

Abstract
Antithrombin is unique among serpin family protein protease inhibitors with respect to the major reactive center loop (RCL) and core conformational changes that mediate allosteric activation of its anticoagulant function by heparin. A critical role for expulsion of the RCL hinge from a native stabilizing interaction with the hydrophobic core in the activation mechanism has been proposed from reports that antithrombin variants that block this change through engineered disulfide bonds block activation. However, the sufficiency of core conformational changes for activation without expulsion of the RCL from the core is suggested by variants that are activated without the need for heparin and retain the native RCL–core interaction. To resolve these apparently conflicting findings, we engineered variants in which disulfides designed to block the RCL conformational change were combined with constitutively activating mutations. Our findings demonstrate that while a reversible constitutive activation can be engineered in variants that retain the native RCL–core interaction, engineered disulfides that lock the RCL native conformation can also block heparin allosteric activation. Such findings support a three-state allosteric activation model in which constitutive activating mutations stabilize an intermediate-activated state wherein core conformational changes and a major activation have occurred without the release of the RCL from the core but with a necessary repositioning of the RCL to allow productive engagement with an exosite. Rigid disulfide bonds that lock the RCL native conformation block heparin activation by preventing both RCL repositioning in the intermediate-activated state and the release of the RCL from the core in the fully activated state.
Graphical Abstract
Among members of the serpin superfamily of protein protease inhibitors, antithrombin, an essential anticoagulant regulator of blood clotting proteases, is unique with respect to the major conformational changes that mediate its allosteric activation by heparin.1 These unique conformational changes have been revealed by X-ray structures of native and heparin-activated antithrombin (Figure 1). Antithrombin is thus the only serpin known to have the hinge of the reactive center loop (RCL) inserted into the hydrophobic core of the major β-sheet, sheet A in the native repressed state, and the RCL expelled from sheet A in the heparin-activated state.2,3,a Moreover, antithrombin is the only one in which major conformational changes in the heparin binding site and core of the serpin take place as a consequence of heparin binding.4 The uniqueness of these structural features among serpins is evident from the classification of antithrombin in its own clade.5
Figure 1.

Conformational changes induced by heparin allosteric activation of antithrombin. X-ray structures of native (PDB entry 1E05), intermediate-activated (PDB entry 1NQ9), and fully activated antithrombin (PDB entry 1E03) are shown highlighting the major secondary structure elements, helix D (blue), sheet A (magenta), and the RCL (yellow), that undergo conformational changes upon allosteric activation with sheet B shown for reference (cyan). The bound heparin pentasaccharide in the two activated states is shown as sticks (red). The native state is characterized by the insertion of the RCL hinge P14 residue (side chain in space-filling representation) into a hydrophobic binding pocket in the middle of sheet A. Major core conformational changes are induced in the intermediate-activated state that result in a partial closing of the middle strands of sheet A but with retention of the RCL hinge–sheet A interaction. Additional conformational changes are induced in the fully activated state that fully close the gap in sheet A and expel the RCL hinge and extend helix D at its C-terminus. The similarity of the RCL conformation beyond the hinge region in all three states is a consequence of the RCL interacting with a second latent antithrombin molecule in the crystal structures and therefore does not reflect the true RCL conformation in solution.
More recent X-ray structures of heparin-activated antithrombin have identified an intermediate activation state in which just the heparin binding site and core conformational changes have occurred (Figure 1).6,7 This state has been presumed to be on the pathway to the fully activated state in which the RCL has been expelled from sheet A. Such structures have raised questions concerning the relative importance of these changes for activation. Do the core conformational changes merely serve to expel the RCL from sheet A with the latter being the critical conformational change that causes activation, or does the insertion of the RCL into the sheet A core prior to activation serve to maintain a low-activity conformation of the serpin body in the absence of heparin with the core conformational changes being most critical for activation? Early studies suggested that expulsion of the RCL from the core was the sine qua non for activation by allowing RCL-bound proteases to productively engage an exosite.8-13 Supporting this view, two reports found that antithrombin variants that lock the native reactive center loop (RCL) conformation through engineered disulfide bonds block RCL expulsion and allosteric activation by heparin.7,14 However, more recent reports have shown that antithrombin variants that are constitutively activated in the absence of heparin yet retain the native RCL inserted conformation can be engineered,15,16 thereby implying that core conformational changes are sufficient for activation with expulsion of the RCL not required for RCL-bound proteases to productively engage the exosite.
By using the same disulfide constraints but on a background of conformationally activating mutations, we now show that while the previous findings are correct, their interpretation of the critical role of the expulsion of the RCL from sheet A for antithrombin activation is not. This has allowed us not only to reconcile all published findings but also to propose a mechanism for antithrombin activation that explains both the need for core conformational change and the role of RCL hinge insertion.
MATERIALS AND METHODS
Proteins and Heparin.
Recombinant antithrombin variants were constructed on an N135Q background to prevent glycosylation of N135 and consequent heparin binding heterogeneity17 as in previous studies.18 Gene mutagenesis was carried out by polymerase chain reaction using specially designed oligonucleotides from Integrated DNA Technologies and Pfu Turbo DNA polymerase from Agilent/Stratagene according to the manufacturer’s instructions. All mutations were confirmed by DNA sequencing. Antithrombin variants with double-cysteine mutations designed to form a disulfide link between the RCL hinge P13 or P14 residue and strand 3 or 5 of sheet A included V375C/S380C, K222C/E381C, L373C/S380C, and E374C/E381C (Figure 2).7,14 A K222C/K139C double-cysteine variant linking adjacent residues in sheet A was engineered as a control. Some of these double-cysteine mutations were combined with mutations at the C-terminal end of helix D previously shown to partially or fully activate antithrombin in the absence of heparin.15,16 These changes included Y131L alone and Y131L combined with changes of residues 132–136 from RKAQK to the helix-promoting residues, EEAAE (designated hD) (Figure 2). The Y131L mutation was also combined with S380W to provide a reporter of the RCL conformational change accompanying heparin allosteric activation.19 Antithrombin variants were expressed in Sf9 insect cells using the baculovirus expression system (Thermo Fisher Scientific/Invitrogen) and purified from culture medium by a combination of heparin affinity and ion exchange chromatography, as previously described.18,20 The protein concentration was determined from the absorbance at 280 nm using an extinction coefficient of 37700 M−1 cm−1,21 except for P14 Trp and Y131L variants in which corrected extinction coefficients were calculated on the basis of the contributions of Trp and Tyr residues to the extinction coefficient.22 Purified human coagulation factors IXa and Xa were purchased from Enzyme Research Laboratories, and purified human thrombin was purchased from Molecular Innovations. Active protease concentrations were determined from standard substrate assays previously calibrated with the active site-titrated enzyme.23 A synthetic heparin pentasaccharide corresponding to the antithrombin binding sequence in heparin sufficient for allosteric activation of antithrombin was provided by Sanofi/Aventis (Toulouse, France). A full-length heparin (50-mer) containing the pentasaccharide binding sequence and capable of activating antithrombin reactions with protease through both allosteric and bridging mechanisms was prepared by size and affinity fractionation of commercial heparin as described previously.24
Figure 2.

Map of mutated antithrombin residues in the X-ray structure of native antithrombin. Shown on the left is the X-ray structure of native antithrombin (PDB entry 1E05) (cyan) with sheet A (magenta), helix D (hD, blue), and the RCL (yellow) highlighted and the residues mutated in this study shown as sticks. The boxed area is magnified on the right to more clearly show the mutated residues. Double-cysteine mutations designed to form disulfide bonds between the RCL P14 (S380) or P13 (E381) residue and sheet A residues, L373, E374, and V375 in strand 5 (s5A) and K222 in strand 3 (s3A), are labeled. A control disulfide between K222 on s3A and K139 on s2A was also engineered. Constitutive activating mutations were in residues Y131–K136 (YRKAQK → LEEAAE) in the loop extending from the C-terminal end of helix D (hD). The RCL P14 hinge residue, S380, was also mutated to Trp to provide a reporter of the RCL conformational change accompanying full heparin activation with or without the additional constitutive activating mutation, Y131L.
SDS–PAGE.
The purity of recombinant antithrombin variants and their ability to form SDS-stable complexes with thrombin were analyzed by SDS–PAGE on 10% polyacrylamide gels under reducing or nonreducing conditions using the Laemmli discontinuous buffer system.25 Reactions of antithrombin variants with thrombin were performed in I = 0.15 sodium phosphate buffer (pH 7.4) (see below) at 25 °C with 0.5–1.1 molar equivalents of the protease relative to serpin for 5–20 min followed by quenching with 40 μM FPR-chloromethylketone. SDS sample buffer was then added, and samples were boiled for 3–4 min prior to electrophoresis. Protein bands were detected by staining with GelCode Blue (Thermo Scientific). The effect of heparin on the substrate reaction of the E374C/E381C antithrombin variant with thrombin and factor Xa was analyzed on a 4–20% gradient gel with the Laemmli buffer system under nonreducing conditions based on the reduced electrophoretic mobility of antithrombin following RCL cleavage under these conditions.26 Estimates of the kcat/KM for thrombin and factor Xa cleavage were made by analyzing cleavage of 2 μM antithrombin variant over a 100-fold range of serial dilutions of the proteases (3–300 nM).
DTDP Assays.
Assays of antithrombin variants for free cysteines with the reagent DTDP was performed essentially as described previously27 in 0.1 M sodium phosphate, 0.2 mM EDTA, 7.5 M guanidine-HCl buffer (pH 7.0). Protein samples (1–6 μM) were mixed with buffer to yield a final concentration of 5 M guanidine-HCl and incubated for 15 min at room temperature to unfold the protein. The DTDP reagent was then added to a final concentration of 170 μM, and then the absorbance scanned between 250 and 400 nm to measure the peak absorbance of the 4-thiopyridone product of the reaction with free sulfhydryl groups at 324 nm relative to a buffer/reagent baseline. Known concentrations of free cysteine were assayed to provide a standard curve for calculation of sulfhydryl groups in antithrombin variants.
Experimental Conditions.
Antithrombin–protease reaction stoichiometries and kinetics were analyzed in I = 0.15, pH 7.4 buffers at 25 °C. Reactions with thrombin were conducted in 20 mM sodium phosphate, 100 mM NaCl, 0.1 mM EDTA, and 0.1% PEG 8000, whereas reactions with factor Xa and factor IXa were performed in 100 mM Hepes, 93.5 mM NaCl, 5 mM CaCl2, and 0.1% PEG 8000.
Inhibition Stoichiometries.
The stoichiometries of antithrombin inhibition of proteases (SI) were determined in the absence and presence of pentasaccharide and full-length heparins by end point assays essentially as in past studies.11,24 Fixed concentrations of protease and heparin (~100 nM) were incubated with increasing molar ratios of antithrombin to protease for a time sufficient to complete the reaction based on measured rate constants and yield >50% inhibition. Residual protease activity was then assayed by dilution of an aliquot into a 100 μM chromogenic or 50 μM fluorogenic substrate solution in assay buffer, and the initial linear absorbance (405 nm) or fluorescence (λex = 380 nm; λem = 440 nm) increase measured for several minutes. Plots of residual protease activity as a function of the molar ratio of antithrombin to protease were linear and yielded the stoichiometry of inhibition from the abscissa intercept. The substrates used were S2238 for thrombin (Chromogenix), Spectrozyme FXa for factor Xa (American Diagnostica), and Pefachrome or Pefafluor for factor IXa (Centerchem). Substrate solutions for factor IXa reactions were made up in assay buffer adjusted to pH 8 and supplemented with 33% ethylene glycol and 10 mM CaCl2 to increase assay sensitivity;28 50–100 μg/mL Polybrene was included in all substrates to neutralize heparin when present.
Kinetics of Antithrombin–Protease Reactions.
The kinetics of antithrombin inhibition of proteases was studied under pseudo-first-order conditions with antithrombin concentrations at least 5-fold greater than that required to fully inhibit protease based on measured inhibition stoichiometries as in previous studies.24,29 For reactions without heparin or in the presence of heparin pentasaccharide concentrations sufficient to fully complex antithrombin and with measured rate constants of ≤104 M−1 s−1, full reaction time courses of the loss of protease activity were measured by assaying residual protease activity in aliquots of reaction mixtures as a function of time as described in measurements of reaction stoichiometries. Progress curves were fit by a single-exponential decay function with a zero end point for thrombin and factor Xa reactions and a non-zero end point for factor IXa reactions to account for a minor fraction of degraded protease that is less susceptible to inhibition30 to obtain the observed pseudo-first-order rate constant (kobs). Apparent second-order association rate constants were calculated by dividing kobs by the concentration of antithrombin. For reactions in the presence of pentasaccharide or full-length heparin that were faster than 104 M−1 s−1, reactions were conducted for a fixed time in the presence of increasing concentrations of heparin at catalytic levels and residual protease activity was then assayed. The loss of protease activity was in this case fit by an exponential function with heparin concentration instead of time as the independent variable. The apparent second-order rate constant was then obtained by dividing the exponential rate constant by the product of the fixed reaction time and the fractional saturation of antithrombin with heparin, the latter calculated on the basis of the KD measured for heparin binding.29 Corrections of apparent second-order association rate constants for the extent of antithrombin reaction along the substrate pathway were made by multiplying by measured SIs to obtain the rate constant reflecting reaction along the inhibitory pathway (ka).31
Heparin Binding.
Binding stoichiometries and dissociation constants (KD) for antithrombin–heparin pentasaccharide interactions were measured by titrations of wild-type and variant antithrombins (100 nM) with the heparin pentasaccharide monitored from the enhancement of tryptophan fluorescence that accompanies heparin binding as in previous studies.24 Titrations were performed in 20 mM sodium phosphate, 0.1 mM EDTA, 0.1% PEG 8000 buffers with either no NaCl (I = 0.05) or 0.10 M NaCl (I = 0.15) (pH 7.4). At the lower ionic strength, binding was essentially stoichiometric due to low nanomolar heparin binding affinities, and thus, fitting of fluorescence titration data at this ionic strength by the equilibrium binding equation provided good fits of the heparin binding stoichiometry and the maximal fluorescence change. Fluorescence titrations performed at a physiological ionic strength (I = 0.15) were fit by the equilibrium binding equation with the stoichiometry fixed at the value determined in I = 0.05 buffer to obtain KD and the maximal fluorescence change.
Fluorescence Spectra.
Protein fluorescence spectra of antithrombin variants were acquired in I = 0.15 sodium phosphate buffer (pH 7.4) at 25 °C from 300 to 400 nm in 5 nm steps with 10 s integrations and 4 nm excitation and 16 nm emission bandwidths on an SLM 8000 spectrofluorometer. Spectra were obtained at least in duplicate in the absence and presence of an ~10-fold molar excess of heparin pentasaccharide to fully complex the inhibitor, averaged, and then corrected for buffer and dilution by heparin (<1%). Stoichiometric heparin binding titrations in I = 0.05 NaPi buffer (pH 7.4) were performed to ensure equivalent concentrations of functional protein. In the case of the S380W/Y131L and Y131L antithrombin variants, corrections for a minor fraction of inactive, presumably latent protein were made on the basis of the indistinguishable fluorescence of native and latent forms of the serpin.32
Melting Temperature.
Thermal denaturation analysis of antithrombin variants was performed over the temperature range of 40–75 °C at 100 nM protein in I = 0.15 sodium phosphate buffer (pH 7.4) by monitoring changes in tryptophan fluorescence that accompany denaturation as described previously.29 Measurements were taken on an SLM 8000 spectrofluorometer at a λex of 280 nm and a λem of 340 nm with temperature regulation by a Peltier-controlled cuvette holder from Quantum Northwest. Melting curves were analyzed by nonlinear regression fitting to the van’t Hoff equation to generate the melting temperature (TM).29
RESULTS
Introduction of Double-Cysteine and Constitutively Activating Mutations in Antithrombin Minimally Affects Reactivity with Thrombin.
To resolve apparently discrepant findings regarding the importance for heparin allosteric activation of antithrombin of the hallmark RCL conformational change accompanying activation (viz. expulsion of the RCL hinge from the hydrophobic core of sheet A), we engineered variants that combined mutations that blocked this conformational change and activation with those that caused constitutive activation without the RCL conformational change. Antithrombin variants with double cysteines designed to link the RCL and sheet A to block the RCL conformational change were thus engineered as previously reported7,14 and combined with mutations at the C-terminal end of helix D previously shown to partially and fully activate antithrombin without heparin and without inducing the RCL conformational change.16 The latter helix-promoting mutations are thought to activate antithrombin by mimicking the extension of helix D, another signature conformational change accompanying heparin allosteric activation, but in a manner that uncouples this change from the RCL conformational change. Controls included variants with just the engineered disulfides or the constitutive activating mutations. Figure 2 shows the locations of the double-cysteine mutations designed to link the RCL hinge P14 or P13 residue to strand 3 or 5 of sheet A as well as the constitutive activating mutations in the loop extending from the C-terminus of helix D. A control disulfide variant linking adjacent s3A and s2A residues, not expected to block the RCL conformational change, was also engineered.
All variants were pure and reactive with thrombin as determined by SDS–PAGE analysis (not shown). All formed SDS-stable complexes with thrombin except for the E374C/E381C variant whose reaction with thrombin resulted in an electrophoretic mobility shift indicative of the preferential RCL cleavage of antithrombin as a substrate (Figure 3). The Y131L/V375C/S380C variant was mostly unreactive with thrombin on SDS–PAGE, suggesting a significant fraction of inactive, presumably latent inhibitor. This was verified by measurement of a high stoichiometry of thrombin inhibition (Table 1). All antithrombin mutants exhibited rates and stoichiometries of thrombin inhibition in the absence and presence of pentasaccharide or full-length heparins (Table 1) indistinguishable or only modestly decreased from those of the wild type, except for the E374C/E381C variant that showed a greatly increased inhibition stoichiometry (SI > 100) consistent with the SDS–PAGE results. Estimation of the kcat/KM for thrombin cleavage of the latter variant was consistent with a wild-type-like reactivity (~104 M−1 s−1) in the rate-limiting Michaelis complex and acylation reaction steps. The reaction of the Y131L/V375C/S380C variant with thrombin was also similar to that of the wild type after correction of apparent rate constants for the fraction of the active inhibitor (Table 1). All variants thus appeared to function normally as inhibitors or substrates of thrombin in native and allosterically activated states and also exhibited near-normal full-length heparin bridging rate enhancements, consistent with the mutations not adversely affecting reactivity with this protease.
Figure 3.

SDS–PAGE analysis of the effect of heparin on the substrate reaction of E374C/E381C antithrombin with thrombin and factor Xa. Reactions of 2 μM E374C/E381C antithrombin with catalytic thrombin (30 nM) or FXa (300 nM) were performed in the absence (lanes 2, 4, and 6) or presence of 2 μM full-length heparin (H50) (lanes 3 and 5) or 4 μM heparin pentasaccharide (H5) (lane 7) for 15 min followed by quenching with a molar excess of tripeptide chloromethylketone inhibitor (FPR for thrombin and EGR for FXa) and analyzed by SDS–PAGE on a 4–20% gradient gel under nonreducing conditions as described in Materials and Methods. Components present in each reaction mixture are indicated below each lane of the gel. The protease concentrations sufficient to produce a minor cleavage in the absence of heparin were determined on the basis of estimates of the kcat/KM for cleavage as described in Materials and Methods. Reactions with catalytic human neutrophil elastase (HNE) (60 nM) in the absence (lane 8) and presence of H50 (lane 9) for 15 min were performed as a control to demonstrate the nearly full reactivity of the variant as a substrate.35 The addition of an equivalent amount of unreacted antithrombin to the latter reaction mixture after quenching with AAPV-chloromethylketone confirmed the identity of the lower-mobility antithrombin band as RCL-cleaved antithrombin (lane 10).
Table 1.
Stoichiometries and Kinetics of Protease Inhibition by Antithrombin Variantsa
| AT variant | SI−H | k a,−H | SIH5 | k a,H5 |
ka,H5/ ka,−H |
SIH50 | k a,H50 |
ka,H50/ ka,H5 |
|---|---|---|---|---|---|---|---|---|
| Thrombin Reactions | ||||||||
| wild type | 1.2 ± 0.1 | (1.1 ± 0.2) × 104 | 1.3 ± 0.1 | (2.3 ± 0.3) × 104 | 2.1 | 1.7 ± 0.1 | (3.4 ± 0.5) × 107 | 1500 |
| V375C/S380C | 1.8 ± 0.2 | (3.8 ± 0.1) × 103 | 2.4 ± 0.3 | (5.0 ± 0.9) × 103 | 1.3 | 2.8 ± 0.1 | (2.2 ± 0.6) × 107 | 4400 |
| L373C/S380C | 1.2 ± 0.1 | (6.4 ± 0.7) × 103 | 1.2 ± 0.1 | (7.7 ± 0.8) × 103 | 1.2 | 1.7 ± 0.1 | (9.2 ± 1.7) × 106 | 1200 |
| K222C/E381C | 1.4 ± 0.1 | (5.3 ± 0.4) × 103 | 2.5 ± 0.1 | (9.0 ± 0.6) × 103 | 1.7 | 14 ± 1 | (4.1 ± 0.4) × 107 | 4600 |
| K222C/K139C | 1.1 ± 0.1 | (1.3 ± 0.2) × 104 | 1.1 ± 0.1 | (8.5 ± 1.2) × 103 | 0.7 | 2.9 ± 0.2 | (2.6 ± 0.3) × 107 | 3100 |
| S380C | 1.0 ± 0.1 | (8.1 ± 0.9) × 103 | – | – | – | 1.5 ± 0.2 | (2.0 ± 0.6) × 107 | 2500b |
| helix D | 1.4 ± 0.1 | (1.1 ± 0.3) × 104 | 1.6 ± 0.1 | (1.3 ± 0.2) × 104 | 1.2 | 1.8 ± 0.1. | (8.5 ± 1.4) × 106 | 650 |
| helix D/L373C/S380C | 1.8 ± 0.2 | (5.9 ± 2.6) × 103 | 2.6 ± 0.1 | (9.1 ± 1.9) × 103 | 1.5 | 3.4 ± 0.2 | (3.0 ± 0.2) × 106 | 330 |
| Y131L | 1.5 ± 0.1 | (1.3 ± 0.2) × 104 | 1.6 ± 0.1 | (2.3 ± 0.2) × 104 | 1.8 | 2.5 ± 0.1 | (2.1 ± 0.3) × 107 | 910 |
| Y131L/V375C/S380C | 60 ± 2 | (7.9 ± 1.0) × 103 | 79 ± 5 | (1.0 ± 0.2) × 104 | 1.3 | 68 ± 7 | (1.3 ± 0.2) × 107 | 1300 |
| S380W | 14 ± 1 | (1.0 ± 0.1) × 104 | 8.8 ± 0.2 | (2.5 ± 0.1) × 104 | 2.5 | 25 ± 1 | ND | – |
| Y131L/S380W | 6.7 ± 0.5 | (9.5 ± 1.1) × 103 | 4.2 ± 0.2 | (1.2 ± 0.1) × 104 | 1.3 | 13 ± 1 | (3.6 ± 0.4) × 106 | 300 |
| Factor Xa Reactions | ||||||||
| wild type | 1.2 ± 0.1 | (4.0 ± 0.8) × 103 | 1.8 ± 0.3 | (1.4 ± 0.3) × 106 | 350 | 2.1 ± 0.1 | (1.8 ± 0.1) × 107 | 13 |
| V375C/S380C | 2.0 ± 0.4 | (5.3 ± 1.3) × 102 | 5.6 ± 0.8 | (1.6 ± 1.0) × 105 | 300 | 5.0 ± 0.3 | (7.0 ± 0.9) × 106 | 44 |
| L373C/S380C | 1.2 ± 0.1 | (6.5 ± 0.7) × 102 | 2.0 ± 0.1 | (2.2 ± 0.3) × 105 | 340 | 2.0 ± 0.2 | (4.0 ± 0.8) × 106 | 18 |
| K222C/E381C | 2.3 ± 0.1 | (4.1 ± 0.4) × 102 | 4.7 ± 0.3 | (7.0 ± 1.8) × 102 | 1.7 | 10 ± 1 | (1.1 ± 0.2) × 105 | 160 |
| K222C/K139C | 1.1 ± 0.1 | (2.6 ± 0.3) × 103 | 2.0 ± 0.1 | (1.5 ± 0.2) × 105 | 58 | 7.7 ± 1.4 | (2.4 ± 0.5) × 106 | 16 |
| S380C | 1.2 ± 0.1 | (1.6 ± 0.2) × 103 | 1.9 ± 0.1 | (6.3 ± 0.7) × 105 | 750 | – | – | – |
| helix D | 1.4 ± 0.1 | (3.1 ± 0.6) × 105 | 2.2 ± 0.3 | (1.0 ± 0.2) × 106 | 3.2 | 5.6 ± 0.8 | (2.4 ± 0.6) × 107 | 24 |
| helix D/L373C/S380C | 2.3 ± 0.1 | (6.9 ± 1.0) × 103 | 4.2 ± 0.3 | (4.0 ± 0.4) × 105 | 58 | 5.1 ± 0.6 | (1.3 ± 0.3) × 106 | 3.3 |
| Y131L | 1.7 ± 0.2 | (4.7 ± 0.9) × 104 | 1.8 ± 0.2 | (8.4 ± 1.6) × 105 | 18 | 1.8 ± 0.2 | (2.6 ± 0.4) × 106 | 3.1 |
| Y131L/V375C/S380C | 65 ± 5 | (1.5 ± 0.3) × 103 | 180 ± 3 | (5.4 ± 0.3) × 105 | 360 | 69 ± 2 | (2.0 ± 0.1) × 106 | 3.7 |
| S380W | 28 ± 2 | (1.3 ± 0.2) × 104 | 82 ± 4 | (6.0 ± 1.0) × 105 | 46 | 55 ± 2 | (2.9 ± 0.4) × 106 | 4.8 |
| Y131L/S380W | 19 ± 1 | (8.9 ± 1.2) × 104 | 32 ± 2 | (5.4 ± 1.0) × 105 | 6.1 | 28 ± 1 | (2.8 ± 0.3) × 106 | 5.2 |
| Factor IXa Reactions | ||||||||
| wild type | 1.0 ± 0.1 | (1.8 ± 0.3) × 102 | 1.0 ± 0.1 | (2.5 ± 0.4) × 104 | 140 | 1.1 ± 0.1 | (6.0 ± 0.8) × 106 | 240 |
| V375C/S380C | ND | (3.9 ± 0.1) × 100c | 5.6 ± 0.4 | (7.8 ± 1.1) × 103 | >360c | 7.3 ± 0.7 | (1.2 ± 0.3) × 107 | 1500 |
| L373C/S380C | ND | (2.1 ± 0.8) × 100c | 1.4 ± 0.1 | (7.6 ± 1.5) × 103 | >2600c | 1.4 ± 0.1 | (4.8 ± 0.9) × 105 | 63 |
| K222C/E381C | ND | (0.2 ± 0.1) × 100c | ND | (0.2 ± 0.1) × 100c | 1 | 2.8 ± 0.2 | (4.5 ± 0.9) × 102 | >800c |
| K222C/K139C | ND | (5.2 ± 0.1) × 10c | 2.0 ± 0.2 | (7.6 ± 1.0) × 103 | 150 | – | – | – |
| S380C | 1.0 ± 0.1 | (2.7 ± 0.2) × 10 | 1.0 ± 0.1 | (1.3 ± 0.1) × 104 | 480 | – | – | – |
| helix D | 1.4 ± 0.1 | (1.8 ± 0.2) × 104 | 1.3 ± 0.4 | (2.9 ± 1.4) × 104 | 1.6 | – | – | – |
| helix D/L373C/S380C | ND | (1.2 ± 0.1) × 10c | 2.3 ± 0.1 | (9.2 ± 0.6) × 103 | >330c | – | – | – |
Stoichiometries of inhibition (SI) and second-order association rate constants (ka) for antithrombin–protease reactions were determined in I = 0.15 sodium phosphate buffer for thrombin reactions or in I = 0.15 Hepes/calcium buffer for FXa and FIXa reactions, all at pH 7.4 and 25 °C in the absence of heparin (−H), in the presence of heparin pentasaccharide (+H5), or in the presence of full-length heparin (+H50), as described in Materials and Methods. All measurements were made at least in duplicate with errors representing the range or standard error. Heparin allosteric activation rate enhancements are indicated by the kH5/k−H ratio, and heparin bridging rate enhancements are indicated by the kH50/kH5 ratio.
The ratio represents ka,H50/ka,−H.
Apparent values of ka because slow reaction rates precluded measurement of SIs. Lower limits of rate enhancements are based on corrections of apparent ka values assuming SIs no greater than those measured in the presence of heparin.
Effects of Double-Cysteine Mutations on Disulfide Formation and Stability.
Surprisingly, upon assaying the variants for free cysteines with the reagent 4,4′-dithiodipyridine (DTDP) under denaturing conditions, we found that neither the V375C/S380C nor L373C/S380C variant had formed significant disulfide based on >80% of the engineered cysteines being free (Table 2). The reliability of the assay was verified by controls showing the absence of free cysteines in wild-type antithrombin and the presence of one free cysteine in the single-cysteine antithrombin variants S380C and R393C (1.02 ± 0.06 cysteines/antithrombin). By contrast, the K222C/E381C, K222C/K139C, and E374C/E381C variants showed minimal free cysteine, consistent with the engineered cysteines in a major fraction of these variants (80–90%) having formed a disulfide bond. We did not have sufficient protein to assay the combined Y131L/V375C/S380C and hD/L373C/S380C variants due to their poorer expression.
Table 2.
Heparin Affinities, Stabilities, and Free Sulfhydryl Contents of Antithrombin Variantsa
| AT variant | KD (nM) | ΔFmax/Fo (%) |
TM (°C) | [−SH]/[AT] |
|---|---|---|---|---|
| Wild type | 2 ± 1 | 44 ± 2 | 56.4 ± 0.3 | 0.04 ± 0.08 |
| V375C/S380C | 90 ± 12 | 9 ± 1 | 58.4 ± 0.2 | 1.52 ± 0.18 |
| L373C/S380C | 81 ± 4 | 19 ± 1 | 58.2 ± 0.2 | 1.57 ± 0.09 |
| S380C | 48 ± 7 | 36 ± 2 | 58.8 ± 0.3 | 1.08 ± 0.07 |
| E374C/E381C | 78 ± 14 | 12 ± 1 | 59.2 ± 0.3 | 0.38 ± 0.12 |
| K222C/E381C | 21 ± 2 | 15 ± 1 | 63.6 ± 0.1 | 0.20 ± 0.12 |
| K222C/K139C | 9 ± 4 | 17 ± 1 | 55.3 ± 0.3 | 0.44 ± 0.19 |
| helix D | 1 ± 1 | 26 ± 2 | 50.6 ± 0.2 | – |
| helix D/L373C/S380C | 1 ± 2 | 7 ± 1 | 52.1 ± 0.1 | – |
Heparin binding affinities (KD) and fluorescence enhancements induced by heparin binding (ΔFmax/Fo) were measured by fluorescence titrations of 100 nM antithrombin variant with heparin pentasaccharide in I = 0.15 sodium phosphate buffer (pH 7.4). Melting temperatures (TM) were determined by thermal denaturation profiles in the same buffer. Free sulfhydryl groups were measured by the DTDP assay in guanidine-HCl buffer as described in Materials and Methods. All measurements were made at least in duplicate with errors representing either the range or standard error.
The melting temperatures of these variants were consistent with stabilization of some but not all by the double-cysteine mutations (Table 2). The V375C/S380C and L373C/S380C variants showed melting temperatures slightly increased (1.6–1.8 °C) versus that of the wild type (56.4 ± 0.3 °C). However, such an increase was similar to that observed with the S380C single mutant and could be ascribed to the stabilizing effect of the cysteine substitution of the RCL hinge P14 serine residue on the interaction with the sheet A hydrophobic core in native antithrombin.29,33 By contrast, the E374C/E381C and K222C/E381C variants exhibited melting temperatures significantly increased by 3 and 7 °C, respectively, relative to that of the wild type, consistent with these surface-exposed cysteines having formed a stabilizing disulfide bond. However, the disulfide bond in the control K222C/K139C variant slightly decreased stability, suggesting that the disulfide in this case introduced conformational strain in the native state.34 The double-cysteine mutations also only slightly increased stability in the hD/L373C/S380C combined variant relative to the hD control, the latter of which exhibited a destabilizing 5 °C decrease in melting temperature.
The formation of a disulfide bond in the K222C/E381C variant was also evidenced in the previous study of this variant by an increased mobility on nonreducing SDS–PAGE relative to the wild type.14 We similarly observed significantly increased mobilities on nonreducing SDS–PAGE of this variant as well as the E374C/E381C variant, whereas the K222C/K139C, V375C/S380C, and L373C/S380C variants showed marginal mobility changes when compared to the wild type on nonreducing SDS–PAGE (not shown).
Heparin Activation Induces Limited RCL Expulsion in Double-Cysteine Variants.
To assess the extent to which the double-cysteine mutations blocked the RCL conformational change, we examined the tryptophan fluorescence changes induced by heparin binding that signal the expulsion of the RCL from the hydrophobic core of sheet A. The heparin pentasaccharide induced tryptophan fluorescence changes in all double-cysteine variants that were significantly reduced (2–4-fold) from that of the wild type (Table 2). Control variants without the double-cysteine mutations, including the helix D variant and an S380C single-cysteine variant, showed closer to normal fluorescence enhancements in agreement with previous reports. Titrations of these changes showed that heparin bound all variants with stoichiometries close to 1 (0.7–1.0) and with 10–40-fold reduced heparin affinities relative to that of the wild type, except for hD and hD/L373C/S380C variants, which bound heparin with indistinguishable nanomolar affinities comparable to that of the wild type. The wild-type-like heparin affinities of the hD variants were consistent with the uncoupling of the conformational activating effects of the helix D mutations on inhibitory activity from comparable effects on heparin affinity.16 Notably, the reduced fluorescence changes and heparin affinities we observed for V375C/S380C and K222C/E381C variants were similar to those previously reported,7,14 despite the fact that the former variant had not formed a disulfide in our study. Thus, all double-cysteine mutations limit the extent of heparin-induced expulsion of the RCL from sheet A and reduce heparin affinity regardless of whether a disulfide forms with the exception of the hD/L373C/S380C variant.
Only Double-Cysteine Variants That Form Disulfides Block Heparin Allosteric Activation.
All variants except for the E374C/E381C variant that reacted as a substrate of FXa showed basal rates of factor Xa inhibition significantly decreased compared to that of the wild type (7–10-fold) (Table 1). Notably, heparin pentasaccharide binding to the V375C/S380C, L373C/S380C, and K222C/K139C variants enhanced the rates of factor Xa inhibition ~60–300-fold to reach similar allosterically activated rates and rate enhancements approaching but still less than those of the wild type. By contrast, binding of the pentasaccharide to the K222C/E381C variant resulted in no significant enhancement of the basal reaction rate. SDS–PAGE analysis showed that the heparin pentasaccharide similarly produced no detectable enhancement of the rate of the substrate reaction of the E374C/E381C variant with FXa (Figure 3). Notably, the reactions with factor Xa of the K222C/E381C and E374C/E381C variants that had formed disulfide bonds were accelerated by a full-length heparin, indicating that while the disulfide bonds had blocked allosteric activation of these variants, they did not affect rate enhancement through the heparin bridging mechanism (Table 1 and Figure 3). Similar findings were made in analyzing the kinetics of inhibition of factor IXa by the double-cysteine variants. All variants thus exhibited major 50–100-fold decreased basal rates of factor IXa inhibition relative to that of the wild type whereas heparin pentasaccharide binding produced rates close to that of the activated wild type and supranormal allosteric activation rate enhancements with the V375C/S380C, L373C/S380C, and K222C/K139C variants but failed to enhance the rate of inhibition of the K222C/E381C variant (Table 1). The V375C/S380C and L373C/S380C double-cysteine variants that had not formed disulfides as well as the control K222C/E139C variant that formed a disulfide outside the RCL thus all appeared to be robustly allosterically activated by the heparin pentasaccharide, whereas the K222C/E381C and E374C/E381C variants that had formed RCL–sheet A disulfides appeared to have lost the ability to be allosterically activated.
Such findings confirmed the report that the K222C/E381C disulfide variant that links the RCL to s3A blocks heparin allosteric activation14 and extended these findings by showing that another E74C/E381C disulfide variant that links the RCL to s5A similarly blocks allosteric activation. The observation that the V375C/S380C double-cysteine variant as well as the L373C/S380C variant did not form a disulfide accounts for their ability to undergo normal allosteric activation rate enhancements. The burial of the s5A V375C and L373C cysteines and RCL hinge P14 cysteine beneath sheet A and the consequent loss of conformational mobility presumably constrain their ability to form a disulfide. By contrast, the free cysteines in the E374C/E381C and K222C/E381C variants are surface-exposed and exhibit greater degrees of conformational freedom, accounting for their propensity to form a disulfide. Interestingly, analysis of the propensity of all the engineered double-cysteine mutants to form a disulfide bond with “Disulfide by Design” software that accounts for the strict geometric requirements of adjacent cysteines forming disulfides36 did not predict any of these pairs of residues to form disulfide bonds in native antithrombin structures (PDB entry 1E05 or 1T1F). However, Cβ–Cβ distances were the shortest for the three variants observed to form disulfides and most comparable to the Cβ–Cβ distances of the three natural disulfides in antithrombin (Table 3). While the previous study of the K222C/E381C variant provided supporting evidence that the engineered cysteine pair did form a disulfide bond based on the DTDP assay and altered mobility on nonreducing SDS–PAGE,14 no comparable evidence in the subsequent study of the V375C/S380C variant was provided to show that the engineered cysteines had formed a disulfide.7 It is nevertheless possible that different folding conditions of the antithrombin variants in our insect cell expression system versus the previous mammalian cell expression system could account for the different propensities of the s5A-P14 double-cysteine variants to form disulfide bonds.
Table 3.
Side Chain Distances of Native and Substituted Cysteines in Native Antithrombin Structuresa
| 1E05 | 1T1F | |||
|---|---|---|---|---|
| AT residues | Cα–Cα | Cβ–Cβ | Cα–Cα | Cβ–Cβ |
| V375–S380 | 4.9 | 6.1 | 4.8 | 5.9 |
| L373–S380 | 6.9 | 6.4 | 6.8 | 6.5 |
| E374–E381 | 5.4 | 5.2 | 5.6 | 5.8 |
| K222–E381 | 5.2 | 4.5 | 5.3 | 4.4 |
| K222–K139 | 5.1 | 4.3 | 5.3 | 4.5 |
| C8–C128 | 5.5 | 4.1 | 5.7 | 3.8 |
| C21–C95 | 5.5 | 4.2 | 5.5 | 3.8 |
| C247–C430 | 6.2 | 3.6 | 6.2 | 3.8 |
Distances (in angstroms) between α-carbons and β-carbons of paired cysteine residues were measured in dimeric (PDB entry 1E05) and monomeric (PDB entry 1T1F) native antithrombin structures with PyMol version 2.2.3.
V375C/S380C and L373C/S380C Double-Cysteine Variants Reverse the Activating Rate Enhancements of Helix D Mutations.
While the V375C/S380C and L373C/S380C double-cysteine variants were found to not form disulfides, they did produce a marked stabilization of the native state similar to that of the S380C variant (Table 2). It was thus interesting to find that when these double-cysteine mutations were combined with mutations in helix D previously shown to allosterically activate antithrombin in the absence of heparin, the activating effects were significantly attenuated (Table 1). The activating effects of the Y131L or hD mutations were thus reduced when combined with the double-cysteine mutations from ~10- and ~100-fold enhancements of the factor Xa basal rates for Y131L and hD mutations alone to ~2- and ~10-fold enhancements in the context of the double-cysteine mutations. The cysteine mutations thus suppressed but did not abolish the constitutive activations. These suppressed activating effects were reversed upon heparin allosteric activation of the variants, which resulted in ~300- and ~60-fold further enhancements of the basal rates of factor Xa inhibition for Y131L and hD combined variants, respectively, to yield full wild-type-like activation of the variants similar to that of the control variants lacking the double cysteines.
Similar RCL Conformational Changes Are Induced by Heparin in Constitutively Activated Antithrombin Variants.
Our previous findings showed that mutations in helix D that activate antithrombin in the absence of heparin did not result in the changes in tryptophan fluorescence that signal allosteric activation, whereas heparin binding did induce these changes with minimal further activation.16 Whereas Trp225 and Trp307 are the major contributors to these changes in wild-type antithrombin,37 an RCL hinge P14 Trp variant was previously shown to directly report the expulsion of the buried RCL P14 Trp from sheet A.19 The P14 Trp fluorescence of the variant could thus be resolved relative to the wild type and be shown to undergo a 17 nm red-shift upon heparin allosteric activation, consistent with the transfer of the P14 Trp from a buried hydrophobic to a solvent-exposed aqueous environment.
We incorporated the P14 Trp substitution into a Y131L variant antithrombin that is partially activated in the absence of heparin to assess whether the activated mutant induces the RCL conformational change. The wild type and Y131L and S380W single mutants served as controls. All mutants were pure and fully reactive with thrombin based on SDS–PAGE analysis (not shown). However, as expected from previous studies, the P14 Trp variants preferentially reacted as substrates of thrombin and formed little or no detectable stable complexes. This was evident from both SDS–PAGE results and the observed stoichiometries of thrombin inhibition that were significantly greater than 1 in contrast to the wild type and Y131L controls that inhibited thrombin with stoichiometries close to 1 (Table 1). The preferred substrate reaction is a consequence of the P14 Trp hindering the RCL conformational change that traps the protease as an SDS-stable acyl–intermediate complex.29 Despite the increased inhibition stoichiometries, all variant antithrombins inhibited thrombin in the absence or presence of pentasaccharide or full-length heparins with rate constants indistinguishable from that of the wild type after correction for the fraction of inhibitor reacting through the substrate pathway (Table 1).
The constitutive activating effect of the Y131L single mutation is evident from the 10-fold enhancement of the basal rate of antithrombin inhibition of factor Xa relative to the wild type. The S380W RCL P14 residue mutation alone also produces an ~3-fold activating effect on the basal rate of antithrombin inhibition of factor Xa, and combining the S380W and Y131L mutations results in a nearly additive ~20-fold enhancement of the basal antithrombin–factor Xa reaction rate, after correction in both cases for the accelerated substrate reaction of these P14 Trp variants (Table 1). Heparin pentasaccharide allosteric activation of the variants further enhanced the factor Xa inhibition rates of the mutants ~20-fold for the Y131L single mutant, ~50-fold for the P14 Trp single mutant, and ~6-fold for the double mutant, resulting in pentasaccharide-activated rates similar to or ≤2-fold greater than that of the wild type. A full-length heparin produced even further ~3–5-fold enhancements in factor Xa inhibition rates of wild-type and mutant antithrombins due to an additional boost in rate by full-length heparin bridging (Table 1).
Titrations of the tryptophan fluorescence changes that report heparin binding and activation of antithrombin showed that heparin bound all variants with high affinities significantly greater than that of the wild type and with stoichiometries close to 1, except for the Y131L single and double mutants that appeared to have a minor fraction of nonfunctional, presumably latent inhibitor. To determine how constitutive activation of antithrombin by the Y131L mutation affected the basal and heparin-activated fluorescence of the P14 Trp reporter of the RCL conformational change, we examined the fluorescence spectra of the variants in the absence and presence of saturating heparin pentasaccharide. A P14 Ala variant was used as a control for the fluorescence contribution of the wild-type Trp residues to account for a minor effect of the wild-type P14 Ser hydroxyl group on this fluorescence (Figure 4A).29 The Y131L mutation did not significantly affect the fluorescence properties in the absence or presence of heparin.b As expected, the intensity of the fluorescence spectrum of the P14 Trp variants was greater than that of the P14A control in the absence or presence of saturating heparin pentasaccharide due to the additional tryptophan (Figure 4B,C).
Figure 4.

Fluorescence spectra of P14 Trp variants and controls with and without heparin pentasaccharide activation and resolution of the P14 Trp spectrum by difference. Fluorescence spectra of control and P14 Trp antithrombin variants were obtained from 300 to 400 nm in the absence and presence of a 10-fold molar excess of heparin pentasaccharide sufficient to fully complex the serpins and normalized relative to the maximum of the control wild-type antithrombin in panel A and the control P14A antithrombin in panels B and C. Corrections for a minor fraction of non-heparin binding protein were made for some variants as described in Materials and Methods. Each spectrum represents the average of spectra obtained in two or three independent experiments. Control spectra of wild-type, P14A, and Y131L antithrombin are shown in panel A, those of P14W and P14A in panel B, and those of P14W/Y131L and P14A in panel C. The spectrum of the P14 Trp in the P14W and P14W/Y131L mutants was obtained by subtraction of the P14A control spectra from the mutant spectra. The P14A control spectra were assumed to provide the most accurate contribution of the fluorescence of the four wild-type Trp residues in both P14 Trp variants because the Ala side chain lacks the side chain hydroxyl group of the wild-type Ser as does the Trp substitution and the Y131L mutation does not significantly perturb the fluorescence of the wild type. Error bars reflecting the standard error or the range of values at the spectral maxima are shown for the controls in panel A and difference spectra in panels B and C.
The fluorescence contribution of the P14 Trp to these changes was resolved by subtracting the fluorescence spectra of the control P14A antithrombin from the spectra of the S380W single or Y131L/S380W double mutant. The earlier study of the P14 Trp single mutant found that the resolved fluorescence spectrum of the P14 Trp exhibits a maximum characteristic of a buried Trp shielded from solvent (λmax = 337 nm) in the native state and that following heparin activation, the P14 Trp undergoes a 17 nm red-shift (λmax = 354 nm), consistent with solvent exposure due to expulsion of the P14 Trp residue from its hydrophobic binding pocket beneath sheet A in the activated state.19 We reproduced this finding with the S380W single mutant, observing a similar shift in the emission peak from 330 to 335 nm in the unactivated mutant to ~350 nm following heparin activation, thereby confirming that the P14 Trp does report the RCL conformational change induced by heparin allosteric activation. Interestingly, the resolved spectrum of the P14 Trp in the context of the Y131L activating mutation was indistinguishable from that in the absence of this mutation with an identical maximum (λmax = 330–335 nm) and intensity, consistent with a similar hydrophobic environment in the native state. Moreover, a comparable large red shift of the fluorescence maximum (λmax = 350–355 nm), consistent with transfer of the tryptophan from a hydrophobic to a solvent-exposed environment, was observed upon heparin activation. Thus, the P14 Trp appears to undergo indistinguishable large changes in environment following heparin activation of both the wild type and the constitutively activated Y131L variant. Constitutive activation of this mutant in the absence of heparin thus does not detectably affect the burial of the RCL hinge in sheet A, and heparin allosteric activation results in a similar expulsion of the RCL hinge from its hydrophobic interaction with sheet A.
DISCUSSION
Heparin allosterically activates antithrombin to rapidly inhibit two target proteases, factor Xa and factor IXa, by inducing global conformational changes in the serpin that allow RCL-bound proteases to productively interact with an exosite.1,38 These conformational changes are induced by the binding of heparin to helix D and transmitted through the hydrophobic core to ultimately expel the RCL hinge from an interaction with sheet A and allow the RCL to extend away from sheet A and the serpin body.7 Whereas an early hypothesis suggested that the latter expulsion of the RCL from sheet A was the essential activating change that enabled the RCL to access the exosite,7-9,14 subsequent mutagenesis studies demonstrated that the exosite was an important determinant of both the native repressed and heparin-activated reactivity of antithrombin with factors Xa and IXa.11,20,39 Such findings implied that expulsion of the RCL from sheet A was not required for a bound protease to engage the exosite and instead suggested that nonproductive and productive modes of interaction of the protease with the exosite in native and activated states accounted for heparin activation.
Recent studies have resolved heparin allosteric activation of antithrombin into a minimally three-step process involving (i) binding of heparin to helix D, (ii) an intermediate activation stage in which global antithrombin conformational changes induced in the heparin binding site and transmitted through the hydrophobic core have taken place, and (iii) a final stage in which the RCL hinge P14 residue is expelled from a hydrophobic interaction with sheet A and the gap in the sheet is closed by an extension of the adjacent helix D where heparin is bound (Figure 5).6,7,29,40 This three-step activation mechanism is supported by X-ray crystal structures that reveal the progressive conformational changes accompanying heparin binding to and activation of native antithrombin in intermediate and fully activated states (Figure 1)3,6,7 as well as rapid kinetic studies of heparin activation.40 Moreover, saturation mutagenesis of the RCL P14 hinge residue has supported a minimal three-step mechanism of activation and suggested that the major activating effect occurs in the intermediate stage with a minor contribution from the final RCL expulsion conformational change.29
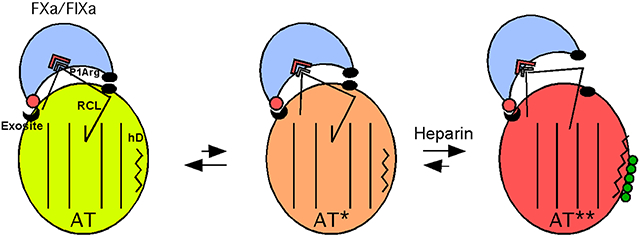
Figure 5.

Three-state model for heparin allosteric activation of antithrombin. Cartoon depiction of the three allosteric states of wild-type antithrombin and their Michaelis complex interactions with FXa or FIXa. In the native state of antithrombin (AT, light green), the RCL hinge is inserted into the center of sheet A (shown as parallel lines) and bound protease (light blue) interacts productively with the RCL P1 Arg residue but nonproductively with an exosite on the serpin body, thereby repressing inhibitory activity. In the intermediate-activated state (AT*, orange), major core conformational changes have occurred with retention of the RCL hinge–sheet A interaction but with repositioning of the RCL to allow a productive interaction of bound protease with the exosite, resulting in a major activation of serpin inhibitory activity. Heparin (green spheres) binding to helix D (zigzag line) induces the fully activated state (AT**, red-orange) in which helix D has been extended and the RCL is expelled from sheet A, causing the bound protease to move away from the serpin body and produce a modest further activation of inhibitory activity due to a loss of repulsive interactions (black ovals) with the serpin body. An allosteric equilibrium exists between native and intermediate-activated states in the absence of heparin with the native state dominant in the wild type. The allosteric equilibrium is shifted to intermediate and fully activated states in the presence of heparin with the fully activated state being dominant. The effects of the helix D and double-cysteine mutations on the allosteric equilibria are depicted below the cartoon. In the absence of heparin, helix D mutations shift the equilibrium to favor the intermediate-activated state whereas buried double-cysteine mutations in the RCL hinge and sheet A that do not form a disulfide shift the equilibrium in favor of the native state. Combining the double-cysteine mutations with helix D mutations attenuates the activating effects of the latter by shifting the allosteric equilibrium back toward the native state. Heparin binding favors the fully activated state for antithrombin variants with helix D or double-cysteine mutations that do not form a disulfide. Double-cysteine mutations that form a disulfide bond that locks the RCL–sheet A interaction block both RCL repositioning in the intermediate-activated state and RCL expulsion in the fully activated state and thereby prevent allosteric activation.
Other studies have supported only a minor role for RCL expulsion in heparin allosteric activation. Thus, antithrombin variants with heparin binding site mutations that selectively block the final activating extension of helix D and expulsion of the RCL from sheet A upon heparin binding, but not the intermediate activating structural changes, nevertheless undergo major activation of their inhibitory activity against factors Xa and IXa upon heparin binding.41,42 Moreover, antithrombin variants with mutations that promote the extension of helix D in the native state are partially or fully constitutively activated to rapidly inhibit factors Xa and IXa in the absence of heparin but have not undergone the intrinsic tryptophan fluorescence changes that report the expulsion of the RCL from sheet A.16
However, other findings have been difficult to interpret in view of this mechanism. Thus, engineered disulfide bonds in antithrombin that link the RCL to sheet A to block the expulsion of the RCL that accompanies heparin allosteric activation of the serpin were found to greatly attenuate the ability of heparin to induce the tryptophan fluorescence changes and completely abrogated the ability of heparin to enhance the rates of factor Xa and IXa inhibition.7,14 Such findings thus seemed to support the importance of RCL expulsion for allosteric activation.
The studies presented here have resolved these disparate findings by engineering antithrombin variants that combined disulfides designed to block the expulsion of the RCL from sheet A or introduced a P14 Trp reporter of RCL expulsion together with constitutively activating mutations. The latter strategy was based on an earlier study showing that an engineered RCL hinge P14 Trp undergoes major changes in fluorescence properties upon heparin allosteric activation, consistent with expulsion of the hinge residue from a hydrophobic binding pocket in sheet A and exposure to solvent.19 We found no evidence that the expulsion of the RCL from sheet A occurred in a constitutively activated Y131L variant with the P14 Trp reporter mutation in the absence of heparin despite the ~20-fold activation of the basal rate of factor Xa inhibition and higher heparin affinity that reflect partial activation. The P14 Trp thus appears to exist in a hydrophobic environment in this highly activated variant indistinguishable from that in the unactivated wild type. Moreover, heparin binding caused a similar large red-shift in the emission spectrum of the P14 Trp as with the control P14 Trp variant, consistent with heparin binding being required to cause the P14 Trp to become solvent-exposed through expulsion of the RCL from sheet A. Such findings suggest that the Y131L mutation partially activates antithrombin in the absence of heparin by shifting the conformational equilibrium between native and intermediate-activated states to favor the intermediate-activated structure in which the RCL remains inserted in sheet A and the intrinsic tryptophans are minimally perturbed (Figure 5).29 The S380W mutation itself partially activates antithrombin in the absence of heparin because the RCL hinge tryptophan substitution for the wild-type serine perturbs the RCL–sheet A interaction, causing the allosteric equilibrium to shift somewhat in favor of the intermediate-activated state. Heparin binding fully activates these constitutively activated variants as evidenced by the fluorescence changes of intrinsic tryptophans that report the expulsion of the RCL from sheet A. In contrast to the four wild-type Trp residues that are far apart and unlikely to be affected by energy transfer,37 the engineered P14 Trp lies close to Trp225 in the sheet A binding pocket. Energy transfer is thus possible in the native and intermediate states when the P14Trp is bound in the pocket but unlikely after the P14 Trp is expelled from the pocket and becomes solvent-exposed. However, the possibility of differential energy transfer in wild-type and partially activated Y131L contexts appears unlikely given that alignment of Trp225 and the P14 residue in native and intermediate structures reveals that the distances and relative orientations of these residues do not change in the intermediate,6,7,43 suggesting that any energy transfer should affect the two resolved P14 Trp spectra similarly.
Our studies have confirmed that the previously reported engineered disulfide that links the RCL P13 residue to s3A does indeed block heparin allosteric activation of antithrombin.14 However, we could not reproduce the finding that a disulfide linking the RCL P14 residue to s5A through either V375 or L373 similarly blocks heparin allosteric activation.7 This appeared to be a consequence of the latter double-cysteine variants not having formed a disulfide. However, a double-cysteine variant linking the RCL P13 residue to s5A through E374 was found to form a disulfide, and like the s3A–P13 disulfide, this disulfide similarly blocked heparin allosteric activation, although as a substrate rather than as an inhibitor of factor Xa. Thus, our results confirm that disulfides that link the RCL to s3A or s5A block heparin allosteric activation.
Rather than providing proof that expulsion of the RCL from sheet A is required for heparin allosteric activation, such findings can more coherently be rationalized in the context of the three-step heparin allosteric activation model in which the major activating effect occurs at the intermediate rather than final activation stage (Figure 5).29 The intermediate activation state thus must require the repositioning of the RCL and the exosite to allow RCL-bound factor Xa or IXa to change from a nonproductive to a productive mode of interaction with the exosite while the RCL hinge remains bound in this state to sheet A. X-ray structures of the intermediate-activated state show that the global conformational changes in the hydrophobic core that characterize this state can be deconstructed into the rotational movement of a major subdomain in which the exosite resides relative to the conformationally mobile RCL and two of three other subdomains.4,7 It is thus anticipated that such conformational changes in the intermediate-activated state will result in a repositioning of the RCL relative to the exosite to allow activation. Because of crystal packing that distorts the RCL conformation in the X-ray structures,3 the true RCL conformation in this state cannot be discerned. It is nevertheless clear that the required repositioning of the RCL in the intermediate state would be affected by the rigid anchoring of the RCL to sheet A by disulfides with greatly restricted conformational freedom36 and could thus be blocked by such disulfides.
Combining the helix D constitutive activating mutations with the strand 5A–P14 double-cysteine mutations that did not form a disulfide was surprisingly found to suppress the constitutive activating effects. This can be understood in the context of the properties of the two strand 5A–P14 double-cysteine variants. These variants thus resemble the P14 Cys single variant with respect to their decreased affinities for heparin and decreased basal rates of FXa and FIXa inhibition. These effects of the P14 Cys substitution were previously shown to reflect the preferential stabilization of the RCL hinge–sheet A interaction and the shift of the allosteric equilibrium in favor of the native state with low heparin affinity and repressed reactivity with factors Xa and IXa.29,33 Interestingly, the more marked decreases in the FXa and FIXa basal inhibition rates by the double-cysteine mutations than the single-cysteine mutation parallel those produced by mutation of critical exosite residues that cause the greatest stabilization of the native state.11 The double-cysteine mutations thus appear to stabilize and shift the allosteric equilibrium toward the native state to a greater extent than the single-cysteine mutation. The double-cysteine mutations would therefore be expected to similarly reverse the constitutive activating effects of the helix D mutations by shifting the allosteric equilibrium in favor of the native state. Our findings are thus in accord with the three-step allosteric activation model (Figure 5). Constitutive activating mutations shift an equilibrium between native and intermediate-activated states in favor of the intermediate state in the absence of heparin, whereas mutations that stabilize the native state shift this equilibrium back toward the native structure. While we did not attempt to combine the constitutive activating mutations with the double-cysteine mutations that did form a disulfide, our findings suggest that if such disulfide links block both intermediate and full activating conformational changes they would be expected to block the constitutive activating effects as well as full heparin activation.
CONCLUSIONS
Our findings reconcile previous apparently contradictory reports regarding the role of core and RCL conformational changes in the mechanism of heparin allosteric activation of antithrombin. We have confirmed that engineered disulfides that block expulsion of the RCL from sheet A also block allosteric activation by heparin and have shown that antithrombin can be constitutively activated by mutations that promote core conformational changes without inducing expulsion of the RCL from sheet A. These findings can be reconciled in the context of a three-step allosteric mechanism of activation now supported by several independent studies.6,7,29,40 In accordance with this mechanism, the major conformational changes induced in the core in the intermediate-activated state are of paramount importance for activation and do not require expulsion of the RCL from sheet A. Because the core conformational changes activate antithrombin by causing a nonproductive interaction of RCL-bound proteases with an exosite to become productive, a repositioning of the RCL and exosite is necessary to accomplish this change. This repositioning can be blocked by rigid disulfide bonds that constrain the conformational mobility of the RCL in the intermediate state. Insertion of the RCL into sheet A is thus compatible with activation and must therefore stabilize both native and intermediate-activated states in the absence of heparin. Only upon heparin binding is the RCL–sheet A interaction destabilized and the allosteric equilibrium shifted in favor of expulsion of the RCL from sheet A. This expulsion results in an additional minor activation due to movement of RCL-bound proteases away from unfavorable interactions with the serpin body.39
Funding
This work was supported by National Institutes of Health Grant R37 HL39888 (to S.T.O.).
ABBREVIATIONS
- RCL
reactive center loop
- FXa
factor Xa
- FIXa
factor IXa
- DTDP
4,4′-dithiodipyridine
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SI
stoichiometry of inhibition
Footnotes
Accession Codes
Human antithrombin, P01008; human prothrombin, P00734; human factor X, P00742; human factor IX, P00740; human neutrophil elastase, P08246.
The authors declare no competing financial interest.
While an X-ray structure of heparin cofactor II has shown evidence of RCL insertion in the native state, the structure was of a dimer in which the RCL comprised the interface with a second latent molecule, raising doubts about whether this represents the solution conformation.44 Moreover, unlike antithrombin, no corroborative evidence in solution for heparin inducing core conformational changes in heparin cofactor II has been reported. Indeed, unpublished nuclear magnetic resonance evidence from one of the authors (P.G.W.G.) indicates the absence of any change in core aromatic residue resonances of the protein upon heparin activation.
In our previous study, small differences in fluorescence relative to that of the wild type were noted for the Y131L variant in native and heparin-activated states.16 More careful analysis of equivalent functional concentrations of protein revealed that such differences were insignificant (Figure 4A).
Contributor Information
Gonzalo Izaguirre, Department of Periodontics, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Richard Swanson, Department of Periodontics, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Ryan Roth, Department of Periodontics, University of Illinois at Chicago, Chicago, Illinois 60612, United States.
Peter G. W. Gettins, Department of Biochemistry and Molecular Genetics, University of Illinois at Chicago, Chicago, Illinois 60612, United States
Steven T. Olson, Department of Periodontics, University of Illinois at Chicago, Chicago, Illinois 60612, United States
REFERENCES
- (1).Gettins PGW, and Olson ST (2016) Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem. J 473, 2273–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Carrell RW, Stein PE, Fermi G, and Wardell MR (1994) Biological implications of a 3Å structure of dimeric antithrombin. Structure 2, 257–270. [DOI] [PubMed] [Google Scholar]
- (3).Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, and Carrell RW (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U. S. A 94, 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Whisstock JC, Pike RN, Jin L, Skinner R, Pei XY, Carrell RW, and Lesk AM (2000) Conformational Changes in Serpins: II. The Mechanism of Activation of Antithrombin by Heparin. J. Mol. Biol 301, 1287–1305. [DOI] [PubMed] [Google Scholar]
- (5).Irving JA, Pike RN, Lesk AM, and Whisstock JC (2000) Phylogeny of the Serpin Superfamily: Implications of Patterns of Amino Acid Conservation for Structure and Function. Genome Res. 10, 1845–1864. [DOI] [PubMed] [Google Scholar]
- (6).Johnson DJD, and Huntington JA (2003) Crystal structure of antithrombin in a heparin-bound intermediate state. Biochemistry 42, 8712–8719. [DOI] [PubMed] [Google Scholar]
- (7).Langdown J, Belzar KJ, Savory WJ, Baglin TP, and Huntington JA (2009) The critical role of hinge-region expulsion in the induced-fit heparin binding mechanism of antithrombin. J. Mol. Biol 386, 1278–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Johnson DJ, Li W, Adams TE, and Huntington JA (2006) Antithrombin-S195A factor Xa-heparin structure reveals the mechanism of antithrombin activation. EMBO J. 25, 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Johnson DJD, Langdown J, and Huntington JA (2010) Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7 A structure of the ternary complex. Proc. Natl. Acad. Sci. U. S. A 107, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Izaguirre G, Swanson R, Raja SM, Rezaie AR, and Olson ST (2007) Mechanism by which exosites promote the inhibition of blood coagulation proteases by heparin-activated antithrombin. J. Biol. Chem 282, 33609–33622. [DOI] [PubMed] [Google Scholar]
- (11).Izaguirre G, Aguila S, Qi L, Swanson R, Roth R, Rezaie AR, Gettins PGW, and Olson ST (2014) Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J. Biol. Chem 289, 34049–34064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Manithody C, Yang L, and Rezaie AR (2002) Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry 41, 6780–6788. [DOI] [PubMed] [Google Scholar]
- (13).Yang L, Manithody C, Olson ST, and Rezaie AR (2003) Contribution of basic residues of the autolysis loop to the substrate and inhibitor specificity of factor IXa. J. Biol. Chem 278, 25032–25038. [DOI] [PubMed] [Google Scholar]
- (14).Langdown J, Johnson DJD, Baglin TP, and Huntington JA (2004) Allosteric activation of antithrombin critically depends upon hinge region extension. J. Biol. Chem 279, 47288–47297. [DOI] [PubMed] [Google Scholar]
- (15).Dela Cruz RG, Jairajpuri MA, and Bock SC (2006) Disruption of a tight cluster surrounding tyrosine 131 in the native conformation of antithrombin III activates it for factor Xa inhibition. J. Biol. Chem 281, 31668–31676. [DOI] [PubMed] [Google Scholar]
- (16).Dementiev A, Swanson R, Roth R, Isetti G, Izaguirre G, Olson ST, and Gettins PGW (2013) The allosteric mechanism of activation of antithrombin as an inhibitor of factor IXa and factor Xa. Heparin-independent full activation through mutations adjacent to helix D. J. Biol. Chem 288, 33611–33619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Turk B, Brieditis I, Bock SC, Olson ST, and Björk I (1997) The Oligosaccharide Side Chain on Asn-135 of α-Antithrombin, Absent in β-Antithrombin, Decreases the Heparin Affinity of the Inhibitor by Affecting the Heparin-Induced Conformational Change. Biochemistry 36, 6682–6691. [DOI] [PubMed] [Google Scholar]
- (18).Izaguirre G, Zhang W, Swanson R, Bedsted T, and Olson ST (2003) Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa dependent on heparin activation of the serpin. J. Biol. Chem 278, 51433–51440. [DOI] [PubMed] [Google Scholar]
- (19).Huntington JA, Olson ST, Fan B, and Gettins PGW (1996) Mechanism of Heparin Activation of Antithrombin. Evidence for Reactive Center Loop Preinsertion with Expulsion upon Heparin Binding. Biochemistry 35, 8495–8503. [DOI] [PubMed] [Google Scholar]
- (20).Izaguirre G, and Olson ST (2006) Residues Tyr253 and Glu255 in strand 3 of β-sheet C of antithrombin are key determinants of an exosite made accessible by heparin activation to promote rapid inhibition of factors Xa and IXa. J. Biol. Chem 281, 13424–13432. [DOI] [PubMed] [Google Scholar]
- (21).Nordenman B, Nyström C, and Björk I (1977) The size and shape of human and bovine antithrombin III. Eur. J. Biochem 78, 195–203. [DOI] [PubMed] [Google Scholar]
- (22).Pace C, Vajdos F, Fee L, Grimsley G, and Gray T (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ersdal-Badju E, Lu A, Peng X, Picard V, Zendehrouh P, Turk B, Björk I, Olson ST, and Bock SC (1995) Elimination of glycosylation heterogeneity affecting heparin affinity of recombinant human antithrombin III by expression of a β-like variant in baculovirus-infected insect cells. Biochem. J 310, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Olson ST, Björk I, and Shore JD (1993) Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 222, 525–560. [DOI] [PubMed] [Google Scholar]
- (25).Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- (26).Björk I, Ylinenjärvi K, Olson ST, and Bock PE (1992) Conversion of antithrombin from an inhibitor of thrombin to a substrate with reduced heparin affinity and enhanced conformational stability by binding of a tetradecapeptide corresponding to the P1 to P14 region of the putative reactive bond loop of the inhibitor. J. Biol. Chem 267, 1976–1982. [PubMed] [Google Scholar]
- (27).Riener CK, Kada G, and Gruber HJ (2002) Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem 373, 266–276. [DOI] [PubMed] [Google Scholar]
- (28).Sturzebecher J, Kopetzki E, Bode W, and Hopfner K-P (1997) Dramatic enhancement of the catalytic activity of coagulation factor IXa by alcohols. FEBS Lett. 412, 295–300. [DOI] [PubMed] [Google Scholar]
- (29).Roth R, Swanson R, Izaguirre G, Bock SC, Gettins PGW, and Olson ST (2015) Saturation mutagenesis of the antithrombin reactive center loop P14 residue supports a three-step mechanism of heparin allosteric activation involving intermediate and fully-activated states. J. Biol. Chem 290, 28020–28036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bedsted T, Swanson R, Chuang Y-J, Bock PE, Björk I, and Olson ST (2003) Heparin and Calcium Ions Dramatically Enhance Antithrombin Reactivity with Factor IXa by Generating New Interaction Exosites. Biochemistry 42, 8143–8152. [DOI] [PubMed] [Google Scholar]
- (31).Gettins P. (2002) Serpin Structure, Mechanism, and Function. Chem. Rev 102, 4751–4803. [DOI] [PubMed] [Google Scholar]
- (32).Richard B, Swanson R, Schedin-Weiss S, Ramirez B, Izaguirre G, Gettins PGW, and Olson ST (2008) Characterization of the conformational alterations, reduced anticoagulant activity, and enhanced antiangiogenic activity of prelatent antithrombin. J. Biol. Chem 283, 14417–14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Huntington JA, and Gettins PGW (1998) Conformational Conversion of Antithrombin to a Fully Activated Substrate of Factor Xa without Need for Heparin. Biochemistry 37, 3272–3277. [DOI] [PubMed] [Google Scholar]
- (34).Pecher P, and Arnold U (2009) The effect of additional disulfide bonds on the stability and folding of ribonuclease A. Biophys. J 141, 21–28. [DOI] [PubMed] [Google Scholar]
- (35).Jordan RE, KIlpatrick J, and Nelson RM (1987) Heparin Promotes the Inactivation of Antithrombin by Neutrophil Elastase. Science 237, 777–780. [DOI] [PubMed] [Google Scholar]
- (36).Craig DB, and Dombkowski AA (2013) Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinf. 14, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Meagher JL, Beechem JM, Olson ST, and Gettins PGW (1998) Deconvolution of the Fluorescence Emission Spectrum of Human Antithrombin and Identification of the Tryptophan Residues That Are Responsive to Heparin Binding. J. Biol. Chem 273, 23283–23289. [DOI] [PubMed] [Google Scholar]
- (38).Olson ST, Richard B, Izaguirre G, Schedin-Weiss S, and Gettins PGW (2010) Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie 92, 1587–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gettins PGW, and Olson ST (2009) Activation of antithrombin as a factor IXa and Xa inhibitor involves mitigation of repression rather than positive enhancement. FEBS Lett. 583, 3397–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schedin-Weiss S, Richard B, and Olson ST (2010) Kinetic evidence that allosteric activation of antithrombin by heparin is mediated by two sequential conformational changes. Arch. Biochem. Biophys 504, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Meagher JL, Olson ST, and Gettins PGW (2000) Critical Role of the Linker Region Between Helix D and Strand 2A in Heparin Activation of Antithrombin. J. Biol. Chem 275, 2698–2704. [DOI] [PubMed] [Google Scholar]
- (42).Belzar KJ, Zhou A, Carrell RW, Gettins PGW, and Huntington JA (2002) Helix D Elongation and Allosteric Activation of Antithrombin. J. Biol. Chem 277, 8551–8558. [DOI] [PubMed] [Google Scholar]
- (43).Johnson DJD, Langdown J, Li W, Luis SA, Baglin TP, and Huntington JA (2006) Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J. Biol. Chem 281, 35478–35486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Baglin T, Carrell R, Church F, Esmon C, and Huntington J (2002) Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc. Natl. Acad. Sci. U. S. A 99, 11079–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]