

Abstract
Background
It is currently unknown if disease severity modifies response to therapy in pulmonary arterial hypertension (PAH). We aimed to explore if disease severity, as defined by established risk-prediction algorithms, modified response to therapy in randomised clinical trials in PAH.
Methods
We performed a meta-analysis using individual participant data from 18 randomised clinical trials of therapy for PAH submitted to the United States Food and Drug Administration to determine if predicted risk of 1-year mortality at randomisation modified the treatment effect on three outcomes: change in 6-min walk distance (6MWD), clinical worsening at 12 weeks and time to clinical worsening.
Results
Of 6561 patients with a baseline US Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL 2.0) score, we found that individuals with higher baseline risk had higher probabilities of clinical worsening but no difference in change in 6MWD. We detected a significant interaction of REVEAL 2.0 risk and treatment assignment on change in 6MWD. For every 3-point increase in REVEAL 2.0 score, there was a 12.49 m (95% CI 5.86–19.12 m; p=0.001) greater treatment effect in change in 6MWD. We did not detect a significant risk by treatment interaction on clinical worsening with most of the risk-prediction algorithms.
Conclusions
We found that predicted risk of 1-year mortality in PAH modified treatment effect as measured by 6MWD, but not clinical worsening. Our findings highlight the importance of identifying sources of treatment heterogeneity by predicted risk to tailor studies to patients most likely to have the greatest treatment response.
Shareable abstract (@ERSpublications)
Disease severity in pulmonary arterial hypertension modifies treatment effect by change in 6-min walk distance but not clinical worsening in a meta-analysis of randomised clinical trials, highlighting the variable performance of surrogate end-points https://bit.ly/3NSJ9Pg
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease characterised by elevated pulmonary arterial pressure and pulmonary vascular resistance resulting in right heart failure and death [1, 2]. Pharmacotherapeutic options have increased over the past two decades and have improved outcomes; however, PAH remains a fatal disease [3]. Despite the US Food and Drug Administration (FDA) approval of 15 drugs, there are limited data regarding which patients might derive more or less benefit from therapy [4]. Randomised clinical trials (RCTs) provide estimates of average treatment effects but they are less able to explore variability of treatment impact between trial participants, termed heterogeneity of treatment effect (HTE). Inferring average clinical benefit for individual patients may therefore be misleading [5].
In many chronic diseases, patients with greater disease severity often achieve greater benefit from effective interventions. Several risk-prediction algorithms derived from PAH registry data identify patients at low, intermediate and high risk of 1-year mortality and have been used increasingly in clinical care [5, 6]. Stratifying patients by illness severity at baseline using established risk-prediction rules could detect clinically important HTE and treatment–covariate interactions [7–9]. This may help guide future trials by identifying patients with the largest treatment response and the clinical care of patients by personalising their treatment plan. However, it is not currently known if predicted risk using these prediction rules modifies treatment effect in PAH.
To address this knowledge gap, we harmonised individual participant data (IPD) from 18 phase 3 RCTs in PAH submitted to the US FDA for regulatory approval. We hypothesised that patients with higher predicted risk of mortality at randomisation would receive greater treatment benefit from active drug versus comparator (placebo).
Study design and methods
Study and patient selection
We received IPD from 28 RCTs of therapies for PAH that were submitted to the US FDA in 2000–2013. We excluded two phase 2 studies, seven open label extension and phase 4 studies, and one study that only included participants with chronic thromboembolic pulmonary hypertension (CTEPH). Our study sample included 18 studies (supplementary table S1) [10–26]. We included all adult patients with a diagnosis of PAH. We excluded 1) patients with a diagnosis of CTEPH and 2) patients who were randomised but never received active therapy (figure 1).
FIGURE 1.
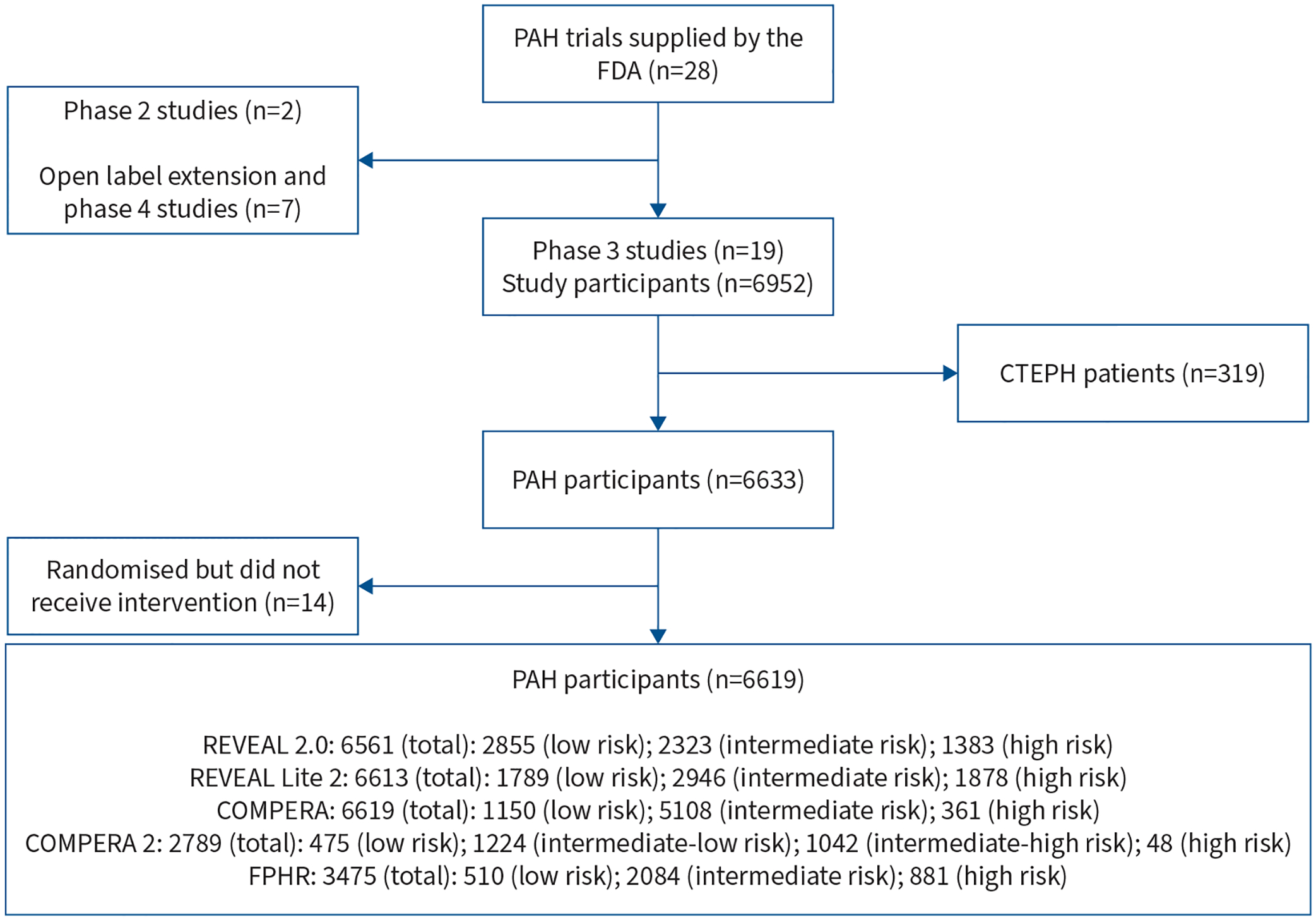
Patient inclusion flowchart. PAH: pulmonary arterial hypertension; FDA: US Food and Drug Administration; CTEPH: chronic thromboembolic pulmonary hypertension; REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management; REVEAL Lite 2.0: abridged version of REVEAL 2.0; COMPERA: Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; COMPERA 2.0: modified four-strata model of COMPERA; FPHR: French Pulmonary Hypertension Registry.
The 18 RCTs studied 10 drugs over five drug classes: 1) endothelin receptor antagonists: bosentan, ambrisentan, sitaxentan and macitentan; 2) soluble guanylate cyclase stimulators: riociguat; 3) phosphodiesterase type 5 inhibitors: sildenafil and tadalafil; 4) prostacyclin analogues: treprostinil and iloprost; and 5) prostacyclin receptor agonists: selexipag. All studies except for the AMBITION trial [12] allocated patients to investigational therapy or placebo. For AMBITION, we considered the monotherapy arms (ambrisentan alone or tadalafil alone) as the “control” arm. For trials that included varying doses of the investigational therapy, we combined all doses into a single active therapy arm.
Data harmonisation has been previously described in detail (supplementary material) [27].
Risk-prediction algorithms
We assessed the risk of 1-year mortality at the screening or randomisation visit using the US Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL 2.0) [28], the abridged version of REVEAL 2.0 (REVEAL Lite 2) [29], Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) [30], the modified four-strata model of COMPERA (COMPERA 2.0) [31] and the invasive French Pulmonary Hypertension Registry (FPHR) score (supplementary material) [31].
Outcomes
6-min walk distance
We defined a change in 6-min walk distance (Δ6MWD) as the difference between measurements at baseline and at 12 or 16 weeks (supplementary material).
Clinical worsening
Clinical worsening (CW) events were defined as any one of the following events: 1) all-cause death, 2) lung transplantation, 3) atrial septostomy, 4) hospitalisation for worsening PAH, 5) discontinuation of study treatment (or study withdrawal) for worsening PAH, 6) initiation of parenteral (intravenous or subcutaneous) prostanoid therapy or 7) decrease of at least 15% of 6MWD from baseline, combined with either i) worsening of World Health Organization (WHO) functional class from baseline or ii) the addition of approved PAH treatment. This composite end-point included clinically important events and could be assessed uniformly across studies. Time-to-event analysis used time expressed in weeks from randomisation to the first CW event. If no CW events occurred, follow-up time was censored at the last visit or end of study participation if the individual did not complete the study. Additionally, we used the 12-week risk of CW as a binary variable.
Statistical analysis
We primarily used a two-stage meta-analysis. For Δ6MWD, we ran linear regression models in each trial with Δ6MWD as the dependent variable and baseline risk (on a continuous scale from the prediction rules) and treatment assignment as the main independent variables without adjusting for additional covariates. We obtained effect estimates, standard errors and confidence intervals in each individual trial. In the second stage, we used the inverse variance weighted random-effects model to allow for between-trial heterogeneity to combine study-specific effect estimates from the first stage to generate summary results and forest plots. The random-effects model was fitted with a restricted maximum likelihood estimation with confidence intervals derived using the Hartung, Knapp, Sidik, Jonkman (HKSJ) approach. We followed the same two-stage approach with models that included an interaction with baseline risk (on the continuous scale) and treatment assignment. We used the Stata package, ipdmetan [32].
For the analysis of CW by 12 weeks, we ran logistic regression models in each of the 18 RCTs separately to obtain odds ratios, standard errors and confidence intervals and combined the effect estimates in the second stage. For time-to-event analysis, we restricted our analysis to three trials, AMBITION, GRIPHON and SERAPHIN, because other trials did not have follow-up beyond 16 weeks. In the first stage, we ran Cox proportional hazards models within each trial with time to CW as the outcome, and baseline risk (continuous scale) and treatment assignment as independent variables to obtain hazard ratios, standard errors and confidence intervals and combined the effect estimates in the second stage using the random-effects model above. We included an interaction term with baseline risk (on the continuous scale) and treatment assignment in each trial separately. In the second stage, the interactions were pooled with the random effects, restricted maximum likelihood estimation and HKSJ approach.
We conducted sensitivity analyses using a one-stage meta-analysis using mixed-effects models for all three outcomes with baseline risk (on the continuous scale), centred at each trial-specific mean with two interaction terms to remove aggregation bias: 1) treatment arm by centre-specific baseline risk in each trial and 2) treatment arm by mean baseline risk across all participants. All one-stage models were unadjusted. Analyses were conducted using Stata/BE 17.0 (StataCorp).
Results
Study population
Out of 6633 participants from phase 3 studies with PAH, 14 did not receive the intervention, leaving 6619 in the study sample (figure 1). Available data allowed for the calculation of risk scores in 6561 (99.1%), 6613 (99.9%), 6619 (100%), 2789 (42.1%) and 3475 (52.5%) at baseline for REVEAL 2.0, REVEAL Lite 2, COMPERA, COMPERA 2.0 and FPHR scores, respectively. We chose to primarily present results for REVEAL 2.0 due to its strong discrimination of risk strata in this patient cohort. Analyses using the other algorithms were similar, unless otherwise described below.
Of the 6561 patients with a calculated REVEAL 2.0 score at baseline, 2855 (44%) were designated as low risk, 2323 (35%) were intermediate risk and 1383 (21%) were high risk (table 1).
TABLE 1.
Baseline characteristics of study participants, stratified by REVEAL 2.0 risk category
| Participants with available data# (n) | Total | Low risk | Intermediate risk | High risk | p-value¶ | |
|---|---|---|---|---|---|---|
| Participants, N | 6561 | 2855 | 2323 | 1383 | ||
| Age, years | 49.2±15.4 | 45.9±14.6 | 49.6±15.3 | 55.5±15.2 | <0.001 | |
| Sex | <0.001 | |||||
| Female | 5143 (78.4) | 2263 (79.3) | 1899 (81.7) | 981 (70.9) | ||
| Male | 1418 (21.6) | 592 (20.7) | 424 (18.3) | 402 (29.1) | ||
| Race | <0.001 | |||||
| American Indian or Alaskan Native | 53 (0.8) | 36 (1.3) | 13 (0.6) | 4 (0.3) | ||
| Asian | 951 (14.5) | 459 (16.1) | 323 (13.9) | 169 (12.2) | ||
| Black or African American | 261 (4.0) | 112 (3.9) | 99 (4.3) | 50 (3.6) | ||
| Other | 53 (0.8) | 27 (0.9) | 17 (0.7) | 9 (0.7) | ||
| Unknown | 726 (11.1) | 315 (11.0) | 296 (12.7) | 115 (8.3) | ||
| White | 4517 (68.8) | 1906 (66.8) | 1575 (67.8) | 1036 (74.9) | ||
| Ethnicity | <0.001 | |||||
| Hispanic or Latino | 686 (10.5) | 350 (12.3) | 236 (10.2) | 100 (7.2) | ||
| Not Hispanic or Latino | 5641 (86.0) | 2448 (85.7) | 1959 (84.3) | 1234 (89.2) | ||
| Unknown | 234 (3.6) | 57 (2.0) | 128 (5.5) | 49 (3.5) | ||
| PAH aetiology | <0.001 | |||||
| Connective tissue disease | 1755 (27.0) | 474 (16.7) | 678 (29.4) | 603 (44.3) | ||
| Congenital heart disease | 511 (7.9) | 313 (11.1) | 164 (7.1) | 34 (2.5) | ||
| Drug and toxin-induced | 118 (1.8) | 63 (2.2) | 45 (2.0) | 10 (0.7) | ||
| HIV associated | 65 (1.0) | 43 (1.5) | 19 (0.8) | 3 (0.2) | ||
| Heritable | 70 (1.1) | 15 (0.5) | 24 (1.0) | 31 (2.3) | ||
| Idiopathic | 3942 (60.7) | 1909 (67.5) | 1364 (59.1) | 669 (49.2) | ||
| Other | 36 (0.6) | 13 (0.5) | 13 (0.6) | 10 (0.7) | ||
| Body mass index, kg·m −2 | 26.9±6.4 | 27.0±6.3 | 26.8±6.6 | 26.8±6.0 | 0.36 | |
| 6MWD, m | 347.2±83.7 | 391.4±62.0 | 333.3±75.4 | 279.2±82.6 | <0.001 | |
| WHO functional class | <0.001 | |||||
| I | 47 (0.7) | 44 (1.5) | 2 (0.1) | 1 (0.1) | ||
| II | 2210 (33.7) | 1625 (56.9) | 481 (20.7) | 104 (7.5) | ||
| III | 4098 (62.5) | 1182 (41.4) | 1776 (76.5) | 1140 (82.5) | ||
| IV | 203 (3.1) | 4 (0.1) | 62 (2.7) | 137 (9.9) | ||
| Laboratory data | ||||||
| BNP, pg·mL−1 | 360 | 262.5±345.8 | 96.6±124.5 | 272.2±249.0 | 578.7±492.7 | <0.001 |
| NT-proBNP, pg·mL−1 | 2431 | 1304.5±2075.5 | 282.7±284.5 | 1315.3±1244.7 | 2832.8±2987.0 | <0.001 |
| eGFR, mL·min−1 | 6372 | 87.7±25.1 | 96.6±21.3 | 84.9±24.7 | 73.8±25.8 | <0.001 |
| Haemodynamic data | ||||||
| Right atrial pressure, mmHg | 4374 | 8.6±5.3 | 7.4±4.5 | 9.0±5.5 | 10.7±6.1 | <0.001 |
| Mean pulmonary arterial pressure, mmHg | 4696 | 52.6±15.5 | 51.0±16.6 | 54.2±15.3 | 53.6±13.2 | <0.001 |
| Pulmonary capillary wedge pressure, mmHg | 4510 | 9.2±3.5 | 9.3±3.5 | 9.2±3.5 | 9.1±3.5 | 0.24 |
| Cardiac index, L·min−1·m−2 | 3838 | 2.4±0.8 | 2.6±0.8 | 2.3±0.7 | 2.2±0.7 | <0.001 |
| Pulmonary vascular resistance, Wood units | 4565 | 11.7±7.0 | 10.5±6.7 | 12.5±7.1 | 13.0±7.0 | <0.001 |
| Treatment arm | <0.001 | |||||
| Placebo | 2629 (40.1) | 1091 (38.2) | 926 (39.9) | 612 (44.3) | ||
| Active treatment | 3932 (59.9) | 1764 (61.8) | 1397 (60.1) | 771 (55.7) |
Data are presented as n (%) or mean±SD, unless otherwise indicated. REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management; PAH: pulmonary arterial hypertension; 6MWD: 6-min walk distance; WHO: World Health Organization; BNP: brain natriuretic peptide; NT-proBNP: N-terminal pro-brain natriuretic peptide; eGFR: estimated glomerular filtration rate.
for laboratory and haemodynamic data;
determined from ANOVA or Chi-square test, as appropriate.
6-min walk distance
The placebo-adjusted treatment effect of Δ6MWD was 22.83 m (95% CI 17.52–28.14 m, p<0.001) when adjusted for baseline REVEAL 2.0. There was no significant association between REVEAL 2.0 score and Δ6MWD after adjustment for treatment assignment (per three-point increment: −0.39 m, 95% CI −6.34–5.56 m; p=0.89) (figure 2a). On the categorical three-tiered risk scale, there was no significant difference in Δ6MWD for each increase in risk category (2.93 m, 95% CI −2.44–8.29 m; p=0.27) when controlling for treatment assignment.
FIGURE 2.
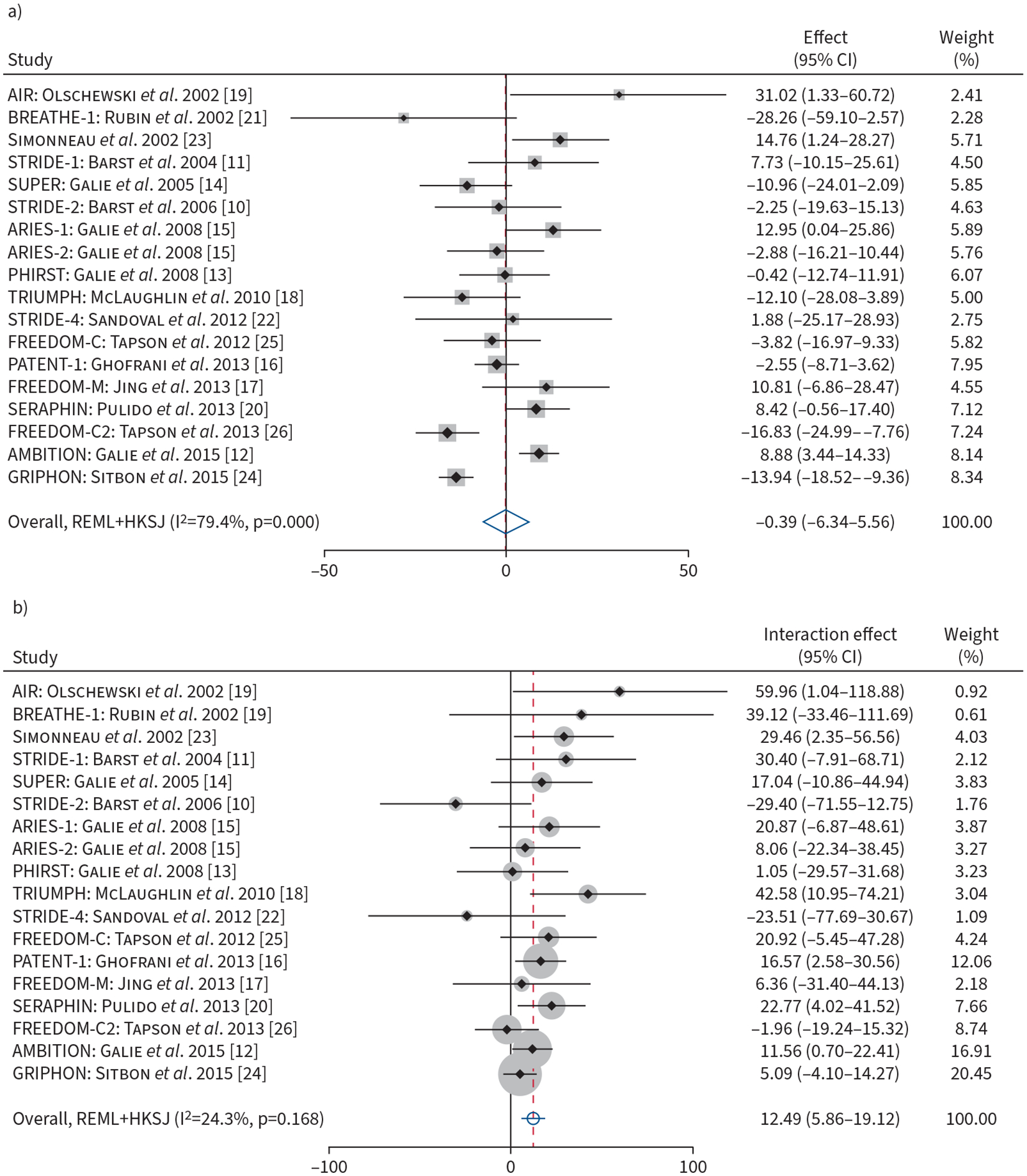
a) Forest plot of association of baseline REVEAL 2.0 risk (on continuous scale, per three-point increment) with change in 6-min walk distance (6MWD) from baseline to end of follow-up. b) Forest plot of the baseline REVEAL 2.0 (on continuous scale, per three-point increment) by treatment interaction terms and change in 6MWD from baseline to end of follow-up. Weights are from random-effects model. REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management; REML: restricted maximum likelihood; HKSJ: Hartung, Knapp, Sidik, Jonkman approach.
Despite the lack of association between the REVEAL 2.0 score or category and Δ6MWD, baseline REVEAL 2.0 score modified the treatment effect on the Δ6MWD. For every three-point higher baseline REVEAL 2.0 score, there was a 12.49 m (95% CI 5.86–19.12 m; p=0.001) greater treatment effect in terms of the Δ6MWD (figures 2b and 3a). When analysed on the categorical scale, for every increase in the baseline REVEAL 2.0 risk category there was an 8.42 m (95% CI 0.82–16.01 m; p=0.03) greater treatment effect. Stratifying by REVEAL 2.0 risk category, individuals who were low risk at baseline derived a placebo-adjusted treatment effect of 15.0 m, compared to 25.7 m for intermediate-risk and 34.0 m for the high-risk groups (figure 3b). There was considerable heterogeneity present among trials for the association of baseline REVEAL 2.0 and Δ6MWD (I2=79.4%) (figure 2a), and low heterogeneity present for the association of baseline REVEAL 2.0 by treatment interaction (I2=24.3%) (figure 2b). Sensitivity analyses using other algorithms are shown in supplementary table S2a. Higher risk predicted by REVEAL Lite 2, COMPERA and FPHR showed significantly greater placebo-adjusted treatment effects; however, risk assessed by COMPERA 2.0 did not modify the treatment effect.
FIGURE 3.
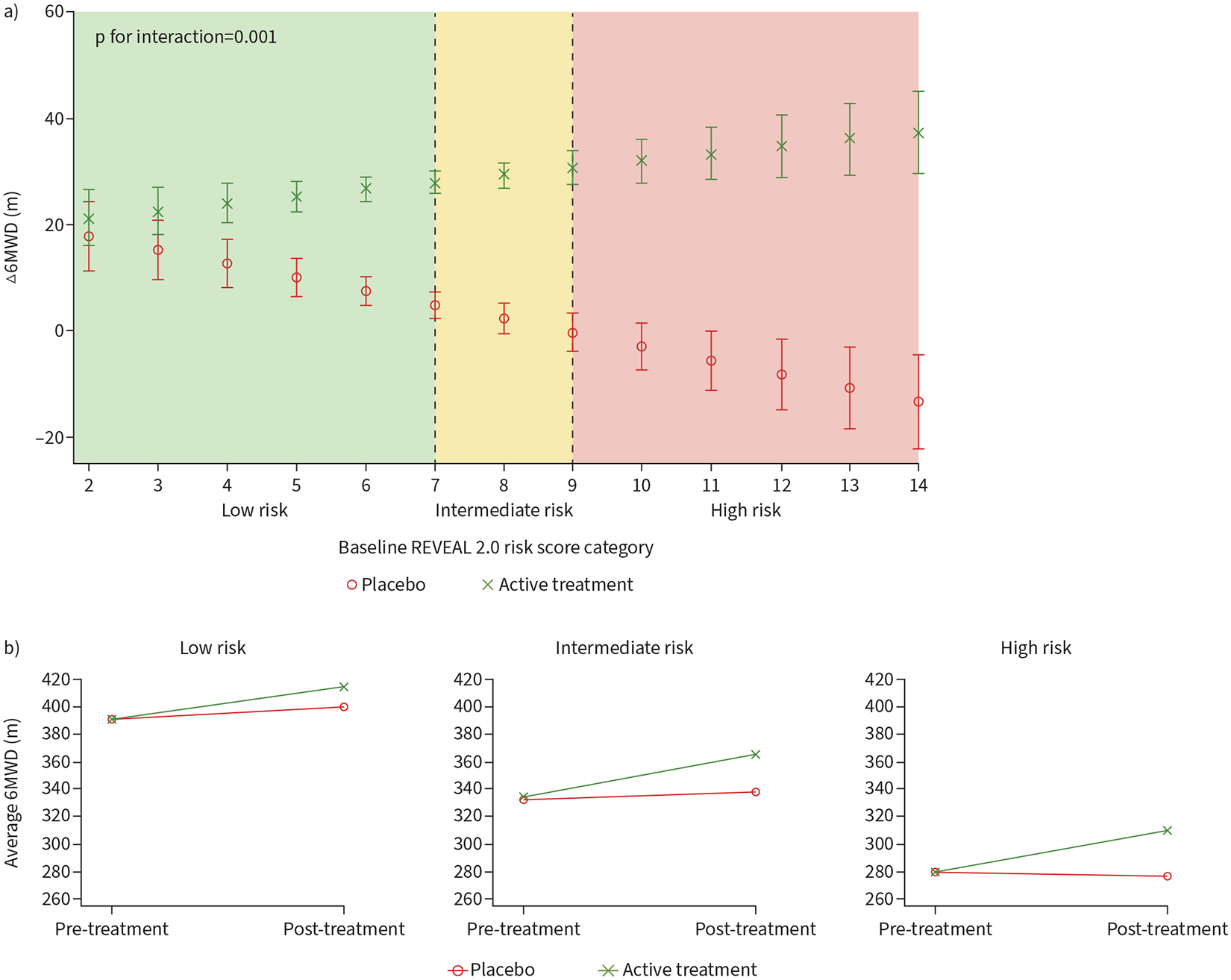
a) Interaction between baseline REVEAL 2.0 risk by treatment on change in 6-min walk distance (6MWD). b) Pre- and post-treatment 6MWD stratified by baseline REVEAL 2.0 risk. REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management.
Clinical worsening at 12 weeks
A total of 1257 individuals had at least one CW event (supplementary table S3). Of these events, 523 (41.6%) occurred at or before 12 weeks. Among the CW events at 12 weeks, 175 (33.5%) occurred in the three event-driven trials (AMBITION, GRIPHON, SERAPHIN).
In all trials, participants assigned to active treatment had lower odds of CW at 12 weeks than those receiving placebo (OR 0.50, 95% CI 0.40–0.63; p<0.001) when adjusted for baseline REVEAL 2.0 score. Each three-point increment on the continuous scale REVEAL 2.0 score was associated with over threefold greater odds of CW at 12 weeks (OR 3.22, 95% CI 2.70–3.83; p<0.001) (figure 4a). Higher risk category (intermediate versus low or high versus intermediate) was associated with a greater than twofold odds of CW at 12 weeks (OR 2.21, 95% CI 1.81–2.70; p<0.001).
FIGURE 4.
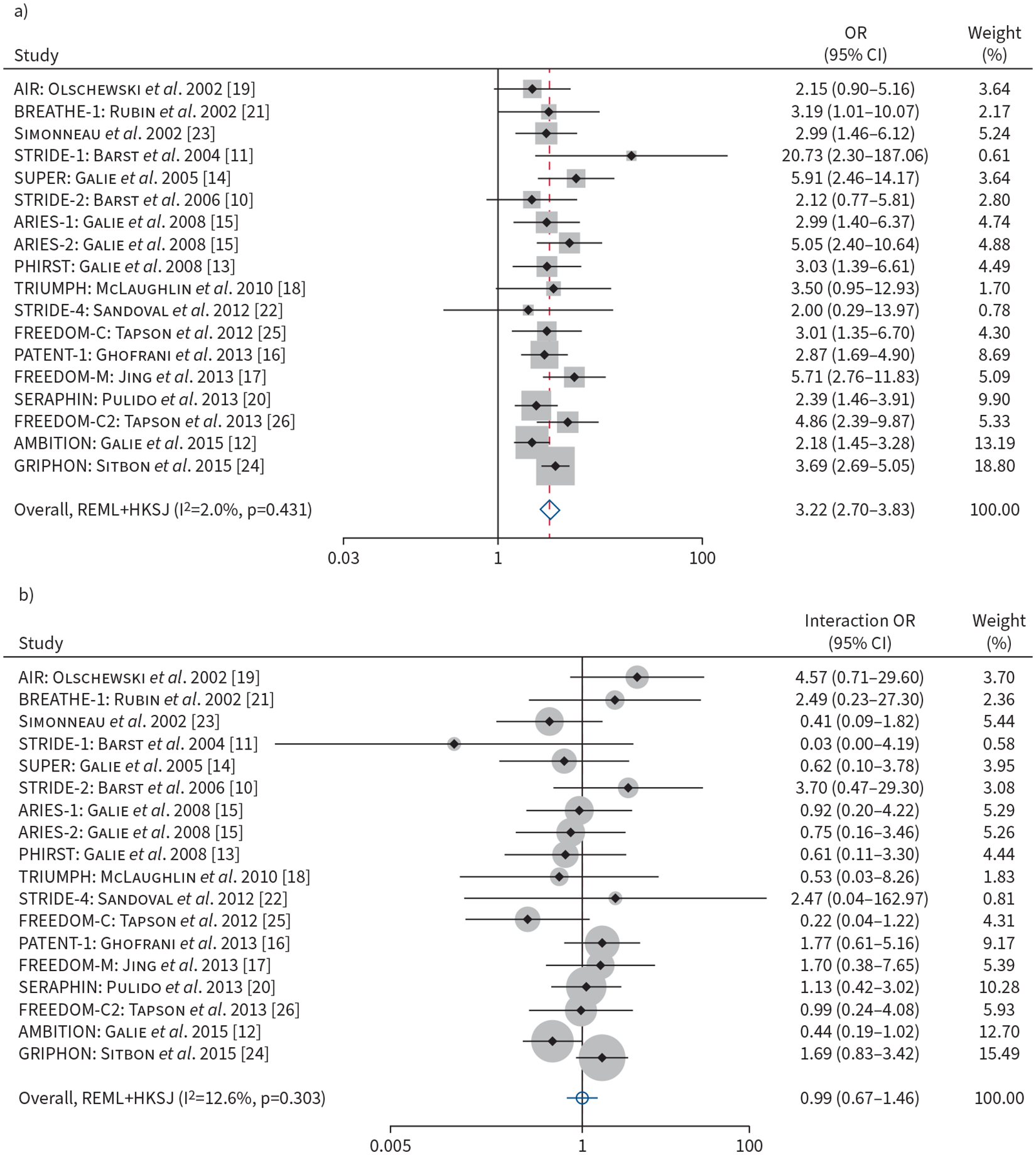
a) Forest plot of association of baseline REVEAL 2.0 risk (on continuous scale, per three-point increment) with odds of clinical worsening at 12 weeks. b) Forest plot of the baseline REVEAL 2.0 (on continuous scale, per three-point increment) by treatment interaction terms and odds of clinical worsening at 12 weeks. Weights are from random-effects model. REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management.
The effect of active treatment on the odds of CW at 12 weeks was not modified by baseline REVEAL 2.0 risk (p for interaction=0.96) (figure 4b). There was no clinically important heterogeneity present among trials for both the association of baseline REVEAL 2.0 risk and CW at 12 weeks (I2=2.0%) (figure 4a) or the association of baseline REVEAL 2.0 risk by treatment interaction (I2=12.6%) (figure 4b). Sensitivity analyses using the remaining algorithms revealed similar findings of increased odds of CW with higher baseline risk. While we did not find significant effect modification of treatment effect on baseline risk using REVEAL Lite 2 or COMPERA 2.0, we found significant interactions with the following scenarios: 1) baseline COMPERA risk by treatment (one-stage analysis only) and 2) baseline FPHR risk by treatment (one- and two-stage analysis). Treatment response was modified by COMPERA score, where a one-point increase of COMPERA score on the continuous scale (higher risk) attenuated the odds of CW at 12 weeks by 47% (p for interaction=0.02) with one-stage analysis. Treatment response was also modified by FPHR score. For our two-stage analysis, every one-point increase of FPHR score on the continuous scale (higher risk) attenuated the odds of CW at 12 weeks by 38% (p for interaction=0.03). With one-stage analysis, every one-point increase of FPHR score on the continuous scale attenuated the odds of CW at 12 weeks by 39% (p for interaction=0.01) (supplementary table S2b).
Time to clinical worsening
We restricted our time-to-event analysis to AMBITION, GRIPHON and SERAPHIN. Participants receiving active treatment had a 41% decrease in the hazard of CW compared to those receiving placebo when adjusted for baseline REVEAL 2.0 score (HR 0.59, 95% CI 0.46–0.77; p<0.001). A three-point increase in the REVEAL 2.0 score on the continuous scale was associated with more than a twofold higher risk of CW (HR 2.18, 95% CI 1.99–2.39; p<0.001) (figure 5a). Higher risk category (intermediate versus low or high versus intermediate) was associated with a doubling of the risk of CW (HR 2.00, 95% CI 1.49–2.69; p<0.001).
FIGURE 5.
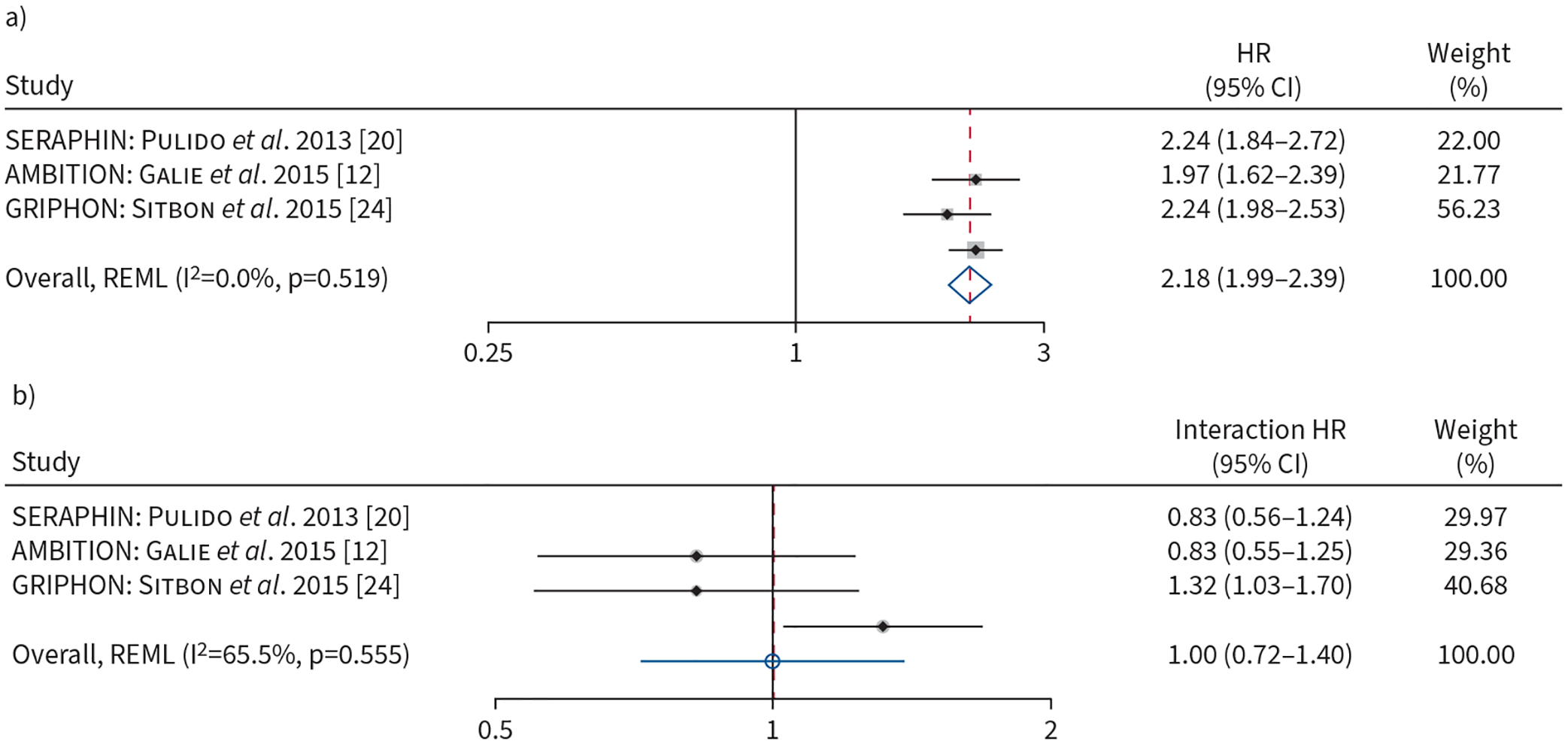
a) Forest plot of association of baseline REVEAL 2.0 risk (on continuous scale, per three-point increment) with time to clinical worsening. b) Forest plot of the baseline REVEAL 2.0 risk (on continuous scale, per three-point increment) by treatment interaction terms and time to clinical worsening. Weights are from random-effects model. REVEAL 2.0: US Registry to Evaluate Early and Long-Term PAH Disease Management.
The association of active treatment with the time-to-CW was not modified by baseline REVEAL 2.0 risk (p for interaction=0.99) (figure 5b). There was no clinically important heterogeneity present among trials for the association of baseline REVEAL 2.0 and time-to-CW (I2=0.0%) (figure 5a), but substantial heterogeneity was present for the association of baseline REVEAL 2.0 risk by treatment interaction (I2=65.5%) (figure 5b). Sensitivity analyses using the other algorithms revealed similar findings (supplementary table S2c).
Discussion
Using IPD meta-analysis across 18 RCTs in PAH, we found that individuals with higher baseline risk as predicted by several prediction rules had higher probabilities of CW but no difference in Δ6MWD when controlling for treatment assignment. However, we did detect a significant interaction of baseline risk score and treatment assignment on Δ6MWD, where participants with higher risk had a greater treatment effect compared to participants with lower risk. This finding was consistent across all risk algorithms except COMPERA 2.0, where there was no significant risk by treatment interaction on Δ6MWD. We found significant HTE on CW by baseline predicted risk using the FPHR score only; the other prediction scores did not significantly modify the treatment effect on CW.
There are several possible explanations for the HTE on Δ6MWD. Patients with lower baseline 6MWD and greater severity of illness may derive greater benefit from drug intervention. A similar effect has been demonstrated in other disease processes whereby sicker individuals derive greater benefit from treatments as compared to healthier individuals [33]. Conversely, a “ceiling effect” of response to therapy for individuals with higher baseline 6MWD raises concern for masking treatment effect, which has resulted in the exclusion of “healthier” individuals from PAH RCTs (baseline 6MWD >450 m) [34]. We demonstrated the ceiling effect phenomenon exists in this PAH RCT cohort, where lower risk patients with higher baseline 6MWD derived a smaller treatment effect compared to higher risk patients with lower baseline 6MWD. These traits are likely shared among many human diseases rather than a specific phenomenon in PAH.
We did not detect significant risk by treatment interactions on CW across the majority of risk-prediction models. This suggests that patients at all strata of predicted risk of death at 1 year gain similar treatment benefit in terms of CW. This finding was consistent across all risk algorithms except for when using the COMPERA risk score (one-stage analysis) and the FPHR risk score. In these three instances, higher baseline risk conferred a greater treatment benefit. However, this HTE may be misleading because several studies lacked the invasive haemodynamic data required for calculation of the FPHR score.
The discrepancy of findings of HTE in terms of Δ6MWD and CW may seem surprising. While Δ6MWD was used as a clinical end-point in PAH trials that led to US FDA approval of several pulmonary vasodilators, its performance as a surrogate for CW events and survival is questionable [35]. Prior studies demonstrated no correlation between Δ6MWD and survival benefit or incidence of clinical events, suggesting there may be a disconnect between the 6MWD as a functional, clinically pertinent end-point and other end-points that reflect short- or long-term disease progression and morbidity [36, 37]. The clinical prediction rules used in this study were all focused on survival at 1 year; risk stratification to identify treatment heterogeneity may require different prediction rules predicated on long-term outcomes.
Furthermore, although we did detect a significant treatment by risk interaction for baseline risk score and treatment assignment on Δ6MWD, the clinical significance of this finding is unknown. The effect on 6MWD resulting from the interaction was smaller than the minimal clinically important difference. The minimal clinically important difference for mean group differences for 6MWD as derived and validated using anchoring to the Medical Outcomes Short Form Physical Component Score was 24 m [38]. Because the additional improvement in Δ6MWD for patients with higher risk is relatively small, the clinical implications remain unclear.
Despite the lack of a significant baseline risk by treatment interaction for CW, patients with more symptoms and greater risk of adverse outcomes likely require more aggressive therapy or treatment escalation if they do not adequately respond.
To our knowledge, this is the first study exploring the heterogeneity of treatment effects by predicted risk of mortality in PAH using a large IPD meta-analysis across 18 RCTs. We also examined interactions using two widely used primary outcomes in PAH, Δ6MWD and CW, and performed both time-to-event analysis and logistic regression for CW. We further performed analyses using several externally validated risk algorithms and a one-stage meta-analytic approach that demonstrated consistent findings.
Out study is not without limitations. First, our analysis only included trials that were submitted to the US FDA for drug approval and did not include other negative or unpublished studies. Many studies did not collect brain natriuretic peptide levels at baseline, which precluded the calculation of COMPERA 2.0 for many participants. Furthermore, our analysis treated all active treatment drug classes and doses as a single active treatment arm. We did not stratify our analysis by drug class or doses, which would reduce sample sizes. It is also possible that different drug classes may have different treatment effect heterogeneity, but our study is not powered to detect treatment class-specific interactions. Although our study used a large IPD cohort, clinically important interactions may not have been detected due to type II error. Certain components of our two-stage analyses resulted in a high degree of heterogeneity between studies, and thus the pooling of these studies should be interpreted with caution. For time-to-event analysis, we only had access to three RCTs with long-term follow-up. The 12-week duration of most trials may be too short to detect meaningful CW outcomes. In addition, the patient populations may have differed across studies. Although the diagnosis of PAH was required, there was variable time between the documented time of diagnosis and randomisation visits. Furthermore, some studies allowed for the use of background therapy, but earlier studies excluded patients on concomitant treatment.
In conclusion, we found that baseline risk predicted CW in our IPD RCT population in PAH. Participants in these RCTs across all risk levels received a similar relative benefit to active therapy in terms of CW events. Individuals with higher baseline risk derived a greater placebo-adjusted treatment effect for Δ6MWD. This study demonstrates that predicted risk has a differential treatment response by outcome in PAH. Treatment heterogeneity should be considered based on the clinical outcome studied.
Supplementary Material
Conflict of interest:
H-M. Pan has received funding support from the National Institutes of Health (T32HL007891). R.L. McClelland has received full-time-equivalent salary support via a subcontract from the University of Pennsylvania. J.S. Fritz has had grants or contracts from United Therapeutics as monies paid to the institution for the conduct of multicentre pulmonary arterial hypertension drug trials. J.H. Holmes has received funding support from the Cardiovascular Medical Research and Education Fund; has received grants or contracts from the National Institutes of Health, University of Florida Juvenile Diabetes Research Foundation and the University of Pavia; has served as a participant on the Clinical Data to Health External Advisory Board and COACH T2D (Columbia University); and has served unpaid leadership or fiduciary roles for the American College of Medical Informatics, American College of Epidemiology and the Artificial Intelligence Society. J. Minhas has received funding support from the National Institutes of Health (T32HL007891) and the American Thoracic Society Early Career Investigator Award. H.I. Palevsky has participated on a data safety monitoring board for studies for pulmonary arterial hypertension sponsored by United Therapeutics. S.M. Kawut has received funding support from the National Institutes of Health (K24HL103844) and the Cardiovascular Medical Research and Education Fund; received consulting fees from Janssen, Morphic and Regeneron; received payment or honoraria from Janssen; contributed to continuing medical education courses through Accredo, Actelion, Aerovate, Bayer, Inari Medical, Merck, United Therapeutics, Janssen, Liquidia and Pfizer; received support for attending meetings from Aerovate; participated in data safety monitoring boards or advisory boards for United Therapeutics, Acceleron, Vivus and Aerovate; participated in leadership or fiduciary roles for the editorial board of the European Respiratory Journal (ended 2022); received stock or stock options from Verve Therapeutics; and received remote monitory equipment from PhysIQ. N. Al-Naamani has received funding support from the National Institutes of Health (K23HL141584) and from the Cardiovascular Medical Research and Education Fund. The remaining authors disclose no potential conflicts of interest.
Support statement:
Support was provided by the Cardiovascular Medical Research and Education Fund (S.M. Kawut), the National Institutes of Health (K24HL103844, S.M. Kawut; K23HL141584, N. Al-Naamani; and T32HL007891, H-M. Pan and J. Minhas). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012; 122: 4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLaughlin VV, Shah SJ, Souza R, et al. Management of pulmonary arterial hypertension. J Am Coll Cardiol 2015; 65: 1976–1997. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009; 119: 2250–2294. [DOI] [PubMed] [Google Scholar]
- 4.Halliday SJ, Hemnes AR. Identifying “super responders” in pulmonary arterial hypertension. Pulm Circ 2017; 7: 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angus DC, Chang CH. Heterogeneity of treatment effect: estimating how the effects of interventions vary across individuals. JAMA 2021; 326: 2312–2313. [DOI] [PubMed] [Google Scholar]
- 6.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022; 43: 3618–3731. [DOI] [PubMed] [Google Scholar]
- 7.Kent DM, Steyerberg E, van Klaveren D. Personalized evidence based medicine: predictive approaches to heterogeneous treatment effects. BMJ 2018; 363: k4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riley RD, Debray TPA, Fisher D, et al. Individual participant data meta-analysis to examine interactions between treatment effect and participant-level covariates: statistical recommendations for conduct and planning. Stat Med 2020; 39: 2115–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott JV, Garnett CE, Kanwar MK, et al. Enrichment benefits of risk algorithms for pulmonary arterial hypertension clinical trials. Am J Respir Crit Care Med 2021; 203: 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006; 47: 2049–2056. [DOI] [PubMed] [Google Scholar]
- 11.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004; 169: 441–447. [DOI] [PubMed] [Google Scholar]
- 12.Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 2015; 373: 834–844. [DOI] [PubMed] [Google Scholar]
- 13.Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009; 119: 2894–2903. [DOI] [PubMed] [Google Scholar]
- 14.Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353: 2148–2157. [DOI] [PubMed] [Google Scholar]
- 15.Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy (ARIES) study 1 and 2. Circulation 2008; 117: 3010–3019. [DOI] [PubMed] [Google Scholar]
- 16.Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013; 369: 330–340. [DOI] [PubMed] [Google Scholar]
- 17.Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation 2013; 127: 624–633. [DOI] [PubMed] [Google Scholar]
- 18.McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010; 55: 1915–1922. [DOI] [PubMed] [Google Scholar]
- 19.Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347: 322–329. [DOI] [PubMed] [Google Scholar]
- 20.Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013; 369: 809–818. [DOI] [PubMed] [Google Scholar]
- 21.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896–903. [DOI] [PubMed] [Google Scholar]
- 22.Sandoval J, Torbicki A, Souza R, et al. Safety and efficacy of sitaxsentan 50 and 100 mg in patients with pulmonary arterial hypertension. Pulm Pharmacol Ther 2012; 25: 33–39. [DOI] [PubMed] [Google Scholar]
- 23.Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800–804. [DOI] [PubMed] [Google Scholar]
- 24.Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015; 373: 2522–2533. [DOI] [PubMed] [Google Scholar]
- 25.Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest 2012; 142: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 26.Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013; 144: 952–958. [DOI] [PubMed] [Google Scholar]
- 27.Min J, Appleby DH, McClelland RL, et al. Secular and regional trends among pulmonary arterial hypertension clinical trial participants. Ann Am Thorac Soc 2022; 19: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest 2019; 156: 323–337. [DOI] [PubMed] [Google Scholar]
- 29.Benza RL, Kanwar MK, Raina A, et al. Development and validation of an abridged version of the REVEAL 2.0 risk score calculator, REVEAL Lite 2, for use in patients with pulmonary arterial hypertension. Chest 2021; 159: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017; 50: 1700740. [DOI] [PubMed] [Google Scholar]
- 31.Hoeper MM, Pausch C, Olsson KM, et al. COMPERA 2.0: a refined four-stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J 2022; 60: 2102311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisher DJ. Two-stage individual participant data meta-analysis and generalized forest plots. Stata J 2015; 15: 369–396. [Google Scholar]
- 33.Sabatine MS, De Ferrari GM, Giugliano RP, et al. Clinical benefit of evolocumab by severity and extent of coronary artery disease: analysis from FOURIER. Circulation 2018; 138: 756–766. [DOI] [PubMed] [Google Scholar]
- 34.Frost AE, Langleben D, Oudiz R, et al. The 6-min walk test (6MW) as an efficacy endpoint in pulmonary arterial hypertension clinical trials: demonstration of a ceiling effect. Vascul Pharmacol 2005; 43: 36–39. [DOI] [PubMed] [Google Scholar]
- 35.Ventetuolo CE, Gabler NB, Fritz JS, et al. Are hemodynamics surrogate end points in pulmonary arterial hypertension? Circulation 2014; 130: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaine S, Simonneau G. The need to move from 6-minute walk distance to outcome trials in pulmonary arterial hypertension. Eur Respir Rev 2013; 22: 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macchia A, Marchioli R, Marfisi R, et al. A meta-analysis of trials of pulmonary hypertension: a clinical condition looking for drugs and research methodology. Am Heart J 2007; 153: 1037–1047. [DOI] [PubMed] [Google Scholar]
- 38.Moutchia J, McClelland RL, Al-Naamani N, et al. Minimal clinically important difference in the six-minute walk distance for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2023; 207: 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.