting beneficial microbes into the holobiont assembly, thereby reducing the chemical environmental phytotoxicity.
Genetic potential for bioprospecting
Hydrolytic dehalogenases, a prominent gene group among the annotated BRENDA-EC42 functions, play a crucial role in economical and environmentally friendly industrial processes and biotechnological applications [68, 95–97]. While extensively studied in soil bacteria commonly exposed to manufactured organohalogens (e.g., herbicides and pesticides) [98], recent marine genomics and metagenomics investigations have revealed microbial hydrolytic dehalogenases with unique functional and structural properties [91, 92, 99]. Lately, the marine haloalkane dehalogenase DmmA, encoded by a metagenomic DNA fragment, and exhibited exceptional substrate specificity in degrading several environmental pollutants that are resistant to other closely related enzymes [100].
By studying specific marine holobionts, we have unveiled a significant blue genetic potential, including putative bacterial haloalkane dehalogenases, among other dehalogenases and halogenases. These discoveries provide a highly helpful resource for future experimental verification. Furthermore, many genes were assigned to different uncultured taxa based on MAG taxonomic classification. The microbial genetic diversity, particularly from novel or poorly known taxa, is currently an added value for the development of synthetic biology to design specific heterologous recombinant DNA circuits to be expressed in engineered microbial platforms and address application challenges outside-the-lab such as bioremediation and bio-based synthesis of chemical building blocks [27, 101]. Expression of heterologous genes in engineered microbial platforms could offer novel solutions to the mineralization of recalcitrant contaminants [102] or organohalide production [26]. In addition, genes encoding halogenases, such as the tryptophan halogenases, are today an emerging opportunity in the integration of synthetic biology and synthetic chemistry (GenoChemetics). This integration allows the fine-tuning of the bioactivity, bioavailability, and reactivity of medicinally and agriculturally relevant aromatic and aliphatic compounds via selective C-H functionalization [25]. This expansion enhances the portfolio of commercially important organohalides [103, 104].
Conclusion
The comprehensive data presented here enriches our understanding on the composition and functions of the macroalgae-associated bacterial community. This work helps to unravel the still largely unknown role of the microbial dark matter. The analysis of specific MAGs encoded functions provides insights into their potential involvement in organohalogen metabolism, offering the possibility to develop innovative halo- and dehalogenation biocatalysts.
Supplementary Information
Additional file 1: Table S1. Sphaerococcus coronopifolius. Table S2. Sequencing Metadata. Table S3. Sphaerococcus coronopifolius. Table S4. High- and medium-quality MAGs. Genome sizes, completness and contaminations estimated are reported. Table S5. GTDB-Tk taxonomic classification of Sphaerococcus coronopifolius MAGs. Table S6. Number of novel candidate taxa emerged after the GTDB-Tk classification of binned prokaryotic MAGs. Table S7. Gene density. Table S9. Annotated BRENDA-EC161 functions* in each single MAG§ and taxonomic affiliation†.
Additional file 2: Fig. S1. Venn diagram based on the number of prokaryotic orders assigned to metagenomic contigs (SSU rRNAs taxonomic analysis). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S2. Classified prokaryotic genera of macroalgal-associated microbiomes based on SSU rRNAs analysis. A, Venn diagram reporting the numbers of shared and unique genera. B, List of shared and unique prokaryotic genera of the 3 macroalgal microbiomes and related taxonomic orders. Orders also assigned to MAGs are in bold. MAGs orders enriched in BRENDA-EC161 functions are highlighted in yellow. Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S3. Prokaryotic phyla assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per Phylum are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S4. Prokaryotic classes assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per class are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S5. Prokaryotic orders assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per order are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S6. Phylogenetic analysis of HsMAG32 by FastTree2. The phylogenetic tree based on alignment similarity of a set of 49 core, universal genes defined by COGs, was developed considering an unsupervised set of 20 closely related genomes available on the public KBase genomes database. HsMAG32 highlighted in yellow is positioned in the Archaea domain. Hs, Halopteris scoparia. The local-bootstrap support values are indicated in red. Table S1. List of 161 complete EC numbers*, corresponding to as many enzymes† characterized to be involved in the cellular metabolism of halogens, available in BRENDA (Braunschweig Enzyme Database). Table S2. BRENDA-EC161* and -EC42‡ functions annotated for each macroalgal metagenome.
Acknowledgements
The authors would like to thank Marta Barbato, assistant professor at the Aarhus University (Denmark), for her contribution to the metagenomic DNA extraction and quantitative PCRs. The authors would also like to thank Sharon Natasha Cox, researcher at the Bari University (Italy), for her contribution to revising the manuscript, and Barbara De Marzo, Annarita Armenise, and Luigi Boccaccio for technical administrative assistance.
Authors’ contributions
A.P. provided conceptual framework, design and project coordination. A.H.E and S.B. contributed ideas on the project. A.H.E. collected the samples. A.P. and A.L. extracted the metagenomic DNA. C.M. performed the metagenomic DNA sequencing. B.F. performed the metagenomic assemblies, the prokaryotic binning steps and the taxonomic SSU profiles. A.P. generated the BRENDA-EC161 database. A.L. performed the functional annotation of contigs and MAGs, and phylogenetic trees. A.P. examined MAGs and metagenomes, and interpreted data. A.L. contributed to examine MAGs and metagenomes. A.P. wrote the paper. A.L., B.F., A.H.E, S.B., E.P. and G.P. participated in the manuscript writing. A.P., S.B., A.H.E., and G.P. revised the manuscript. A.P. and G.P. supervised the study. All authors approved the final version of the manuscript.
Funding
This work was supported by INMARE (H2020-BG-04-2014, GA 634486), the Italian ELIXIR FOE (CUP B53C22000690005), the Blue Bioeconomy Pact with the project Algae Vertical N° C6449 5664-00000026 funded by the Portuguese Plan of Recovery and Resilience (PRR) and the European Union and by Portuguese national funds from FCT — Foundation for Science and Technology — through projects UIDB/04326/2020, UIDP/04326/2020 and LA/P/0101/2020 awarded to CCMAR, as well as contract CEECINST/00114/2018 to AHE.
The funder had no role in the study design, data collection, and interpretation, or the decision to submit the work for publication.
Availability of data and materials
Raw sequencing data and metadata are available at the following link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA915238?reviewer=v47i8nc2gqedcte9p627rqgl7a.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 2.Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Marine natural products. Nat Prod Rep. 2015;32:116–211. doi: 10.1039/C4NP00144C. [DOI] [PubMed] [Google Scholar]
- 3.Almaabadi AD, Gojobori T, Mineta K. Marine metagenome as a resource for novel enzymes. Genomics Proteomics Bioinformatics. 2015;13:290–5. doi: 10.1016/j.gpb.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zakem EJ, Polz MF, Follows MJ. Redox-informed models of global biogeochemical cycles. Nat Commun. 2020;11:5680. doi: 10.1038/s41467-020-19454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer-Cifuentes IE, Werner J, Jehmlich N, Will SE, Neumann-Schaal M, Öztürk B. Synergistic biodegradation of aromatic-aliphatic copolyester plastic by a marine microbial consortium. Nat Commun. 2020;11:5790. doi: 10.1038/s41467-020-19583-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paoli L, Ruscheweyh H-J, Forneris CC, Hubrich F, Kautsar S, Bhushan A, et al. Biosynthetic potential of the global ocean microbiome. Nature. 2022;607:111–118. doi: 10.1038/s41586-022-04862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hugenholtz P, Skarshewski A, Parks DH. Genome-based microbial taxonomy coming of age. Cold Spring Harb Perspect Biol. 2016;8:a018085. doi: 10.1101/cshperspect.a018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez-R LM, Gunturu S, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, et al. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018;46:W282–W288. doi: 10.1093/nar/gky467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frioux C, Singh D, Korcsmaros T, Hildebrand F. From bag-of-genes to bag-of-genomes: metabolic modelling of communities in the era of metagenome-assembled genomes. Comput Struct Biotechnol J. 2020;18:1722–1734. doi: 10.1016/j.csbj.2020.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data. 2018;5:170203. doi: 10.1038/sdata.2017.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–1542. doi: 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 13.Wilkins LGE, Leray M, O’Dea A, Yuen B, Peixoto RS, Pereira TJ, et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 2019;17:e3000533. doi: 10.1371/journal.pbio.3000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan S, Harder T, Burke C, Steinberg P, Kjelleberg S, Thomas T. The seaweed holobiont: understanding seaweed-bacteria interactions. FEMS Microbiol Rev. 2013;37:462–476. doi: 10.1111/1574-6976.12011. [DOI] [PubMed] [Google Scholar]
- 15.Glasl B, Robbins S, Frade PR, Marangon E, Laffy PW, Bourne DG, et al. Comparative genome-centric analysis reveals seasonal variation in the function of coral reef microbiomes. ISME J. 2020;14:1435–1450. doi: 10.1038/s41396-020-0622-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigel BL, Miranda KK, Fogarty EC, Watson AR, Pfister CA. Functional insights into the kelp microbiome from metagenome-assembled genomes. mSystems. 2022;7:e0142221. doi: 10.1128/msystems.01422-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goecke F, Labes A, Wiese J, Imhoff J. Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser. 2010;409:267–299. doi: 10.3354/meps08607. [DOI] [Google Scholar]
- 18.Maximilien R, de Nys R, Holmstrom C, Gram L, Givskov M, Crass K, Kjelleberg S, Steinberg PD. Chemical mediation of bacterial surface colonisation by secondary metabolites from the red alga Delisea pulchra. Aquat Microb Ecol. 1998;15:233–246. doi: 10.3354/ame015233. [DOI] [Google Scholar]
- 19.Ohsawa N, Ogata Y, Okada N, Itoh N. Physiological function of bromoperoxidase in the red marine alga, Corallina pilulifera: production of bromoform as an allelochemical and the simultaneous elimination of hydrogen peroxide. Phytochemistry. 2001;58:683–692. doi: 10.1016/S0031-9422(01)00259-X. [DOI] [PubMed] [Google Scholar]
- 20.Nightingale PD, Malin G, Liss PS. Production of chloroform and other low molecular weight halocarbons by some species of macroalgae. Limnol Oceanogr. 1995;40:680–689. doi: 10.4319/lo.1995.40.4.0680. [DOI] [Google Scholar]
- 21.Paul NA, de Nys R, Steinberg PD. Seaweed-herbivore interactions at a small scale: direct tests of feeding deterrence by filamentous algae. Mar Ecol Prog Ser. 2006;323:1–9. doi: 10.3354/meps323001. [DOI] [Google Scholar]
- 22.Lane AL, Nyadong L, Galhena AS, Shearer TL, Stout ES, Parry RM, Kwasnik M, Wang MD, Hay ME, Fernandez FM, Kubanek J. Desorption electrospray ionization mass spectrometry reveals surface-mediated antifungal chemical defence of a tropical seaweed. Proc Natl Acad Sci USA. 2009;106:7314–7319. doi: 10.1073/pnas.0812020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gribble GW. Biological activity of recently discovered halogenated marine natural products. Mar Drugs. 2015;13:4044–4136. doi: 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, Moore BS. Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem Rev. 2017;117:5619–5674. doi: 10.1021/acs.chemrev.6b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowe C, Molyneux S, Sharma SV, Zhang Y, Gkotsi DS, Connaris H, et al. Halogenases: a palette of emerging opportunities for synthetic biology–synthetic chemistry and C-H functionalisation. Chem Soc Rev. 2021;50:9443–9481. doi: 10.1039/D0CS01551B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calero P, Volke DC, Lowe PT, Gotfredsen CH, O’Hagan D, Nikel PI. A fluoride-responsive genetic circuit enables in vivo biofluorination in engineered Pseudomonas putida. Nat Commun. 2020;11:5045. doi: 10.1038/s41467-020-18813-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cros A, Alfaro-Espinoza G, De Maria A, Wirth NT, Nikel PI. Synthetic metabolism for biohalogenation. Curr Opin Biotechnol. 2022;74:180–193. doi: 10.1016/j.copbio.2021.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Atashgahi S, Sánchez-Andrea I, Heipieper HJ, van der Meer JR, Stams AJM, Smidt H. Prospects for harnessing biocide resistance for bioremediation and detoxification. Science. 2018;360:743–746. doi: 10.1126/science.aar3778. [DOI] [PubMed] [Google Scholar]
- 29.Maucourt B, Vuilleumier S, Bringel F. Transcriptional regulation of organohalide pollutant utilisation in bacteria. FEMS Microbiol Rev. 2020;44:189–207. doi: 10.1093/femsre/fuaa002. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen B-AT, Hsieh J-L, Lo S-C, Wang S-Y, Hung C-H, Huang E, et al. Biodegradation of dioxins by Burkholderia cenocepacia strain 869T2: role of 2-haloacid dehalogenase. J Hazard Mater. 2021;401:123347. doi: 10.1016/j.jhazmat.2020.123347. [DOI] [PubMed] [Google Scholar]
- 31.Mapelli F, Marasco R, Rolli E, Barbato M, Cherif H, Guesmi A, Ouzari I, Daffonchio D, Borin S. Potential for plant growth promotion of rhizobacteria associated with Salicornia growing in tunisian hypersaline soils. Biomed Res Int. 2013;2013:248078. doi: 10.1155/2013/248078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrero B, Madriñán H, Vieites JM, Espiñeira MJ. Rapid identification of seaweeds in food products by PCR combined with ALF-RFLP and FINS methodologies. Agric Food Chem. 2010;58:11586–11592. doi: 10.1021/jf103464b. [DOI] [PubMed] [Google Scholar]
- 33.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fosso B, Santamaria M, Marzano M, Alonso-Alemany D, Valiente G, Donvito G, et al. BioMaS: a modular pipeline for Bioinformatic analysis of Metagenomic AmpliconS. BMC Bioinformatics. 2015;16:203. doi: 10.1186/s12859-015-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu Z, Gu L, Eils R, Schlesner M, Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: the United States department of energy systems biology knowledgebase. Nat Biotechnol. 2018;36:566–569. doi: 10.1038/nbt.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 42.Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38:1079–1086. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 43.Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. Poon AFY, editor. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 46.Weinberger F, Friedlander M, Hoppe HG. Oligo-agars elicit an oxidative burst in Gracilaria conferta (Rhodophyta) J Phycol. 1999;35:747–755. doi: 10.1046/j.1529-8817.1999.3540747.x. [DOI] [Google Scholar]
- 47.Palmer CJ, Anders TL, Carpenter LJ, Küpper FC, McFiggans G. Iodine and halocarbon response of Laminaria digitata to oxidative stress and links to atmospheric new particle production. Environ Chem. 2005;2:282–290. doi: 10.1071/EN05078. [DOI] [Google Scholar]
- 48.Thapa HR, Lin Z, Yi D, Smith JE, Schmidt EW, Agarwal V. Genetic and biochemical reconstitution of bromoform biosynthesis in Asparagopsis lends insights into seaweed reactive oxygen species enzymology. ACS Chem Biol. 2020;15:1662–1670. doi: 10.1021/acschembio.0c00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nylund GM, Cervin G, Persson F, Hermansson M, Steinberg PD, Pavia H. Seaweed defence against bacteria: a poly-brominated 2-heptanone from the red Alga Bonnemaisonia Hamifera inhibits bacterial colonisation. Mar Ecol Prog Ser. 2008;369:39–50. doi: 10.3354/meps07577. [DOI] [Google Scholar]
- 50.Paul NA, de Nys R, Steinberg PD. Chemical defence against bacteria in the red Alga Asparagopsis armata: linking structure with function. Mar Ecol Prog Ser. 2006;306:87–101. doi: 10.3354/meps306087. [DOI] [Google Scholar]
- 51.Greff S, Zubia M, Genta-Jouve G, Massi L, Perez T, Thomas OP. Mahorones, highly brominated cyclopentenones from the red Alga Asparagopsis taxiformis. J Nat Prod. 2014;77:1150–1155. doi: 10.1021/np401094h. [DOI] [PubMed] [Google Scholar]
- 52.McConnell O, Fenical F. Halogen chemistry of the red alga Asparagopsis. Phytochemistry. 1977;15:367–374. doi: 10.1016/0031-9422(77)80067-8. [DOI] [Google Scholar]
- 53.Woolard F, Moore RE, Roller PP. Halogenated acetic and acrylic acids from the red alga Asparagopsis taxiformis. Phytochemistry. 1979;18:617–620. doi: 10.1016/S0031-9422(00)84271-5. [DOI] [Google Scholar]
- 54.Zanolla M, Altamirano M, Carmona R, De la Rosa J, Souza-Egipsy V, Sherwood A, Tsiamis K, Barbosa AM, Muñoz AR, Andreakis N. Assessing global range expansion in a cryptic species complex: insights from the red seaweed genus Asparagopsis (Florideophyceae) J Phycol. 2018;54:12–24. doi: 10.1111/jpy.12598. [DOI] [PubMed] [Google Scholar]
- 55.Kinley RD, Martinez-Fernandez G, Matthews MK, de Nys R, Magnusson M, Tomkins NW. Mitigating the carbon footprint and improving productivity of ruminant livestock agriculture using a red seaweed. J Clean Prod. 2020;259:120836. doi: 10.1016/j.jclepro.2020.120836. [DOI] [Google Scholar]
- 56.Florez JZ, Camus C, Hengst MB, Buschmann AH. A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front Microbiol. 2017;8:2561. doi: 10.3389/fmicb.2017.02561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trias R, García-Lledó A, Sánchez N, López-Jurado JL, Hallin S, Bañeras L. Abundance and composition of epiphytic bacterial and archaeal ammonia oxidizers of marine red and brown macroalgae. Appl Environ Microbiol. 2012;78:318–325. doi: 10.1128/AEM.05904-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Tang X, Mo Z, Mao Y. Metagenome-assembled genomes from Pyropia haitanensis microbiome provide insights into the potential metabolic functions to the seaweed. Front Microbiol. 2022;13:857901. doi: 10.3389/fmicb.2022.857901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roth F, Broman E, Sun X, Bonaglia S, Nascimento F, Prytherch J, et al. Methane emissions offset atmospheric carbon dioxide uptake in coastal macroalgae, mixed vegetation and sediment ecosystems. Nat Commun. 2023;14:42. doi: 10.1038/s41467-022-35673-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aires T, Serrão EA, Engelen AH. Host and environmental specificity in bacterial communities associated to two highly invasive marine species (genus Asparagopsis) Front Microbiol. 2016;7:559. doi: 10.3389/fmicb.2016.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grigorian E, Groisillier A, Thomas F, Leblanc C, Delage L. Functional characterization of a L-2-haloacid dehalogenase from Zobellia galactanivorans DsijT suggests a role in haloacetic acid catabolism and a wide distribution in marine environments. Front Microbiol. 2021;12:725997. doi: 10.3389/fmicb.2021.725997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu R, Wei X, Song W, Wang L, Cao J, Wu J, et al. Novel chloroflexi genomes from the deepest ocean reveal metabolic strategies for the adaptation to deep-sea habitats. Microbiome. 2022;10:75. doi: 10.1186/s40168-022-01263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Latham J, Brandenburger E, Shepherd SA, Menon BRK, Micklefield J. Development of halogenase enzymes for use in synthesis. Chem Rev. 2018;118:232–269. doi: 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]
- 64.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci USA. 2011;108:14288–14293. doi: 10.1073/pnas.1101591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paul C, Pohnert G. Production and role of volatile halogenated compounds from marine algae. Nat Prod Rep. 2011;28:186–195. doi: 10.1039/C0NP00043D. [DOI] [PubMed] [Google Scholar]
- 66.Kladi M, Vagias C, Roussis V. Volatile halogenated metabolites from marine red algae. Phytochem Rev. 2004;3:337–366. doi: 10.1007/s11101-004-4155-9. [DOI] [Google Scholar]
- 67.Chovancová E, Kosinski J, Bujnicki JM, Damborský J. Phylogenetic analysis of haloalkane dehalogenases. Proteins. 2007;67:305–316. doi: 10.1002/prot.21313. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Xiang Q, Zhou Q, Xu J, Pei D. Mini review: advances in 2-haloacid dehalogenases. Front Microbiol. 2021;12:758886. doi: 10.3389/fmicb.2021.758886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giese B, Laturnus F, Adams FC, Wiencke C. Release of volatile iodinated C1–C4 hydrocarbons by marine macroalgae from various climate zones. Environ Sci Technol. 1999;33:2432–2439. doi: 10.1021/es980731n. [DOI] [Google Scholar]
- 70.Kurata A, Kurihara T, Kamachi H, Esaki N. 2-Haloacrylate reductase, a novel enzyme of the medium chain dehydrogenase/reductase superfamily that catalyzes the reduction of a carbon-carbon double bond of unsaturated organohalogen compounds. J Biol Chem. 2005;280:20286–20291. doi: 10.1074/jbc.M414605200. [DOI] [PubMed] [Google Scholar]
- 71.Whitfield FB, Helidoniotis F, Shaw KJ, Svoronos D. Distribution of bromophenols in species of marine algae from eastern Australia. J Agric Food Chem. 1999;47:2367–2373. doi: 10.1021/jf981080h. [DOI] [PubMed] [Google Scholar]
- 72.Nunes N, Ferraz S, Valente S, Barreto MC, de Carvalho MP. Biochemical composition, nutritional value, and antioxidant properties of seven seaweed species from the Madeira Archipelago. J Appl Phycol. 2017;29:2427–2437. doi: 10.1007/s10811-017-1074-x. [DOI] [Google Scholar]
- 73.Gao H, Wang Y, Guo Z, Liu Y, Wu Q, Xiao J. Optimization of ultrasound-assisted extraction of phenolics from Asparagopsis taxiformis with deep eutectic solvent and their characterization by ultra-high-performance liquid chromatography-mass spectrometry. Front Nutr. 2022;9:1036436. doi: 10.3389/fnut.2022.1036436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xun L, Belchik SM, Xun R, Huang Y, Zhou H, Sanchez E, Kang C, Board PG. S-Glutathionyl-(chloro)hydroquinone reductases. A novel class of glutathione transferases. Biochem J. 2010;428:419–427. doi: 10.1042/BJ20091863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meux E, Prosper P, Ngadin A, Didierjean C, Morel M, Dumarçay S, Lamant T, Jacquot JP, Favier F, Gelhaye E. Glutathione transferases of Phanerochaete chrysosporium: a S-glutathionyl-P-hydroquinone reductase belongs to a new structural class. J Biol Chem. 2011;286:9162–9173. doi: 10.1074/jbc.M110.194548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lam LK, Zhang Z, Board PG, Xun L. Reduction of benzoquinones to hydroquinones via spontaneous reaction with glutathione and enzymatic reaction by S-glutathionyl-hydroquinone reductases. Biochemistry. 2012;51:5014–5021. doi: 10.1021/bi300477z. [DOI] [PubMed] [Google Scholar]
- 77.Flores-Chaparro CE, Chazaro Ruiz LF, Alfaro de la Torre MC, Huerta-Diaz MA, Rangel-Mendez JR. Biosorption removal of benzene and toluene by three dried macroalgae at different ionic strength and temperatures: algae biochemical composition and kinetics. J Environ Manage. 2017;193:126–35. doi: 10.1016/j.jenvman.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 78.Zhang C, Lu J, Wu J. Adsorptive removal of polycyclic aromatic hydrocarbons by detritus of green tide algae deposited in coastal sediment. Sci Total Environ. 2019;670:320–327. doi: 10.1016/j.scitotenv.2019.03.296. [DOI] [PubMed] [Google Scholar]
- 79.Lebrero R, Ángeles R, Pérez R, Muñoz R. Toluene biodegradation in an algal-bacterial airlift photobioreactor: influence of the biomass concentration and of the presence of an organic phase. J Environ Manage. 2016;183:585–593. doi: 10.1016/j.jenvman.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 80.Zhou J-L, Yang L, Huang K-X, Chen D-Z, Gao F. Mechanisms and application of microalgae on removing emerging contaminants from wastewater: a review. Bioresour Technol. 2022;364:128049. doi: 10.1016/j.biortech.2022.128049. [DOI] [PubMed] [Google Scholar]
- 81.Pelkonen O, Vainio H. Spectral interactions of a series of chlorinated hydrocarbons with cytochrome P-450 of liver microsomes from variously-treated rats. FEBS Lett. 1975;51:11–14. doi: 10.1016/0014-5793(75)80843-X. [DOI] [PubMed] [Google Scholar]
- 82.Poulos TL. Cytochrome P450: molecular architecture, mechanism, and prospects for rational inhibitor design. Pharm Res. 1988;5:67–75. doi: 10.1023/A:1015920931701. [DOI] [PubMed] [Google Scholar]
- 83.Castro CE, Yokoyama W, Belser NO. Biodehalogenation: active site versus enzymatic and whole cell rates with cytochromes P-450. Environ Toxicol Chem. 1989;8:13–18. [Google Scholar]
- 84.Raucy JL, Kraner JC, Lasker JM. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit Rev Toxicol. 1993;23:1–20. doi: 10.3109/10408449309104072. [DOI] [PubMed] [Google Scholar]
- 85.Guengerich FP. Cytochrome P450 proteins and potential utilization in biodegradation. Environ Health Perspect. 1995;103:25–28. doi: 10.1289/ehp.95103s425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Allocati N, Federici L, Masulli M, Di Ilio C. Glutathione transferases in bacteria. FEBS J. 2009;276:58–75. doi: 10.1111/j.1742-4658.2008.06743.x. [DOI] [PubMed] [Google Scholar]
- 87.Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/bj3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sousa JCG, Barbosa MO, Ribeiro ARL, Ratola N, Pereira MFR, Silva AMT. Distribution of micropollutants in estuarine and sea water along the Portuguese coast. Mar Pollut Bull. 2020;154:111120. doi: 10.1016/j.marpolbul.2020.111120. [DOI] [PubMed] [Google Scholar]
- 89.Cunha SC, Ferreira R, Marmelo I, Vieira LR, Anacleto P, Maulvault A, Marques A, Guilhermino L, Fernandes JO. Occurrence and seasonal variation of several endocrine disruptor compounds (pesticides, bisphenols, musks and UV-filters) in water and sediments from the estuaries of Tagus and Douro Rivers (NE Atlantic Ocean coast) Sci Total Environ. 2022;838:155814. doi: 10.1016/j.scitotenv.2022.155814. [DOI] [PubMed] [Google Scholar]
- 90.Fairchild JF, Ruessler DS, Carlson AR. Comparative sensitivity of five species of macrophytes and six species of algae to atrazine, metribuzin, alachlor, and metolachlor. Environ Toxicol Chem. 1998;17:1830e1834. doi: 10.1002/etc.5620170924. [DOI] [Google Scholar]
- 91.Li A, Shao Z. Biochemical characterization of a haloalkane dehalogenase DadB from Alcanivorax dieselolei B-5. PLoS One. 2014;9:e89144. doi: 10.1371/journal.pone.0089144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kunka A, Damborsky J, Prokop Z. Haloalkane dehalogenases from marine organisms. Methods Enzymol. 2018;605:203–251. doi: 10.1016/bs.mie.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 93.Laturnus F, Giese B, Wiencke C, Adams FC. Low-molecular-weight organoiodine and organobromine compounds released by polar macroalgae – the influence of abiotic factors. Fresenius J Anal Chem. 2000;368:297–302. doi: 10.1007/s002160000491. [DOI] [PubMed] [Google Scholar]
- 94.Rolli E, Vergani L, Ghitti E, Patania G, Mapelli F, Borin S. ‘Cry-for-help’ in contaminated soil: a dialogue among plants and soil microbiome to survive in hostile conditions. Environ Microbiol. 2021;23:5690–5703. doi: 10.1111/1462-2920.15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagata Y, Ohtsubo Y, Tsuda M. Properties and biotechnological applications of natural and engineered haloalkane dehalogenases. Appl Microbiol Biotechnol. 2015;99:9865–9881. doi: 10.1007/s00253-015-6954-x. [DOI] [PubMed] [Google Scholar]
- 96.Xiang Y, Li S, Rene ER, Lun X, Zhang P, Ma W. Detoxification of fluoroglucocorticoid by Acinetobacter pittii C3 via a novel defluorination pathway with hydrolysis, oxidation and reduction: performance, genomic characteristics, and mechanism. J Hazard Mater. 2023;452:131302. doi: 10.1016/j.jhazmat.2023.131302. [DOI] [PubMed] [Google Scholar]
- 97.Shan Y, Yu W, Shen L, Guo X, Zheng H, Zhong J, et al. Conjugation with inulin improves the environmental stability of haloalkane dehalogenase DhaA. Enzyme Microb Technol. 2021;149:109832. doi: 10.1016/j.enzmictec.2021.109832. [DOI] [PubMed] [Google Scholar]
- 98.Sarma H, Joshi SJ. Metagenomics combined with stable isotope probe (SIP) for the discovery of novel dehalogenases producing bacteria. Bull Environ Contam Toxicol. 2022;108:478–484. doi: 10.1007/s00128-020-03004-7. [DOI] [PubMed] [Google Scholar]
- 99.Mazur A, Prudnikova T, Grinkevich P, Mesters JR, Mrazova D, Chaloupkova R, et al. The tetrameric structure of the novel haloalkane dehalogenase DpaA from Paraglaciecola agarilytica NO2. Acta Crystallogr D Struct Biol. 2021;77:347–356. doi: 10.1107/S2059798321000486. [DOI] [PubMed] [Google Scholar]
- 100.Buryska T, Babkova P, Vavra O, Damborsky J, Prokop Z. A haloalkane dehalogenase from a marine microbial consortium possessing exceptionally broad substrate specificity. Appl Environ Microbiol. 2018;84:e01684–e1717. doi: 10.1128/AEM.01684-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brooks SM, Alper HS. Applications, challenges, and needs for employing synthetic biology beyond the lab. Nat Commun. 2021;12:1390. doi: 10.1038/s41467-021-21740-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gong T, Xu X, Che Y, Liu R, Gao W, Zhao F, et al. Combinatorial metabolic engineering of Pseudomonas putida KT2440 for efficient mineralization of 1,2,3-trichloropropane. Sci Rep. 2017;7:7064. doi: 10.1038/s41598-017-07435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roy AD, Grüschow S, Cairns N, Goss RJM. Gene expression enabling synthetic diversification of natural products: chemogenetic generation of pacidamycin analogs. J Am Chem Soc. 2010;132:12243–12245. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]
- 104.Sharma SV, Tong X, Pubill-Ulldemolins C, Cartmell C, Bogosyan EJA, Rackham EJ, et al. Living GenoChemetics by hyphenating synthetic biology and synthetic chemistry in vivo. Nat Commun. 2017;8:229. doi: 10.1038/s41467-017-00194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Sphaerococcus coronopifolius. Table S2. Sequencing Metadata. Table S3. Sphaerococcus coronopifolius. Table S4. High- and medium-quality MAGs. Genome sizes, completness and contaminations estimated are reported. Table S5. GTDB-Tk taxonomic classification of Sphaerococcus coronopifolius MAGs. Table S6. Number of novel candidate taxa emerged after the GTDB-Tk classification of binned prokaryotic MAGs. Table S7. Gene density. Table S9. Annotated BRENDA-EC161 functions* in each single MAG§ and taxonomic affiliation†.
Additional file 2: Fig. S1. Venn diagram based on the number of prokaryotic orders assigned to metagenomic contigs (SSU rRNAs taxonomic analysis). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S2. Classified prokaryotic genera of macroalgal-associated microbiomes based on SSU rRNAs analysis. A, Venn diagram reporting the numbers of shared and unique genera. B, List of shared and unique prokaryotic genera of the 3 macroalgal microbiomes and related taxonomic orders. Orders also assigned to MAGs are in bold. MAGs orders enriched in BRENDA-EC161 functions are highlighted in yellow. Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S3. Prokaryotic phyla assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per Phylum are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S4. Prokaryotic classes assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per class are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S5. Prokaryotic orders assigned by GTDB-Tk to macroalgal MAGs. Number of MAGs assigned per order are indicated in Sc, At and Hs columns. Percentage is also indicated (upper bar chart). Sc, Sphaerococcus coronopifolius; At, Asparagopsis taxiformis and Hs, Halopteris scoparia. Fig. S6. Phylogenetic analysis of HsMAG32 by FastTree2. The phylogenetic tree based on alignment similarity of a set of 49 core, universal genes defined by COGs, was developed considering an unsupervised set of 20 closely related genomes available on the public KBase genomes database. HsMAG32 highlighted in yellow is positioned in the Archaea domain. Hs, Halopteris scoparia. The local-bootstrap support values are indicated in red. Table S1. List of 161 complete EC numbers*, corresponding to as many enzymes† characterized to be involved in the cellular metabolism of halogens, available in BRENDA (Braunschweig Enzyme Database). Table S2. BRENDA-EC161* and -EC42‡ functions annotated for each macroalgal metagenome.
Data Availability Statement
Raw sequencing data and metadata are available at the following link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA915238?reviewer=v47i8nc2gqedcte9p627rqgl7a.