

Abstract
Infinium Methylation BeadChips are widely used to profile DNA cytosine modifications in large cohort studies for reasons of cost-effectiveness, accurate quantification, and user-friendly data analysis in characterizing these canonical epigenetic marks. In this work, we conducted a comprehensive evaluation of the updated Infinium MethylationEPIC v2 BeadChip (EPICv2). Our evaluation revealed that EPICv2 offers significant improvements over its predecessors, including expanded enhancer coverage, applicability to diverse ancestry groups, support for low-input DNA down to one nanogram, coverage of existing epigenetic clocks, cell type deconvolution panels, and human trait associations, while maintaining accuracy and reproducibility. Using EPICv2, we were able to identify epigenome and sequence signatures in cell line models of DNMT and SETD2 loss and/or hypomorphism. Furthermore, we provided probe-wise evaluation and annotation to facilitate the use of new features on this array for studying the interplay between somatic mutations and epigenetic landscape in cancer genomics. In conclusion, EPICv2 provides researchers with a valuable tool for studying epigenetic modifications and their role in development and disease.
INTRODUCTION
In higher-order eukaryotic species, DNA cytosine modifications, including 5-methylcytosine [1] and 5-hydroxymethylcytosine [2], are extensively implicated in gene regulation and other cellular processes. Analysis of cytosine modifications uncovers principles of chromatin dynamics and epigenetic dysregulation in human development and disease [3]. Knowledge of the genome-wide cytosine modification profile reveals cell identity [4], cell pathological state [5] and mitotic history [6] and holds therapeutic and diagnostic promises in medicine [7].
Illumina’s Infinium technology-based DNA methylation microarray assays have been one of the most widely used technologies for epigenome studies in humans [8] (Fig 1A), and more recently, mice [9] and other mammalian species [10]. This technology is based on bead-bound 50nt target-specific oligonucleotides that hybridize with bisulfite converted genomic DNA. The methylation detection is achieved using one of two Infinium chemistries. Infinium-I chemistry involves two bead types - one targeting the methylated cytosine, and the other targeting the unmethylated cytosine. Infinium-II chemistry only uses one bead type and distinguishes the two methylation states using a color-discriminating single-base extension [8]. Compared to other genome-wide methylation assays [11] such as high-throughput bisulfite sequencing, Infinium Methylation BeadChips are more cost effective [12], quantitative [13] and user-friendly with many well-maintained, standard-compliant community software tools from the bioinformatics community [14]. More than 100,000 samples profiled by HumanMethylation450 BeadChips (HM450) have been deposited to the Gene Expression Omnibus (GEO).
Figure 1:
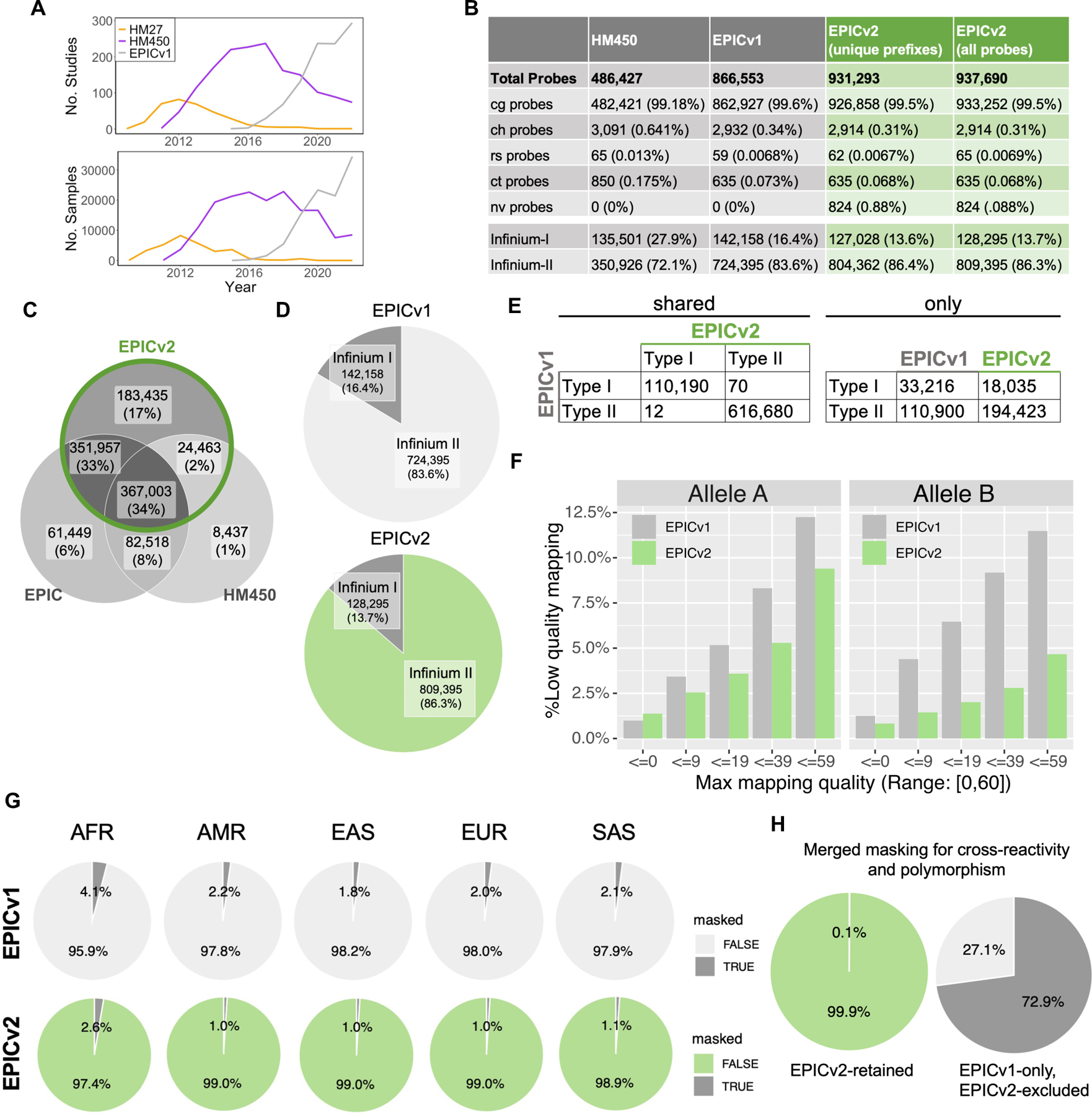
Enhanced probe mapping and applicability of EPICv2 in diverse human populations. (A) Total number of Infinium DNA methylation BeadChip studies and deposited datasets in GEO. (B) Probe counts for HM450, EPICv1, EPICv2, and unique prefix counts for EPICv2. Abbreviations: “cg”: CpG cytosine methylation probes; “ch”: non-CG cytosine methylation probes; “rs”: common SNP probes; “nv”: probes for somatic mutations found in cancer; and “ct”: quality control probes. (C) Venn diagram illustrating the percentage of EPICv2 probes retained from predecessor arrays. (D) Infinium-I and Infinium-II chemistry ratios for EPICv1 and EPICv2 probes. EPICv2 data is from all probes, same as panel B. (E) Infinium-I and Infinium-II chemistry ratios for shared EPICv1-v2 probes and exclusive EPICv1/EPICv2 probes. (F) Mapping quality of EPICv1 and EPICv2 probes, differentiated by allele A and allele B. (G) Proportion of probes masked due to ancestry-specific SNP overlaps. Abbreviations: AFR, African population; AMR, Admixed American; EAS, East Asian; EUR, European; SAS, South Asian. (H) Percentage of probes with cross-reactivity and sequence polymorphism influence issues, comparing shared EPICv1-EPICv2 and EPICv1-only probes.
Human Infinium BeadChip assays have evolved over multiple generations, ranging from HM27 [15], HM450 [8], EPIC [16], to the most recent EPICv2. Each succeeding generation embodies a more comprehensive coverage of the human genome and more versatile probe designs compared to the previous ones. Many expansions parallel improvements in understanding of DNA methylation biology. For example, DNA methylation was originally known best for its role in epigenetic silencing at gene promoters [17, 18]. Therefore, the first Infinium array, the HM27 only included ~27,000 probes to query promoter CpG methylation. HM450 expands HM27 to include probes to query gene body CpG methylation, leading to a growing appreciation of gene body methylation in gene expression regulation [19]. The EPIC array, released in 2015, expanded most significantly on cis-regulatory elements [16], reflecting an increasing recognition of these enhancers carrying a tissue-specific methylation signature [20].
Genome coverage expansion comes with a better understanding of Infinium chemistry and its non-canonical usage in practice. Most notably, it was discovered that Infinium-I probe out-of-band channel signal can be co-opted for parameterizing background subtraction [21] and detection p-value calculation [22]. Similarly, Infinium-I probe extension switch can be used de facto as SNP probes for inferring subject ethnicity [23]. Total signal intensities can be used to infer copy number variations [24], and more recently fractions of cells from different species [9]. Technical confounders that significantly influence probe hybridization and extension have also been better understood [25, 26]. In addition, the array has also been shown to work with other base conversion methods to query 5mC and 5hmC [27–29].
The Infinium BeadChip EPICv2 was recently launched by Illumina. As the cost benefit between array and sequencing technologies is being narrowed, we are critically curious whether this update to the Infinium array platform will bring advantage in other aspects. This study provides a critical evaluation of its probe design, genome coverage, quantitative performance, and practical use in large cohort studies, with a focus on comparing it to the previous human DNA methylation BeadChip Arrays. We specifically assess its performance with low input, probe mappability, susceptibility to sequence polymorphisms, and the utility of newly added probes targeting somatic mutations. Additionally, we examine the technical performance of replicate probes, their coverage on existing epigenetic clocks and cell type deconvolution panels, and their potential for identifying EWAS discoveries. The study also investigates whether the newly added probes can accurately resolve cell identity.
RESULTS
EPICv2 has improved probe mapping and utility in diverse human populations.
EPICv2 features a larger probe count than its predecessors, HM450 and EPICv1, with 937,690 probes compared to 486,427 and 866,552, respectively. Like HM450 and EPICv1, EPICv2 probes predominantly target CpG cytosine methylation (“cg” probes), with a smaller fraction targeting non-CpG cytosine methylation (“ch” probes), common SNPs (“rs” probes), and quality controls (“ct” probes) [16] (Fig 1B). Over 99% of the probes in EPICv2 target CpG cytosine methylation, while the numbers of probes for non-CpG methylation, common human single-nucleotide polymorphisms (SNPs), and control probes are comparable to those found in HM450 and EPICv1. Additionally, EPICv2 incorporates 824 new probes that specifically target recurrent somatic mutations found in cancer (“nv” probes) (Fig 1B). EPICv2 retains a high percentage of cg probes from its predecessors, with 83% of EPICv1 probes and 81% of HM450 probes retained (Fig 1C). Notably, 24,463 cg probes from HM450 that were not present in EPICv1 are reintroduced in EPICv2. Additionally, 183,435 new cg probes were added, representing 20% of the total cg probes in the EPICv2 array. The array uses the same chemistry as previous generations of Infinium BeadChips, with a similar ratio of Infinium-I and Infinium-II probes (Fig 1D).
The shared probes between EPICv1 and EPICv2 largely maintain Infinium probe design. Only a small number of probes changed, with 70 Infinium-I probes switching to Infinium-II chemistry and 12 Infinium-II probes switching to Infinium-I in the EPICv2 update (Fig 1E). The number of deleted Infinium-I probes exceeded the number of added probes, leading to a lower proportion of Infinium-I probes in EPICv2 (Fig. S1A). EPICv2 contains fewer probes with poor mapping to GRCh38 compared to EPICv1 (Fig 1F). Infinium-I probes have two alleles whose sequences map consistently to enable accurate methylation calling (Fig S1B). In addition, fewer probes are subject to direct influence by ancestry-specific genetic variation (Fig 1G). However, AFR is still subject to such direct influence more than other ethnicity groups, consistent with the higher genetic diversity of the African population (Fig 1G). Of the probes deleted in EPICv2, 72.9% were found to have issues with cross-reactivity or direct influence from sequence polymorphism (Fig 1H). In contrast, only 0.1% of the retained probes were affected by these factors. These improvements in probe design and selection result in a more accurate and reliable assessment of DNA methylation patterns across diverse human populations.
EPICv2 generates highly reproducible data between sample and probe replicates.
We evaluated the correlation of methylation measurements between technical replicates of various human cell lines using the EPICv2 platform. Technical replicates refer to bisulfite-converted DNA samples from the same cell line that were processed in separate batches on the EPICv2 platform. The cell lines used in this study included GM12878 (B-cell-derived), LNCaP (prostate cancer-derived), K562 (lymphoblast cells), and HCT116 (colorectal carcinoma). For HCT116, two distinct clones were analyzed in two different laboratories to assess technical reproducibility.
Our findings showed that methylation measurements between technical replicates on the EPICv2 platform were highly correlated (Fig. 2A). The Spearman’s rank correlation coefficient (rho) between technical replicates was significantly higher than that between non-replicates (Fig. 2B). We found a lower inter-cell line correlation for EPICv2-added probes, indicating that these additional probes exhibit improved discrimination of cell identities (Fig. 2C). Furthermore, the observed differences in measurements between technical replicates were not affected by probe types (cg, ch, rs, nv) (Fig. S2A).
Figure 2:
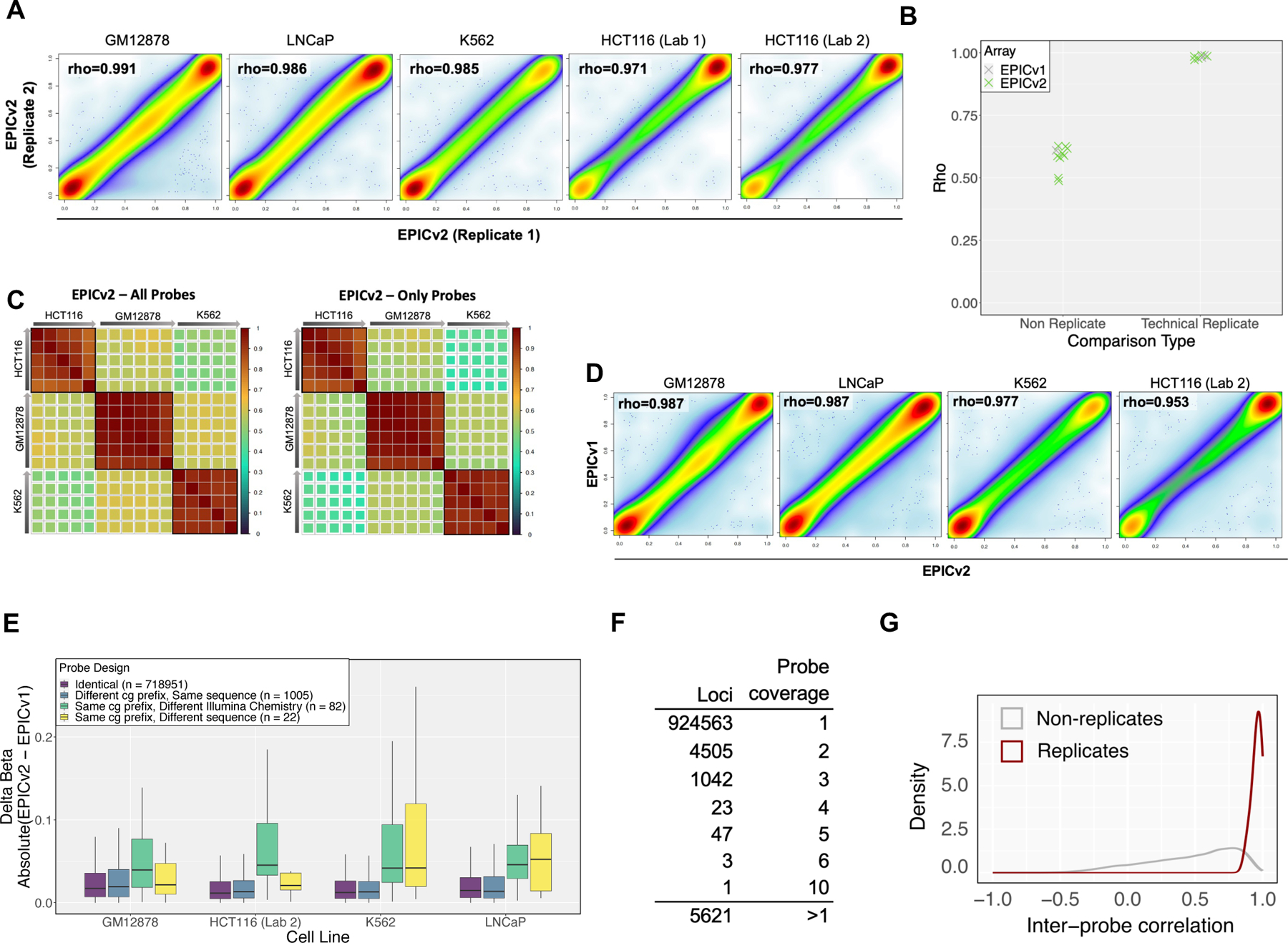
Assessment of EPICv2 reproducibility between sample replicates and replicate probes. (A) Methylation measurement correlations between technical replicates of GM12878, LNCaP, K562, and HCT116 cell lines. (B) Comparison of Spearman’s rank correlation coefficients (rho) between technical and non-technical replicates. (C) Lower inter-cell line correlation for newly added EPICv2 probes, indicating increased discriminatory power. Arrows represent DNA input from high to low. (D) Methylation measurement correlation using EPICv1 and EPICv2 on four human cell lines. (E) Comparison of EPICv1-EPICv2 design switches and probes with identical sequences in both platforms. (F) Number of loci with multiple probe replication coverage. (G) Correlations among replicate probes compared to non-replicate probes, emphasizing probe design robustness.
To determine whether EPICv2 produces consistent data compared to EPICv1, we measured the correlation of EPICv2 methylation measurements with EPICv1 measurements performed on the same bisulfite-converted cell line DNA samples. We observed that EPICv1 and EPICv2 generated highly correlated results on shared probes (Fig 2D), with the Spearman’s rho between EPICv1 and EPICv2 measurements performed on the same cell line being higher than that between different cell lines (Fig S2B, S2C). Among the 727,232 shared probes, 82 probes underwent Infinium chemistry changes, and 22 probes had different sequences due to strand choice switches (Fig S2E). Probes with altered designs exhibited slightly higher methylation differences than probes with identical sequences in EPICv2 and EPICv1 (Fig 2E). When integrating EPICv1 and EPICv2 data for analysis, caution should be taken interpreting subtle methylation differences from these probes.
Unlike EPICv1 and HM450, EPICv2 adopts the recent mouse methylation BeadChip’s probe naming convention to accommodate more flexible probe designs and replicates. EPICv2 probe IDs consist of a prefix and a suffix [9]. The prefix uniquely identifies the 122-mer template DNA, reminiscent of probe ID names in EPICv1 and HM450 probes. The suffixes indicate the Watson or Crick strand to which the probe will hybridize, the strand where cytosine deamination will occur, the Infinium chemistry type (1 or 2), and an enumerating replicate index for multiple versions of the same design. Replicate probes share the same probe name prefixes but have different suffixes, targeting the same 122-mer in various ways (different strands or Infinium chemistries) (Fig S2D). Of the 5,483 replicate probes in EPICv2, 5,222 have the same Infinium chemistry. A small fraction has different Infinium chemistry, strand preference, or both (Fig S2E), resulting in 5,621 loci with multiple probe coverage (Fig 2F). Most correlations among replicate probes are close to 1, significantly higher than non-replicate probes (Fig 2G), emphasizing the probe design’s robustness and validating alternative designs. Interestingly, signal intensities do not decrease in probes with a higher number of replicates (Fig S2D), suggesting that replicate probes do not interfere with each other’s hybridization under standard processing conditions.
EPICv2 reveals DNA methylation dynamics in models of epigenetic modifiers.
We evaluated the accuracy of EPICv2 by comparing its measured DNA methylation levels with those obtained from whole genome bisulfite sequencing (WGBS) on GM12878, LNCaP, and K562 cells. We found that the EPICv2-WGBS correlation on the same cell lines is much higher than between different cell lines (Fig 3A). The Spearman’s correlation between EPICv2 beta value and WGBS methylation fractions on the same cell lines are 0.854, 0.874, and 0.866, respectively (Fig 3B). Differences in cell culture conditions may contribute to slightly lower WGBS correlation, as a similar correlation (~0.89) is seen when running EPICv2 on DNA from different HCT116 cells at two different labs (Fig S3A). Compared to WGBS, EPICv2 data exhibits a shift towards intermediate values due to the effect of residual signal background (Fig 3B, Fig S3B).
Figure 3:
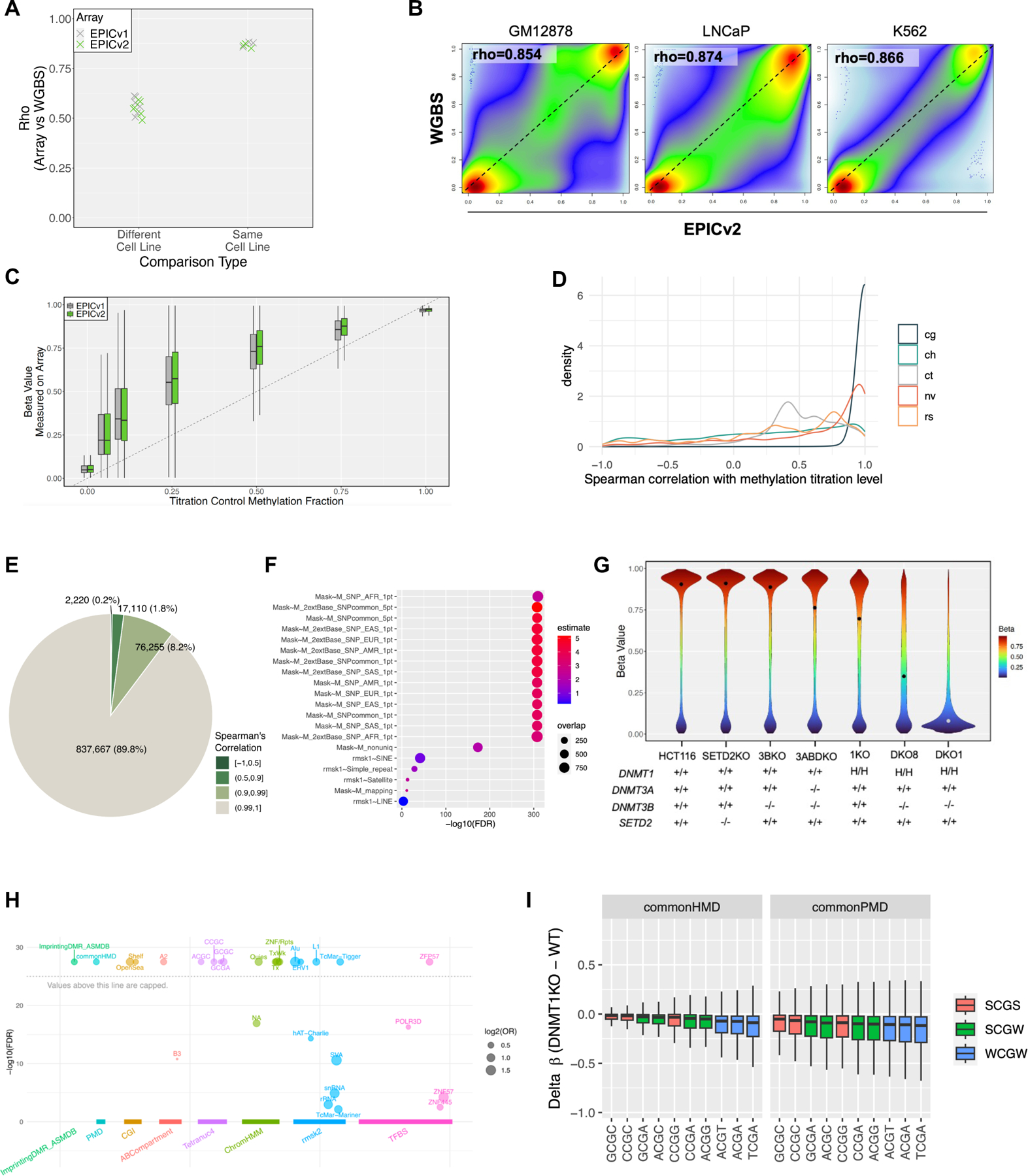
EPICv2 reveals DNA methylation variation in wild type cell lines and cell line models of epigenetic modifiers. (A) EPICv2-WGBS correlation between the same and different cell lines. (B) Comparison of EPICv2-measured DNA methylation level with whole genome bisulfite sequencing (WGBS) on GM12878, LNCaP and K562. (C) Comparison of EPICv2 accuracy on cell line DNA of known titrated methylation fractions. (D) Correlation of EPICv2 probes with known DNA methylation level in titration experiment. (E) Distribution of probes by correlation with titrated fraction. (F) Functional analysis of poorly correlated probes. (G) Drop of global DNA methylation levels in HCT116-derived cell lines with mutated or deleted DNMTs and/or SETD2. (H) Enrichment of CpGs that retain DNA methylation in DKO1 cells. (I) Sequence context of loss of DNA methylation in DNMT1KO cells, stratified by common partially methylated domains (PMDs) vs. common highly methylated domains (HMDs), and CpGs flanked by A/T (W) or G/C (S).
We also evaluated the accuracy of EPICv2 on cell line DNA with known titrated methylation fractions (Fig 3C). The order of the genome-wide median DNA methylation levels was consistent with the titrated methylation fractions. However, samples titrated to intermediate methylation levels were associated with greater variance. The genome-wide median methylations deviate from the titration fractions towards the higher end. In this experiment, EPICv2 produced comparable accuracy to EPICv1 (Fig 3C). The systematic deviation is likely due to signal background influence, as noted in previous generations of Infinium arrays [9]. We utilized the titration data to explore the utility of each probe in measuring DNA methylation. As expected, most cg-probes produced beta values that were highly correlated with titration (Fig 3D). However, non-CG (ch) probes, SNP (rs) probes, SNV (nv) probes, and control (ct) probes were more random in correlation with titration. This is consistent with the fact that our methylation control titrated methylation level of only CpG cytosines but not non-CG cytosines or somatic mutations. Overall, 89.8% of the EPICv2 probes had a Spearman’s correlation >0.99 with the titrated fraction (Fig 3E) and 98% of probes >0.9 (Fig 3E). However, 2,220 (~0.2%) cg probes did not display a strong correlation with titration (<0.5). Probes with high correlation were associated with high (close to 1) β- value effect size, while those with poor correlation were associated with small (often <0.5) effect sizes (Fig S3H).
We conducted functional analysis of the poorly correlated probes and found that they were enriched in sequence polymorphisms, poor probe mapping, and co-localization with repetitive genomes such as simple repeats, satellite, and retrotransposable elements (Fig 3F). Therefore, we recommend masking those probes for analysis (see Availability). These results demonstrate that EPICv2 is an accurate tool for measuring DNA methylation, and that most of its probes are highly correlated with titration fractions. But caution must be taken to mask residual poor mapping, non-unique mapping and influence from sequence polymorphisms.
To assess the ability of EPICv2 to capture biological variations, we generated methylation profiles of HCT116 cell lines with hypomorphic DNMT1 (DNMT1ΔE2−5) [30] or knockout for rest of DNMTs or SETD2 using EPICv2 (Fig 3G). These cell lines carried homozygous mutations to DNMT1, DNMT3A, DNMT3B, and SETD2 (see Fig S3F for details). Our analysis revealed a dramatic drop in global methylation level in the hypomorphic DNMT1-DNMT3B knock out (DKO1 and DKO8) cell lines, followed by DNMT1KO (DNMT1ΔE2−5) and DNMT3A-DNMT3BKO, while SETD2KO and DNMT3BKO showed the least reduction of global DNA methylation levels (Fig 3G). DKO1 shows more reduction in DNA methylation compared to DKO8 cells consistent with prior report [31]. Notably, CpGs that retain DNA methylation in DKO1 cells are enriched for imprinting-associated differentially methylated regions (DMRs), RNA polymerase III binding, and transposable elements such as Alu, ERV1, and LINE-1 (Fig 3H). Similarly, the loss of DNA methylation in DNMT1KO cells primarily affects common partially methylated domains and CpGs flanked by A/T (W) (Fig 3I). These results demonstrate the ability of EPICv2 to capture biologically relevant changes in DNA methylation levels in response to genetic modifications.
EPICv2 generates informative DNA methylome from as low as one nanogram input DNA
DNA methylation is seeing extensive applications in liquid biopsy-based diagnostics. However, clinical samples such as plasma cfDNA are often limited in quantity. To determine the performance of EPICv2 in lower input ranges and facilitate its use in clinical applications, we profiled diluted DNA as well as DNA extracted from a specific number of cells determined by flow sorting (Fig 4A). We found that probe success rates decreased as the amount of input DNA dropped, but it continued to remain higher than 50% for 1ng input DNA (Fig 4A, 4B). The lower success rates observed in K562 and HCT116 cancer cell lines compared to GM12878 may be due to aneuploidy and genomic deletion (Fig 4B). Technical replicates become less reproducible at lower input, with correlation coefficients of 0.93 and 0.91 observed for 5,000 and 500 cells, respectively, compared to >0.98 for higher input (Fig 4C). Nonetheless, data from low input DNA remained highly correlated with 250ng DNA (Fig 4D, 4E, S4A) and EPICv1 data (Fig S4B). However, the correlation between high input and 1ng DNA input samples decreased to 0.92 with a more dichotomized beta value distribution, likely due to the allelic nature of DNA methylation in every cell and the higher chance of allelic dropout in samples of limited cell numbers.
Figure 4:
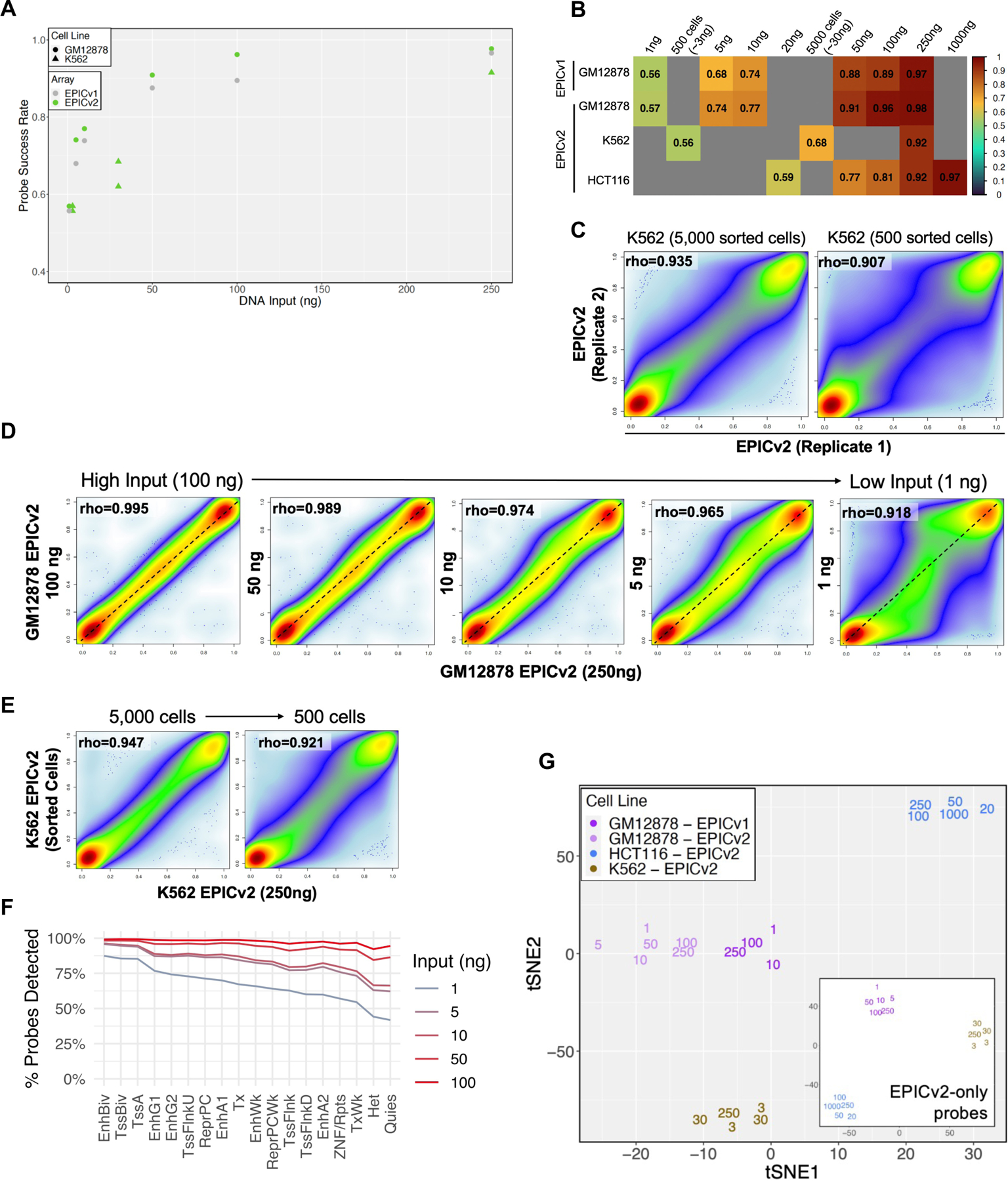
EPICv2 performance at low input ranges. Scatter plot (A) and heatmap (B) illustrating probe success rates for various input amounts and cell lines. (C) Correlation coefficient between replicates comparing 500 sorted cells (left) and 5000 sorted cells (right). (D) Correlation between low input (from 100ng to 1ng) and 250ng DNA input samples. (E) Correlation between low input (5000 and 500 sorted cells) and 250ng DNA input samples. (F) Distribution of probe detection success rate across genomic regions for different input amounts. (G) tSNE analysis of beta values for low and high input samples, using all probes or only probes added in EPICv2 (subpanel). Labeled the number corresponds to the input amount (ng). Input amounts from sorted cells are estimated assuming 6pg DNA per cell.
Investigation of probes that lost detection in lower input samples revealed that quiescent or heterochromatic regions were more likely to lose detection, whereas bivalent transcription start sites and enhancers were most resistant to detection failure (Fig 4F). This disparity is likely due to the high difference in CpG density between quiescent and bivalent regions. Interestingly, low input samples maintained global methylome similarity with higher input samples, as evidenced by tSNE analysis (Fig 4G). This clustering pattern is also seen with just EPICv2-added probes (Fig 4G subpanel).
EPICv2 covers CpGs essential for epigenetic clocks and cell type deconvolution.
We conducted a comprehensive annotation of the probes in EPICv2 and their coverage across the epigenome. The results showed that EPICv2-added probes were more enriched in enhancer elements while being depleted in quiescent regions and heterochromatin. On the other hand, EPICv2-deleted probes were enriched in CpG islands, constitutively active transcription start sites, and bivalent promoters, as well as repetitive elements (Fig 5A, S5A). This suggests that EPICv2 places greater emphasis on the regulatory genome while losing coverage on promoters with less DNA methylation variation (Fig S5A, S5B). These changes in probe set allow for capture of greater variability of DNA methylation change across different physiological and pathological conditions.
Figure 5:
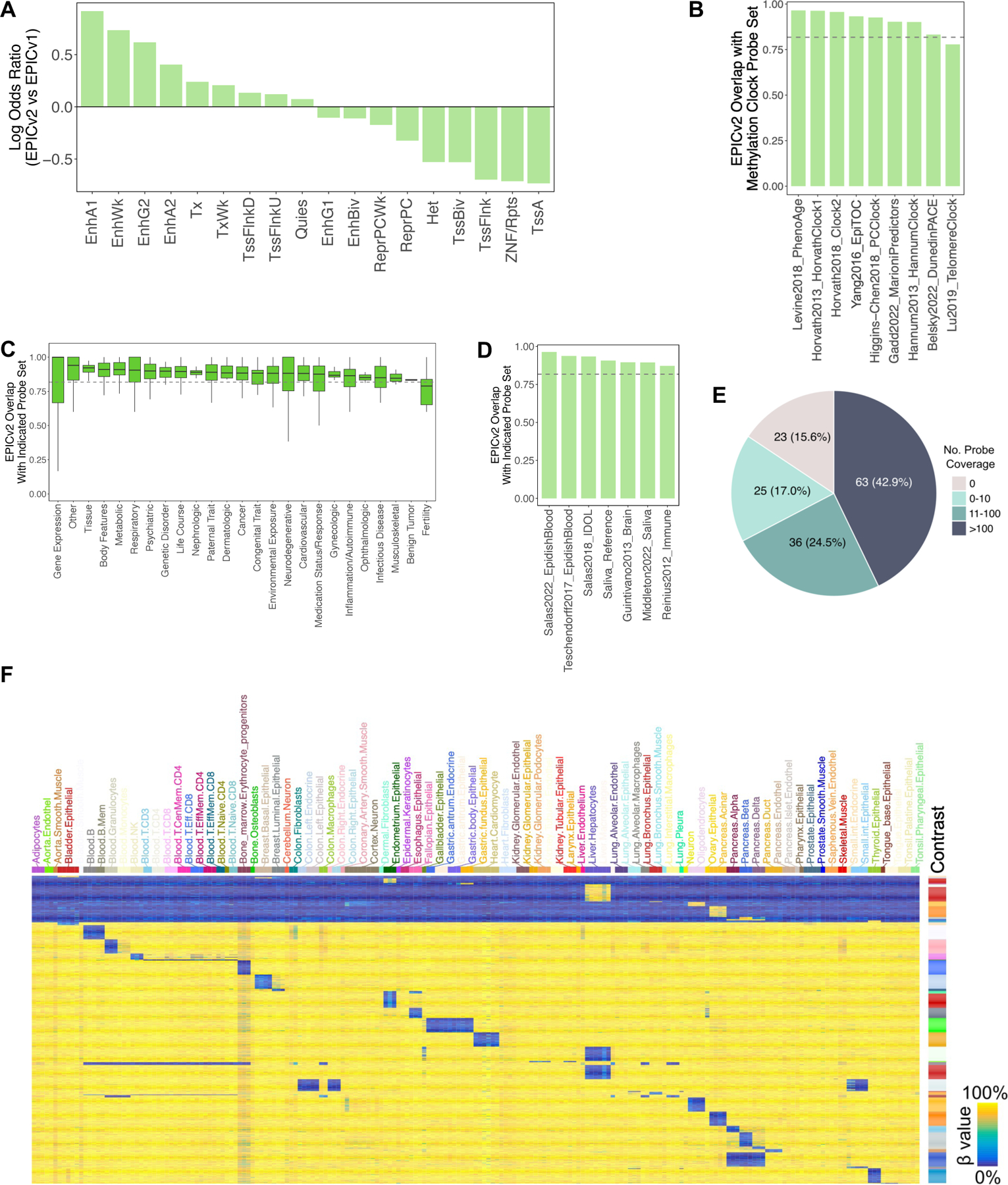
Annotation of EPICv2 probes for epigenetic clocks and cell type deconvolution panels. (A) Odds ratios of EPICv2-vs-EPICv1 across different ChromHMM features (ENCODE v2, Methods). (B) Probe coverage comparison between EPICv2 and previous human methylation BeadChips for various applications including methylation clocks (B), epigenome-wide association studies (EWAS) (C), and cell-type deconvolution panels (D). Dashed line represent expected overlap from random probe selection for deletion. (E) Number of distinct pairwise contrasts covered by EPICv2 probes based on WGBS-derived human cell type panels. (F) Percentage of contrasts covered by varying numbers of probes, showcasing the EPICv2’s ability to capture signature of diverse cell types.
On the chromatin compartment level, EPICv2 covers 2–4% of CpGs in each compartment (as defined in [32]) (Fig S5C). EPICv2 gains coverage on all compartments except B4, which was previously enriched by EPICv1 due to the presence of KRAB-ZNF genes (Fig S5D, S5E). The CpG island coverage ratio remains largely the same between EPICv1 and EPICv2 (Fig S5F). Compared to EPICv1, EPICv2 is more evenly distributed in the genome, being less enriched in CpG islands but also less depleted in CpG open seas (Fig S5G). EPICv2-added probes are enriched in CpG shores but not in CpG island itself compared to the genome average (Fig S5H).
Previous generations of Infinium Methylation BeadChips have been widely used to construct epigenetic clocks, cancer classifiers, and to study the epigenome-wide association of common human diseases/traits. In designing the EPICv2 array, an important goal was to ensure that these biomarkers remain available for future arrays without disruption of practical applications. We evaluated nine human methylation clocks, seven cell-type deconvolution panels, and 26 human trait groups previously studied for DNA methylation association. Our analysis showed that EPICv2 effectively retained most probes from previous epigenetic clocks, with the exception of telomere clocks (Fig 5B). Most epigenetic clocks showed higher-than-random capture rates. Infinium arrays have also been used successfully to discover DNA methylation variations associated with human traits in epigenome-wide association studies (EWAS). We found that most previous EWAS hits are still retained in EPICv2 (Fig 5C). Gene expression-associated CpGs tend to be most preserved, underscoring the regulatory relevance of the retained CpGs. The only depleted group is fertility-related CpGs.
Another powerful application of methylation is in cell type deconvolution. We surveyed seven reference panels and found that EPICv2 covers these panels better than random selection (Fig 5D). Using the latest WGBS-based human cell type panels [33], we calculated the number of distinct contrasts covered by EPICv2 probes. We found that only 15.6% of the contrasts were not covered by EPICv2, and 43% of the contrasts were covered by more than 100 probes, suggesting that EPICv2 can robustly query cell type composition in the corresponding cell types (Fig 5E, 5F).
EPICv2 enables joint epigenome- somatic mutation analysis in cancer.
EPICv2 added 824 probes to detect somatic mutations in human cancers (identified by nv probe ID prefixes) [34] (Fig 6A). Unlike cytosine methylation, rs and nv probes use Infinium chemistry to query sequence variations (Fig 1B). Most nv probes are designed with Infinium-I chemistry. Multiple probes can target mutations on the same site with each probe for a different alternative allele. 163 loci were targeted twice and 92 sites were targeted three times for different alternative alleles. 66 probes are designed with Infinium-II chemistry, and they can target one alternative allele. The nv probes target 59 unique genes, with the TP53 gene being the most targeted gene (113 times) (Fig 6B, S6A). Most of the TP53 mutations are missense mutations and are located in DNA binding domains and tetramerization motif of the protein (Fig 6C). We tested the nv probes on HCT116 cell lines, which is known to contain a KRAS G13D mutation [35]. Distinctive allele frequency readings were observed for KRAS G13D compared to other cell lines (Fig 6D). Some null-calls showed an intermediate reading in other cell lines, likely due to suboptimal hybridization and extension, rather than a heterozygous genotype. This is likely due to internal CpGs in the probe sequence. Probes with more than one or two CpGs within 10bp of the 3’-end are associated with lower total intensities (Fig 6E). In general, NV probes are more susceptible to detection failure compared to cg-, ch-, and rs- probes (Fig 6F). This is also supported by correlation of nv probe reading with known methylation fraction in control samples with titrated methylation levels (Fig S6C). Additionally, EPICv2 can also detect copy number alterations. As proof of concept, we identified the loss of 9p deletion and 22 amplifications in K562 cells, which are linked to its signature BCR-ABL1 fusion, and 2p and 13q21 deletion in LNCaP cells (Fig 6G, 6H).
Figure 6:
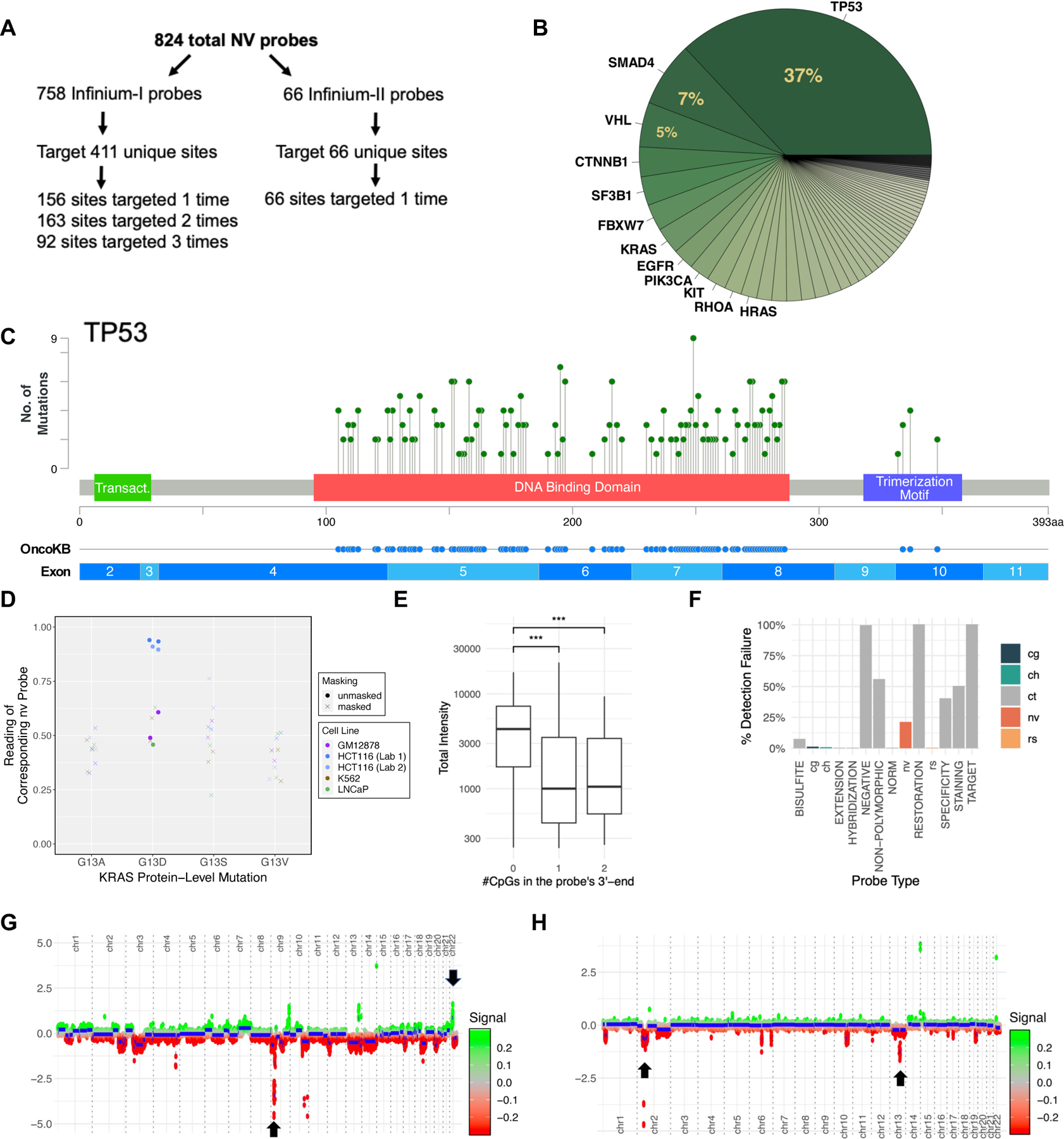
EPICv2 facilitates somatic mutation analysis in cancer. (A) Distribution of nv probes among different Infinium design types. (B) Pie chart displaying genes targeted by nv probes. (C) Location of TP53 mutations targeted by nv probes. (D) EPICv2 reading of probes targeting KRAS G13 mutations in HCT116 cells. The following probes query the displayed mutations: nv-GRCh38-chr12–25245347-25245347-C-A_BC11 (G13V), GRCh38-chr12–25245347-25245347-C-T_BC11 (G13D), GRCh38-chr12–25245348-25245348-C-T_BC11 (G13S), GRCh38-chr12–25245347-25245347-C-G_TC11 (G13A); (E) Effect of the number of CpGs within 10bp of the 3’-end on total intensities of nv probes. (F) Detection failure rate comparison between nv probes and other probe types. (G) Copy number profile of K562 cells, showing chromosome 9 deletion and chromosome 22 amplification. (H) Copy number profile of LNCaP cells, showing chromosome 2 and 13 deletions.
DISCUSSION
The Infinium DNA methylation BeadChip has been highly successful in genome-wide methylation assays for human cohort studies. We comprehensively investigated the latest member of the Infinium array family, EPICv2, which introduces novel features to improve technical performance and jointly interrogate genetic and epigenetic variations in cancer genomics.
Firstly, the updated probe ID system accommodates probe replicates to measure methylation levels of the same CpG dinucleotide, adhering to the new nomenclature from the recent mouse array. This system differentiates replicate probes based on top versus bottom strand, bisulfite-converted versus opposite strand, and Infinium-I versus -II chemistry. An index is employed to distinguish full replicates when no other design differences exist. This enhancement allows for increased design flexibility to bypass neighboring single-nucleotide polymorphism influences and suboptimal probe sequence choices. We confirmed the congruence of these alternative designs in generating DNA methylation readings (Fig 2F). However, residual methylation differences exist between replicate probes of varying designs, warranting further investigation (Fig 2G). Particularly, these design variations could introduce uncertainties affecting the performance of machine learning models, such as those used for cancer classification and age prediction, especially when trained using data from earlier generations of the technology. To cope with this, we introduced informatics solutions to resolve multiple replicate probe measurements, enabling integrative analysis with existing EPIC and HM450 data.
Secondly, EPICv2 introduces a new probe category, the nv probes, targeting recurrent cancer somatic mutations. Probes targeting common human genetic polymorphisms have proven useful for identifying sample swaps and inferring subject ancestry. The nv probes employ a similar design principle to assess the presence of somatic mutations. Although untested in primary human tumor samples, we successfully identified the KRAS G13D mutation in HCT116 cells. However, nv probes tend to exhibit lower signal intensity due to uncertain methylation states of internal CpGs, which can affect hybridization and extension. Our benchmark annotates these probes while suggesting potential improvements. The addition of nv probes in EPICv2 may enable a multi-omics cell count deconvolution and tumor purity analysis based on both DNA methylation and somatic mutations.
Thirdly, EPICv2 implements a significant change to the bulk probe content based on its predecessor, with the removal of 143,967 EPIC probes and addition of 207,898 probes, including 24,463 reintroduced HM450 probes. Despite these modifications, EPICv2 retains 83% (95% of the high-quality) probes of EPICv1 and 81% of HM450 probes, ensuring backward compatibility. Our analysis indicates that most CpGs from existing epigenetic clocks and cell type deconvolution panels are preserved in EPICv2. Many probes deleted in EPICv2 were previously identified as having mapping issues or overlap with common human genetic polymorphisms [23]. Their deletion improves EPICv2’s technical robustness and applicability across diverse human populations. The added probes are significantly enriched in enhancer elements, shifting the array content toward regulatory genome with variable DNA methylation levels. A recent independent validation study [36] that described an EPICv2 application to human primary normal and cancer tissue samples align with our observations.
Finally, we utilized EPICv2 to examine properties common to all Infinium BeadChip technologies in greater detail. Notably, we found that EPICv1 can profile low input samples with adequate sensitivity and accuracy due to the isothermal amplification in Infinium BeadChip protocols. Despite the recommended 250ng input, we obtained valuable data from as little as 1ng DNA and DNA extracted from 500 sorted cells. These findings expand EPICv2’s applicability to scenarios with limited DNA quantity, such as cell-free DNA or saliva samples. Furthermore, we validated EPICv2’s effectiveness in detecting copy number alterations and uncovered DNA methylation dynamics in DNMT mutant cell lines, corroborating epigenome and sequence signatures found in DNMT1KO, DKO1, and DKO8 cells.
While these advancements in the field are indeed remarkable, it is crucial to recognize that EPICv2 does still carry certain inherent constraints that are prevalent across previous iterations of Infinium technologies. For instance, when juxtaposed against titrated methylation levels, the beta value readouts by EPICv2 may deviate from DNA methylation fractions (Fig. 3C) as expected from background signal tempering and residual dye bias. It is also easy to see that EPICv2 cannot accurately capture completely unmethylated and fully methylated methylation levels due to the presence of residual signal background (Fig. S3C).
Furthermore, the success of probe hybridization is contingent upon the robust assumption of the underlying sequence and could be vulnerable to genetic variations, be they somatic mutations in cancer or polymorphic genetic alterations within the human population. Even with meticulous array annotation, certain artifacts owing to undetected sequence variations might still prove challenging to identify. For instance, nearly 20,000 (2%) probes are associated with suboptimal correlation with methylation titration (Fig 3F), some of which lack identifiable causes such as overlap with sequence variations. To mitigate the potential for misinterpretation, our probe annotation leveraging both sequence-based computational predictions and empirical data, as documented in our available resources (see Availability). Overall, our study offers practical guidance, essential annotations, and valuable insights for employing this updated Infinium BeadChip technology.
MATERIAL & METHODS
Cell cultures
GM12878, K562 (CCL-243), and LNCaP (CRL-1740), cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The K562 is cultured in Iscove’s Modified Dulbecco’s Medium (30–2005, ATCC), 10% Fetal Bovine Serum (FBS) (45000–736, Gibco), and 1% penicillin/streptomycin (15140122, Gibco). The LNCaP was cultured in Roswell Park Memorial Institute Medium (RPMI-1640) (30–2001, ATCC), 10% FBS, and 1% penicillin/streptomycin (15140122, Gibco). GM12878 cells were cultured with RPMI-1640 (72400047, Invitrogen), and 15% Fetal Bovine Serum (Gibco, 45000–736), 1% GlutaMAX™ (Gibco, 35050061), and 1% penicillin/streptomycin (15140122, Gibco). All cells were maintained in a 37°C incubator with 5% CO2 and cultured at a 75 cm2 culture flask (Fisher, BD353136). HCT116 cells from Van Andel Institute (Lab 2) was cultured as previously described [37].
Cell flow sorting and low-input DNA testing
5 × 106 cell pellets were resuspended in 50 μL of 0.1 μg/1 mL of 4,6-Diamidino-2-phenylindole (DAPI) (D9542–5MG, Sigma-Aldrich) in 1 mL of Phosphate-buffered saline (PBS) (10010023, Life Technology,). Cells were filtered by a Falcon Cell Strainer Snap Cap (352235, Falcon). DAPI-negative cells (500 and 5,000) from K562 were sorted and collected into 96-well plates pre-loaded with 10 μL of 1X M-Digestion Buffer (D5020–9, Zymo Research) using a BD FACSAriaTM Fusion cell sorter (BD Biosciences) using a 100 μm nozzle. The other low-input cell line DNA samples were obtained by diluting extracted cell line DNA after Qubit quantification.
DNA extraction and DNA bisulfite conversion
Cells were harvested by centrifugation at 100 G for 5 min at room temperature and washed twice using PBS (10010023, Gibco). Cells were incubated with 500 μL of lysis buffer (10 mM Tris pH 8.0, 300 mM NaCl, 5 mM EDTA pH 8.0, 0.5% SDS, and distilled water) and 10 μL of Proteinase K (P8107S, NEB) for 2 hours at 55°C, and genomic DNA was purified using phenol:chloroform:isoamyl alcohol mixture (P3803–100ML, Sigma-Aldrich) and isopropanol precipitation with GlycoBlue (AM9515, Invitrogen). DNA was resuspended in 200 μl of 1M Tris buffer pH 8.0. For array analysis with 500 and 5000 cells, the DNA was resuspended in 46 μl of the 1M Tris buffer. 1 μl of the extracted DNA was quantified using Qubit 4.0 Fluorometer (Invitrogen) using the dsDNA HS Assay Kit (Q33231, Invitrogen). HCT116 DNA from Van Andel Institute was extracted as previously described [38]. Bisulfite conversion was performed using the EZ DNA methylation kit (D5001, Zymo Research) according to the manufactural protocol with the specified modifications for Illumina Infinium Methylation Assay. We maximized array input as 10 μL following Lee et al. (in submission).
SETD2KO and DNMTs KO cell lines DNA
HCT116 derivative cell lines 1KO, 3BKO, 3ABDKO, DKO1, and DKO8 were obtained from Dr. Stephen Baylin’s laboratory. SETD2KO cell line was generated from HCT116 using CRISPR-Cas9 Lentivirus. All these cell lines were cultured in McCoy’s 5A medium supplemented with 10% FBS and 1% penicillin/streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. Genomic DNA was extracted as described above.
EPICv2 Infinium BeadChip data preprocessing
Preprocessing, quality control, and analysis of the Infinium MethylationEPIC v2 array IDATs files were processed using the SeSAMe package [22]. The standard openSesame workflow is employed to process raw signal data to beta values. Briefly, the openSesame workflow first calculated probe detection P value using the pOOBAH algorithm, which leverages the fluorescence of out-of-band (OOB) probes. It then performed normalization using noob, which uses OOB probes to perform a normal exponential deconvolution of fluorescent intensities, followed by a dye bias correction using the dyeBiasNL function. Signal intensities were then summarized into beta values using the getBetas function. Probes are optionally collapsed to cg-numbers using getBetas function with the collapseToPfx=TRUE option.
Public datasets
BS-seq datasets for GM12878, LNCaP and K562 cells were downloaded from GEO using the following accession: GSM5649439, GSM2308596, and GSE86832. Only CpGs with sequencing depths greater than or equal to 10 are considered in analysis. The EPICv2 A1 manifest were downloaded from manufacturer’s website (https://support.illumina.com/content/dam/illumina-support/documents/downloads/productfiles/methylationepic/MethylationEPIC%20v2%20Files.zip). CpGs associated with human traits from 1067 EWAS studies were downloaded from the EWAS catalog [39] and EWAS atlas databases [40]. Each study and its associated significant probes were grouped into one of 26 major categories according to the trait examined. Each major category was then intersected with the EPICv2 manifest to assess the proportion of probe retention. Epigenetic clock and cell type deconvolution panels were manually curated from prior studies (Supplemental Table S1).
Probe masking and manifest annotation
Human SNP and ancestry information were downloaded from dbSNP (version 20180418) [41]. Gene models for probe annotation (both version 41 and version 36 for backward compatibility) was downloaded from GENCODE [42]. Probe sequences were mapped to GRCh38 human genome assembly using BISCUIT. Consensus ChromHMM segmentation was derived from 833 ENCODE ChromHMM calls from ENCODE version 2 [43]. Cancer somatic mutations were annotated using cBioPortal mutation mapper [44]. SNP influence on probe functions was predicted using InfiniumManifestAnnotator (https://github.com/zhou-lab/InfiniumManifestAnnotator) [45].
Supplementary Material
ACKNOWLEDGEMENT
We thank Peter S Choi for providing cell culture assistance.
FUNDING
The NIH/NIGMS (grant R35GM146978 awarded to W.Z.). It was also supported by W.Z.’s startup fund at Children’s Hospital of Philadelphia and research sponsorship by FOXO Bioscience.
Footnotes
Competing interests
WZ received BeadChips from Illumina Inc. to conduct this analysis. NR is an employee of Illumina Inc.
Availability of data and materials
Informatics support for EPICv2 is implemented in SeSAMe (version 1.17.9+) available through Bioconductor (https://bioconductor.org/packages/release/bioc/html/sesame.html). EPICv2 probe annotations, including masking, titration correlation and functional link information, are available at the following annotation website (http://zwdzwd.github.io/InfiniumAnnotation). All EPICv2 and EPIC data produced in this study is available through GEO under the accession GSE228820.
BIBLIOGRAPHY
- 1.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607. [DOI] [PubMed] [Google Scholar]
- 4.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou W, Dinh HQ, Ramjan Z, Weisenberger DJ, Nicolet CM, Shen H, et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet. 2018;50:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392:777–86. [DOI] [PubMed] [Google Scholar]
- 8.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. [DOI] [PubMed] [Google Scholar]
- 9.Zhou W, Hinoue T, Barnes B, Mitchell O, Iqbal W, Lee SM, et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genomics. 2022;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arneson A, Haghani A, Thompson MJ, Pellegrini M, Kwon SB, Vu H, et al. A mammalian methylation array for profiling methylation levels at conserved sequences. Nat Commun. 2022;13:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. [DOI] [PubMed] [Google Scholar]
- 12.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou L, Ng HK, Drautz-Moses DI, Schuster SC, Beck S, Kim C, et al. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci Rep. 2019;9:10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teschendorff AE, Relton CL. Statistical and integrative system-level analysis of DNA methylation data. Nat Rev Genet. 2018;19:129–47. [DOI] [PubMed] [Google Scholar]
- 15.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. [DOI] [PubMed] [Google Scholar]
- 16.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Genet. 1997;13:444–9. [DOI] [PubMed] [Google Scholar]
- 18.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–74. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989;3:612–9. [DOI] [PubMed] [Google Scholar]
- 21.Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W, Triche TJ, Laird PW, Shen H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;46:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017;45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136:181–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hop PJ, Zwamborn RAJ, Hannon EJ, Dekker AM, van Eijk KR, Walker EM, et al. Cross-reactive probes on Illumina DNA methylation arrays: a large study on ALS shows that a cautionary approach is warranted in interpreting epigenome-wide association studies. NAR Genom Bioinform. 2020;2:lqaa105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson KC, Houseman EA, King JE, von Herrmann KM, Fadul CE, Christensen BC. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat Commun. 2016;7:13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon O, Macisaac JL, Tindula G, Kobor MS, Eskenazi B, Holland N. 5-Hydroxymethylcytosine in cord blood and associations of DNA methylation with sex in newborns. Mutagenesis. 2019;34:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Lee MK, Perreard L, Kelsey KT, Christensen BC, Salas LA. Navigating the hydroxymethylome: experimental biases and quality control tools for the tandem bisulfite and oxidative bisulfite Illumina microarrays. Epigenomics. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egger G, Jeong S, Escobar SG, Cortez CC, Li TWH, Saito Y, et al. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci USA. 2006;103:14080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Carvalho DD, Sharma S, You JS, Su S-F, Taberlay PC, Kelly TK, et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012;21:655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Nanni L, Sungalee S, Zufferey M, Tavernari D, Mina M, et al. Systematic inference and comparison of multi-scale chromatin sub-compartments connects spatial organization to cell phenotypes. Nat Commun. 2021;12:2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature. 2023;613:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alves S, Castro L, Fernandes MS, Francisco R, Castro P, Priault M, et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget. 2015;6:30787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noguera-Castells A, García-Prieto CA, Álvarez-Errico D, Esteller M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics. 2023;18:2185742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, et al. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS ONE. 2009;4:e8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battram T, Yousefi P, Crawford G, Prince C, Sheikhali Babaei M, Sharp G, et al. The EWAS Catalog: a database of epigenome-wide association studies. Wellcome Open Res. 2022;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, Zou D, Li Z, Gao R, Sang J, Zhang Y, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019;47:D983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding W, Kaur D, Horvath S, Zhou W. Comparative epigenome analysis using Infinium DNA methylation BeadChips. Brief Bioinformatics. 2023;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Informatics support for EPICv2 is implemented in SeSAMe (version 1.17.9+) available through Bioconductor (https://bioconductor.org/packages/release/bioc/html/sesame.html). EPICv2 probe annotations, including masking, titration correlation and functional link information, are available at the following annotation website (http://zwdzwd.github.io/InfiniumAnnotation). All EPICv2 and EPIC data produced in this study is available through GEO under the accession GSE228820.