

Abstract
There are approximately 250 million people chronically infected with hepatitis B virus (HBV) worldwide. Although HBV is often integrated into the host genome and promotes hepatocarcinogenesis, vulnerability of HBV integration in liver cancer cells has not been clarified. The aim of our study is to identify vulnerability factors for HBV‐associated hepatocarcinoma. Loss‐of‐function screening was undertaken in HepG2 and HBV‐integrated HepG2.2.15 cells expressing SpCas9 using a pooled genome‐wide clustered regularly interspaced short palindromic repeats (CRISPR) library. Genes whose guide RNA (gRNA) abundance significantly decreased in HepG2.2.15 cells but not in HepG2 cells were extracted using the MAGeCK algorithm. We identified four genes (BCL2L1, VPS37A, INSIG2, and CFLAR) that showed significant reductions of gRNA abundance and thus potentially involved in the vulnerability of HBV‐integrated cancer cells. Among them, siRNA‐mediated mRNA inhibition or CRISPR‐mediated genetic deletion of INSIG2 significantly impaired cell proliferation in HepG2.2.15 cells but not in HepG2 cells. Its inhibitory effect was alleviated by cotransfection of siRNAs targeting HBV. INSIG2 inhibition suppressed the pathways related to cell cycle and DNA replication, downregulated cyclin‐dependent kinase 2 (CDK2) levels, and delayed the G1‐to‐S transition in HepG2.2.15 cells. CDK2 inhibitor suppressed cell cycle progression in HepG2.2.15 cells and INSIG2 inhibition did not suppress cell proliferation in the presence of CDK2 inhibitor. In conclusion, INSIG2 inhibition induced cell cycle arrest in HBV‐integrated hepatoma cells in a CDK2‐dependent manner, and thus INSIG2 might be a vulnerability factor for HBV‐associated liver cancer.
Keywords: CDK2, CRISPR/Cas, HBV, liver cancer, pooled library screen
CRISPR loss‐of‐function screen identified INSIG2 as a candidate for the vulnerability in hepatitis B virus‐integrated liver cancer cells.

Abbreviations
- cccDNA
covalently closed circular DNA
- CDK2
cyclin‐dependent kinase 2
- CRISPR
clustered regularly interspaced short palindromic repeat
- gRNA
guide RNA
- HBc
HBV core protein
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV/HCV
hepatitis B virus/hepatitis C virus
- HBx
HBV X protein
- HCC
hepatocellular carcinoma
- KD/KO
knockdown/knock out
- NC
negative control
- pgRNA
pregenomic RNA
- WB
western blot
1. INTRODUCTION
It is estimated that 250 million people are infected with HBV, of which more than 30 million have developed chronic hepatitis. 1 , 2 Once HBV infects the human body, it remains in hepatocytes in the form of cccDNA, and the mechanisms of cccDNA formation and degradation are currently largely unknown, making complete elimination from the body difficult with current treatment. In addition, it is often clinically experienced that HCC develops from the noncirrhotic liver in HBV‐infected patients, while HCC mostly occurs from the cirrhotic liver in HCV‐infected patients. This is thought to be due to the fact that HBV promotes hepatocarcinogenesis not only through chronic liver inflammation and subsequent liver fibrosis but also through its genome integration into the host. 3 Indeed, extensive studies revealed a variety of oncogenic roles of HBV integration into the host genome such as TERT activation and chromosomal rearrangements in HCC patients. 4 Meanwhile, although several molecular‐targeted therapies, including immunotherapies, have become available for advanced HCC patients, 5 no specific treatment for HBV‐associated liver cancer has been investigated to date.
The CRISPR/Cas9 system was reported in 2012, 6 facilitating various genetic modifications. The CRISPR/Cas9 system, which consists of the Cas9 enzyme and a gRNA, is able to knock out any gene of interest in eukaryotic cells by precisely cutting DNA, based on a gRNA‐defined target sequence, and inducing double‐strand breaks. We have recently reported the utility of this technology as a potential novel therapy against HBV by directly cleaving and degrading cccDNA. 7 In addition, the CRISPR/Cas system has been recently used as a powerful tool of large‐scale loss‐of‐function genetic screening, named as pooled CRISPR library screen. 8 By designing gRNAs for entire human genes and introducing a pooled gRNA library into any Cas9‐expressing human cells with selective phenotypic pressure, it is possible to identify genes involved in any biological process of interest such as cell proliferation, survival, and drug resistance in a high‐throughput manner. We have previously undertaken multiple pooled CRISPR library screens and discovered genes involved in various biological phenotypes including the vulnerability of acute myeloid leukemia cells, intraperitoneal metastasis of ovarian cancer, and regorafenib‐resistance of HCC cells. 9 , 10 , 11 In particular, the discovery of cancer‐specific vulnerabilities is useful for the development of less‐toxic therapeutics targeting the Achilles' heel of cancer cells.
In this study, to identify the vulnerability of HBV‐integrated liver cancer, we undertook a whole‐genome pooled CRISPR library screen in liver cancer cell lines with or without HBV integration. We have identified and validated INSIG2 as the vulnerability of HBV‐integrated liver cancer cells. Our study could shed light on new therapies targeting the vulnerability of HBV‐associated liver cancer.
2. MATERIALS AND METHODS
2.1. Cell culture
HepG2 cells were purchased from the Japanese Cancer Resources Bank and grown in DMEM supplemented with 10% FBS (26140; Gibco). HepG2.2.15 cells (provided by Professor Matsuura at Osaka University) are a derivative of HepG2 cells and carry HBV DNA integrated into the host chromosome. This cell line produces a variety of HBV (genotype D)‐specific mRNAs and secrete HBsAg, HBeAg, and Dane particles. 12 HepG2.2.15 cells were grown in DMEM with 10% FBS and 250 μg/mL G418 (Nacalai Tesque Inc.). These cell lines were confirmed to be free from mycoplasma contamination.
2.2. Lentivirus production
The whole genome CRISPR/Cas library contained 113,526 gRNAs targeting 18,731 genes (approximately 7 gRNAs per gene) and was propagated by electroporation. Cas9‐blast vector was purchased from Addgene (#52962). To produce lentivirus, the library or Cas9‐blast vector, psPAX2 (Addgene) and pVSVg (Addgene) in OptiMEM (Thermo Fisher Scientific) were mixed with Lipofectamine 2000 (Thermo Fisher Scientific). The mixture was added to HEK293 cells. Three days later, the supernatant was collected and stored at −80°C.
2.3. Generation of Cas9‐positive HepG2 and HepG2.2.15 cells
HepG2 cells and HepG2.2.15 cells were plated 1 day before the lentivirus infection. Next day, we changed the medium to serum‐free medium containing 8 μg/mL polybrene (Merck Millipore) with lentivirus containing LentiCas9‐blast. From the next day, HepG2 and HepG2.2.15 cells were cultured in medium containing 5 and 3 μg/mL blasticidin (Fujifilm Wako Pure Chemical Corporation), respectively. Subsequently, multiple single clones were obtained by limiting the dilution method, and the expression of Cas9 in these cells was determined by WB analysis.
2.4. In vitro whole‐genome pooled CRISPR library screen
To insert one gRNA into one cell, we determined the MOI for 20% infection efficiency in HepG2 and HepG2.2.15 cells, which were 0.2 and 0.3, respectively. A total of 250 million Cas9‐expressing HepG2 or HepG2.2.15 cells were transduced with CRISPR library at 0.2 or 0.3 MOI, respectively. DNA was collected soon after the completion of puromycin selection (D2) and after 2 weeks culture (D14). We maintained the cell number more than 500‐fold of library size over the experimental course to maintain the library complexity in the transduced cells. Genomic DNA was extracted using the Blood & Cell Culture DNA Maxi Kit (Qiagen) according to the manufacturer's protocol with RNase treatment. Guide RNA sequences integrated into the gDNA were amplified by Illumina‐adapted forward and reverse indexed PCR primers following the protocol from the Broad Institute 13 and purified using a PCR purification kit (Qiagen). The purified amplicons were multiplexed and sequenced in Illumina HiSeq. The frequency of each gRNA in HepG2 and HepG2.2.15 at D2 and D14 was calculated. The MAGeCK algorithm was used to identify genes (false discovery rate < 0.05) whose abundance changed significantly between two groups. 14
2.5. siRNA transfection
We designed siRNAs targeting the HBV genome but not the human genome based on the previous report (Table S1). 15 , 16 All other siRNA oligonucleotides were purchased from Thermo Fisher Scientific (Table S2). According to the manufacturer's protocol, the siRNAs were transfected using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific). Three to four days after transfection, knockdown efficiency was assessed by quantitative RT‐PCR.
2.6. RNA isolation and real‐time PCR
The RNA was extracted from cells using the RNeasy Mini Kit (Qiagen) according to the manufacturer's protocol. For the analysis of pgRNA, DNase (RNase‐Free DNase Set; Qiagen) was used to remove genomic DNA from the isolated RNA. The isolated RNA was reverse transcribed using ReverTra Ace qPCR RT Master Mix (Toyobo). Quantitative PCR was carried out using the QuantStudio 6 Flex Standard RT‐PCR system with TaqMan Gene Expression Assay probes (Thermo Fisher Scientific). The probe list is shown in Table S3, and the following primer set was used for detection of pgRNA: forward, 5′‐TGTCCTACTGTTCAAGCCTCCAA‐3′ and reverse, 5′‐GAGAGTAACTCCACAGTAGCTCCAA‐3′.
2.7. Cell proliferation assay
Cell proliferation was assessed using WST‐1 reagent (Takara Bio). WST‐1 reagent was added to each well and incubated for 1 h at 37°C. The absorbance was measured at 450 and 650 nm (reference) using a microplate reader SH‐9000Lab (Corona).
2.8. Generation of CRISPR‐mediated KO cell lines
The gRNAs targeting BCL2L1 and INSIG2 (Table S4) were cloned into a lentiviral gRNA expression vector (lentiGuide‐Puro; #52963, Addgene). Lentivirus containing lentiGuide‐Puro expressing NC/BCL2L1 gRNA or INSIG2 gRNA was generated and transduced into HepG2‐Cas9 cells or HepG2.2.15‐Cas9 cells.
2.9. Western blot analysis
The western blot procedure has been described previously. 17 We used lysis buffer with a phosphatase inhibitor (Nacalai Tesque, Inc.) and a protease inhibitor (Nacalai Tesque, Inc.) to isolate protein. We used anti‐HBc Ab clone 7B2 18 and the following Abs to immunodetect each band (Table S5). Each band level was measured by using ImageJ software (version 1.52) and normalized to β‐actin band levels.
2.10. Cell cycle assay
Cells were collected with 0.05% trypsin, and cell cycle was evaluated according to the manufacturer's protocol using the Cycletest Plus DNA Reagent Kit (Becton Dickinson). The number of cells in each phase was assessed by using a FACSCanto flow cytometer.
2.11. Plasmid DNA transfection
FLAG‐HBx plasmid DNA was provided by Professor Motoyuki Otsuka (Okayama University). Myc‐INSIG2 plasmid DNA was designed by Vector Builder. According to the manufacturer's protocol, the plasmids were transfected using Lipofectamine 2000 reagent (Thermo Fisher Scientific). Protein expression was confirmed by WB analysis.
2.12. Immunoprecipitation
Primary Ab was mixed with cell lysate and slowly rotated at 4°C for 90 min. Protein G Sepharose (Abcam) was added and rotated at 4°C for an additional 60 min, then the beads were washed five times with lysis buffer. Immunoprecipitates were extracted by adding SDS sample buffer and heating at 95°C for 5 min.
2.13. Statistical analysis
All data are presented as the means ± SEM. Comparisons between two groups were carried out by unpaired Student's t‐test or one‐way ANOVA in GraphPad Prism 6 software. A value of p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Pooled CRISPR library screen identified four gene candidates related to vulnerability of HBV‐integrated liver cancer cells
To search for the vulnerability of HBV‐integrated liver cancer cells, we decided to perform CRISPR dropout screens using the HepG2 cells and their HBV‐integrated derivative cells (HepG2.2.15). We lentivirally transduced a Cas9 expression vector into these cells and generated stable single clones that express Cas9 (HepG2‐Cas9 cells and HepG2.2.15‐Cas9 cells) (Figure 1A). They were transduced with the pooled lentiviral CRISPR library containing 113,526 gRNAs targeting 18,731 human genes. In transduction, the concentration of lentivirus was adjusted to introduce one gRNA per cell in each of the HepG2‐Cas9 and HepG2.2.15‐Cas9 cells. Transduced cells were collected soon after 2‐day puromycin selection (D2) and after subsequent 2‐week culture (D14). Isolated DNAs from these cells were analyzed by NGS to determine gRNA abundance. There were 533 and 131 genes whose gRNA levels significantly decreased in HepG2‐Cas9 cells and HepG2.2.15‐Cas9 cells, respectively, at D14 compared to at D2 (Figure 1B). Among 93 dropout genes common in both cell lines, 15 genes were essential for cell survival, which were statistically significant enriched (p < 0.05 by χ2‐test), suggesting the reliability of the dropout screen. Based on three criteria: (1) gRNA abundance in HepG2.15 cells at D14 was more than two times lower than that at D2, (2) gRNA abundance in HepG2 cells at D14 was less than a 2‐fold difference compared to that at D2, and (3) gRNA abundance in HepG2.15 cells at D14 was significantly lower than that in HepG2 cells at D14 (p < 0.05), we selected four genes (BCL2L1, INSIG2, CFLAR, and VPS37A) as candidates for the vulnerability of HBV‐integrated cells among 38 genes only dropout in HepG2.2.15‐Cas9 cells (Figure 1C). The abundance of most of the gRNAs targeting these four genes was reduced in HepG2.2.15 cells compared to HepG2 cells (Figure 1D).
FIGURE 1.
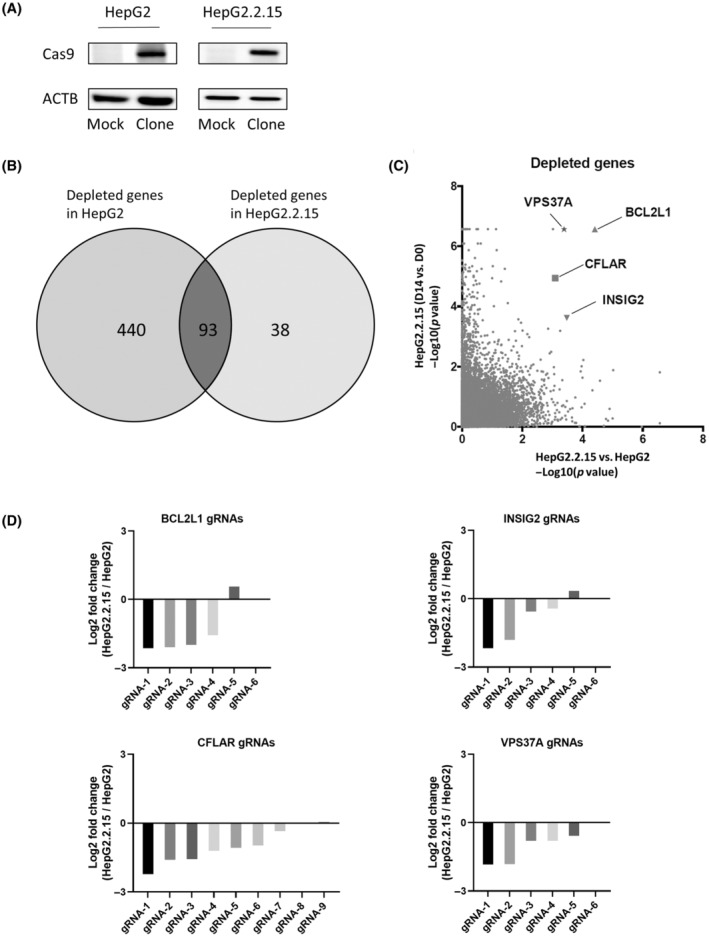
Pooled clustered regularly interspaced short palindromic repeat (CRISPR) library screen Identifies four gene candidates related to the vulnerability of hepatitis B virus (HBV)‐integrated liver cancer cells. (A) Western blot (WB) analysis of Cas9 protein levels in the HepG2 and HepG2.2.15 cell lines. Clone, Cas9‐transduced monoclonal cells; Mock, Cas9‐nontransduced cells. (B) MAGeCK analysis showed significantly depleted (false discover rate <0.05) genes compared to the beginning of the culture both in HepG2 cells and HepG2.2.15 cells. (C) Negative log10 p value for each gene. Horizontal axis shows depletion in HepG2.2.15 cells compared to HepG2 cells at day 14. Vertical axis shows depletion in HepG2.2.15 cells at day 14 compared to day 2. (D) Log2 fold change values of each guide RNA (gRNA) targeting gene (BCL2L1, INSIG2, VPS37A, and CFLAR) in HepG2.2.15 cells compared to those in HepG2 cells. Note that the fold change of gRNA6 in BCL2L1, INSIG2, and VPS37A were not calculated. Abbreviations: ACTB, β‐actin.
3.2. Deletion of BCL2L1 and INSIG2 specifically reduces cell proliferation in HBV‐integrated liver cancer cells
To validate the results of our CRISPR screen, we first examined the effect of KD of four candidate genes by siRNA on cell proliferation in HepG2 and HepG2.2.15 cells. For each gene, two siRNAs were used, and KD efficiency was confirmed (Figure S1). Among the four genes, KD of BCL2L1 and INSIG2 did not affect the cell proliferation in HepG2 cells but significantly suppressed the cell proliferation in HepG2.2.15 cells (Figure 2A). We also deleted either BCL2L1 or INSIG2 by CRISPR and established three KO lines of each gene in HepG2 and HepG2.2.15 cells (Figure 2B,C). Following KO of BCL2L1 and INSIG2, the cell proliferation was not affected in HepG2 cells but was significantly suppressed in HepG2.2.15 cells (Figure 2D,E). These data suggested that BCL2L1 and INSIG2 might be involved in the vulnerability of HBV‐integrated cells.
FIGURE 2.
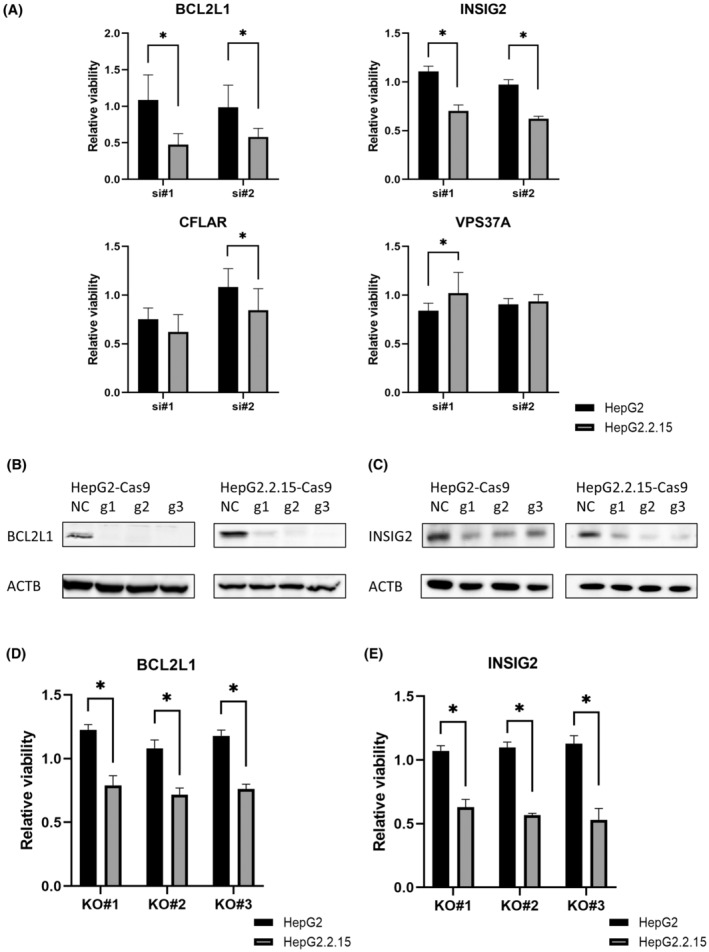
Deletion of BCL2L1 and INSIG2 specifically reduces cell proliferation in hepatitis B virus‐integrated liver cancer cells. (A) Cell viability was measured by WST‐1 assays 4 days after knockdown with siRNA (ratio to negative control [NC], *p < 0.05). (B) Western blot (WB) analysis of BCL2L1 in HepG2‐Cas9 cells and HepG2.2.15 cells after transfection with gBCL2L1. (C) WB analysis of INSIG2 in HepG2‐Cas9 cells and HepG2.2.15 cells after transfection with gINSIG2. (D) Cell viability in HepG2/HepG2.2.15‐gBCL2L1‐Knock Out (KO) cells at 6 days after seeding (ratio to NC, *p < 0.05). (E) Cell viability in HepG2/HepG2.2.15‐gINSIG2‐KO cells at 6 days after seeding (ratio to NC, *p < 0.05). Abbreviations: ACTB, β‐actin; g1/g2/g3, gRNA‐1/gRNA‐2/gRNA‐3.
3.3. Vulnerability induced by INSIG2 inhibition in HBV‐integrated liver cancer cells partly dependent on presence of HBV
Next, we evaluated whether the vulnerability mediated by BCL2L1 and INSIG2 was dependent on the presence of HBV in HepG2.2.15 cells. To this end, we designed siRNA that inhibit all HBV transcripts including 3.5 kb (HBc, pgRNA), 2.4 kb (Large S), 2.1 kb (Small S), and 0.9 kb (HBx). We first confirmed that HBV siRNA significantly suppressed HBsAg, HBeAg, HBc proteins, and intracellular pgRNA levels in HepG2.2.15 cells (Figure 3A). While the reduction of cell proliferation by BCL2L1 siRNA was not affected by the presence of HBV siRNA in HepG2.2.15 cells (Figure 3B), HBV siRNA partially rescued the reduction of cell proliferation by INSIG2 siRNA (Figure 3C), suggesting that the vulnerability induced by INSIG2 inhibition in HepG2.2.15 cells was, at least in part, dependent on the presence of HBV.
FIGURE 3.
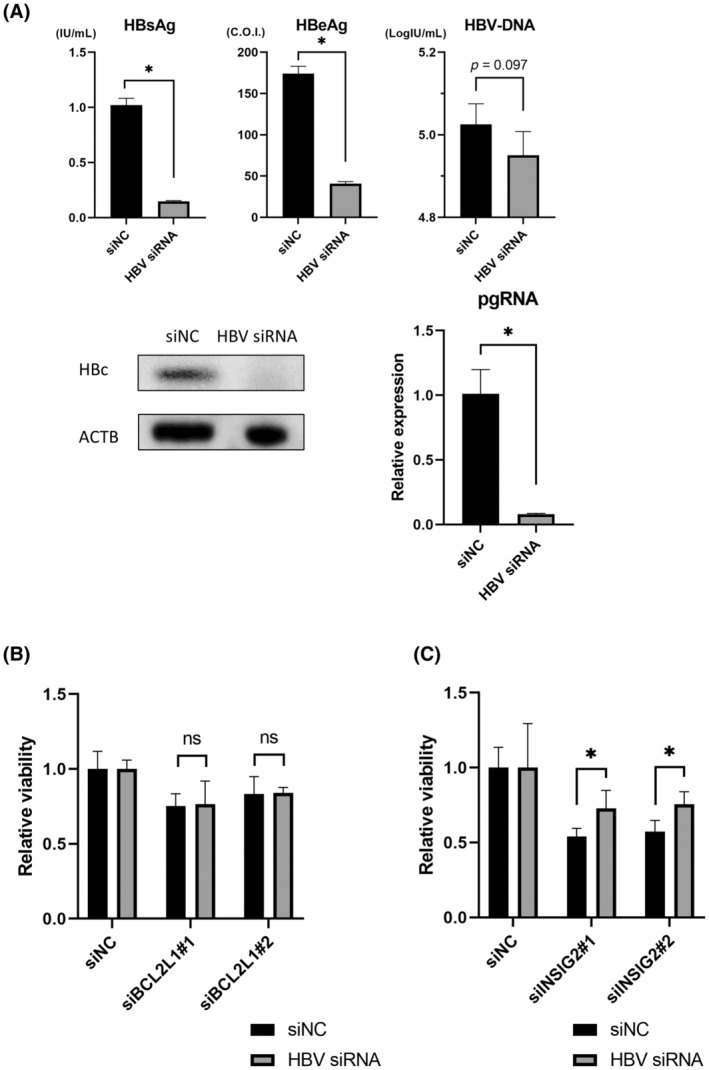
Vulnerability induced by INSIG2 inhibition in hepatitis B virus (HBV)‐integrated liver cancer cells is partly dependent on the presence of HBV. (A) Supernatant hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) levels, HBV core (HBc) protein levels, and intracellular pregenomic RNA (pgRNA) levels in HepG2.2.15 cells 6 days after transfection with HBV siRNA. (*p < 0.05). (B) Cell viability was measured by WST‐1 assays 4 days after transfection with siBCL2L1 and/or HBV siRNA (ratio to negative control [NC], *p < 0.05). (C) Cell viability was measured by WST‐1 assays 4 days after transfection with siINSIG2 and/or HBV siRNA (ratio to NC, *p < 0.05). Abbreviations: ACTB, β‐actin; ns, not significant.
3.4. INSIG2 inhibition delays cell cycle, leading to decrease in HBV‐integrated liver cancer cell proliferation
Next, we undertook RNA sequencing to further study the mechanism of the vulnerability caused by INSIG2 inhibition in HepG2.2.15 cells. Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that the cholesterol biosynthesis pathway was activated following INSIG2 inhibition (Figure 4A), which was consistent with the previous report. 19 Meanwhile, INSIG2 inhibition induced significant downregulation of the pathways involved in the cell cycle and DNA replication in HepG2.2.15 cells (Figure 4A). We thus evaluated the effect of INSIG2 inhibition on the cell cycle. We used nocodazole (inhibitor of microtubule polymerization) to induce mitotic arrest. Twenty‐four hours after nocodazole treatment, the majority of the HepG2 cells had clustered at G2/M phase regardless of the presence or absence of INSIG2 siRNA (Figure 4B). In contrast, the number of cells at G2/M phase was different between NC and INSIG2 siRNA groups in HepG2.2.15 cells, and most of the cells treated with INSIG2 siRNA had remained at G1/S phase (Figure 4C). These results suggested that INSIG2 inhibition delayed G1/S transition only in HBV‐integrated cells. Next, we undertook G1/S phase synchronization by double thymidine block. After this treatment, most cells had clustered at G1/S phase regardless of the presence or absence of INSIG2 siRNA in both HepG2 and HepG2.2.15 cells. After the removal of thymidine, cell cycle progressed in most of the cells in HepG2 cells regardless of the presence or absence of INSIG2 siRNA (Figure 4D). In contrast, in HepG2.2.15 cells, cell cycle progressed in most of the cells treated with NC siRNA but did not in cells treated with INSIG2 siRNA (Figure 4E). Collectively, the number of cells at S and G2/M phase was lower in HepG2.2.15 cells with INSIG2 siRNA than those with NC siRNA (Figure 4E,F). These data further suggested that INSIG2 inhibition delayed cell cycle at G1/S phase, leading to the decrease in cell proliferation in HepG2.2.15 cells.
FIGURE 4.
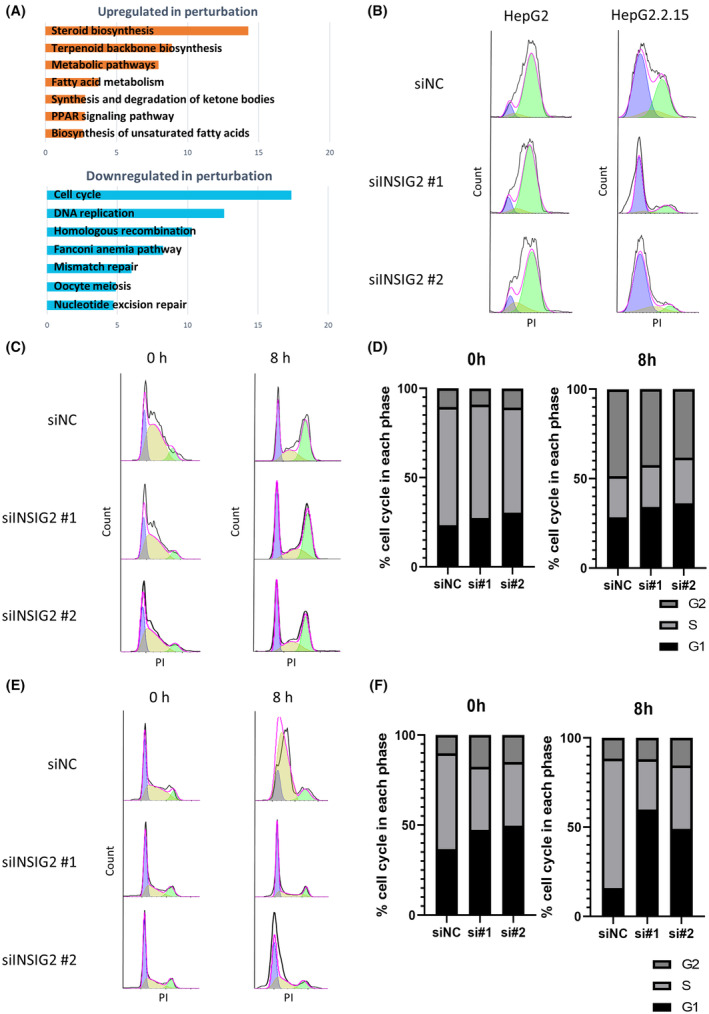
INSIG2 inhibition delays cell cycle, leading to a decrease in cell proliferation in hepatitis B virus‐integrated liver cancer cells. (A) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis results. Horizontal axis indicates the negative log10 p value for each term. (B) To synchronize G2/M, cells were treated with nocodazole (400 ng/mL) after knockdown (KD) with control siRNA (siNC)/INSIG2. Then, HepG2/HepG2.2.15 cells were assessed by flow cytometry. (C‐F) To synchronize G1/S, cells were treated by double thymidine block (2 mM). Then, cells were assessed by flow cytometry immediately and 8 h after release. (C) HepG2 cells after KD with siNC/INSIG2. (D) HepG2 cell population ratio at each stage of cell cycle. (E) HepG2.2.15 cells after KD with siNC/INSIG2. (F) HepG2.2.15 cell population ratio at each stage of cell cycle. Abbreviations: PI, propidium iodide; PPAR, peroxisome proliferator‐activated receptor.
3.5. CDK2 involved in vulnerability caused by INSIG2 inhibition in HBV‐integrated liver cancer cells
To explore the molecules involved in the vulnerability caused by INSIG2 inhibition in HepG2.2.15 cells, we examined the proteins known to be involved in the cell cycle and found that INSIG2 inhibition significantly reduced protein levels of CDK2, known to be involved in G1/S phase progression, 20 in HepG2.2.15 cells but not in HepG2 cells (Figure 5A). Indeed, CDK2 inhibition using CVT‐313, a specific inhibitor of CDK2, inhibited G1/S transition in HepG2.2.15 cells and cell proliferation was suppressed (Figure 5B–D). In addition, in the presence of CDK2 inhibitor, INSIG2 inhibition did not suppress cell proliferation in HepG2.2.15 cells (Figure 5E). These findings suggested that CDK2 might be involved in the vulnerability caused by INSIG2 inhibition only in HepG2.2.15 cells.
FIGURE 5.
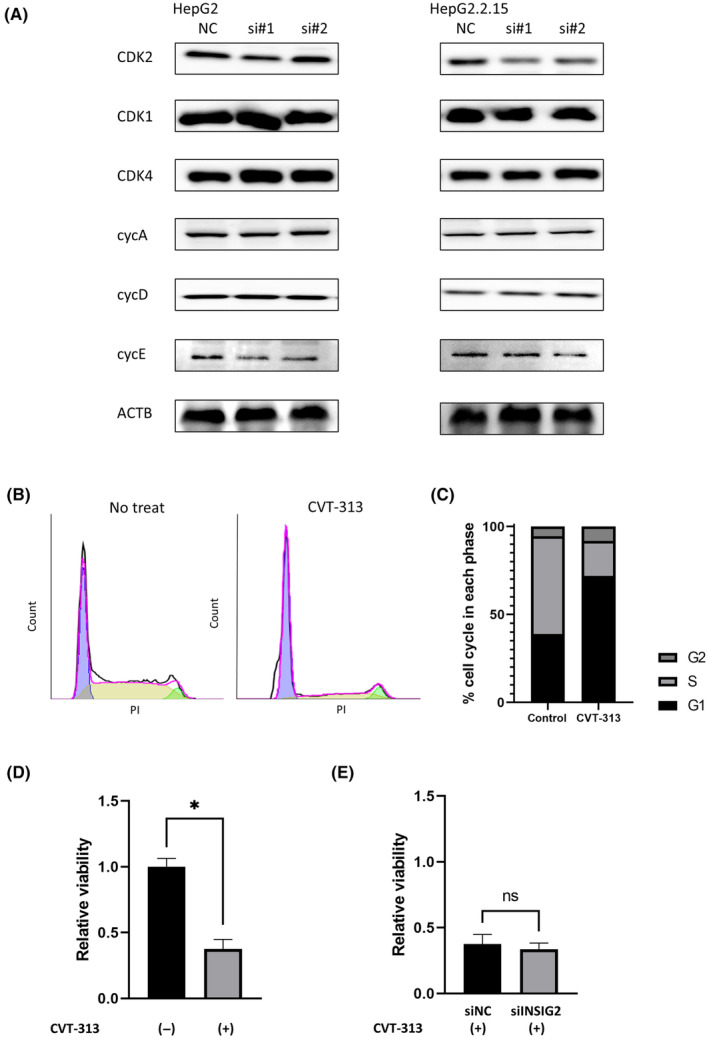
Cyclin‐dependent kinase 2 (CDK2) is involved in the vulnerability caused by INSIG2 inhibition in hepatitis B virus‐integrated liver cancer cells. (A) Western blot analysis of G1/S cell cycle factors in HepG2‐Cas9 cells and HepG2.2.15 cells after transfection with control siRNA (siNC)/siINSIG2. (B) Alteration of cell cycle by CVT‐313 treatment. After 24 h of treatment (left) with no CVT‐313 or (right) with CVT‐313 (5 μM), CDK2 inhibition by CVT‐313 resulted in cell cycle arrest at the G1/S transition. (C) Cell population ratio at each stage of cell cycle is shown. (D) Cell viability was measured by WST‐1 assays 4 days after CVT‐313 (5uM) treatment in HepG2.2.15 cells (ratio to no treatment [no treat], *p < 0.05). (E) Cell viability was measured by WST‐1 assays 4 days after CVT‐313 treatment and transfection with siNC/siINSIG2 (ratio to no treat). Abbreviations: ACTB, β‐actin; cycA/D/E, cyclinA/D/E; ns, not significant; PI, propidium iodide.
3.6. INSIG2 binds to HBx
Finally, we investigated the molecular mechanisms of the dependency of CDK2 levels on INSIG2 in HepG2.2.15. we sought the interaction between INSIG2 and HBx, one of the HBV coding proteins shown to regulate the cell cycle in G1/S phase 21 and bind to CDK2. 22 We introduced the expression plasmids of Myc‐tagged INSIG2 and FLAG‐tagged HBx proteins in the cells (Figure 6A). Western blot analysis using samples precipitated with FLAG Ab showed the binding of INSIG2 to HBx (Figure 6B). Collectively, our findings suggested that the INSIG2 and HBx proteins might make a complex with CDK2, which drives the cell cycle progression in HBV‐integrated liver cancer cells (Figure 6C).
FIGURE 6.
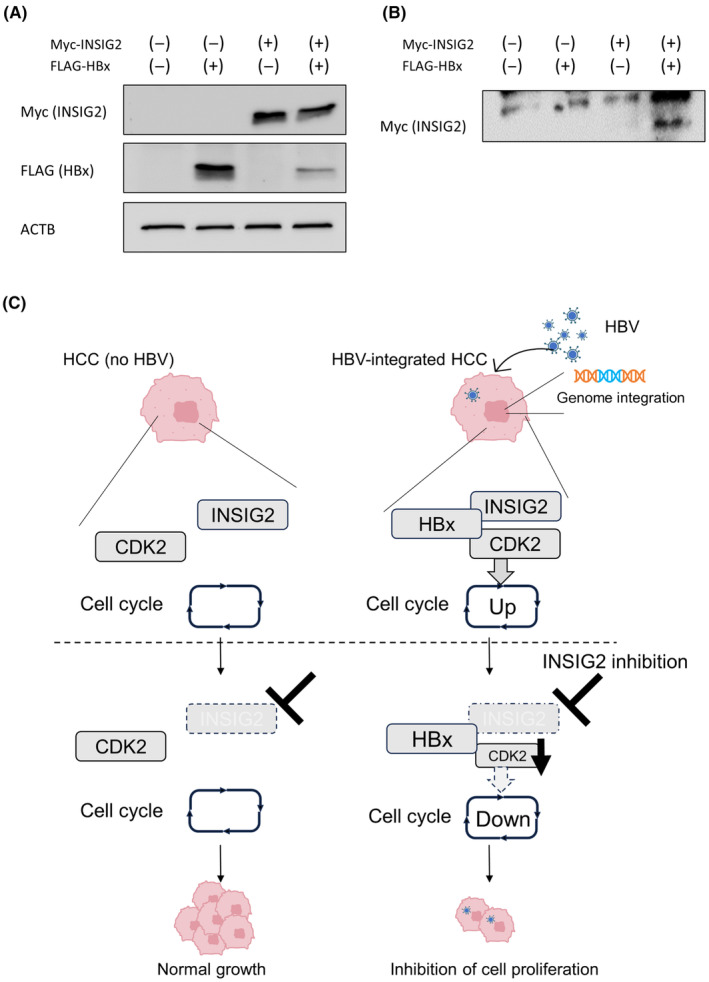
INSIG2 affects cyclin‐dependent kinase 2 (CDK2) through hepatitis B X protein (HBx). (A) Western blot (WB) analysis of extracts from cells transfected with Myc‐INSIG2 and/or FLAG‐HBx, using MYC and FLAG tag Abs. (B) After cells were transfected with MYC‐INSIG2 and/or FLAG‐HBx, immunoprecipitation with FLAG tag Ab was carried out. WB analysis using Myc Tag Ab. (C) Schematic diagram of this study. INSIG2 and CDK2 may be molecularly bound through HBx in hepatitis B virus (HBV)‐integrated cancer cells, creating the dependency of INSIG2 on their cell proliferation. Abbreviations: ACTB, β‐actin; HCC, hepatocellular carcinoma.
4. DISCUSSION
Synthetic lethality is a phenomenon in which a single manipulation allows cell survival, but two or more simultaneous manipulations leads to cell death. This approach has been applied to cancer therapy and exploits the specific vulnerability of cancer cells, which can be drug targets that selectively affect cancer cells. 23 The first successful treatment based on this approach was the use of poly ADP‐ribose polymerase (PARP) inhibitors for patients deficient in the homologous recombination pathway. 24 This discovery has further driven the search for other synthetic lethal targets in a variety of cancer types and large‐scale genetic screens using CRISPR/Cas9 or RNAi have been recently performed to this end. 25 Regarding liver cancer, while several CRISPR/Cas9 library screens have been reported, most of them including ours aimed to identify the targets that synergized with chemotherapy including sorafenib, regorafenib, and lenvatinib. 11 , 26 , 27 , 28 Only a few specific cancer cell vulnerabilities, including ATRX‐mutated cells, and under the hypoxic or glutamine‐depleted states, have been reported 29 , 30 , 31 and the vulnerability in HBV‐integrated cancer cells has not been reported to date. Hepatitis B virus is an oncogenic DNA virus and is often integrated into the host genome of the infected cells, which induces genomic instability and abnormal expression of oncogenes and tumor suppressor genes, thereby promoting hepatocarcinogenesis. 3 In addition, integrated viral DNA produces mutated and/or truncated HBx, HBsAg, and HBcAg proteins, high expression of which induces endoplasmic reticulum and mitochondrial stress responses and promotes the occurrence of HCC. In the present study, we hypothesized that HBV integration promotes malignant transformation, but HBV‐integrated cancer cells also require the fitness to high cellular stress for the cells to survive, which can be the vulnerability of HBV‐integrated cancer cells. We thus undertook the genome‐wide CRISPR KO screen to identify the specific vulnerability of the HBV‐integrated HepG2.2.15 cells compared to its parental HepG2 cells. 12
In our screen, we identified INSIG2 as involved in the vulnerability in HBV‐associated liver cancer. INSIG2 has been reported to bind to the SREBP and SCAP complexes, thereby inhibiting SREBP transport to the Golgi apparatus and suppressing sterol synthesis. 32 Recently, the involvement of INSIG2 in de novo lipogenesis, steatosis, and nonalcoholic steatohepatitis, has been reported. 19 , 33 In the study of HBV integration sites in HCC patients, one such site was shown to be the intergenic region of INSIG2. 34
In the present study, inhibition of INSIG2 specifically reduced cell proliferation in HBV‐integrated cells but not in non‐HBV‐associated cells. In addition, INSIG2‐mediated inhibition of cell proliferation was partly canceled by suppression of HBV transcription. These findings suggested that the vulnerability induced by INSIG2 inhibition in HBV‐integrated liver cancer cells is partly dependent on the presence of HBV.
Further analysis revealed that INSIG2 inhibition induced cell cycle arrest at G1/S phase in HBV‐integrated cells. Mechanistically, we found that INSIG2 inhibition suppressed CDK2 in HBV‐integrated cells. CDK2 is a member of the cyclin‐dependent kinase family, which forms a complex with cyclin E and is known to be essential for the transition from G1 to S phase. 35 , 36 Indeed, we showed that CDK2 inhibition suppressed cell proliferation and INSIG2 inhibition did not suppress cell proliferation in the presence of CDK2 inhibitor. These findings suggested that CDK2 could be involved in the vulnerability caused by INSIG2 inhibition in HBV‐integrated cells.
Hepatitis B virus‐related proteins are known to be involved in the cell cycle. 37 , 38 , 39 For example, expression of HBx protein has been shown to promote progression to S phase in vitro, 24 and HBx protein has been reported to be involved in cancer progression by binding with p53, preventing nuclear entry and promoting the transition to S phase. 40 Furthermore, HBx was known to bind to CDK2 proteins. 22 We found that INSIG2 also bound to HBx protein, suggesting the complex formation among CDK2, INSIG2, and HBx protein. Although we are not able to show the further detailed molecular mechanisms of how this complex affects the CDK2 stability in HBV‐integrated cells, this might explain, in part, the dependency of HBV‐integrated cells on CDK2 for their cell proliferation.
In conclusion, our CRISPR loss‐of‐function screen identified INSIG2 as a candidate for the vulnerability in HBV‐integrated liver cancer cells.
AUTHOR CONTRIBUTIONS
Makoto Fukuoka: Writing – original draft. Takahiro Kodama: Writing – review and editing. Kazuhiro Murai: Methodology. Hayato Hikita: Resources. Emi Sometani: Validation. Jihyun Sung: Validation. Akiyoshi Shimoda: Validation. Satoshi Shigeno: Resources. Daisuke Motooka: Methodology. Akira Nishio: Resources. Kunimaro Furuta: Methodology. Tomohide Tatsumi: Resources. Kosuke Yusa: Methodology. Tetsuo Takehara: Supervision.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Figure S1.
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
ACKNOWLEDGEMENTS
This work was supported by the Japan Agency for Medical Research and Development (AMED) under grant number JP23ama221410 (T.K.), JP23fk0210110 (T.K.), JP23fk0210131 (T.K.), JP23fk0310501 (H.H.), JP23fk0310512 (H.H. and T.Tak.), and JP23fk0310524 (T.K.).
Fukuoka M, Kodama T, Murai K, et al. Genome‐wide loss‐of‐function genetic screen identifies INSIG2 as the vulnerability of hepatitis B virus‐integrated hepatoma cells. Cancer Sci. 2024;115:859‐870. doi: 10.1111/cas.16070
REFERENCES
- 1. Trepo C, Chan HLY, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053‐2063. [DOI] [PubMed] [Google Scholar]
- 2. Organization WH . Guidelines for the prevention, care and treatment of persons with chronic hepatitis B infection. 2015. [PubMed]
- 3. Jiang Y, Han QJ, Zhao HJ, Zhang J. The mechanisms of HBV‐induced hepatocellular carcinoma. J Hepatocell Carcinoma. 2021;8:435‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peneau C, Imbeaud S, La Bella T, et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut. 2022;71:616‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6 [DOI] [PubMed] [Google Scholar]
- 6. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murai K, Kodama T, Hikita H, et al. Inhibition of nonhomologous end joining‐mediated DNA repair enhances anti‐HBV CRISPR therapy. Hepatol Commun. 2022;6:2474‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koike‐Yusa H, Li YL, Tan EP, Velasco‐Herrera MD, Yusa K. Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol. 2014;32:267‐273. [DOI] [PubMed] [Google Scholar]
- 9. Tzelepis K, Koike‐Yusa H, De Braekeleer E, et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016;17:1193‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kodama M, Kodama T, Newberg JY, et al. In vivo loss‐of‐function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2017;114:E7301‐E7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kodama T, Suemura S, Myojin Y, et al. Crispr loss‐of‐function screen identifies the hippo SIGNALLING pathway AS the mediator of regorafenib efficacy in hepatocellular carcinoma. Hepatology. 2019;70:1183A‐1184A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sells MA, Chen ML, Acs G. Production of hepatitis‐B virus‐particles in Hep‐G2 cells transfected with cloned hepatitis‐B virus‐DNA. Proc Natl Acad Sci U S A. 1987;84:1005‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo YC, Wang J, Benedict B, et al. Targeting CDC7 potentiates ATR‐CHK1 signaling inhibition through induction of DNA replication stress in liver cancer. Genome Med. 2021;13:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Xu H, Xiao TF, et al. MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Konishi M, Wu CH, Wu GY. Inhibition of HBV replication by siRNA in a stable HBV‐producing cell line. Hepatology. 2003;38:842‐850. [DOI] [PubMed] [Google Scholar]
- 16. Javanbakht H, Mueller H, Walther J, et al. Liver‐targeted anti‐HBV single‐stranded oligonucleotides with locked nucleic acid potently reduce HBV gene expression in vivo . Mol Ther Nucleic Acids. 2018;11:441‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamai T, Hikita H, Fukuoka M, et al. SIRT1 enhances hepatitis virus B transcription independent of hepatic autophagy. Biochem Biophys Res Commun. 2020;527:64‐70. [DOI] [PubMed] [Google Scholar]
- 18. Lee AR, Lim KH, Park ES, et al. Multiple functions of cellular FLIP are essential for replication of hepatitis B virus. J Virol. 2018;92:e00339‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng H, Qin H, Liao M, et al. CD36 promotes de novo lipogenesis in hepatocytes through INSIG2‐dependent SREBP1 processing. Mol Metab. 2022;57:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brooks EE, Gray NS, Joly A, et al. CVT‐313, a specific and potent inhibitor of CDK2 that prevents neointimal proliferation. J Biol Chem. 1997;272:29207‐29211. [DOI] [PubMed] [Google Scholar]
- 21. Lei YM, Xu X, Liu HL, et al. HBx induces hepatocellular carcinogenesis through ARRB1‐mediated autophagy to drive the G(1)/S cycle. Autophagy. 2021;17:4423‐4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherji A, Janbandhu VC, Kumar V. HBx‐dependent cell cycle deregulation involves interaction with cyclin E/A‐cdk2 complex and destabilization of p27Kip1. Biochem J. 2007;401:247‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Setton J, Zinda M, Riaz N, et al. Synthetic lethality in cancer therapeutics: the next generation. Cancer Discov. 2021;11:1626‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim KH, Seong BL. Pro‐apoptotic function of HBV X protein is mediated by interaction with c‐FLIP and enhancement of death‐inducing signal. EMBO J. 2003;22:2104‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurata M, Yamamoto K, Moriarity BS, Kitagawa M, Largaespada DA. CRISPR/Cas9 library screening for drug target discovery. J Hum Genet. 2018;63:179‐186. [DOI] [PubMed] [Google Scholar]
- 26. Wei L, Lee D, Law CT, et al. Genome‐wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for sorafenib resistance in HCC. Nat Commun. 2019;10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin HJ, Shi YP, Lv YY, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595:730‐734. [DOI] [PubMed] [Google Scholar]
- 28. Xu F, Tong M, Tong CSW, et al. A combinatorial CRISPR‐Cas9 screen identifies Ifenprodil as an adjunct to sorafenib for liver cancer treatment. Cancer Res. 2021;81:6219‐6232. [DOI] [PubMed] [Google Scholar]
- 29. Yang CX, Lee D, Zhang MS, et al. Genome‐wide CRISPR/Cas9 library screening revealed dietary restriction of glutamine in combination with inhibition of pyruvate metabolism as effective liver cancer treatment. Advanced . Science. 2022;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bao MHR, Yang CX, Tse APW, et al. Genome‐wide CRISPR‐Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021;34:34. [DOI] [PubMed] [Google Scholar]
- 31. Liang JB, Zhao H, Diplas BH, et al. Genome‐wide CRISPR‐Cas9 screen reveals selective vulnerability of ATRX−mutant cancers to WEE1 inhibition. Cancer Res. 2020;80:510‐523. [DOI] [PubMed] [Google Scholar]
- 32. Yabe D, Brown MS, Goldstein JL. Insig‐2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element‐binding proteins. Proc Natl Acad Sci U S A. 2002;99:12753‐12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim JY, Wang LQ, Sladky VC, et al. PIDDosome‐SCAP crosstalk controls high‐fructose‐diet‐dependent transition from simple steatosis to steatohepatitis. Cell Metab. 2022;34:1548‐1560.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li WY, Cui XF, Huo Q, et al. Profile of HBV integration in the plasma DNA of hepatocellular carcinoma patients. Curr Genomics. 2019;20:61‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vandenheuvel S, Harlow E. Distinct roles for cyclin‐dependent kinases in cell‐cycle control. Science. 1993;262:2050‐2054. [DOI] [PubMed] [Google Scholar]
- 36. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 37. Goldstein JL, DeBose‐Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35‐46. [DOI] [PubMed] [Google Scholar]
- 38. Benn J, Schneider RJ. Hepatitis‐B virus HBX protein deregulates cell‐cycle checkpoint controls. Proc Natl Acad Sci U S A. 1995;92:11215‐11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X, Huo BN, Liu J, Huang X, Zhang SY, Feng T. Hepatitis B virus X reduces hepatocyte apoptosis and promotes cell cycle progression through the Akt/mTOR pathway in vivo. Gene. 2019;691:87‐95. [DOI] [PubMed] [Google Scholar]
- 40. Ueda H, Ullrich SJ, Gangemi JD, et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet. 1995;9:41‐47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Table S1.
Table S2.
Table S3.
Table S4.
Table S5.