

Abstract
CPE (Clostridium perfringens enterotoxin) is the major virulence determinant for C. perfringens type F food poisoning, the second most common bacterial food-borne illness in the UK and USA. After binding to its receptors, which include particular human claudins, the toxin forms pores in the cell membrane. The mature pore apparently contains a hexamer of CPE, claudin and, possibly, occludin. The combination of high binding specificity with cytotoxicity has resulted in CPE being investigated, with some success, as a targeted cytotoxic agent for oncotherapy. In this paper, we present the X-ray crystallographic structure of CPE in complex with a peptide derived from extracellular loop 2 of a modified, CPE binding Claudin-2, together with high-resolution native and pore-formation mutant structures. Our structure provides the first atomic resolution data on any part of a claudin molecule and reveals that claudin’s CPE binding fingerprint (NPLVP), is in a tight turn conformation and binds, as expected, in CPE’s C-terminal claudin binding groove. The leucine and valine residues insert into the binding groove while the first residue, asparagine, tethers the peptide via an interaction with CPE’s aspartate 225 and the two prolines are required to maintain the tight turn conformation. Understanding the structural basis of the contribution these residues make to binding will aid in engineering CPE to target tumour cells.
Keywords: enterotoxin, claudin, receptor, complex, structure
Graphical abstract
Introduction
Clostridium perfringens is a Gram-positive anaerobic spore forming bacterium that is ubiquitous in nature. It can secrete an array of toxins that vary from strain to strain. This toxin diversity allows C. perfringens to cause a range of diseases including human food poisoning, gas gangrene, human enteritis necroticans, and enterotoxaemia in livestock.
CPE (Clostridium perfringens enterotoxin) is the major virulence determinant for C. perfringens type-F food poisoning (formerly type A food poisoning) 1. This type of food poisoning is a significant problem across the globe, ranking as the second most common bacterial food-borne illness in both the USA and UK. The Centers for Disease Control and Prevention (CDC) in the USA estimates that nearly 1 million cases of C. perfringens type F food poisoning occur annually in the USA (CDC, Estimates of Foodborne Illness in the United States. 2011 11 June, 2012; Available from: http://www.cdc.gov/foodborneburden/2011-foodborne-estimates.html). It results from consumption of food contaminated with C. perfringens strains that produce CPE in the intestines. Two observations confirm that CPE is the responsible toxin. Firstly, inactivation of the cpe gene eliminated the enteric pathogenicity of a food poisoning strain in animal models and this attenuation was reversible by complementation 2. Secondly, ingestion of purified CPE by human volunteers resulted in the gastro-enteritis (GI) symptoms of type F food poisoning 3.
CPE is also associated with cases of hospital- and community-acquired AAD (antibiotic-associated diarrhoea) and SD (sporadic diarrhoea), which are more serious that typical cases of type-F food borne illness 4. While the media focuses on AAD and SD caused by strains of Clostridium difficile, CPE is also responsible for a significant fraction of hospital-acquired enteric illnesses 5; 6. CPE has also been linked with some veterinary GI diseases 7 and, controversially, sudden infant death syndrome 8.
CPE differs from other C. perfringens toxins in that it is not a secreted toxin but rather is released at the completion of sporulation upon lysis of the mother cell at which time CPE can represent up to 15% of dry cell mass 9. This membrane-interacting toxin is a 319-residue polypeptide chain of approximately 35 kDa molecular weight 10. It can bind to several mammalian cell types via human Claudin-3 or −4 11, which belong to the large claudin family of 20–27 kDa tight junction (TJ) proteins 12; 13; 14. Other human claudins have also been shown to bind CPE, though with lower affinity 15. The high specificity of this interaction has resulted in CPE being identified as a potential biotechnology agent, and, in particular as an oncotherapeutic 16; 17; 18; 19. Understanding the basis for this specificity will result in the potential to alter which claudins recognise CPE and therefore target different cell-types, as described in Takahashi et al20.
Cytotoxicity and cell death, which occur within 30 minutes of CPE exposure at 37 °C 21, result from alterations in membrane permeability caused by pore formation 11. In mammalian cells, CPE forms two large complexes named CH-1 and CH-2 22; 23. These two CPE complexes are both SDS-resistant and their stoichiometry and mass have been the subject of continued study, with recent estimates indicating sizes of approximately 425–500 kDa and 550–660 kDa, for CH-1 and CH-2 respectively. These complexes apparently consist of a CPE hexamer, claudins and, for CH-2, another tight junction protein named occludin 24.
Winkler et al 25 reported that specificity for Claudin-3 and −4 is provided via a fingerprint sequence (149NPLVP153) in extracellular loop 2 (ECL2) of the claudin. While the exact sequence only occurs in mouse Claudin-3, amino acids in position 3, 4 and 5 of the fingerprint can be conservatively substituted. Introduction of this fingerprint by mutation of two residues was sufficient to turn a peptide derived from non-CPE binding mouse Claudin-2 into a peptide that bound CPE better than the wild-type CPE-binding mouse Claudin-4. Mutation of the fingerprint residues reduces binding of claudin to CPE (Table 1). The involvement of residues outside the fingerprint is not ruled out and other claudins lacking a complete fingerprint are reported to bind CPE, including human Claudin-8 and −14 15. However, these peptides bind to the toxin with reduced affinity, and their affinity for CPE is increased by introducing the canonical fingerprint residues. CPE has been shown to interact with claudin via its C-terminal domain (residues 194–319 26). In particular, tyrosines 306, 310 and 312 and leucine 315 in CPE are important for claudin-binding 27; 28 (Table 1). The structure of the C-terminal domain in isolation has been determined to 1.75 Å 29 and the residues associated with claudin-binding reside at the base of a cleft in the C-terminal protein surface. However, the C-terminal domain in isolation is insufficient for cytotoxicity. In the absence of the C-terminal domain (residues 200-319), CPE is not cytotoxic and cannot form active pores 26; 29. In addition, alanine-scanning mutagenesis identified aspartate 48 (D48A) as a key residue for large complex formation 30, while the deletion of up to the first 44 residues from native CPE results in a slight increase in cytotoxicity 31.
Table 1.
CPE and Claudin-4 mutations affecting the interaction between them.
| (A) CPE mutations affecting activity | ||||
|---|---|---|---|---|
| Mutation | Methods used | Approx. Cytotoxicity as % wild-type activity | Comment | Ref. |
|
| ||||
| Wild-type | 100 | - | ||
|
| ||||
| Full-length CPE | ||||
|
| ||||
| Asp48Ala/As n/Glu |
86Rb release | <1 | 30 | |
| Ile51Ala | 86Rb release | <1 | 30 | |
| Tyr306Ala | LDH release competition/TEER | 64/64 | 28 | |
| Tyr310Ala | LDH release competition/TEER | 72/94 | 28 | |
| Tyr312Ala | LDH release competition/TEER | 73/100 | 28 | |
| Leu315Ala | LDH release competition/TEER | 70/45 | 28 | |
|
| ||||
| C-terminal domain only | Claudin-4 binding as % wild-type | |||
|
| ||||
| Leu223Ala | Pull-down | 100 | Claudin-3 binding 20% | 34 |
| Arg227Ala | Pull-down | 80 | Claudin-3 binding 10% | 34 |
| Leu254Ala | Pull-down | 80 | Claudin-3 binding 30% | 34 |
| Asp284Ala | Pull-down | 10 | Claudin-3 binding 100% | 34 |
| (B) Claudin extracellular loop 2 mutations affecting C-CPE binding -numbering as for Claudin-2 ECL2 | ||||
| Mutation | Methods used | Approx. Binding as % wild-type activity | Comment | Ref. |
|
| ||||
| Wild-type | 100 | - | ||
|
| ||||
| Claudin 3 | ||||
|
| ||||
| Asn149Asp | Pull-down | 5 | Mouse ECL-2 peptide | 25 |
| Leu151Ala | Pull-down | 5 | Mouse ECL-2 peptide | 25 |
| Asn149Asp/Ar g158Tyr |
125I-CPE binding/TTC cytotoxicity |
1 | Human full-length | 39 |
| Asn149Asp | Pull-down | 50% | Mouse full-length | 34 |
|
| ||||
| Claudin 4 | ||||
|
| ||||
| Asn149Asp | Cytotoxicity | <1 | Human full-length | 40 |
Previously, we (and others 32; 33) have determined the structure of full-length CPE (Figure 1). In all the crystal structures solved to date32; 33, and in those in this paper, the CPE forms an intimate trimer, whose interface has the characteristics of a biological significant interface, though this trimer has not yet been shown to have any functional significance. These structures show that residues implicated in claudin binding (Table 1), including Tyr 306, 310 and 312 form a pocket on the protein surface. All of these claudin-binding pockets are accessible and on the same side of the trimer. More recent studies on the C-terminal domain have identified a number of residues on the opposite edge of this pocket as also important for binding, including Ser 256, Ile 258 and Val 259 34 (Table 1).
FIGURE 1.

Structure of full-length CPE. Cartoon representation of (A) the monomer, coloured pale green and pink for the oligomerisation domain and light cyan for the C-terminal claudin binding domain. The membrane-inserting residues are highlighted in red. (B) The trimer seen in the crystal. The monomers are coloured green, maroon and violet, the claudin-binding pockets are highlighted by drawing of likely claudin-binding residues as spheres.
The full-length structure showed that CPE is a member of the aerolysin-like β pore-forming toxin (βPFT) family. βPFTs are a group of cytotoxic proteins with divergent structures and sequences that are characterized by their common ability to permeabilize cell membranes and ultimately to cause cell-death 35. They all have structurally related oligomerisation domains, but unrelated receptor-binding domains. Despite their diverse sequences, these toxins have a common mode of action in that they each possess an amphipathic stretch of residues that, on binding to the appropriate cell–surface receptors, forms a β-hairpin that contributes to the membrane-spanning oligomeric β-barrel of the pore. CPE differs slightly from the rest of the group in that the amphipathic residues (residues 81–106) that have been identified as the β-hairpin pore-forming residues 23; 36 adopt a helical conformation in the soluble form, while these residues more normally fold into a β-hairpin in other members of the group.
At high concentrations, CPE is able to form cation-preferring pores in pure lipid bilayers in the absence of receptor-claudin or other proteins 37. Electric current measurements across lipid bilayers containing CPE pores show that it preferentially transports small positive ions, with ions larger than approximately 6.0 Å unable to pass through the CPE pore 37.
In this paper we present a number of structures. Firstly that of CPE with the N-terminal 37 residues deleted (ΔN37CPE). This construct is stable and easier to crystallise than full-length CPE, the N-terminal 37 residues are disordered in all the full-length structures solved to date, and removal of up to the first 44 amino-acids of CPE causes a 2–3 fold increase in toxicity 31. We then look at the structure of the ΔN37CPE with the mutation Asp48Ala introduced and show that the inactivity of this mutant is not due to any major conformational change. Finally, we present the structure of CPE with bound peptide derived from the ECL2 of mouse claudin 2 (CPE-CLD2), containing two mutations required for CPE-binding. The CPE-CLD2 structure shows that the fingerprint sequence is required to form an unusual tight turn conformation in order to allow optimal interaction with the CPE binding pocket, and for the first time provides atomic resolution information on the interaction between CPE and claudin.
Results
ΔN37CPE
ΔN37CPE has increased activity compared to full-length CPE and is more stable and easier to purify, concentrate and crystallise, so it has been used for all the studies described here. In order to distinguish changes induced by the loss of 37 N-terminal residues, we first determined the structure of the ΔN37CPE in isolation. ΔN37CPE crystals diffracted to a resolution of 1.9 Å and were in spacegroup C2 with cell dimensions a=191.7, b=128.3, c=137.2 Å, β=133.8°. The structure was solved by molecular replacement using full-length CPE (PDBID 2XH6)32 as a model and revealed six copies of the CPE monomer in the asymmetric unit (asu) arranged in two trimers with a solvent content of 58% (v/v). After refinement, the final R/Rfree was 17.6/19.7%. Further statistics are listed in Table 2 and the co-ordinates deposited with PDB ID 3ZIX. The overall structure of the ΔN37CPE monomer is unchanged from that of full-length CPE, in which the N-terminal 34 amino acids were disordered and not visible in the electron density map 32. In the three structures described in this paper, there are in total 27 crystallographically independent copies of the CPE monomer, due to high levels of non-crystallographic symmetry. Table 3 lists the mean and standard deviation for the all Cα-atom RMSD (root mean square deviation) between each copy of the monomer in the asu for one structure and each copy in a second. Table 3 shows that the CPE monomers in ΔN37CPE are essentially unchanged from those in the full-length CPE.
Table 2.
Data collection and refinement statistics (Molecular replacement)
| DN37CPE | DN37CPE-D48A | CPE-CLD2 complex | ||
|---|---|---|---|---|
|
| ||||
| Data collection | ||||
| Space group | C2 | C2 | C2 | |
| Cell dimensions | ||||
| a, b, c (Å) | 191.7, 128.3, 137.2 | 190.6, 128.0, 136.4 | 369.6, 100.3, 265.4 | |
| α, β, γ (°) | 90.0, 133.8, 90.0 | 90.0, 133.8, 90.0 | 90.0, 119.7, 90.0 | |
| Resolution (Å) | 47.0–1.9 (1.941.90)* | 98.4–1.9 (1.94–1.9) |
20.0–3.4 (3.5–3.4) | |
| Rsym or Rmerge | 6.5 (122.0) | 14.6 (144.5) | 20.7 (77.8) | 17.4 (64.3) |
| I/σI | 17.42 (1.57) | 7.42 (1.26) | 5.55 (1.05 | 7.7 (1.7) |
| Completeness (%) | 98.2 (95.1) | 99.2(94.8) | 81.9 (47.4) | 53.7 (9.7) |
| Redundancy | 7.66 (7.34) | 7.32 (6.9) | 3.4 (1.9) | 4.0 (2.65) |
| Refinement | ||||
| Resolution (Å) | 47–1.9 | 49.23–1.9 | 20–3.4 | |
| No. reflections | 185641 | 185455 | 96758 | |
| Rwork/ Rfree | 17.6/19.7 | 17.5/19.6 | 20.4/24.0 | |
| No. atoms | ||||
| Protein | 13415 | 13397 | 34326 | |
| Ligand/ion | 1947 | 1870 | 0 | |
| Water | 1762 | 1685 | 0 | |
| B-factors | ||||
| Protein | 44.4 | 48.0 | 117.9 | |
| Ligand/ion | 59.9 | 63.0 | n/a | |
| Water | 52.8 | 56.5 | n/a | |
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.010 | 0.0096 | 0.010 | |
| Bond angles (°) | 1.09 | 1.09 | 1.37 | |
Number of crystals for each structure should be noted in footnote.
Values for the highest resolution shell are given in parentheses.
Table 3.
Mean and standard deviation for the Ca Root Mean Square Deviation (RMSD) between each copy of the molecule in Structure 1 and Structure 2. The number of noncrystallographic symmetry-related copies in each structure is indicated in brackets. CPE: Clostridium perfringens enterotoxin. DN37CPE: CPE with the N-terminal 37 residues deleted. DN37CPE-D48A: DN37CPE with Asp48 mutated to Ala. CPE-CLD2: DN37CPE complexed with a peptide derived from mouse Claudin-2 extracellular loop II.
| (a) Monomers | ||||
|---|---|---|---|---|
| Å | Full-length CPE (3) | DN37CPE (6) | DN37CPE-D48A (6) | CPE-CLD2 (15) |
| CPE-CLD2 (15) |
0.81 ± 0.21 | 0.72 ± 0.1 | 0.72 ± 0.09 | 0.26 ± 0.1 |
| DN37CPE-D48A (6) | 0.68 ± 0.1 | 0.34 ± 0.11 | 0.38 ± 0.07 | |
| DN37CPE (6) | 0.70 ± 0.09 | 0.36 ± 0.09 | ||
| Full-length CPE (3) | 0.56 ± 0.13 | |||
| (b) Trimers | ||||
| Å | Full-length CPE (1) | DN37CPE (2) | DN37CPE- D48A (2) | CPE-CLD2 (5) |
| CPE-CLD2 (5) |
1.04 ± 0.03 | 1.1 ± 0.05 | 1.2 ± 0.09 | 0.35 ±0.1 |
| DN37CPE-D48A (2) | 0.99 ± 0.05 | 0.15 ± 0.02 | 0.61 (no SD) | |
| DN37CPE (2) | 0.99 ± 0.003 | 0.57 (no SD) | ||
| Full-length CPE (1) | N/A | |||
There are two significant areas of difference between ΔN37CPE and full-length CPE. These differences are present in all chains, and are illustrated in Figure 2. Firstly, though the first N-terminal residue corresponding to the CPE sequence in ΔN37CPE is residue 38, removal of the His-tag added to the sequence for purification left four non-native residues (sequence Gly34-Ala-Met-Gly37) remaining N-terminal to the first native CPE amino-acid. These four residues are ordered and visible in the electron density map for the ΔN37CPE structure, and adopt a different conformation from the native residues 34–37 in the full-length CPE structure (sequence Asn34-Ser-Asn-Leu37). In ΔN37CPE the N-terminus extends towards the C-terminal domain, this would be sterically prohibited in full-length CPE, and in the full-length structure residues 34–37 extend into the solvent. It is likely that the presence of the disordered N-terminal peptide extending out towards the C-terminal domain results sterically hinders the binding of the C-terminal domain to claudin and that this reason its removal results in a slightly more active toxin.
FIGURE 2.

Differences between the structure of full-length CPE and ΔN37CPE (A) at the N-terminus, the N-terminal domain of full-length CPE is shown in maroon that for ΔN37CPE in pale green. (B) P191 peptide-flip. The full-length CPE conformation is shown in maroon, and symmetry related molecules in the ΔN37CPE crystal packing in semi-transparent grey. It is evident that the full-length conformation clashes with these residues. The ΔN37CPE is in pale green, red and cyan and does not clash.
The second difference between the two structures is that Pro191 adopts a cis conformation in full-length CPE while it is trans in ΔN37CPE (Figure 2b). The change in conformation results in additional movement in the surrounding chain. This section of CPE is solvent-exposed and mediates symmetry interactions in each copy of ΔN37CPE. The cis conformation of full-length CPE extends further into solvent, and, if maintained in the ΔN37CPE crystal form, would result in a steric clash. Given the small activation energy required to change the conformation of proline, it is likely this steric clash is the cause of the difference.
ΔN37CPE TRIMER
Full-length CPE forms a trimer in all crystal forms reported to date 32; 33. The six copies of ΔN37CPE in the asu of this crystal structure are arranged in two similar trimers. The all Cα-atom RMSD between these two trimers and that in the full-length CPE is 0.99 Å (supplementary Figure 1 and Table 3b) in both cases. This is larger than the mean RMSD between monomers (Table 3a) due to a movement of the C-terminal domain of one monomer in the trimer and the N-terminal domain of a second monomer with which it is interacting. The result is a reduction of space between monomers close to the likely location of the 34 disordered residues in full-length CPE. It is probably the loss of these residues that allows the more tightly packed trimer to form, providing further evidence of the steric hindrance to interactions with other molecules caused by the disordered N-terminal peptide.
ΔN37CPE-D48A
Introduction of the mutation Asp48Ala into CPE has previously been shown to prevent pre-pore formation and thereby eliminate mature pore formation and cytotoxicity 23; 30. Determination of the full-length CPE structure showed this residue is solvent exposed and distant from both the predicted claudin-binding site in the C-terminus and from the amphipathic residues (81–106) expected to insert into the membrane 32. We determined the structure of ΔN37CPE-D48A to identify if the mutation induced any structural changes that would explain the loss of cytotoxicity. ΔN37CPE-D48A crystallised in the same conditions as for ΔN37CPE, the crystals diffracted to 1.9 Å, had the same spacegroup, C2, as the ΔN37CPE crystals and very similar cell dimensions of a=190.6, b= 128.0, c=136.4 Å, β=133.8. Structure solution by molecular replacement revealed a similar arrangement of six CPE monomers in two trimers as for the ΔN37CPE crystals. The R/Rfree after refinement was 17.5/19.6%, further statistics are listed in Table 2 and the co-ordinates deposited with PDB ID 3ZIW. The mutant ΔN37CPE-D48A monomer and trimer are similar to that of ΔN37CPE as shown by the mean all Cα-atom RMSD between different copies (Table 3). However, there is clear negative difference electron density indicating the loss of the aspartate sidechain at residue 48 (Figure 3). A recent X-ray structure of another aerolysin-like toxin, Eisenia fetida Lysenin 38 has identified a sphingomyelin binding site on one edge of the N-terminal domain. However, when the molecules are compared, this site is on the opposite side of the domain from Asp48. Therefore, no conclusions about the loss of activity in CPE-D48A mutant can be reached from our high-resolution structure. Oligomerisation and membrane-insertion will induce conformation in CPE and the Asp48Ala mutation may affect these conformation changes. Alternatively Asp48 may form part of the oligomerisation interface. Further studies are required to identify the role of this residue in CPE cytotoxicity.
FIGURE 3.
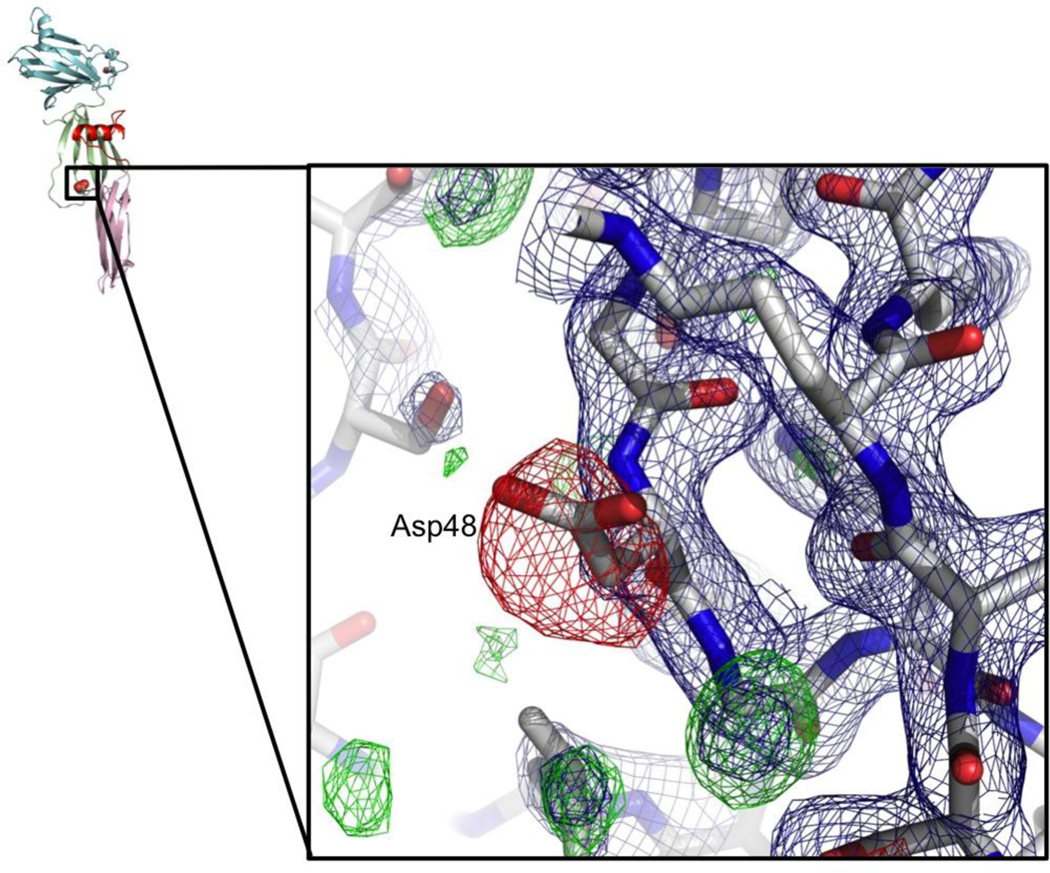
Difference density for D48A, contoured at +3.0 (green) and −3.0 (red) rms, phases calculated following initial rigid-body refinement. Final 2Fo-Fc map for D48A mutant, contoured at 1.5 rms in blue. Asp 48 side-chain conformation in Δ37CPE shown as sticks with thin dark grey bonds.
PEPTIDE-BOUND STRUCTURE (CPE- CLD2 ECL2 VARIANT)
Co-crystallisation of ΔN37CPE with a peptide derived from a variant mouse Claudin-2 ECL2 with two residue changes to increase CPE binding affinity 25 resulted in a new crystal form.
Many authors have shown that the composition of ECL2 is important for CPE binding, using both chimeric claudins (with ECL2 from one claudin exchanged for that from another)39 and site-directed mutations34; 40 in full-length claudin. Winkler et al 25 showed that using peptides corresponding to the ECL2 sequence in surface plasmon resonance experiments gave results that reflected binding and cytotoxicity assays using full-length claudin expressed in mammalian cells. Wild-type mouse claudin-2 (UniProt ID O88552) does not bind CPE 25. However, mutating Asp149 to Asn and Ser155 to Ala resulted in a ECL2 peptide that bound C-CPE more tightly than the ECL2 peptide derived from a Claudin known to bind CPE. The importance of residue 149 being Asn for CPE binding mutation has been shown by many researchers 25; 34; 39; 40 (Table 1). The mutations result in a CPE binding fingerprint identical to that in CPE-binding claudins and thus the results seen here are expected to be applicable to CPE-claudin interactions in full-length native CPE. ΔN37CPE was incubated overnight with this peptide (141HGILRDFYNPLVPDAMKFEI160).
Following incubation, crystallisation trials resulted in crystals diffracting to 3.4 Å. Data analysis revealed a spacegroup of C2 once again, but this time with cell dimensions of a=369.6, b=100.3, c=265.4 Å and β=119.7 °. Structure solution revealed 15 molecules in the asu arranged in five trimers, with a solvent content of 70 %. The presence of both high NCS and high solvent content resulted in more detailed maps than is normally expected at this resolution. Refinement gave an R/Rfree of 20.4/24.0 %, with further statistics provided in Table 2 and the co-ordinates deposited with PDB ID 4P5H. All the monomers and trimers in the asu are essentially identical to each other and similar to the monomer and trimers seen in the higher resolution ΔN37CPE structure, to which they were restrained during refinement (Table 3a and b).
CONFORMATION OF CLD2 IN THE CPE-CLD2 CRYSTALS
Difference electron density maps calculated using phases from the initial molecular replacement showed clear difference electron density (more than three times the root-mean-square electron density) at the cleft in the C-terminal domain that has been previously associated with claudin binding 29, Figure 4A. The presence of two prolines in the CPE-binding fingerprint of claudin allowed easy identification of these residues in the binding pocket. In addition, the residues in claudin that are most important for CPE binding have been identified in a number of studies 25; 34; 40. Several of these studies went on to mutate CPE residues they hypothesised interacted with the mutated claudin residues and showed this restored function. These studies allowed us to assign sidechains to claudin residues based on the CPE residues they interacted with. Figure 4C shows the final 2Fo-Fc map, while the interactions between the protein and peptide are illustrated in Figure 4B and D. Strong density is present in each of the 15 binding grooves for the residues of the mutant CLD2-derived peptide that have been shown to be essential for CPE-binding 25, in particular the fingerprint residues 149–153 (NPLVP), which can only tolerate conservative mutations, discussed below. As the CLD2-derived peptide extends away from CPE and into solvent the NCS relationship between the copies breaks down, and in most cases it has not been possible to reconstruct its entire length. Residues leading away from the fingerprint towards N-and C-terminii can clearly be seen to be adopting a helical conformation, but NCS between CLD2 copies breaks down as distance from the groove increases, resulting in a loss of electron density map quality, so that we have been unable to build these helical parts of the peptide into the electron density map. We will therefore concentrate our discussion on the fingerprint residues (149–153) and well-ordered mutant CLD2-derived peptide residues either side that interact with CPE. In the remainder of the results and discussion, residues from the CLD2-derived peptide will be prefaced with cld, while those from CPE will be prefaced cpe for clarity.
FIGURE 4.

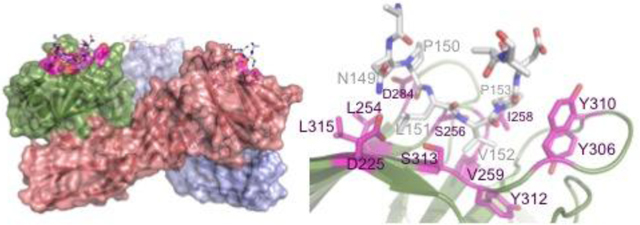
(A) Difference map showing typical density (contoured in green at 3.0 rms) for claudin-derived peptide for a typical one of the 5 trimers in the peptide-bound asymmetric unit. Trimer is shown as cartoon representation. (B) One of the 5 trimers in the asymmetric unit showing CPE as cartoon representation with semi-transparent surface representation and the final claudin-derived peptide in stick representations, with peptide interacting residues in magenta. (C) Final refined 2Fo-Fc electron density map contoured at 1.5 rms in dark blue with final refined coordinates shown as sticks, protein with green bonds and claudin derived peptide with white coloured bonds (D) Stick representation of final refined peptide-bound structure, coloured as (B) with cpe residues that interact with claudin coloured magenta and labelled.
The formation of the CPE-CLD2 complex buries on average, 390 Å2 of CPE’s solvent-accessible surface in each monomer. The peptide is tethered at either end by possible electrostatic interactions, in particular between cldAsn149 and cpeAsp225 and cldAsp154 and cpeTyr310. However, the interaction is overwhelming hydrophobic in nature (Figure 4D), with cldVal152 and cldPro153 packing into first identified binding pocket 29. cldVal152 packs against cpeIle258, cpeVal259 and cpeTyr312 while cldPro153 stacks against cpeTyr306 and cpeTyr310. cldLeu151 is inserted into the pocket recently identified by Veshnyakova et al 34, including residues cpeLeu254 and cpeLeu315, confirming their analysis of the importance of these residues.
There is a constriction in the claudin-binding groove of CPE, as a result of the sidechain of cpeSer313 protruding into it, that separates the originally identified pocket formed by residues including cpeTyrosines 306, 310 and 312 from the newly identified and slightly shallower pocket containing cpeLeu254 and 315. This constriction defines the orientation of cldVal152 needed for insertion into CPE’s claudin-binding groove. The required orientation is achieved by presentation of these two residues (cldLeu151 and cldVal152) at the apex of a tight turn. Of the five residues in the claudin NPLVP fingerprint associated with CPE binding (cld residues 149–153), both cldPro150 and cldPro153 are required to form the tight turn that allows cldLeu151 and cldVal152 to insert into the correct pockets in CPE’s claudin binding groove, with cldPro150 adopting a cis conformation.
Discussion
EFFECT OF N-TERMINAL RESIDUES ON CPE CYTOTOXICITY
It has been shown that the deletion of the N-terminal 37 residues from CPE results in 2–3-fold increased cytotoxicity 31. Previously determined structures of CPE have shown that the N-terminal 37 residues of CPE are disordered 32; 33. The structure of ΔN37CPE presented here reveals that the absence of these residues has no significant effect on the rest of the CPE structure. However the presence of the disordered residues in full-length CPE can be inferred from the changes in orientation between the N- and C-terminal domains and the change in conformation of the ordered N-terminal residues between the full-length and ΔN37CPE structures. The presence of this 37-residue disordered peptide inhibits CPE oligomerization, as has been shown by Kokai-Kun & McClane, 1997 26, probably through steric hindrance of claudin receptor binding.
IMPLICATIONS OF THE CPE-CLD2 COMPLEX STRUCTURE
While the mutated mouse Claudin-2 derived peptide used in these studies binds more tightly than other peptides, it has been reported that cell-lines expressing mouse Claudin-3, −4, −7 or −8 or human claudin-4 are all highly sensitive to CPE exposure 39, and peptides derived from ECL2 of mouse Claudins-3, −6, −7, −9 and −14 bind to CPE 25 with various levels of affinity. Claudin peptides with a 5-residue fingerprint of the form NP[L/M][V/T/L][P/A] show the highest affinity for CPE. Asparagine at position 1 of the fingerprint is required to formed the necessary stabilizing interaction with cpeAsp225 and proline at position 2 is required as this residue contains a cis peptide. The next two residues are required to be medium-sized hydrophobic residues, to fit into the pockets on the CPE C-terminal domain surface, and finally the residue at position 5 in the fingerprint is required to form a tight-turn, but does not adopt a cis conformation, so that either a Proline or Alanine residue can be substituted at this point.
Additional residues in claudin are also involved in the interaction, for example, Claudin-8 can still bind CPE, all be it with reduced affinity, despite lacking a complete CPE-binding fingerprint 15. In Claudin-8, the fingerprint residues have the sequence NSIVN and it should be noted that replacement of the Serine for the correct fingerprint residue, Proline, significantly increases the affinity of Claudin-8 for CPE. The same study 41 showed that Claudin-14 also binds CPE, at reduced affinity, compared to Claudin-3 and −4, despite having a complete CPE-binding fingerprint. It highlighted the mutation of cldAsp146, N-terminal to the fingerprint to Asn in Claudin-14, and showed that restitution of the more common Asp at this position restored CPE-binding affinity. cldAsp146 is not visible in these electron density maps so that we are not able to comment on the likely reason for this.
Takahashi et al 20 introduced mutations into a fragment of CPE (residues 116–319) called C-CPE, to create a broad-spectrum claudin-binding protein. One of the mutations was cpeSer313His. The larger residue will likely completely obscure the cpeTyr306/310/312 binding pocket, which we have shown confers specificity on CPE. The loss of this pocket therefore effectively removes binding specificity. The remaining mutations were at CPE residues 304, 305, 307 and 309, and introduced a number of positively-charged residues. These residues are outside of the specificity-conferring pocket and have likely formed an entirely new, predominantly electrostatic, recognition site for claudins in general.
The binding of a claudin peptide to CPE was modelled and the predicted binding orientation was tested by introducing mutations into both CPE and the claudin loop in a study published by Veshnyakova et al 34. Our CPE-CLD2 complex structure shows the peptide bound in the pocket in the opposite orientation to that which their modelling suggested (with the pockets containing cldLeu151 and cldVal152 swapped). However, the orientation of the cldLeu152 in the smaller pocket as observed by X-ray crystallography leaves sufficient room to accommodate the Leu152Phe mutation that their model suggested it could not. In addition, Veshnyakova et al 34 showed that the mutation cldAsn149Asp reduced binding to the C-terminal domain of CPE. Their model proposed that cldAsn149 and cpeAsp284 would be interacting, but complementary mutations of these two residues failed to rescue Claudin binding. In contrast, in our structure, cpeAsp284 is positioned so that it is only able to interact with Claudin peptide mainchain so could not rescue cldAsn149Asp binding, while cpeAsp225 is interacting with cldAsn149, explaining why mutation to Asp is deleterious.
Kimura et al 39 noted that the claudin-binding groove of CPE has a negative electrostatic charge. They altered the electrostatic charge in ECL2 and showed that increasing the electronegativity in this loop decreased binding. However, we show that one of the mutations they made (Asn149Asp), which has been shown by many others to eliminate CPE binding in normally CPE-sensitive claudins, has a direct interaction with CPE. The other mutation, Arg158Tyr, is further from the peptide-binding groove in our structure and is not ordered. However, we note that others have shown that introduction of a charged residue at this point does not alter C-CPE binding in the absence of the Asn149Asp mutation25, and that the Claudin-2 ECL2-derived peptide we have used in these studies has the hydrophobic, Phe, in this position. Therefore it seems likely that work provides further evidence that Asn149 is essential for CPE binding rather than electrostatics being of primary importance in the CPE-claudin interaction.
IMPLICATIONS FOR PORE INSERTION
1). BOUND POLYETHYLENE GLYCOL MOLECULES
We have identified a number of bound polyethylene glycol (PEG) molecules in both the ΔN37CPE and ΔN37CPE-D48A structures. There are 6 well-ordered PEG molecules in each structure at each of the interfaces between molecules in both trimers (Figure 5). These molecules have direct interactions with the amphipathic sequence that has been shown to be involved in membrane insertion (residues 81–106). In addition there are less well-ordered PEG molecules bound to the C-terminal domain on the same face as residues known to be important for claudin-binding. PEG is an amphipathic molecule and may be mimicking the recognition of phospholipid headgroups following the initial binding of CPE to cell membrane surfaces.
FIGURE 5.

Cartoon representation of one of the trimer from the ΔN37CPE crystal form. N-terminal domain in lighter shades, C-terminal domain darker. Bound polyethylene glycol (PEG) molecules are shown in atom-coloured spheres (carbon grey). The location of the PEG molecules is similar in both trimers in the Δ37CPE asymmetric unit (asu) and in both trimers in the D48A asu. The claudin-binding pockets of the CPE molecules are indicated by bond representation of cpeTyr306, 310 and 312, coloured the same as each C-terminal domain.
II). CPE PACKING ARRANGEMENT IN THE CRYSTAL
The packing of the CPE-CLD2 complex in the asymmetric unit is very interesting as it hints at a role for cpeAsp48. The five trimers in each asymmetric unit are packed in a helix with a 72° turn between each successive trimer, resulting in the 5-trimer repeat (supplementary Figure 2). The ΔN37CPE-D48A mutant structure described above showed that the mutation cpeAsp48Ala does not affect the protein fold. The ΔN37CPE-D48A structure, and previous CPE structures 32; 33, have shown cpeAsp48 is solvent exposed and distant from the claudin-binding pockets in the same monomer and trimer, from the recently identified lipid binding site on another aerolysin-like βPFT, lysenin, 38 and from the likely membrane-inserting residues 81–106. However, cpeAsp48 in one trimer is just 6 Å from the claudin-derived peptide bound to the adjacent trimer in the helix (Figure 6A and B). While in the peptide-bound structure the membrane-inserting residues 81–106 are buried, and therefore a conformation change will be necessary to form the prepore, the importance of cpeAsp48 for both cytotoxicity and maintaining the helical interactions seen in the crystal suggests that the helix may represent an initial claudin-bound form of CPE. In tight junctions within a cell-cell contact claudin has been shown to form a ‘strand and groove’ architecture. However, the protein assembly within the tight junctions is so tightly packed by the trans-interactions between the extracellular loops of the claudin molecules that CPE cannot penetrate into this structure 25. The latter work showed that CPE associates to claudin oligomers at the cell membrane outside the tight junctions. Based on this, and the CPE trimer helix described above, a scheme of the proposed interaction is depicted in figure 6C.
FIGURE 6.
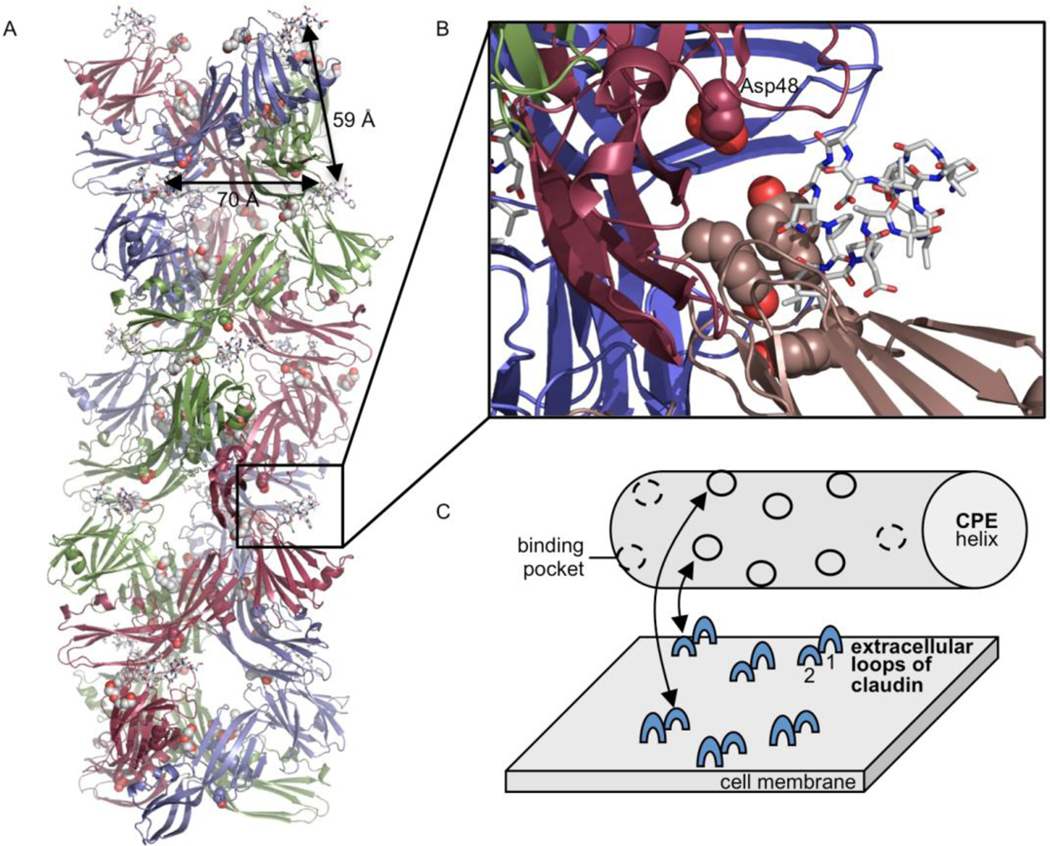
(A) Cartoon representation of trimer helix in the asymmetric unit, with claudin-derived peptides shown as sticks and Asp48 and Tyr306, 310 and 312 as atom spheres. The location of PEG as seen in the ΔN37CPE structure are shown as yellow sticks. Distances between claudin binding sites both within the trimer and between adjacent trimers in the helix are indicated. (B) Close up of (A), with monomer to which peptide is bound coloured pale pink and monomer from the adjacent trimer coloured maroon. (C) Scheme of the potential interaction between CPE trimer helix and claudin oligomers and the membrane surface of a claudin expressing cell outside of cell-cell contacts.
IN CONCLUSION
The results we present here show for the first time the conformation of a peptide derived from claudin ECL2 bound to CPE. In addition to the valuable information it provides about the molecular determinants of the highly specific interaction between CPE and Claudin-3 and −4, our work provides the first experimental atomic resolution data for any claudin, and shows that ECL2 has an unusual conformation that is important for its interaction with CPE. We have presented here three structures of CPE mutants, determined from crystals grown in conditions different to those of the first full-length CPE structures. There are several copies of the CPE monomer in each of the asymmetric units and in all cases they are contained in the same intimate trimer seen in the initial structures. While this trimer is certainly not the membrane active form, its persistence in all atomic resolution structures of CPE suggests that it may have some biological relevance, for example in pore formation. Finally, the unusual packing of the CPE-CLD2 complex in the crystals reveals an interaction of cpeAsp48 with claudin-derived peptide bound to the claudin-binding pocket of a CPE molecule in a different trimer.
Materials and Methods
CLONING
The full length CPE gene was PCR amplified from C. perfringens strain 8–6 and cloned into pGEX2T. Using this template a truncated CPE construct was generated (residues 37–319). A ΔN37CPE construct was created from this using forward (NcoI) 5’-CATGCCATGGGCAGTGATGGATTATATG-3’ and reverse (XhoI) 5’-CCGCTCGAGTTAAAATTTTTGAAATAATATTG-3’ oligonucleotide primers (synthesised by MWG Eurofins) with which a polymerase chain reaction (PCR) for 25 cycles was performed. The reaction product was run on an agarose gel and a gel extraction (Qiagen) protocol was followed. The vector pHis 42 and the PCR product were digested with NcoI and XhoI before ligating the two together. Positive clones were sequenced to ensure no mutations were introduced. The resultant construct had a recombinant Tobacco Etch Virus (rTEV) cleavable His6-Tag on the N-terminus.
D48A MUTANT
Using the ΔN37CPE construct, aspartate 48 was mutated to alanine using the QuickChange method (Agilent Technologies). Primers 5’-GTAATAGATAAAGGAGCTGGTTGGATATTAGGGGAACC-3’ and 5’-GGTTCCCCTAATATCCAACCAGCTCCTTTATCTATTAC-3’ were utilised to introduce the mutation.
PROTEIN EXPRESSION AND PURIFICATION
Protein was expressed in E.coli BL21 (DE3) cell-line. Cells were grown to an OD600 of 0.7±0.1 at 37 °C with vigorous shaking, before cooling at room temperature for 30 minutes. Cells were induced using IPTG to a final concentration of 0.5mM and grown overnight at 26 °C shaking at 170 RPM. Cells were pelleted and stored at −20 °C.
Cell pellets were re-suspended on ice in ice-cold lysis buffer (50 mM Tris-HCl pH7.5, 500 mM NaCl and protease inhibitor cocktail (Roche Bioscience). Re-suspended cells were lysed by passing through a homogeniser twice to ensure complete cell lysis. Cell lysate was sonicated for three 30 s bursts with a 1 min rest period between each burst. The insoluble fraction was removed by centrifugation at 18,000 RPM using a Sorvall RC+ with a F21A-8×50y rotor at 4 °C.
The resultant supernatant was filtered through a 0.44 μm (Millipore) to remove any remaining insoluble material. The supernatant was loaded onto a HisTrap (GE Healthcare) 5 ml column and eluted with a stepwise gradient of imidazole (4 %, 8 % and finally a gradient up to 100% of 500 mM in a buffer also containing 20 mM Tris HCl pH 7.5 and 500 mM NaCl). The fractions containing CPE were pooled and dialysed overnight at 4 °C in 20 mM Tris-HCl pH 7.5, 150 mM NaCl and 2 mM β-mercaptoethanol. ΔN37CPE was also dialysed in the presence of rTEV to remove the His6-tag in a weight ratio of 25:1 (ΔN37CPE: rTEV). The resultant solution was passed through a HisTrap (GE Healthcare) 5 ml column and the flowthrough was collected. The flowthrough was concentrated using a 10 kDa MW cut off concentrator (Millipore) and loaded onto a Superdex S200 size exclusion column. The gel filtration column was washed with a buffer containing 20mM Tris-HCl pH7.5, 150 mM NaCl and 1 mM DTT. Fractions containing CPE were pooled and concentrated to 20 mg/ml, their purity was verified by native gel and the protein was then stored at −80 °C.
CRYSTALLISATION
ΔN37CPE, ΔN37CPE-D48A and CPE-CLD2 were screened for crystallisation conditions using Hampton Research Crystal Screen I and II, JCSG and PACT, promising conditions were optimised using the hanging-drop technique with 2 μL 20 mg/ml protein and 1 μL mother liquor, the well contained 100 μL mother liquor. Crystals of ΔN37CPE and ΔN37CPE-D48A grew at 16 °C from mother liquor containing 25% (w/v) polyethylene glycol, average mw 1500 Da (PEG 1500) buffered by 100 mM SPG buffer (succinic acid, sodium dihydrogen phosphate and glycine in the ratio 2:7:7) at pH6.0. For CPE-CLD2, protein was incubated overnight at 4°C with 1 mM peptide (sequence: HGILRDFYNPLVPDAMKFEI) supplied by Biosynthesis (Lewisville, Texas, USA) before setting up crystallisation trials as described above with mother liquor containing 22 % (v/v) 2-methyl-2,4-pentanediol and 200 mM ammonium acetate buffered by 100 mM sodium citrate, pH 5.7. Prior to freezing, crystals were cryoprotected by soaking in mother liquor supplemented by 15% (v/v) PEG 400. The peptide was taken from ECL2 of mouse Claudin-2 141–168 which does not bind CPE, however, two serines were replaced by an asparagine and an alanine (underlined in the sequence above) recreating the CPE binding fingerprint from claudin-3 25.
DATA COLLECTION, PHASING AND REFINEMENT
All datasets were collected at the European Synchrotron Radiation Facility (Grenoble, France) on beamline ID23–2 (ΔN37CPE), ID29 (ΔN37CPE-D48A and CPE-CLD2) and indexed, integrated and scaled with XDS software suite 43. Both ΔN37CPE and ΔN37CPE-D48A belonged to the same crystal symmetry and diffracted to 1.9 Å. The spacegroup was C2 and the cell dimensions were a=191.7, b= 128.3, c=137.2 Å, β=133.8° for ΔN37CPE and a=190.6, b=128.0, c=136.4 Å, β=133.8° for ΔN37CPE-D48A. ΔN37CPE was solved by molecular replacement with Phaser 44 using a monomer from our original CPE structure 32 (PDBID 2XH6) as a model. It revealed that there were 6 molecules in the asymmetric unit (asu), corresponding to a Matthews Volume of 2.9 Å3/Da and a solvent content of 58% (v/v). The mutant ΔN37CPE-D48A data is isomorphous with the ΔN37CPE data, so the refined ΔN37CPE co-ordinates were subjected to Rigid Body refinement against the ΔN37CPE-D48A data with Buster version 1.10.0 45 resulting in both R-factor and Rfree of 27.6%. Data processing statistics are listed in Table 2.
The CPE-CLD2 complex crystallised in a different form, also spacegroup C2, but with cell dimensions a=369.6, b= 100.3, c=265.4 Å, β=119.7°. The data were integrated and scaled with XDS 43 to resolution of 3.4 Å. The CPE-CLD2 complex structure was solved by molecular replacement with Phaser 44 and a trimer of the refined ΔN37CPE co-ordinates, and gave 15 CPE monomers in the asu with a Matthew’s Volume of 4 Å3/Da and a solvent content of 70%. Statistics are listed in Table 2.
Following structure solution, all three models were refined with Buster 45 with NCS restraints 46 and rebuilt in Coot 47. In the case of the lower resolution CPE-CLD2 dataset restraints to the higher resolution native structure, ΔN37CPE, were also employed 46. The final R/Rfree was 17.6/19.7 % for ΔN37CPE, 17.5/19.6% for ΔN37CPE-D48A and 20.4/24.0% for CPE-CLD2. Further refinement statistics are provided in Table 2.
Supplementary Material
Highlights.
Clostridium perfringens enterotoxin is a toxin and potential oncotherapeutic.
We present the structure of a CPE-receptor analogue (modified Claudin-2) complex.
Claudin extracellular loop-2 adopts an unusual tight turn to bind to CPE.
Understanding this interaction is vital to develop CPE’s biopharmaceutical potential.
Acknowledgements
The authors wish to acknowledge MRC for funding CEN (project G0700051), the Wellcome Trust for funding CGS and AKB (project: WT089618MA) and NIH for grant R37 AI19844–30 supporting BMc.
Footnotes
Accession Numbers
The structures described in this paper have been submitted to the protein databank with the following IDs: ΔN37CPE – 3ZIX; ΔN37CPE-D48A – 3ZIW; CPE-CLD2 – 4P5H
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adak GK, Long SM & O’Brien SJ. (2002). Trends in indigenous foodborne disease and deaths, England and Wales: 1992 to 2000. Gut 51, 832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarker MR, Carman RJ & McClane BA. (1999). Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol Microbiol 33, 946–58. [DOI] [PubMed] [Google Scholar]
- 3.Skjelkvale R & Uemura T. (1977). Experimental Diarrhoea in human volunteers following oral administration of Clostridium perfringens enterotoxin. J Appl Bacteriol 43, 281–6. [DOI] [PubMed] [Google Scholar]
- 4.Carman RJ. (1997). Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8, S46–S48. [Google Scholar]
- 5.Brett MM, Rodhouse JC, Donovan TJ, Tebbutt GM & Hutchinson DN. (1992). Detection of Clostridium perfringens and its enterotoxin in cases of sporadic diarrhoea. J Clin Pathol 45, 609–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mpamugo O, Donovan T & Brett MM. (1995). Enterotoxigenic Clostridium perfringens as a cause of sporadic cases of diarrhoea. J Med Microbiol 43, 442–5. [DOI] [PubMed] [Google Scholar]
- 7.Songer JG. (1996). Clostridial enteric diseases of domestic animals. Clin Microbiol Rev 9, 216–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murrell TG, Ingham BG, Moss JR & Taylor WB. (1987). A hypothesis concerning Clostridium perfringens type A enterotoxin (CPE) and sudden infant death syndrome (SIDS). Med Hypotheses 22, 401–13. [DOI] [PubMed] [Google Scholar]
- 9.McClane B. (2007). Clostridium perfringens. In Food Microbiology: Fundamentals and Frontiers 3rd edit. (Doyle MP & Beuchat LR, eds.), pp. 423–444. ASM Press, Washington, DC, USA. [Google Scholar]
- 10.Czeczulin JR, Hanna PC & McClane BA. (1993). Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect Immun 61, 3429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClane BA. (2001). The complex interactions between Clostridium perfringens enterotoxin and epithelial tight junctions. Toxicon 39, 1781–91. [DOI] [PubMed] [Google Scholar]
- 12.Tsukita S, Furuse M & Itoh M. (2001). Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2, 285–93. [DOI] [PubMed] [Google Scholar]
- 13.Van Itallie CM & Anderson JM. (2006). Claudins and epithelial paracellular transport. Annu Rev Physiol 68, 403–29. [DOI] [PubMed] [Google Scholar]
- 14.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J & Blasig IE. (2008). Structure and function of claudins. Biochim Biophys Acta 1778, 631–45. [DOI] [PubMed] [Google Scholar]
- 15.Shrestha A & McClane B. (2012). Human Claudins-8 and -14 are Receptros Capable of Conveying the Cytotoxic Effects of Clostridium perfringens Enterotoxin. mBio. 2013 Jan 15;4(1):e00594–12. doi: 10.1128/mBio.00594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashimi SM, Yu S, Alqurashi N, Ipe DS & Wei MQ. (2013). Immunotoxin-mediated targeting of claudin-4 inhibits the proliferation of cancer cells. Int J Oncol 42, 1911–8. [DOI] [PubMed] [Google Scholar]
- 17.Lal-Nag M, Battis M, Santin AD & Morin PJ. (2012). Claudin-6: a novel receptor for CPE-mediated cytotoxicity in ovarian cancer. Oncogenesis 1, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neesse A, Hahnenkamp A, Griesmann H, Buchholz M, Hahn SA, Maghnouj A, Fendrich V, Ring J, Sipos B, Tuveson DA, Bremer C, Gress TM & Michl P. (2013). Claudin-4targeted optical imaging detects pancreatic cancer and its precursor lesions. Gut 62, 1034–43. [DOI] [PubMed] [Google Scholar]
- 19.Gao Z & McClane BA. (2012). Use of Clostridium perfringens Enterotoxin and the Enterotoxin Receptor-Binding Domain (C-CPE) for Cancer Treatment: Opportunities and Challenges. J Toxicol 2012, 981626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi A, Saito Y, Kondoh M, Matsushita K, Krug SM, Suzuki H, Tsujino H, Li X, Aoyama H, Matsuhisa K, Uno T, Fromm M, Hamakubo T & Yagi K. (2012). Creation and biochemical analysis of a broad-specific claudin binder. Biomaterials 33, 3464–74. [DOI] [PubMed] [Google Scholar]
- 21.McClane BA & Wnek AP. (1990). Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect Immun 58, 3109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh U, Van Itallie CM, Mitic LL, Anderson JM & McClane BA. (2000). CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J Biol Chem 275, 18407–17. [DOI] [PubMed] [Google Scholar]
- 23.Smedley JG 3rd, Uzal FA & McClane BA. (2007). Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect Immun 75, 2381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson SL, Smedley JG 3rd, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM & McClane BA. (2007). Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell Microbiol 9, 2734–55. [DOI] [PubMed] [Google Scholar]
- 25.Winkler L, Gehring C, Wenzel A, Muller SL, Piehl C, Krause G, Blasig IE & Piontek J. (2009). Molecular determinants of the interaction between Clostridium perfringens enterotoxin fragments and claudin-3. J Biol Chem 284, 18863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kokai-Kun JF & McClane BA. (1997). Determination of functional regions of Clostridium perfringens enterotoxin through deletion analysis. Clin Infect Dis 25 Suppl 2, S165–7. [DOI] [PubMed] [Google Scholar]
- 27.Harada M, Kondoh M, Ebihara C, Takahashi A, Komiya E, Fujii M, Mizuguchi H, Tsunoda S, Horiguchi Y, Yagi K & Watanabe Y. (2007). Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem Pharmacol 73, 206–14. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi A, Komiya E, Kakutani H, Yoshida T, Fujii M, Horiguchi Y, Mizuguchi H, Tsutsumi Y, Tsunoda S, Koizumi N, Isoda K, Yagi K, Watanabe Y & Kondoh M. (2008).Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem Pharmacol 75, 1639–48. [DOI] [PubMed] [Google Scholar]
- 29.Van Itallie CM, Betts L, Smedley JG 3rd, McClane BA & Anderson JM. (2008). Structure of the claudin-binding domain of Clostridium perfringens enterotoxin. J Biol Chem 283, 268–74. [DOI] [PubMed] [Google Scholar]
- 30.Smedley JG 3rd & McClane BA. (2004). Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect Immun 72, 6914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kokai-Kun JF & McClane BA. (1997). Deletion analysis of the Clostridium perfringens enterotoxin. Infect Immun 65, 1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Briggs DC, Naylor CE, Smedley JG 3rd, Lukoyanova N, Robertson S, Moss DS, McClane BA & Basak AK. (2011). Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J Mol Biol 413, 138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y,Sugita-Konishi Y, Yamamoto S, Karatani H & Horiguchi Y. (2011). Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J Biol Chem 286, 19549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veshnyakova A, Piontek J, Protze J, Waziri N, Heise I & Krause G. (2012). Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/−4 protein suggests structural modifications of the toxin to target specific claudins. J Biol Chem 287, 1698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderluh G & Lakey JH. (2008). Disparate proteins use similar architectures to damage membranes. Trends Biochem Sci 33, 482–90. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Theoret JR, Shrestha A, Smedley JG 3rd & McClane BA. (2012). Cysteine-Scanning Mutagenesis Supports the Importance of Clostridium perfringens Enterotoxin Amino Acids 80 to 106 for Membrane Insertion and Pore Formation. Infect Immun 80, 4078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardy SP, Ritchie C, Allen MC, Ashley RH & Granum PE. (2001). Clostridium perfringens type A enterotoxin forms mepacrine-sensitive pores in pure phospholipid bilayers in the absence of putative receptor proteins. Biochim Biophys Acta 1515, 38–43. [DOI] [PubMed] [Google Scholar]
- 38.De Colibus L, Sonnen AF, Morris KJ, Siebert CA, Abrusci P, Plitzko J, Hodnik V, Leippe M, Volpi E, Anderluh G & Gilbert RJ. (2012). Structures of Lysenin Reveal a Shared Evolutionary Origin for Pore-Forming Proteins And Its Mode of Sphingomyelin Recognition. Structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimura J, Abe H, Kamitani S, Toshima H, Fukui A, Miyake M, Kamata Y, Sugita-Konishi Y, Yamamoto S & Horiguchi Y. (2010). Clostridium perfringens enterotoxin interacts with claudins via electrostatic attraction. J Biol Chem 285, 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson SL, Smedley JG 3rd & McClane BA. (2010). Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect Immun 78, 505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shrestha A & McClane BA. (2013). Human Claudin-8 and -14 Are Receptors Capable of Conveying the Cytotoxic Effects of Clostridium perfringens Enterotoxin. mBio. 2013 Jan 15;4(1):e00594–12. doi: 10.1128/mBio.00594-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheffield P, Garrard S & Derewenda Z. (1999). Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif 15, 34–9. [DOI] [PubMed] [Google Scholar]
- 43.Kabsch W. (2010). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ. (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller PA, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C & Womack TO. (2011). Buster 1.10.0 edit. Global Phasing Ltd, Cambridge, UK. [Google Scholar]
- 46.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C & Bricogne G. (2012). Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68, 368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG & Cowtan K. (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.