

Abstract
Orecchioni et al. have identified a role for the olfactory receptor Olfr2 and its human ortholog OR6A2 in atherosclerosis. Olfr2 on vascular macrophages binds octanol, a product of lipid peroxidation, and activates the NLR family pyrin domain containing 3 (NLRP3) inflammasome and interleukin-1β secretion, driving atherosclerosis pathology. Inhibitors of OR6A2 may represent a promising therapy for atherosclerosis.
Atherosclerosis is a chronic inflammatory disease of the arterial vasculature and is caused by dysregulated lipid metabolism and persistent maladaptive immune responses. Accumulation of normal and modified lipoproteins by macrophages leads to foam cell formation in the arterial wall and is associated with activation of innate immune receptors, including NOD-like and Toll-like receptors (NLRs and TLRs), leading to detrimental inflammation and plaque progression [1]. Macrophages in atherosclerotic arteries are highly heterogeneous and encompass multiple subsets with pro-inflammatory and anti-inflammatory properties [2]. A recent study by McArdle et al. used the fluorescent reporter Cx3cr1GFP/+;Cd11cYFPApoe−/− mouse model of atherosclerosis to provide an in-depth characterization of macrophage subsets [3]. Interestingly, while GFP+ macrophage subsets expressed genes enriched in regressing plaques, YFP+ subsets expressed genes enriched in progressing plaques, in particular olfactory receptors and their signal transduction genes.
Olfactory receptors (ORs) are G protein-coupled chemoreceptors (GPCRs) that mainly localize to sensory organs and play critical roles in the detection of odorants and other chemical signals. Growing evidence demonstrates that these chemoreceptors are also expressed in non-olfactory tissues [4], including immune cells. For example, Li et al. found eight ORs expressed in both airway and pulmonary macrophages that function in activation and migration [5]. Here, Orecchioni et al. identified expression of the olfactory receptor Olfr2 in mouse vascular macrophages [6]. They examined mRNA expression of Olfr2 in aortas of Apoe−/− mice and observed a significant upregulation compared to wild-type (WT) mice. Using confocal microscopy and flow cytometry, they confirmed that Olfr2 was mainly expressed in a subset of vascular macrophages in Apoe−/− mice and OlfrGFP mice, consistent with previous findings [3]. The authors also investigated expression of the human ortholog OR6A2 (olfactory receptor 6A2) from a transcriptional dataset of human carotid plaques (BiKE dataset, GSE21545). In line with their findings in mouse aortas, OR6A2 expression increased with plaque macrophage content and O6A2 protein localized to human aortic macrophages.
Olfr2 signals through the Gα subunit and activation leads to cyclic adenosine monophosphate (cAMP) production by adenylate cyclase (Adcy3). To investigate the signal cascade of Olf2, the authors examined changes in cAMP levels triggered by its endogenous ligand octanal. Increased cAMP in response to octanal stimulation was sharply reduced in Adcy3+/− mouse bone marrow-derived macrophages (BMDMs). In Olfr2−/− mice, the authors confirmed loss of Olfr2 protein in vascular macrophages and in the aortic root. While octanal significantly increased Ca2+ flux in BMDMs, Ca2+ flux was reduced with Olfr2 inhibition.
Inflammasome activation in macrophages, in particular activation of the NLRP3 inflammasome (NLR family pyrin domain containing 3), plays a critical role in atherosclerosis. TLR signaling primes the transcription of inflammasome components, and secondary stimuli then induce release of IL-1β [7]. To study the role of Olfr2 in NLRP3 inflammasome activation, the investigators carried out transcriptional analysis of octanal-treated BMDMs and found that oxidative stress pathways were activated by Olfr2 [6]. Octanal triggered production of mitochondrial and cytosolic reactive oxygen species (ROS) in WT BMDMs, and this was significantly reduced in Olfr2−/− BMDMs. Because ROS activate NLRP3, the authors then confirmed that secretion of IL-1β and IL-1α strongly increased with octanal. This increase could be blocked with the Olfr2 antagonist citral or with genetic ablation of Olfr2. In addition, pharmacologic or genetic inhibition of the Nlrp3 inflammasome also reduced Olfr2-mediated IL-1β secretion. In human monocyte-derived macrophages (hMDMs), octanal-induced increases in Ca2+ flux and IL-1β secretion were reduced with OR6A2 inhibition. Together, these data demonstrate that octanal triggers Olfr2-mediated production of ROS, resulting in the activation of the NLRP3 inflammasome and release of IL-1β in mouse and human macrophages.
The authors then investigated the source of endogenous octanal in vivo [6]. Plasma levels of octanal were increased in WT mice fed a high-fat western diet (WD) and even more significantly in Apoe−/− mice fed a WD where plasma concentrations reached ~9 μM, a value close to the EC50 of octanol for Olfr2 in vitro [8]. Interestingly, octanal was not derived from the diet or the microbiome, but rather was generated by lipid peroxidation of oleic acid. In plasma from 196 human subjects, octanal levels positively correlated with total cholesterol, non-high-density lipoprotein cholesterol (non-HDL) and triglycerides, strengthening the translational relevance of octanal/OR6A2 signaling.
Additional in vivo validation included examination of Apoe−/− mice fed a WD and supplementary administration of octanal; this led to even higher plasma octanal levels that associated with striking increases in aortic atherosclerotic plaques, tumor necrosis factor (TNF) and IL-1β plasma levels. Conversely, Apoe−/− mice treated with the Olfr2 antagonist citral showed a significant reduction in atherosclerotic lesion size. Additional genetic experiments further confirmed the detrimental function of Olfr2 signaling. In a series of experiments, irradiated Ldlr−/− mice received either Olfr2−/− or WT bone marrow; Ldlr−/− mice with Olfr2−/− bone marrow showed smaller atherosclerotic lesions en face and in aortic root compared to those reconstituted with WT bone marrow. In addition, Olfr2−/− recipients did not display octanal treatment-induced exacerbation of aortic lesions, supporting the conclusion that octanal-mediated Olfr2 signaling plays a pathogenic role in atherosclerosis.
The work by Klaus Ley’s group and colleagues identifies expression of the mouse olfactory receptor Olfr2 and its human ortholog OR6A2 in vascular macrophages [6]. Olfr2 activation mediates the production of ROS and NLRP3-dependent IL-1β production in both mouse and human macrophages. The authors demonstrate that genetic and pharmacological targeting Olfr2 significantly reduces atherosclerotic plaques in mice, as diagrammed in Figure 1. Of note, Olfr2 inhibition did not alter lipid levels, regardless of the reduction in NLPR3 activation or amelioration of aortic plaques. This finding is consistent with a similar study demonstrating that systemic IL-1β inhibition lowers the rate of recurrent cardiovascular events, which was also independent of a lipid-level lowering effect [9]. This study uncovers a novel mechanism of atherosclerotic progression mediated by Olfr2-mediated activation of the NLRP3 inflammasome and suggests that inhibiting OR6A2 may be a promising therapeutic target for atherosclerosis.
Figure 1. Olfactory receptor 2 in vascular macrophages activates NLRP3-dependent IL-1β production in atherosclerosis.
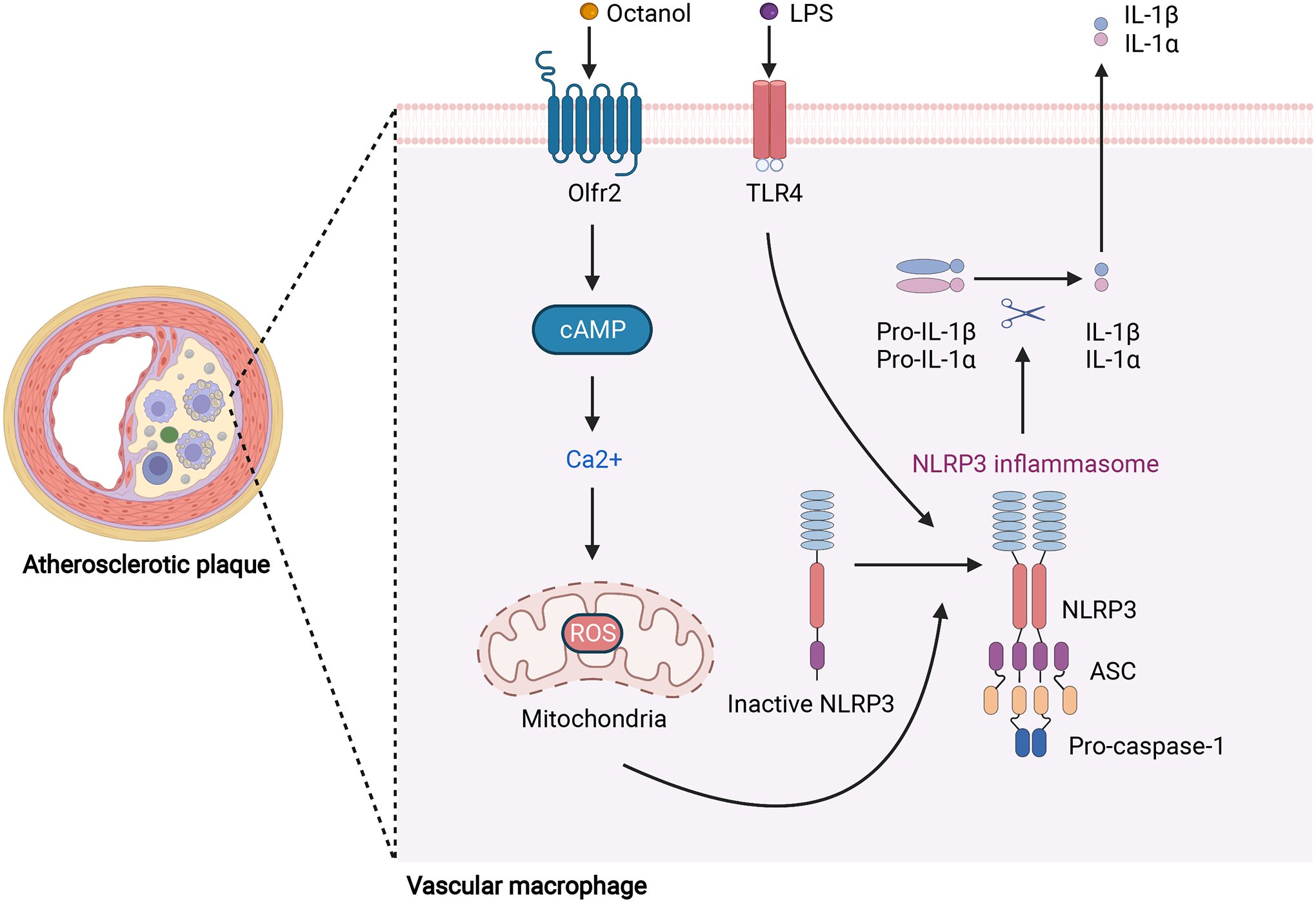
Accumulation of lipoproteins by macrophages leads to foam cell formation in the arterial wall, a hallmark of the atherosclerotic plaque. Olfr2 is expressed in vascular macrophages. Upon activation by its ligand octanol, Olfr2 binds to Gα and induces cAMP production and Ca2+ flux, triggering production of mitochondrial and cytosolic ROS. After priming by LPS via TLR4 activation, ROS serve as the second signal to activate the NLRP3 inflammasome and induce secretion of IL-1β and IL-1α, increasing the inflammatory response and atherosclerotic plaque progression [6]. Abbreviations: Olfr2, olfactory receptor 2; TLR4, Toll-like receptor 4; LPS, lipopolysaccharide; cAMP, cyclic adenosine monophosphate; ROS: reactive oxygen species; NLRP3: NLR family pyrin domain containing 3; ASC, apoptosis-associated speck-like protein containing a CARD; IL-1β, interleukin-1β; IL-1α, interleukin-1α. This figure was created with BioRender.com.
Lipid accumulation in the arterial intima is a fundamental cause of atherosclerotic lesion formation. Lipid-lowering therapies not only resolve lipoprotein modification, but also attenuate macrophage inflammation. The endogenous Olfr2 agonist octanal is a product of lipid peroxidation and has been detected in oxidized LDL (oxLDL) [10], suggesting that local, accumulating oxLDL could be an important source of octanal. Therefore, it will be interesting to investigate whether the combination of lipid lowering agents and Olfr2 inhibitors might have additive benefits in atherosclerosis.
Acknowledgements
The authors are funded by RF1AG053001K, RF1AG070839, 1P30 AG066515, and the American Heart Foundation/Allen Frontiers Award (KIA). KIA is a Chan Zuckerberg Biohub investigator.
Reference
- 1.Back M et al. (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16, 389–406. 10.1038/s41569-019-0169-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore KJ et al. (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13, 709–721. 10.1038/nri3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McArdle S et al. (2019) Migratory and Dancing Macrophage Subsets in Atherosclerotic Lesions. Circ Res 125, 1038–1051. 10.1161/CIRCRESAHA.119.315175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrer I et al. (2016) Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease. Front Aging Neurosci 8, 163. 10.3389/fnagi.2016.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li JJ et al. (2013) Activation of olfactory receptors on mouse pulmonary macrophages promotes monocyte chemotactic protein-1 production. PLoS One 8, e80148. 10.1371/journal.pone.0080148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orecchioni M et al. (2022) Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science 375, 214–221. 10.1126/science.abg3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swanson KV et al. (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y et al. (2014) Aldehyde recognition and discrimination by mammalian odorant receptors via functional group-specific hydration chemistry. ACS Chem Biol 9, 2563–2571. 10.1021/cb400290u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridker PM et al. (2017) Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 10.Thomas CE et al. (1994) Multiple lipid oxidation products in low density lipoproteins induce interleukin-1 beta release from human blood mononuclear cells. J Lipid Res 35, 417–427 [PubMed] [Google Scholar]