

Abstract
Background and Aims:
Bile acids trigger a hepatic inflammatory response, causing cholestatic liver injury. Runt-related transcription factor-1 (RUNX1), primarily known as a master modulator in hematopoiesis, plays a pivotal role in mediating inflammatory responses. However, RUNX1 in hepatocytes is poorly characterized, and its role in cholestasis is unclear. Herein, we aimed to investigate the role of hepatic RUNX1 and its underlying mechanisms in cholestasis.
Approach and Results:
Hepatic expression of RUNX1 was examined in cholestatic patients and mouse models. Mice with liver-specific ablation of Runx1 were generated. Bile duct ligation and 1% cholic acid diet were used to induce cholestasis in mice. Primary mouse hepatocytes and the human hepatoma PLC/RPF/5-ASBT cell line were used for mechanistic studies. Hepatic RUNX1 mRNA and protein levels were markedly increased in cholestatic patients and mice. Liver-specific deletion of Runx1 aggravated inflammation and liver injury in cholestatic mice induced by bile duct ligation or 1% cholic acid feeding. Mechanistic studies indicated that elevated bile acids stimulated RUNX1 expression by activating the RUNX1-P2 promoter through JAK/STAT3 signaling. Increased RUNX1 is directly bound to the promotor region of inflammatory chemokines, including CCL2 and CXCL2, and transcriptionally repressed their expression in hepatocytes, leading to attenuation of liver inflammatory response. Blocking the JAK signaling or STAT3 phosphorylation completely abolished RUNX1 repression of bile acid–induced CCL2 and CXCL2 in hepatocytes.
Conclusions:
This study has gained initial evidence establishing the functional role of hepatocyte RUNX1 in alleviating liver inflammation during cholestasis through JAK/STAT3 signaling. Modulating hepatic RUNX1 activity could be a new therapeutic target for cholestasis.
INTRODUCTION
Cholestasis is characterized by the excessive accumulation of bile acids (BAs) in the liver.[1] Chronic cholestasis progresses to liver fibrosis, cirrhosis, and eventually liver failure.[1] Although the exact mechanisms of BA-induced cholestatic liver injury remain to be elucidated, BA plays an important role in this injury by promoting the production of chemokines (eg, Ccl2, Cxcl2) in hepatocytes.[2–5] JAK/STAT3 signaling mediates inflammatory responses in several human diseases, such as cancer.[6,7] STAT3 signaling has been shown to be activated in cholestatic animal models, and its activation attenuates sepsis-induced liver injury in rats,[8,9] suggesting that it has a protective role in liver inflammatory response. However, it is not known whether JAK/STAT3 signaling mediates BA-induced hepatic inflammation in cholestasis.
Runt-related transcription factor (RUNX) 1, also known as acute myeloid leukemia 1, belongs to the RUNX protein family and is highly conserved in vertebrates.[10] RUNX1 is expressed in a broad range of cells and organs, including hematopoietic stem cells (eg, macrophages and T lymphocytes) and nonhematopoietic tissues (eg, liver, heart, and brain).[11,12] Its expression is regulated by 2 separate promoters: RUNX1-P1 and RUNX1-P2.[13] The RUNX1-P1 promoter was identified in hematopoietic stem cells, whereas the RUNX1-P2 promoter was mostly activated in nonhematopoietic tissues, including the liver.[11,12]
RUNX1 functions as a key transcriptional factor in regulating the expression of genes involved in embryonic development, hematopoiesis, angiogenesis, tumorigenesis, immune response, and especially the inflammatory response.[12,14] It is mainly reported in acute myeloid leukemia and other cancers.[15,16]
Recently, Tang et al.[17] demonstrated that RUNX1 inhibited the activation of NF-κB signaling in respiratory epithelial cells by binding to IKKβ to form protein complexes in the cytoplasm. Genetic deletion of RUNX1 in alveolar epithelial cells also enhances pulmonary inflammation after lipopolysaccharide (LPS) treatment,[17] indicating that RUNX1 acts to repress the inflammatory response.
In contrast, knockdown of RUNX1 in endothelial cells impedes hepatic inflammation in a mouse model of NASH[18]; however, the details of the mechanistic role of RUNX1 in NASH remain unclear. Consistent with this view, Kaur et al.[19] showed that RUNX1 in liver sinusoidal endothelial cells induces angiogenesis and the expression of adhesion molecules (eg, PECAM1 and VCAM1) that correlate with the severity of NASH, thus suggesting that RUNX1 functions as a positive regulator of inflammation. The discrepancy of RUNX1 function in these studies suggests that RUNX1 may play different roles in different cells/tissues and diseases.
Recently, after re-analysis of our single-cell RNA sequencing (scRNA-seq) data in human livers,[20] we found that RUNX1 is widely expressed in all liver cells and that its expression was substantially upregulated in hepatocytes and some immune cells from patients with primary biliary cholangitis (PBC). Previous studies have characterized the role of RUNX1 in liver nonparenchymal cells, including endothelial cells, stellate cells, and immune cells such as dendritic cells.[18,21,22] However, RUNX1 is poorly characterized in liver parenchymal cells, and its expression, functional role, and regulatory mechanism in cholestatic liver diseases have not yet been explored. Therefore, in the current study, we aimed to delineate the functional role of hepatic RUNX1 and the underlying mechanisms in cholestasis.
We found that RUNX1 repressed the inflammatory response stimulated by BA induction of chemokines Ccl2 and Cxcl2 through the JAK/STAT3 signaling in hepatocytes. Genetic ablation of Runx1 reduced cholestatic liver injury in murine models of cholestasis. These findings advance our understanding of RUNX1’s function in the liver and potentially provide a basis for targeting RUNX1 and its relevant signaling pathways in the treatment of cholestasis.
MATERIALS AND METHODS
Human liver tissue collection
This study involving human subjects was carried out in accordance with the Declaration of Helsinki (2013) and the Declaration of Istanbul (2018) of the World Medical Association. Approval for the study protocol was obtained from the Institutional Ethical Review Board at Southwest Hospital affiliated with Army Medical University (Chongqing, China). In the current study, patients were recruited from the Institute of Hepatobiliary Surgery and the Department of Gastroenterology at Southwest Hospital. All patients signed a written informed consent form before the start of the study.
Liver tissues (n = 18) were obtained from patients with obstructive cholestasis (OC) undergoing pancreatoduodenectomy resection with curative intent for suspected pancreatic or periampullary malignancy as described previously.[23] Control liver tissues from patients without cholestasis were also collected in those undergoing resection of liver metastases (n = 11, 6 hepatic hemangioma, and 5 rectal metastases). All collected liver samples were immediately cut into small pieces and fixed in 4% paraformaldehyde or stored in liquid nitrogen. The characteristics of the study subjects are summarized in Supplemental Table 1 (http://links.lww.com/HEP/A63).
For scRNA-seq analysis, a liver biopsy was obtained in patients with PBC (n = 5) with no history of ursodeoxycholic acid treatment and diagnosed by elevated serum alkaline phosphatase levels and positive antimitochondrial antibodies.[20] Autoimmune hepatitis and other liver diseases were excluded by liver histology. Accordingly, control surgical liver samples were obtained from 4 patients with no evidence of PBC, cholestasis, viral hepatitis, or autoimmune hepatitis.[20] The detailed clinical characteristics of these patients for the scRNA-seq analysis are listed in Supplemental Table 2 (http://links.lww.com/HEP/A63).
Liver specimens were also obtained from patients with primary sclerosing cholangitis (PSC) or PBC (n = 16 in each group) and healthy controls (n = 6), and acquired from the Liver Tissue Cell Distribution System at the University of Minnesota (funded by National Institutes of Health Contract # HSN276201200017C), as described previously,[24] and were used for the TaqMan quantitative PCR analysis.
Generation of liver-specific Runx1 knockout (Runx1 cKO) mice and Abcb4 knockout mice
Runx1tm1Tani/J and Alb-re mice were purchased from the Jackson Laboratory (JAX stock #008772, and stock #003574). In the Runx1tm1Tani/J mice (also known as Runx1flox/flox), exon 4 of the Runx1 (runt-related transcription factor-1) gene is flanked by loxP sites. When crossed with a Cre recombinase-expressing strain, this strain can be used to eliminate the tissue-specific expression of Runx1 gene. Runx1flox/flox mice were crossed with AlbCre/+ mice to generate Runx1flox/flox/AlbCre/+ (Runx1 cKO) mice. Mice were genotyped by PCR, primers for Runx1: forward: 5′-GCGTTCCAAGTCAGTTGTAAGCC-3′; reverse: 5′-CTGCATTTGTCCCTTGGTTGACG-3′. The PCR product of Runx1 floxed allele is 550 bp and the PCR product of the wild-type (WT) allele is 362 bp (Supplemental Figure 1A, http://links.lww.com/HEP/A63). Primers for Alb-Cre, forward: 5′-TGGCAAACATACGCAA GGG-3′; reverse: 5′-CGGCAAACGGACAGAAGCA-3′. The PCR product of Alb-Cre positive mice is 450 bp, and no PCR product is observed in Alb-Cre negative mice. Finally, Runx1 liver-specific knockout mice were further confirmed with western blot and multiplex immunofluorescence (IF) analyses (Supplemental Figure 1B, C, http://links.lww.com/HEP/A63).
Abcb4 knockout mice (Abcb4-KO, C57BL/6J background) were developed by Shanghai Model Organisms Center Inc. as we previously reported.[25] Briefly, a 411 bp deletion in exon 3 resulted in a frameshift mutation of Abcb4 and inactivated the Abcb4 gene.
Experimental animals
All studies involving the use of experimental animals were conducted according to the guidelines of the Animal Care and Use Committee at Southwest Hospital (Chongqing, China). The study protocol was approved by the Institutional Animal Care and Use Committee (Army Medical University).
Male 7-week-old C57BL/6J mice were purchased from the Center of Laboratory Animals of Southwest Hospital (Chongqing, China). Upon arrival, mice were housed and acclimatized for 1 week before the experiments to allow them to adapt to their new environment.
For the 7-day bile duct ligation (BDL)-induced mouse model of cholestasis, Runx1flox/flox mice and Runx1 cKO mice were divided into 4 groups: a sham-operation group and a BDL group (with n = 5 and n = 7, respectively, in each group). For the 14-day 1% cholic acid (CA)-induced cholestasis mouse model, Runx1flox/flox mice and Runx1 cKO mice were each divided into 2 groups: a control diet group (n = 5 for each genotype) and a 1% CA diet group (n = 7 for Runx1flox/flox group; n = 5 for Runx1 cKO group).
For the LPS-induced mouse model of cholestasis, WT mice were injected i.p. with 4 mg/kg body weight of LPS or saline for 6 hours (n = 5 in each group). For the 3,5-diethoxycarbonyl-1, 4-dihydrocollidine–induced mouse model of cholestasis, WT mice were fed 0.1% 3,5-diethoxycarbonyl-1, 4-dihydrocollidine or a control diet for 14 days (n = 5 in each group). For the Abcb4 genetic deficiency-induced mouse model of cholestasis, WT mice and Abcb4-KO mice at 8 weeks of age were divided into 2 groups (n = 15 in each group).
All mice were fasted overnight before being sacrificed. Serum was collected and immediately stored at −80°C until analyses were performed. Biochemistry tests were carried out by the Department of Clinical Laboratory Medicine at Southwest Hospital (Chongqing, China). The liver tissues were collected and perfused with PBS) to flush out blood and then immediately cut into small pieces and rapidly frozen in liquid nitrogen until the analyses were done.
Preparation and treatment of primary mouse hepatocytes
Primary mouse hepatocytes were isolated from 10- and 20-week-old mice (WT, Runx1flox/flox, or Runx1 cKO) using collagenase perfusion as previously described.[25] Isolated hepatocytes were cultured in 5% fetal bovine serum–Williams’ Medium E and subsequently treated with 100 μM taurocholic acid (TCA; Sigma-Aldrich) for 12 or 24 hours. Finally, whole-cell lysates were collected as described previously[25,26] for subsequent real-time quantitative PCR and western blot analyses.
Plasmid construction, transfection, and stable cell lines
The human hepatoma PLC/PRF/5 cell line (ATCC) and PLC/PRF/5-ASBT cells were used, as described previously.[23] The pGL3-STAT3 plasmid (STAT3 o/e) was generated by Hunan Fenghui Biotechnology Co. Ltd.
RUNX1 promoter luciferase reporter assays
The pGL3-basic vector was used to generate reporter constructs containing the RUNX1-P1 promoter or the RUNX1-P2 promoter (Fenghbio), as reported and described previously.[23,27] The truncated forms (−944, −778, −308 to +600) of the RUNX1-P2 promoter (Supplemental Figure 2A, http://links.lww.com/HEP/A63) were produced by their primer pairs (Supplemental Table 3, http://links.lww.com/HEP/A63). The pGL3–308/+600 was used to generate pGL3-RUNX1-P2 308MUT harboring mutations in the key motif of the potential STAT3 response element within the RUNX1-P2 promoter region (Supplemental Figure 2B, http://links.lww.com/HEP/A63 and Supplemental Table 3, http://links.lww.com/HEP/A63).
These constructs were co-transfected with pGL3-STAT3 o/e or a control (CTR) vector into PLC5/PRF/5-ASBT cells. After 24 hours, the cells were treated with DMSO or 100 μM TCA for 12 hours. The harvested cells were lysed using 1× passive lysis buffer, and the luciferase activity was measured using the Dual Luciferase Assay kit (Cat#E1910; Promega Corp.) according to the protocol described previously.[23]
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the Magna ChIPA/G Chromatin Immunoprecipitation Kit (Cat#17–10086; Millipore) according to the manufacturer’s instructions. Soluble chromatins were prepared from cultured PLC/PRF/5-ASBT cells or liver tissues of human subjects or experimental mice. The chromatins were immunoprecipitated using STAT3-specific, RUNX1-specific, or NF-κB p65-specific antibodies (Supplemental Table 4, http://links.lww.com/HEP/A63). The primer sequences and the sizes of the amplicon are listed in Supplemental Tables 5 and 6 (http://links.lww.com/HEP/A63). The ChIP assays were performed as previously described.[23]
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was extracted from cells or tissues using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. Reverse transcription and real-time quantitative PCR were performed, as we reported previously.[23,26] The TaqMan probes (Life Technologies Corp.) and SYBR primers used in this study are listed in Supplemental Table 7 (http://links.lww.com/HEP/A63).
Western blot analysis
Total liver tissue homogenates, whole-cell lysates, and nuclear extracts were prepared as previously described.[23,25] Protein samples (20 μg per lane) were separated by 12% or 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (0.22 μm). Next, the membranes were blocked using 5% nonfat milk for 1 hour at room temperature. Then, the blots were cut according to the loaded standard markers. After being incubated with primary antibodies at 4°C for 12 hours, the blots were incubated with horseradish peroxidase–conjugated secondary antibodies at 37°C for 1 hour and visualized with electrochemiluminescence. Finally, these blots were scanned and analyzed using ImageJ software. The sources and their dilutions of the primary antibodies are listed in Supplemental Table 4 (http://links.lww.com/HEP/A63).
Liver histology
Liver sections were stained with hematoxylin and eosin and Sirius Red, as previously described.[23,25] Liver histological assessments were performed blindly by expert pathologists.
IF and multiplex IF analyses
IF was performed as previously described.[26,28] Multiplex IF were performed in human or mouse liver sections, as described previously,[20,29] using a 4-color multiple fluorescent immunohistochemical staining kit (Cat# abs50012/abs50028; Absin) according to the manufacturer’s instructions. CK19 or HNF4α acted as specific biomarkers for cholangiocytes or hepatocytes, respectively.[30,31] The dilutions of the primary antibodies used in this study are presented in Supplemental Table 4, http://links.lww.com/HEP/A63.
Statistical analysis
All data were statistically analyzed using GraphPad Prism software (version 7.0). Comparisons between the 2 groups were determined using an independent-samples t test or Mann-Whitney U test. Multigroup comparison was determined using ordinary 1-way ANOVA or the Kruskal-Wallis test. All experiments were repeated at least 3 times, and the resulting data were expressed as mean ± SD. p < 0.05 was considered to be statistically significant.
RESULTS
Hepatic RUNX1 expression was markedly increased in cholestatic patients and mouse models of cholestasis
Re-analysis of our 3′-scRNA-seq data from human control and PBC livers[20] revealed that RUNX1 is widely expressed in liver cells, including hepatocytes, cholangiocytes, endothelial cells, and immune cells (Supplemental Figure 3, http://links.lww.com/HEP/A63). Strikingly, RUNX1 expression was substantially upregulated in hepatocytes of PBC patients when compared with hepatocytes of control patients, while moderate increase of RUNX1 was also seen cholangiocytes of PBC livers when compared with control livers. Of note, the basal level of RUNX1 in hepatocytes was substantially lower than in cholangiocytes (Supplemental Figure 3, http://links.lww.com/HEP/A63).
To confirm this observation, we further analyzed RUNX1 expression in liver samples from patients with OC, PSC, and PBC. As shown in Figure 1A and B, hepatic RUNX1 expression in OC patients was significantly increased at the mRNA and nuclear protein levels (2.7- and 4.8-fold, respectively), when compared with the controls (Figure 1A, B). Multiplex IF analysis further confirmed that the levels of RUNX1 protein in hepatocytes were markedly elevated in OC livers compared with the control liver specimens (Figure 1C). The increased mRNA expression of RUNX1 was also seen in human liver tissues from patients with PBC or PSC when compared with healthy controls (Supplemental Figure 4, http://links.lww.com/HEP/A63), confirming elevated expression of RUNX1 in human cholestasis.
FIGURE 1.

RUNX1 mRNA and protein levels in the liver tissues of cholestatic patients and mouse models of cholestasis. Relative levels of RUNX1 mRNA (A) and nuclear RUNX1 protein (B) in liver tissues of patients with OC (n = 18) and control (CTR) patients (n = 11). C, controls; O, obstructive cholestasis. *p < 0.05 versus controls. (C) Multiplex immunofluorescence labeling of RUNX1 (red color), HNF4α (purple color, a specific marker for hepatocytes), and CK19 (green color, a specific marker for cholangiocytes) protein in a human OC liver and control. Bile ducts highly expressed RUNX1 protein in both control and OC livers (white arrows). Relative levels of Runx1 mRNA transcripts (D) and nuclear Runx1 protein (E) in liver tissues of sham-operated mice (n = 5) and BDL mice (n = 7). *p < 0.05 versus sham-operated mice. Abbreviations: OC, obstructive cholestasis; RUNX1, runt-related transcription factor-1.
To verify whether the hepatic expression of Runx1 is also altered in cholestatic mouse models, we analyzed its expression in mice after BDL, 1% CA-fed, 0.1% 3,5-diethoxycarbonyl-1, 4-dihydrocollidine diet, i.p. LPS injection, or Abcb4 deficiency. As shown in Figure 1D and E, hepatic Runx1 was significantly elevated in a BDL-induced mouse model of cholestasis compared with WT controls, with 5.7- and 7.5-fold increases in its mRNA and protein levels, respectively. Similar results were observed in other murine models of cholestasis induced by the 1% CA diet, 0.1% 3,5-diethoxycarbonyl-1, 4-dihydrocollidine diet, i.p. LPS injection, and Abcb4-KO (Supplemental Figure 5, http://links.lww.com/HEP/A63). Collectively, these findings provide multiple lines of evidence showing that hepatic RUNX1 expression is markedly induced by cholestasis in human subjects and experimental animals.
However, in contrast to what were seen in human bile ducts (white arrows, Figure 1C and Supplemental Figure 3, http://links.lww.com/HEP/A63), our scRNA-seq data analysis from WT mouse livers demonstrated that the basal expression of Runx1 in mouse cholangiocytes was very low level (Supplemental Figure 6, http://links.lww.com/HEP/A63), and its protein expression in mouse bile ducts was still presented at a low level after BDL for 7 days (white arrows, Supplemental Figure 1C, http://links.lww.com/HEP/A63). This observation indicated that the expression of RUNX1 in human cholangiocytes respond differently from its counterparts in mouse.
Liver-specific deletion of Runx1 aggravated liver inflammation and cholestatic liver injury in mouse models of cholestasis induced by BDL and a 1% CA diet
To delineate the functional role of hepatic RUNX1 in cholestasis, we generated liver-specific Runx1 knockout (Runx1 cKO) mice (Supplemental Figure 1, http://links.lww.com/HEP/A63), and induced cholestasis using BDL and a 1% CA diet.
Liver histological assessment indicated that liver inflammation, necrosis, and fibrosis scores were significantly higher in Runx1 cKO mice than in control (Runx1flox/flox) mice after BDL, whereas bile duct proliferation was similar between the 2 groups (Figure 2). Moreover, serum biochemistry tests were consistent with histological findings.
FIGURE 2.
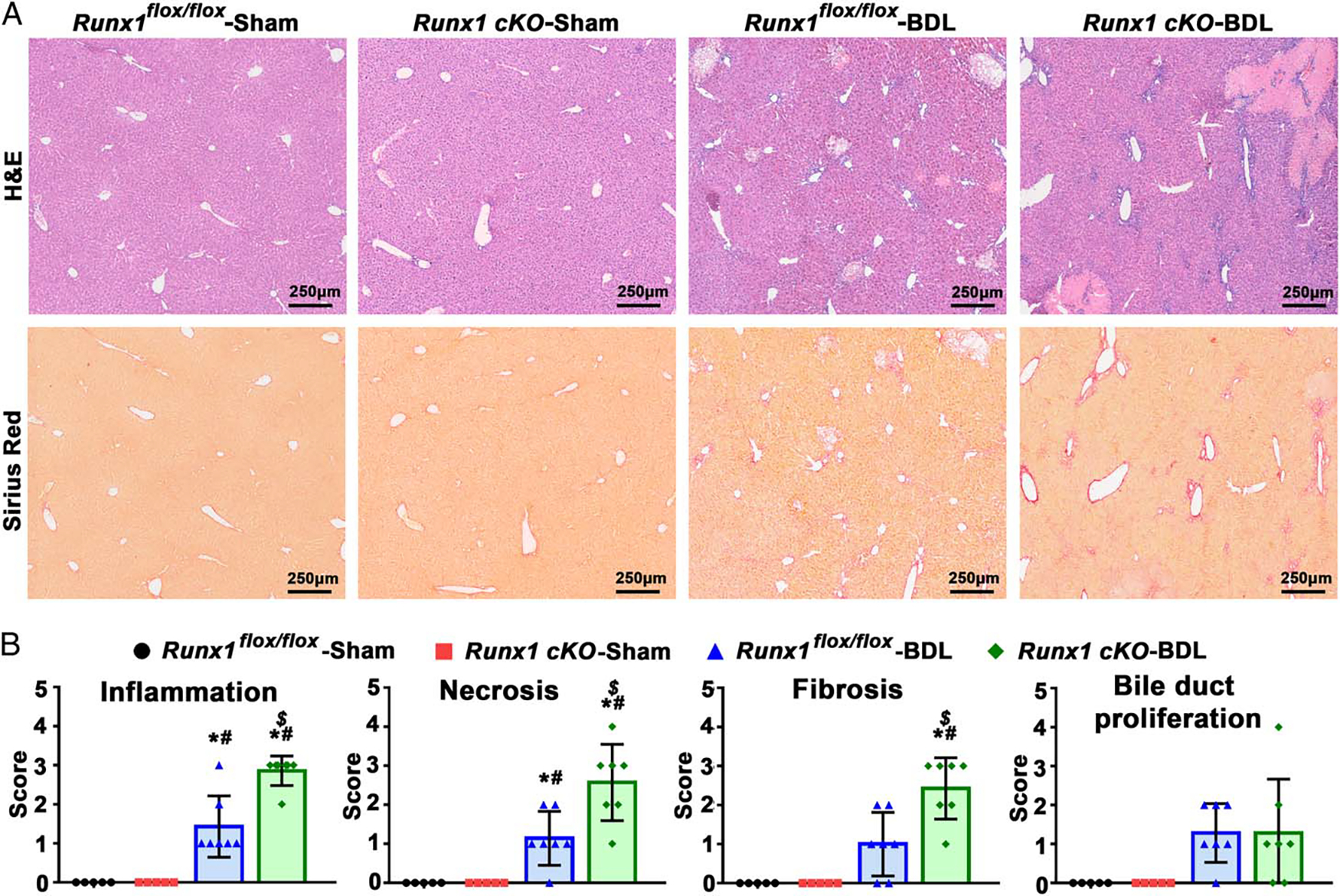
Effects of liver-specific deletion of Runx1 on liver inflammation and injury in a BDL-induced mouse model of cholestasis. (A) H&E staining and Sirius Red staining in BDL mouse livers. (B) Liver histologic assessments, including scores for inflammation, necrosis, fibrosis, and bile duct proliferation. The histologic assessments were blinded and assessed by expert pathologists. N = 5 for each genotype in the sham-operation group; N = 7 for each genotype in the BDL group. *p < 0.05 versus sham-Runx1flox/flox mice; #p < 0.05 versus sham-Runx1 cKO mice; $p < 0.05 versus BDL-Runx1flox/flox mice. Abbreviations: BDL, bile duct ligation; H&E, hematoxylin and eosin.
As demonstrated in Supplemental Table 8, http://links.lww.com/HEP/A63, the levels of alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, total bile salts, and total bilirubin were markedly elevated in Runx1 cKO mice compared with the control mice after BDL. Similar changes in liver function tests were also observed on the 14th day following a 1% CA diet in Runx1 cKO mice (Supplemental Table 9, http://links.lww.com/HEP/A63).
Together, our findings demonstrated that genetic deletion of Runx1 in the liver aggravated hepatic inflammation and liver injury in cholestasis, suggesting that the cholestasis-induced hepatic RUNX1 protein plays a protective role in cholestasis.
Liver-specific ablation of Runx1 enhanced liver neutrophil infiltration and chemokine Ccl2 and Cxcl2 expression in BDL-induced cholestatic mice
Previous studies from us and others indicated that elevated levels of BA in hepatocytes stimulated the expression of chemokines, including Ccl2 and Cxcl2, which attract neutrophils and other inflammatory cells, resulting in an inflammatory response in cholestasis.[2–5]
Therefore, we examined the infiltration of inflammatory cells (eg, neutrophils and macrophages), the expression of chemokines (eg, Ccl2 and Cxcl2), and proinflammatory cytokines (eg, TNFα, IL-1β, and IL-6) in Runx1 cKO mice and Runx1flox/flox mice after BDL. Real-time quantitative PCR analysis demonstrated marked increases in hepatic mRNA levels of inflammatory cell markers Mpo (neutrophils), F4/80, Cd11b (macrophages), Cd3e, Cd8a (T cells), and Cd11c (dendritic cells), chemokines Ccl2 and Cxcl2, and proinflammatory cytokines TNFα, IL-1β and IL-6, in all BDL mice compared with their sham-operated mice, as we reported previously[2] (Figure 3A, B and Supplemental Figure 7, http://links.lww.com/HEP/A63).
FIGURE 3.

Effects of liver-specific ablation of Runx1 on liver neutrophil infiltration and expression of chemokines Ccl2 and Cxcl2 in a BDL-induced mouse model of cholestasis. (A) Relative levels of neutrophil markers Mpo and Elane mRNA transcripts. (B) Relative levels of chemokines Ccl2 and Cxcl2 mRNA transcripts. (C) Relative levels of Mpo, Cxcl2, and Ccl2 protein in the sham-operation group (n = 5 in each genotype) and BDL group (n = 7 in each genotype). *p < 0.05 versus sham-Runx1flox/flox mice; #p < 0.05 versus sham-Runx1 cKO mice; $p < 0.05 versus BDL-Runx1flox/flox mice. (D) IF labeling of Mpo in mouse liver sections in red and cell nucleus in blue (DAPI). Abbreviations: BDL, bile duct ligation; IF, immunofluorescence; Mpo, a specific marker for neutrophils.
Notably, hepatic mRNA and protein levels of neutrophil markers Mpo and/or Elane were significantly higher in Runx1 cKO mice than in Runx1flox/flox mice after BDL (Figure 3A, C). IF labeling of hepatic Mpo further confirmed that hepatic neutrophils were markedly increased in BDL-Runx1 cKO mice when compared with BDL-Runx1flox/flox mice (Figure 3D). However, real-time quantitative PCR and IF labeling of liver sections revealed no significant alterations in hepatic expression of other inflammatory cell markers F4/80, Cd11b (macrophages), Cd3e, Cd8a (T cells), and Cd11c (dendritic cells) between BDL-Runx1flox/flox mice and BDL-Runx1 cKO mice (Supplemental Figure 7A–D, http://links.lww.com/HEP/A63). These findings supported the conclusion that liver-specific deletion of Runx1 increased hepatic infiltration of neutrophils but not macrophages and other inflammatory cells in these cholestatic mice.
Interestingly, hepatic mRNA and protein levels of neutrophil-attracting Ccl2 and Cxcl2 were also dramatically increased in BDL-Runx1 cKO mice when compared with BDL-Runx1flox/flox mice (Figure 3B, C). In addition, the hepatic mRNA transcript levels of the proinflammatory cytokines IL-1β, IL-6, and TNFα were significantly higher in Runx1 cKO mice than Runx1flox/flox mice after BDL (Supplemental Figure 7E, http://links.lww.com/HEP/A63), indicating that liver-specific Runx1 deficiency promotes liver inflammation in cholestasis. These findings suggest that hepatic RUNX1 plays an important role in modulating the hepatic inflammatory response in cholestasis.
BA-induced RUNX1 expression decreases the expression of chemokines CCL2 and CXCL2 in hepatocytes
To determine whether BAs directly induce RUNX1 expression in the cholestatic liver, we first analyzed BA composition in the serum of BDL mice. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis revealed that serum levels of conjugated BAs and unconjugated BAs were dramatically elevated in BDL mice, with 293.2- and 11.7-fold increases, respectively, compared with sham-operated control mice (Supplemental Table 10, http://links.lww.com/HEP/A63).
Because we observed elevated expression of RUNX1 in hepatocytes from both humans and mice, we next asked whether BAs directly alter its expression. To address this question, we then treated primary mouse hepatocytes and the hepatoma PLC/PRF/5-ASBT cell line with conjugated BAs, including TCA, taurochenodeoxycholate acid, glycocholic acid, glycochenodexycholic acid, and taurodeoxycholate acid. Real-time quantitative PCR analyses demonstrated that treatment with BAs significantly increased the mRNA levels of RUNX1 and the 2 chemokines (CCL2 and CXCL2) (Figure 4A, B).
FIGURE 4.

BAs stimulated RUNX1 expression to decrease the expression of chemokines CCL2 and CXCL2 in hepatocytes. Conjugate BAs (including 100 μM TCA, taurochenodeoxycholate acid, glycocholic acid, glycochenodexycholic acid, and taurodeoxycholate acid) significantly increased RUNX1 (A) and chemokines CCL2 and CXCL2 expression (B) at the mRNA levels in PLC/PRF/5-ASBT cell line. *p < 0.05 versus control (DMSO) group (n = 3). (C) Overexpression of the RUNX1 protein abolished BA-induced CCL2 and CXCL2 mRNA expression. *p < 0.05 versus control (DMSO) group; #p < 0.05 versus the TCA-treated group (n = 3). (D) Deletion of Runx1 enhanced BA-induced Ccl2 and Cxcl2 mRNA expression in primary mouse hepatocytes. *p < 0.05 versus DMSO-treated-Runx1flox/flox group; #p < 0.05 versus DMSO-treated Runx1 cKO group; $p < 0.05 versus TCA-treated Runx1flox/flox group (n = 3). TCA induced RUNX1 mRNA levels (E) and nuclear protein (F) in a dose-dependent manner in PLC/PRF/5-ASBT cell line. *p < 0.05 versus DMSO group; #p < 0.05 versus 10 μM TCA group, n = 3. Chromatin immunoprecipitation assays including both real-time quantitative PCR (G) and semiquantitative PCR (H) analyses showed that TCA induced RUNX1 binding to its response element on the CCL2 or CXCL2 promoter in a dose-dependent manner in PLC/PRF/5-ASBT cell line. (I) Hepatic expression of Runx1 nuclear protein in 1% CA-fed mice (n = 7) and chow-fed mice (n = 5). *p < 0.05 versus chow diet group. (J) The activities of hepatic Runx1 binding to Ccl2 or Cxcl2 promoter in 1% CA-fed mice and chow-fed mice (n = 3 in each group). H2O, blank; H3, positive control; IgG, negative control. *p < 0.05 versus control group. Abbreviations: TCA, taurocholic acid; RUNX1, runt-related transcription factor-1.
Interestingly, overexpression of the RUNX1 protein in PLC/PRF/5-ASBT cells abolished BA-induced CCL2 and CXCL2 expression (Figure 4C). Conversely, the deletion of Runx1 enhanced BA-induced Ccl2 and Cxcl2 expression in primary mouse hepatocytes when compared with cells from WT mice (Figure 4D). Furthermore, treatment with a conjugated BA, TCA, increased RUNX1 mRNA and nuclear protein expression (Figure 4E, F) and enhanced the activities of RUNX1 binding to the promoters of CCL2 (ChIP site −258 to −248) or CXCL2 (ChIP site −1935 to −1925) (Figure 4G, H) in a dose-dependent manner.
Next, we examined Runx1 expression and its binding activities to the Ccl2 or Cxcl2 promoters in cholestatic liver tissues. As expected, hepatic expression of Runx1 nuclear protein and its binding activities to the Ccl2 (ChIP site −180 to −170) or Cxcl2 (ChIP site −1479 to −1469) promoter were significantly increased in both 1% CA-fed mice and BDL mice when compared with their corresponding control mice (Figures 1E, 4I, J and Supplemental Figure 8, http://links.lww.com/HEP/A63). These in vitro and in vivo study findings supported the notion that elevated BA-induced RUNX1 expression and binding to the promoter regions of CCL2 or CXCL2 in hepatocytes, thus leading to a reduction in the expression of the 2 chemokines.
BAs stimulated the nuclear expression of STAT3 and enhanced the binding activity of STAT3 to the RUNX1 promoter in hepatocytes
RUNX1 gene expression is regulated by the RUNX1-P1 promoter in hematopoietic stem cells or the RUNX1-P2 promoter in nonhematopoietic tissues, including the liver.[11,12] Luciferase reporter assays demonstrated that conjugated BAs significantly increased the activity of the RUNX1-P2 promoter without altering the RUNX1-P1 promoter activity (Figure 5A) in PLC/PRF/5-ASBT cells.
FIGURE 5.

BAs induced RUNX1 expression through the nuclear STAT3-induced RUNX1-P2 promoter in hepatocytes. (A) Conjugated BAs, such as TCA, GCA, and GCDCA, significantly increased the RUNX1-P2 but not RUNX1-P1 promoter activity in the human hepatoma PLC/PRF/5-ASBT cell line. *p < 0.05 versus control (DMSO) group, n = 3. (B) Conjugated BAs, including TCA, TCDCA, GCA, GCDCA, and TCDCA, markedly stimulated nuclear STAT3 protein expression in the PLC/PRF/5-ASBT cell line. *p < 0.05 versus control (DMSO) group, n = 3. (C) PLC/PRF/5-ASBT cells were transiently transfected with 3 different lengths of the RUNX1-P2 promoter with or without co-transfected STAT3 to identify the transcriptional regulatory site. *p < 0.05. (D) The RUNX1-P2 promoter activity induced by STAT3 overexpression was completely abolished by mutations in the key motif of the STAT3 response element in the RUNX1-P2. *p < 0.05. Conjugated BA and TCA significantly stimulated nuclear STAT3 protein expression in PLC/PRF/5-ASBT cell line. (E) TCA-induced nuclear STAT3 protein expression in a dose-dependent manner in the PLC/PRF/5-ASBT cell line. Chromatin immunoprecipitation assays including both real-time quantitative PCR (F) and semiquantitative PCR (G) methods demonstrated that TCA induced the binding activity of STAT3 to the RUNX1 promoter in a dose-dependent manner in PLC/PRF/5-ASBT cell line. *p < 0.05 versus DMSO group; #p < 0.05 versus 10 μM TCA group. (H) Hepatic Stat3 nuclear protein is increased in 1% CA-fed mice (n = 7) compared with chow-fed mice (n = 5). *p < 0.05 versus chow diet group. (I) The activities of hepatic Runx1 binding to the Runx1 promoter in 1% CA-fed mice and chow-fed mice (n = 3 in each group). H2O, blank; H3, positive control; IgG, negative control. *p < 0.05 versus control group. Abbreviations: BA, bile acid; CA, cholic acid; GCA, glycocholic acid; GCDCA, glycochenodexycholic acid; TCA, taurocholic acid; TCDA, taurodeoxycholate acid; TCDCA, taurochenodeoxycholate acid.
Next, in silico analysis of the promoter region of RUNX1-P2 (http://jaspar.genereg.net) identified a putative STAT3 response element (Supplemental Figure 9, http://links.lww.com/HEP/A63). Interestingly, Western-blotting analysis showed that conjugated BAs induced the nuclear expression of STAT3 protein in PLC/PRF/5-ASBT cells (Figure 5B).
We, therefore, prepared 3 luciferase reporter constructs with insertion of different fragments of the RUNX1-P2 promoter: −944 to +600, −778 to +600, or −308 to +600), into the pGL3-basic vector. We found that overexpression of STAT3 significantly increased the activity of the RUNX1-P2 promoter (region: −308 to +600) in PLC/PRR/5-ASBT (Figure 5C), while this effect was completely abrogated by a mutant RUNX1-P2 promoter (region: −308 to +600 in the STAT3 response element) (Figure 5D).
Furthermore, TCA increased the nuclear expression of STAT3 (Figure 5E) and enhanced the activity of STAT3 binding to the RUNX1-P2 promoter (ChIP site 3 located −18 to −8) (Figure 5F, G) in a dose-dependent manner in PLC/PRF/5-ASBT cells. Next, we examined the hepatic Stat3 nuclear protein and its binding activities to the Runx1 promoter in cholestatic mouse models. As expected, hepatic expression of Stat3 nuclear protein and its binding activities to the Runx1 promoter (ChIP site −1137 to −1128) were significantly increased in both 1% CA-fed mice and BDL mice when compared with their corresponding controls (Figure 5H, I and Supplemental Figure 10, http://links.lww.com/HEP/A63). These in vitro and in vivo data suggest that BA induces RUNX1 expression by stimulating STAT3 activity in the RUNX1-P2 promoter in hepatocytes.
BAs activated JAK-STAT3 signaling to increase the expression of RUNX1 and the binding activities of RUNX1 to Ccl2 and Cxcl2 promoters in hepatocytes
To investigate how BAs activate JAK/STAT3 signaling in hepatocytes, we examined their expression and phosphorylation in BA-treated cells. As illustrated in Figure 6A and B, conjugated BAs (including TCA, taurochenodeoxycholate acid, glycocholic acid, glycochenodexycholic acid, and taurodeoxycholate acid) induced phosphorylation of JAK and STAT3 in PLC/PRR/5-ASBT cells in a dose-dependent manner without affecting total JAK and STAT3 protein expression.
FIGURE 6.

BAs induced activation of JAK/STAT3 signaling and increased the expression of RUNX1 and its binding to the CCL2 and CXCL2 promoters in hepatocytes. (A) Conjugated BAs, including TCA, TCDCA, GCA, GCDCA, and TCDA, increased JAK1/2 and STAT3 phosphorylation but not total JAK1 and STAT3 protein in PLC/PRF/5-ASBT cell line. n = 3, *p < 0.05 versus DMSO group. (B) TCA induced JAK1/2 and STAT3 phosphorylation in a dose-dependent manner in PLC/PRF/5-ASBT cell line. *p < 0.05 versus DMSO group; #p < 0.05 versus 10 μM TCA group (n = 3). Ruxolitinib, a selective inhibitor of JAK signaling, abolished JAK1/2 and STAT3 phosphorylation (C) and markedly decreased nuclear STAT3 and RUNX1 protein levels in PLC/PRF/5-ASBT cell line (D). n = 3, *p < 0.05 versus DMSO group; #p < 0.05 versus 100 μM TCA group. Specific STAT3-binding peptide (APTSTAT3–9R) blocked STAT3 phosphorylation (E) and markedly reduced nuclear expression of RUNX1 protein in PLC/PRF/5-ASBT cell line (F). n = 3, *p < 0.05 versus DMSO group; #p < 0.05 versus 100 μM TCA group. (G) ChIP assays (real-time quantitative PCR method) demonstrated that ruxolitinib significantly abolished TCA-induced activities of RUNX1 binding to the CCL2 or CXCL2 promoter in PLC/PRF/5-ASBT cell line. (H) ChIP assays (real-time quantitative PCR method) displayed that APTSTAT3–9R abolished TCA-induced activities of RUNX1 binding to the CCL2 or CXCL2 promoter in PLC/PRF/5-ASBT cell line. *p < 0.05 versus DMSO group; #p < 0.05 versus 100 μM TCA group. Abbreviations: BA, bile acid; ChIP, chromatin immunoprecipitation; GCA, glycocholic acid; GCDCA, glycochenodexycholic acid; TCA, taurocholic acid; TCDA, taurodeoxycholate acid; TCDCA, taurochenodeoxycholate acid.
Interestingly, inhibition of the JAK1/2 and STAT3 phosphorylation with ruxolitinib, a selective inhibitor of JAK signaling, significantly reduced nuclear expression of STAT3 and RUNX1 protein levels in PLC/PRR/5-ASBT cells (Figure 6C, D). Moreover, blockage of STAT3 phosphorylation using APTSTAT3–9R, a specific STAT3-binding peptide, also significantly decreased the levels of nuclear RUNX1 protein and the activities of RUNX1 binding to CCL2 (−258 to −248) or CXCL2 (−1935 to −1925) promoters (Figure 6E–H). These data indicated that the activation of JAK/STAT3 signaling stimulated the expression of RUNX1, which in turn repressed the expression of CCL2 or CXCL2 in hepatocytes.
Hepatic JAK-STAT3 signaling was activated, and the binding activities of STAT3 to the RUNX1 promoter or RUNX1 to the CCL2/CXCL2 promoter were enhanced in OC patients
Based on the findings in hepatocytes and cholestatic mice, we questioned whether the JAK/STAT3 signaling pathway could be involved in the regulation of hepatic RUNX1 expression in human cholestasis? As shown in Figure 7A and B, the phosphorylation of hepatic JAK and STAT3 and nuclear STAT3 protein expression were significantly elevated in OC patients when compared with controls, indicating that JAK-STAT3 signaling was also activated in human cholestasis.
FIGURE 7.

Examinations of JAK/STAT3 signaling and binding activities of STAT3 to the RUNX1 promoter and RUNX1 to the CCL2 and CXCL2 promoter in patients with obstructive cholestasis. (A) Hepatic relative levels of JAK and STAT3 phosphorylation were remarkably elevated in patients with obstructive cholestasis (n = 18) compared with control patients (n = 11). *p < 0.05 versus control patients. (B) The relative levels of nuclear STAT3 protein were significantly increased in human obstructive cholestatic livers (n = 18) compared with control livers (n = 11). *p < 0.05 versus control patients. (C) Chromatin immunoprecipitation assays (real-time quantitative PCR method) demonstrated that the activity of STAT3 binding to RUNX1 promoter (Site 3) was significantly increased in liver tissues of patients with obstructive cholestasis compared with control patients, consistent with the results in the taurocholic acid–treated PLC/PRF/5-ASBT cell line. n = 4 in each group. (D) Chromatin immunoprecipitation assays (real-time quantitative PCR method) validated that the activities of RUNX1 binding to CCL2 and CXCL2 promoter were significantly increased in human obstructive cholestatic livers. n = 4 in each group. Relative hepatic levels of chemokine CCL2 and CXCL2 transcripts (E) and neutrophil markers ELANE and MPO transcripts (F) were significantly induced in obstructive cholestatic patients (n = 18) compared with control patients (n = 11). Abbreviation: RUNX1, runt-related transcription factor-1.
Increased activity of hepatic STAT3 binding to the RUNX1 promoter (−18 to −8) was also observed in OC patients (Figure 7C and Supplemental Figure 2C, http://links.lww.com/HEP/A63), together with induced hepatic RUNX1 expression (Figure 1A–C). Furthermore, RUNX1 nuclear protein binding to the CCL2 (−258 to −248) or CXCL2 (−1935 to −1925) promoter was also increased in human OC livers (Figure 7D and Supplemental Figure 11, http://links.lww.com/HEP/A63), along with elevated mRNA levels of chemokines CCL2 and CXCL2 (Figure 7E).
Real-time quantitative PCR analysis demonstrated that the mRNA levels of hepatic neutrophil markers ELANE and MPO were also markedly upregulated in human OC livers compared with control livers, consistent with the increases in above chemokine expression (Figure 7F). Altogether, these data suggest that JAK-STAT3 signaling involves hepatic RUNX1-mediated liver inflammation in human cholestasis.
DISCUSSION
In the current study, we examined the functional role and regulatory mechanism of the transcriptional factor RUNX1 in hepatocytes during cholestasis. The novel findings are summarized as follows: (1) hepatic RUNX1 expression was markedly increased in cholestatic livers from both patients (OC, PSC, and PBC) and animal models (Figure 1 and Supplemental Figures 3–5, http://links.lww.com/HEP/A63); (2) liver-specific ablation of Runx1 remarkably aggravated the BA-induced inflammatory response and cholestatic liver injury after BDL and 1% CA feeding in mice (Figures 2 and 3, and Supplemental Tables 8–10, http://links.lww.com/HEP/A63); and (3) overexpression of RUNX1 in hepatocytes repressed the BA-stimulated hepatic inflammatory response through activation of the JAK/STAT3 signaling pathway, which was further verified in liver tissues from OC patients (Figures 4–7). These findings demonstrate a functional role of hepatic RUNX1 in cholestasis and reveal its underlying regulatory mechanism that attenuates liver inflammation in these disorders (Figure 8).
FIGURE 8.

Schematic diagram illustrating the proposed mechanism by which hepatocyte RUNX1 attenuates BA-induced hepatic inflammation in human cholestasis. Initially, excessive accumulation of intrahepatic BA triggers a hepatic inflammatory response that initiates cholestatic liver injury.[2,24] Meanwhile, elevated intrahepatic BAs activate the JAK/STAT3 signaling pathway to stimulate RUNX1 expression in hepatocytes. RUNX1 attenuates hepatic inflammation and cholestatic liver injury by directly binding to CCL2 and CXCL2 promoters in the nuclei to suppress CCL2 and CXCL2 expression. Abbreviations: BA, bile acid; RUNX1, runt-related transcription factor-1.
IA recent study indicated that RUNX1 enhances inflammation in hepatic endothelial cells, which correlates with NASH severity.[19] Huang et al.[21] also demonstrated that the expression of Runx1 is elevated in hepatic stellate cells, and its silencing ameliorates liver fibrosis in CCl4-treated mice. Similarly, reduced expression of Runx1 in dendritic cells attenuates bacteria-induced liver injury.[22] Thus, these studies suggest the role that RUNX1 regulates inflammation and fibrosis in these nonparenchymal cells and tissues.
However, our scRNA-seq analysis of human livers indicated that RUNX1 was widely expressed in all liver cells, including hepatocytes, cholangiocytes, endothelial cells, mesenchymal cells, and blood cells (Supplemental Figure 3, http://links.lww.com/HEP/A63). Because the role of RUNX1 in liver nonparenchymal cells has been characterized in previous studies,[18,21,22] we determined the functional role of RUNX1 in liver parenchyma during cholestasis in this study. Using liver-specific Runx1 knockout mouse model, we demonstrated that hepatic RUNX1 exerted an anti-inflammatory effect in cholestasis. This is supported by multiple lines of evidence. First, liver-specific deletion of Runx1 significantly increased cholestatic liver injury and inflammation in vivo in murine models and in vitro in BA-stimulated chemokine expression in hepatocyte cultures (Figures 2–4). Second, overexpression of RUNX1 in hepatocytes repressed BA induction of chemokine expression (Figure 4).
Notably, RUNX1 protein was highly expressed in normal human cholangiocytes and moderately increased in cholestasis (Figure 1C and Supplemental Figure 3, http://links.lww.com/HEP/A63), suggesting that the cholangiocyte RUNX1 may also play a role in physiological and cholestatic conditions in humans. However, our scRNA-seq data indicated that the expression of Runx1 in mouse cholangiocytes was at a very low level in WT mice (Supplemental Figure 6, http://links.lww.com/HEP/A63). Multiplex IF analysis further confirmed that its protein expression in mouse bile ducts was still at a low level after BDL for 7 days (white arrows, Supplemental Figure 1C, http://links.lww.com/HEP/A63), indicating that RUNX1 expression in cholangiocytes has species specificity. However, it remains to be determined the functional role of RUNX1 in cholangiocytes in response to cholestasis. In the future bile duct–specific knockout or overexpression of RUNX1 transgenic mice may help to answer this question. Moreover, we also observed the different expression patterns of RUNX1 in immune cells between mouse and humans (Supplemental Figures 3 and 6, http://links.lww.com/HEP/A63). Therefore, the functional role of RUNX1 in immune cells under cholestasis also needs further investigation.
Recently, we demonstrated that when BAs accumulate in the murine liver, chemokines Ccl2 and Cxcl2 are induced in hepatocytes and initiate hepatic inflammation and cholestatic liver injury[2] (as confirmed in the current study), along with a dramatic elevation of hepatic RUNX1 (Figures 1 and 4A–F and Supplemental Figure 4, http://links.lww.com/HEP/A63). Further mechanistic studies revealed that RUNX1 inhibited the BA-induced inflammatory response by activating the JAK/STAT3 signaling pathway (Figure 8).
Interestingly, in respiratory epithelial cells, RUNX1 binds to IKKβ in the cytoplasm and competitively impedes the activation of NF-κB signaling.[17] However, the NF-κB signaling was not altered in BDL-Runx1 cKO mice compared with BDL-Runx1flox/flox mice (Supplemental Figure 12, http://links.lww.com/HEP/A63). Furthermore, the activities of NF-κB p65 binding to the Ccl2 or Cxcl2 promoter showed no differences between the 2 groups (Supplemental Figure 13C, E, http://links.lww.com/HEP/A63), as well as in control and OC patients (Supplemental Figure 13D, F, http://links.lww.com/HEP/A63). These data suggest that when RUNX1 attenuates liver inflammation it is independent of the NF-κB signaling, as we previously reported.[2]
In addition, some of us previously reported that conjugated BAs induced the nuclear translocation of the transcription factor, NFAT, to stimulate chemokine expression (eg, Ccl2) in cholestasis.[24] Intriguingly, here we demonstrated that conjugated BAs induced hepatocyte RUNX1 expression by JAK-STAT3 signaling to repress hepatic expression of chemokine and proinflammatory cytokines under cholestasis (Figure 8), suggesting that RUNX1 might act as a feedback repressor to prevent excessive liver inflammation during cholestasis. However, whether RUNX1 represses hepatic inflammation by interfering with the activity of NFAT and how BAs activate JAK/STAT3 in hepatocytes during cholestasis remains to be determined.
In conclusion, this study demonstrated that hepatocyte RUNX1 is markedly upregulated in cholestasis through JAK/STAT3 signaling. This upregulation counteracts inflammation-mediated cholestatic liver injury by repressing chemokine CCL2 and CXCL2 expression. Hence, these findings add a new mechanism to the complex pathogenesis of cholestasis and suggest that hepatocyte RUNX1 and its relevant signaling pathways might be new therapeutic targets for the treatment of cholestasis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr Ruilin Sun (Shanghai Model Organisms Center Inc., Shanghai, China) and team members (Cholestatic Liver Diseases Center, Southwest Hospital, Third Military Medical University, Chongqing, China) for their technical assistance.
Funding information
Supported by grants from the National Natural Science Foundation of China (92268110, 81922012), the Outstanding Youth Foundation of Chongqing (cstc2021jcyj-jqX0005), the Project of Chongqing Universities Innovation Research/Outstanding Medical Research Group (2021cqspt01 and 4246ZO1), and the Science Foundation of Southwest Hospital and Army Medical University (2017YQRC-01, XZ-2019-505-001, and XZ-2019-505-069), China.
Abbreviations:
- BA
bile acid
- BDL
bile duct ligation
- CA
cholic acid
- ChIP
chromatin immunoprecipitation
- GCA
glycocholic acid
- GCDCA
glycochenodexycholic acid
- IF
immunofluorescence
- LPS
lipopolysaccharide
- OC
obstructive cholestasis
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- RUNX1
runt-related transcription factor-1
- scRNA-seq
single-cell RNA sequencing
- TCA
taurocholic acid
- TCDA
taurodeoxycholate acid
- TCDCA
taurochenodeoxycholate acid
- WT
wild-type
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.hepjournal.com.
REFERENCES
- 1.Poupon R, Chazouilleres O, Poupon RE. Chronic cholestatic diseases. J Hepatol. 2000;32:129–40. [DOI] [PubMed] [Google Scholar]
- 2.Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. 2017;2:e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien KM, Allen KM, Rockwell CE, Towery K, Luyendyk JP, Copple BL. IL-17A synergistically enhances bile acid-induced inflammation during obstructive cholestasis. Am J Pathol. 2013;183:1498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology. 2003;38:355–63. [DOI] [PubMed] [Google Scholar]
- 6.Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. 2020;72:486–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu EH, Zheng ZN, Xiao CX, Liu X, Lin XQ. IL-22 relieves sepsis-induced liver injury via activating JAK/STAT3 signaling pathway. J Biol Regul Homeost Agents. 2020;34:1719–27. [DOI] [PubMed] [Google Scholar]
- 9.Xu G, Dai M, Zheng X, Lin H, Liu A, Yang J. Cholestatic models induced by lithocholic acid and alphanaphthylisothiocyanate: different etiological mechanisms for liver injury but shared JNK/STAT3 signaling. Mol Med Rep. 2020;22:1583–93. [DOI] [PubMed] [Google Scholar]
- 10.Appleford PJ, Woollard A. RUNX genes find a niche in stem cell biology. J Cell Biochem. 2009;108:14–21. [DOI] [PubMed] [Google Scholar]
- 11.Sroczynska P, Lancrin C, Kouskoff V, Lacaud G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood. 2009;114:5279–89. [DOI] [PubMed] [Google Scholar]
- 12.Telfer JC, Rothenberg EV. Expression and function of a stem cell promoter for the murine CBFalpha2 gene: distinct roles and regulation in natural killer and T cell development. Dev Biol. 2001;229:363–82. [DOI] [PubMed] [Google Scholar]
- 13.Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, et al. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995;23:2762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang X, Sun L, Wang G, Chen B, Luo F. RUNX1: a regulator of NF-κB signaling in pulmonary diseases. Curr Protein Pept Sci. 2018;19:172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35:934–46. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Lai Q, He C, Fang Y, Yan Q, Zhang Y, et al. RUNX1 promotes tumour metastasis by activating the Wnt/beta-catenin signalling pathway and EMT in colorectal cancer. J Exp Clin Cancer Res. 2019;38:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang X, Sun L, Jin X, Chen Y, Zhu H, Liang Y, et al. Runt-related transcription factor 1 regulates LPS-induced acute lung injury via NF-kappaB signaling. Am J Respir Cell Mol Biol. 2017;57:174–83. [DOI] [PubMed] [Google Scholar]
- 18.Tripathi DM, Rohilla S, Kaur I, Siddiqui H, Rawal P, Juneja P, et al. Immunonano-lipocarrier-mediated liver sinusoidal endothelial cell-specific RUNX1 inhibition impedes immune cell infiltration and hepatic inflammation in murine model of NASH. Int J Mol Sci. 2021;22:8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaur S, Rawal P, Siddiqui H, Rohilla S, Sharma S, Tripathi DM, et al. Increased expression of RUNX1 in liver correlates with NASH Activity Score in patients with non-alcoholic steatohepatitis (NASH). Cells. 2019;8:1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Li Y, Xiao JT, Wang HW, Guo Y, Mao XR, et al. Unique DUOX2+ACE2+ small cholangiocytes are pathogenic targets of primary biliary cholangitis. Nat Commun. 2022. doi: 10.1038/s41467-022-34606-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Li Y, Xu D, Zhang X, Zhou X. RUNX1 regulates SMAD1 by transcriptionally activating the expression of USP9X, regulating the activation of hepatic stellate cells and liver fibrosis. Eur J Pharmacol. 2021;903:174137. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Wang Q, Wang B, Gu Y, Yu H, Yang W, et al. Inhibition of EZH2 ameliorates bacteria-induced liver injury by repressing RUNX1 in dendritic cells. Cell Death Dis. 2020;11:1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Q, Zhang X, Zhang L, Cheng Y, Zhao N, Li F, et al. Solute carrier organic anion transporter family member 3A1 is a bile acid efflux transporter in cholestasis. Gastroenterology. 2018;155:1578–92.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai SY, Yu D, Soroka CJ, Wang J, Boyer JL. Hepatic NFAT signaling regulates the expression of inflammatory cytokines in cholestasis. J Hepatol. 2021;74:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Li L, Zhao N, Pan Q, Zhang L, Xie Q, et al. A novel role for interleukin 32 in cholestasis. Clin Transl Med. 2021;11:e594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chai J, Cai SY, Liu X, Lian W, Chen S, Zhang L, et al. Canalicular membrane MRP2/ABCC2 internalization is determined by Ezrin Thr567 phosphorylation in human obstructive cholestasis. J Hepatol. 2015;63:1440–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pozner A, Goldenberg D, Negreanu V, Le SY, Elroy-Stein O, Levanon D, et al. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol. 2000;20:2297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chai J, He Y, Cai SY, Jiang Z, Wang H, Li Q, et al. Elevated hepatic multidrug resistance-associated protein 3/ATP-binding cassette subfamily C 3 expression in human obstructive cholestasis is mediated through tumor necrosis factor alpha and c-Jun NH2-terminal kinase/stress-activated protein kinase-signaling pathway. Hepatology. 2012;55:1485–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan Q, Luo G, Qu J, Chen S, Zhang X, Zhao N, et al. A homozygous R148W mutation in semaphorin 7A causes progressive familial intrahepatic cholestasis. EMBO Mol Med. 2021;13:e14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang T, Poenisch M, Khanal R, Hu Q, Dai Z, Li R, et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J Hepatol. 2021;75:1420–33. [DOI] [PubMed] [Google Scholar]
- 31.Hargrove L, Kennedy L, Demieville J, Jones H, Meng F, DeMorrow S, et al. Bile duct ligation-induced biliary hyperplasia, hepatic injury, and fibrosis are reduced in mast cell-deficient Kit (W-sh) mice. Hepatology. 2017;65:1991–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.