

Abstract
Opioid use disorder is a public health crisis that causes tremendous suffering for patients as well as substantial social and economic costs for society. There are currently available treatments for patients with opioid use disorder, but they remain intolerable or ineffective for many. Thus the need to develop new avenues for therapeutics development in this space is great. Substantial work in models of substance use disorders, including opioid use disorder, demonstrates that prolonged exposure to drugs of abuse leads to marked transcriptional and epigenetic dysregulation in limbic substructures. It is widely believed that these changes in gene regulation in response to drugs are a key driving factor in the perpetuation of drug taking and seeking behaviors. Thus, development of interventions that could shape transcriptional regulation in response to drugs of abuse would be of high value. Over the past decade there has been a surge in research demonstrating that the resident bacteria of the gastrointestinal tract, collectively the gut microbiome, can have tremendous influence on neurobiological and behavioral plasticity. Previous work from our group and others has demonstrated that alterations in the gut microbiome can alter behavioral responses to opioids in multiple paradigms. Additionally, we have previously reported that depletion of the gut microbiome with antibiotics markedly shifts the transcriptome of the nucleus accumbens following prolonged morphine exposure. In this manuscript we present a comprehensive analysis of the effects of the gut microbiome on transcriptional regulation of the nucleus accumbens following morphine by utilizing germ-free, antibiotic treated, and control mice. This allows for detailed understanding of the role of the microbiome in regulating baseline transcriptomic control, as well as response to morphine. We find that germ-free status leads to a marked gene dysregulation in a manner distinct to adult mice treated with antibiotics, and that altered gene pathways are highly related to cellular metabolic processes. These data provide additional insight into the role of the gut microbiome in modulating brain function and lay a foundation for further study in this area.
Keywords: Microbiome, opioid, gut-brain, RNA-sequencing, WGCNA
Introduction
Opioid use disorder (OUD) is a chronic psychiatric condition marked by increasing and out of control drug intake despite negative consequences, frequent periods of abstinence and relapse, and too often, fatal overdose. Despite the tremendous attention that OUD has received in recent years from the medical and research communities, the rates of overdose and relapse have only continued to increase in recent years (Hedegaard et al., 2018). While there are currently available pharmacotherapies to reduce the burden of OUD, these treatments remain ineffective or intolerable for too many; societal stigma, ineffective medical training, and healthcare disparities result in a substantial number of patients unsuccessfully abstaining from opioid use (Kreek et al., 2019; Pessar et al., 2021; Dydyk et al., 2022). Identification of novel targets for development of therapeutics is a critical need for the field.
While the precise neurobiology underlying substance use disorders remains to be fully understood, there is a robust body of research demonstrating a key role for transcriptomic and epigenetic effects underlying behavioral responses to opioids and other drugs of abuse. Prolonged exposure to drugs of abuse leads to transcriptomic changes in key limbic nuclei that can persist long after the last drug exposure (McClung and Nestler, 2003; Robison and Nestler, 2011; Eipper-Mains et al., 2013). These changes in gene expression are associated with functional changes in critical brain regions and can drive drug taking and seeking behaviors. Recent work has attributed the persistent effects of drug use to changes in activity of transcription factors, as well as epigenetic writers and erasers, which alter the propensity of genes to be transcribed in response to a stimulus (Renthal et al., 2009; Walker et al., 2015; Nestler et al., 2016; Browne et al., 2020). The bulk of this mechanistic work has been performed in animal models of OUD and other substance use disorders; however similar effects have also been demonstrated in human patients with OUD (Egervari et al., 2017).
Most research examining transcriptional and epigenetic regulation in models of OUD and other substance use disorders has focused on the central nervous system (CNS). However, recent research suggests peripheral factors can play a key role in maintaining CNS transcriptional homeostasis. Work over the past 10 years has determined that the population of bacteria that occupy the intestinal tract, collectively called the gut microbiome, is an important regulator of the transcriptional landscape in the brain. Germ-free (GF) mice that are raised in a fully sterile environment with no microbiome have markedly altered transcriptional patterns in multiple brain regions, including the frontal cortex and amygdala (Gacias et al., 2016; Hoban et al., 2016, 2018; Chu et al., 2019). Specifically, the microglia of GF mice have significantly different transcriptomic and epigenetic profiles, which are at least partially reversible by colonization with a normal microbiome (Erny et al., 2015; Thion et al., 2018). This effect is not just seen in mice that have never had a microbiome. Adult mice treated with broad spectrum antibiotics (Abx) to reduce the bulk and complexity of the microbiome have altered transcriptional regulation in multiple brain regions as well. For example, single nucleus sequencing of Abx treated mice shows regulation of numerous genes in all neuronal and glial cell types in the prefrontal cortex (Chu et al., 2019).
Importantly, work from our group and others has identified robust interactions between the microbiome and behavioral and transcriptional response to opioids. Numerous reports have found that depletion of the microbiome alters development of tolerance to opioids and opioid withdrawal symptoms (Lee et al., 2018; Wang et al., 2018; Zhang et al., 2019; Jalodia et al., 2022). We recently reported that an Abx cocktail significantly reduces locomotor sensitization and conditioned place preference for morphine across a wide dose range (Hofford et al., 2021). Depletion of the microbiome with Abx also enhances self-administration of both oxycodone and fentanyl (Hofford et al., 2022; Simpson et al., 2022). In our recent work, we have found significant transcriptional interactions in the nucleus accumbens (NAc) between microbiome status and morphine treatment. Microbiome depletion without drug exposure only produced modest changes in gene expression in the NAc, but combined microbiome depletion and repeated morphine treatment robustly dysregulated transcriptome-wide gene expression (Hofford et al., 2021). Behavioral and transcriptional effects of microbiome depletion were largely reversible via supplementation with a cocktail of microbiome derived metabolites, the short chain fatty acids (SCFAs), suggesting these bacterial byproducts as potential mechanisms of this gut-brain signaling.
While our previous study provided critical information about how manipulations of the microbiome in adult animals can affect transcriptional regulation in response to opioids, mice treated with Abx still have some populations of antibiotic resistant bacteria in the gut, and growth of non-bacterial microorganisms can be affected by depletion of colonic bacteria. Here, we present an analysis of the NAc transcriptome from GF mice treated with saline or morphine for seven days to provide insight into the effects of lifelong total depletion of a microbiome on transcriptional regulation in this key limbic reward structure. Given our previous findings of robust microbiome-induced transcriptional changes in this region (Hofford et al., 2021), and the extensive literature showing the importance of the NAc in driving addiction-like behaviors (Robison and Nestler, 2011), the NAc seemed an appropriate target for more in depth analysis. These new analyses are integrated with the transcriptomic analyses from our previous study to identify patterns of gene expression induced by long-term GF status versus depletion of the microbiome in early adulthood. We find that there are marked changes in the transcriptome of GF mice compared to conventional mice after saline treatment, but show that GF mice have only modest changes in gene expression following morphine. These findings provide critical additional information about the role of the microbiome in modulating gene expression in the CNS, including across development.
Materials and Methods
Animals
For this analysis, the conventionally raised mice were described in the published manuscript by Hofford et al. in 2021, and thus all methods for conventional mice are identical to that manuscript (Hofford et al., 2021). The conventionally raised C57BL/6J male mice (7–9 weeks old, Jackson Laboratories) were group-housed in a humidity and temperature-controlled, pathogen-free colony facility on a 24 hour light/dark cycle (lights on at 7AM and lights off at 7PM). All studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals under an approved protocol from the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee.
Germ-free (GF) male C57BL/6J (7–9 weeks old) mice were bred in-house at the Mount Sinai Microbiome Translational Center and were maintained in flexible vinyl isolators throughout the experiment. Germ-free status was confirmed throughout the study via confirmed negative culturing of aerobic and anaerobic bacteria and negative 16S PCR per facility protocols. Germ-free mice were age-matched to the conventionally raised mice. Both conventionally raised mice and GF mice were provided food and water, ad libitum. All mice received the same standardized mouse chow and water from the same source, but food and water for germ-free mice were sterilized prior to administration.
Preparation of antibiotic drink solutions
Cages (4–5 mice/cage) were randomly assigned to H2O or Abx. The Abx drink solution was comprised of vancomycin (0.5 mg/mL; Chem-Impex International #00315), bacitracin (0.5 mg/mL; Research Products International #B3200025), pimaricin (1.2 μg/mL; Infodine Chemical #7681-93-8), and neomycin (2 mg/mL; Fisher Scientific #BP266925). Mice were administered their respective drink solution (Abx or H2O) for two weeks prior to beginning experimental procedures and remained on their respective solution for the duration of the experiment.
Morphine
Morphine sulfate was kindly provided by the NIDA drug supply program from the National Institute on Drug Abuse. Morphine sulfate was diluted in saline and injected subcutaneously at a dose of 20 mg/kg. For GF mice, the morphine solution was 0.2μm filtered to sterilize prior to administration.
Morphine ELISA
To ensure morphine concentrations administered were equivalent between conventionally raised mice and germ-free mice, the total concentration of morphine used in for all experiments was quantified using a morphine ELISA kit (Abnova #KA0935). All samples were run in duplicate per manufacturer’s instructions with the plate being read at 450nm and 650nm 30 minutes after addition of the reaction stop solution. The values from each solution were normalized to the control (unfiltered) injectate.
Morphine treatment and tissue processing
H2O, Abx, and GF mice were given daily injections of 20 mg/kg morphine or saline (s.c.) (n=5–6/microbiome status/treatment) for seven days. Twenty-four hours following the final injection, mice were rapidly decapitated. Germ free mice were removed from their isolator in a sterile filtered cage immediately prior to dissection. Brains were removed from mice and the nucleus accumbens (NAc) was identified by the anterior commissure. Punches of the NAc were obtained using a 14-gauge blunt needle and snap-frozen on dry ice. Samples were stored at −80°C until RNA extraction. RNA was extracted using TRIzol reagent (Thermo Fisher #15596026) according to manufacturer’s instructions. RNA integrity and purity was determined using an Agilent Bioanalyzer 2100 instrument and Nanodrop (OD 260/280 ratio). cDNA was generated using the SMARTer cDNA kit (Clonetech Laboratories # 634925) from total RNA. cDNA was fragmented using Bioruptor (Diagenode) and illumina-ready, indexed sequencing libraries were made using SPRIworks HT Reagent Kit (Beckamn Coulter, Inc. # B06938). Library quality and quantity were determined using an Agilent Bioanalyzer 2100. Samples were pooled and run on two lanes of an Illumina HiSeq 2500 using the PE100 rapid-mode setting. Data quality were checked using FASTQC (Babraham Institute, Cambridge, UK).
Differential expression analysis of H2O, Abx, and GF NAc samples
The raw FASTQ file for each sample was aligned to the Mus musculus 10 reference genome (NCBI) with the STAR package v2.4.0 (Dobin et al., 2013). Estimation of transcript abundance measures as fragments per kilobase mapped (FPKM) was then performed using Cufflinks from the Tuxedo protocol. Z-scored FPKM values were used for the generation of all heatmaps. For pairwise comparisons aligned read counts were analyzed for differential gene expression using the Biojupies analysis package with default settings (Torre et al., 2018). Statistical significance was set at a threshold of FDR-corrected p value of less than 0.05. Lists of significantly differentially expressed genes were analyzed for gene ontology pathways using G:Profiler (Raudvere et al., 2019).
Raw and processed RNAseq data files can be accessed using the Gene Expression Omnibus (GEO: GSE229978).
Weighted gene co-expression network analysis (WGCNA)
We performed weighted correlation with individual sample weights determined with the ‘signed hybrid’ network (where negatively correlated genes are assumed not connected). The soft thresholding power was determined using scale-free topology of each sample as a fit index. From the determined scale of independence and mean connectivity calculated, we used a soft thresholding power of eight to perform WGCNA. To identify unique modules, a one-step network was constructed using blockwise modules constructed with unsigned topological overlap matrices. To identify distinct modules, we utilized the Dynamic Tree Cut method. Of note, only module eigengenes that reached a threshold of 0.99 or above were included for subsequent network analysis. The edge and node data for all modules were exported to the external R package cytoHubba (Chin et al., 2014) to determine significant hub objects. Topological analysis methods, including Degree, Edge Percolated Component, Maximal Neighborhood Component (MNC), Density of Maximum Neighborhood Component (DMNC), Maximal Clique Centrality (MCC), and six centralities (Bottleneck, EcCentricity, Closeness, Radiality, Betweenness, and Stress). The top 25 eigengenes with the (MCC) scores for a specific module were visualized using Cytoscape software to highlight important hubs and for ease of visualization (Shannon et.al. 2003).
To determine the relationship between modules, the Pearson correlation coefficients between module eigengenes was calculated. Similarly, the relationship between individual eigengenes and microbiome status with treatment (H2O / Abx / GF + Saline / Morphine) were calculated using a Pearson correlation. Heatmaps and pathway analyses for WGCNA figures included all genes assigned to a specific module.
Figure Preparation
Individual panel graphs were created using GraphPad Prism. Two-way ANOVAs were also performed utilizing Graphpad Prism with post-hoc tests to perform additional pairwise comparison for any measures with significant main effects or interactions. Heatmaps were made utilizing Morpheus software from the Broad Institute (https://software.broadinstitute.org/Morpheus). All heatmaps have rows and columns clustered with unsupervised Euclidean distance hierarchical clustering with samples grouped by treatment. Color values for all heatmaps are z-scored FPKM values for the associated genes. Venn diagrams were made using BioVenn (Hulsen et al., 2008). Topological network diagrams were made as described above. The schematic in Figure 1 was made using BioRender with full permission to publish.
Figure 1 – Schematic for experimental design.
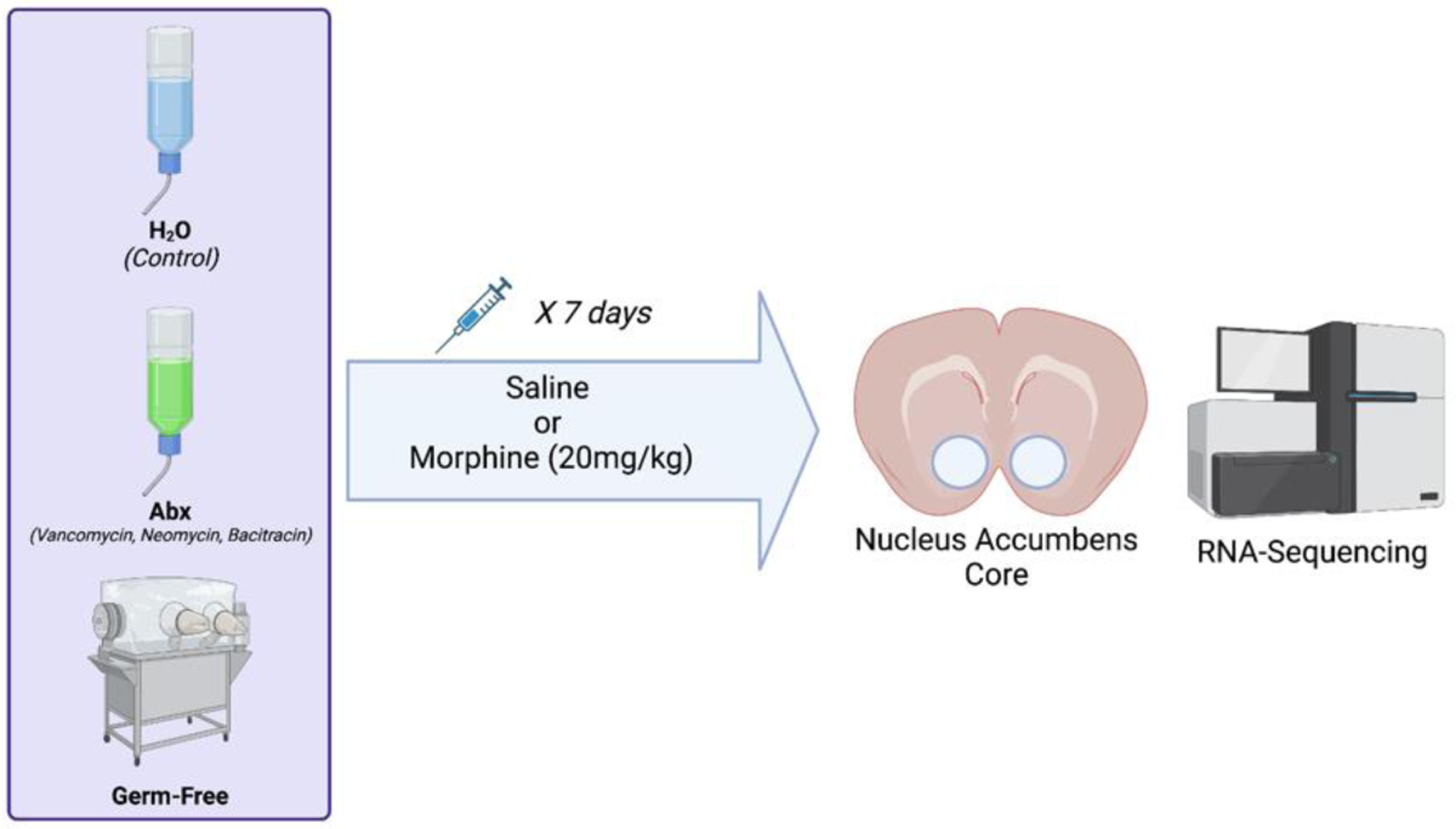
Mice for these experiments were conventionally colonized and maintained on untreated water (H2O), conventionally raised and treated with broad spectrum antibiotics for two weeks prior to injections (Abx), and those that were born and maintained fully germ-free throughout their lifespan (GF). Mice from each group received seven daily injections of saline or morphine (20mg/kg) and were euthanized 24 hours after the final injection, and the nucleus accumbens (NAc) dissected out for RNA isolation and transcriptional profiling.
Results
For these experiments mice were grouped into three distinct microbiome status groups (Fig. 1). These were conventionally raised mice on control water (H2O), conventionally raised mice treated with broad spectrum antibiotics for two weeks prior to the start of morphine treatment and continued on antibiotics throughout (Abx), and a third group that was germ-free from birth (GF). Mice in all groups were treated with daily injections of saline or high dose (20mg/kg) morphine for seven days. Germ-free mice were injected daily inside the gnotobiotic isolator, and their germ-free status was monitored throughout. Twenty four hours after the final injection the nucleus accumbens core (NAc) was dissected and processed for RNA sequencing. Of note, an analysis comparing the data from the H2O and Abx groups was previously published by our group (Hofford et al., 2021). As such, the focus of this manuscript will be analyses of germ-free animals compared to conventionally raised +/− Abx, as well as a more holistic gene coexpression network analysis.
Pairwise differential expression analysis
To assess effects of chronic germ-free status on gene expression in the nucleus accumbens we first compared saline injected germ-free mice (GF-Sal) to conventionally raised standard H2O treated mice (H2O-Sal). Baseline gene expression in germ-free mice, was markedly different from conventionally raised control mice (Fig. 2A - left), with ~4,000 genes being differentially expressed in the germ-free group. To determine molecular pathways that were altered by germ-free status we then performed gene ontology analysis using the G:Profiler software package (Raudvere et al., 2019). In Fig. 2A – right we highlight some pathways of interest demonstrating that germ-free mice have marked alterations in genes related to cellular metabolism (protein metabolic process & biosynthetic process) as well as robust changes in genes related to nuclear and cytosolic function. The full list of significantly altered GO terms is available as Table S1.
Figure 2 – Comparisons of conventionally raised mice with germ-free.
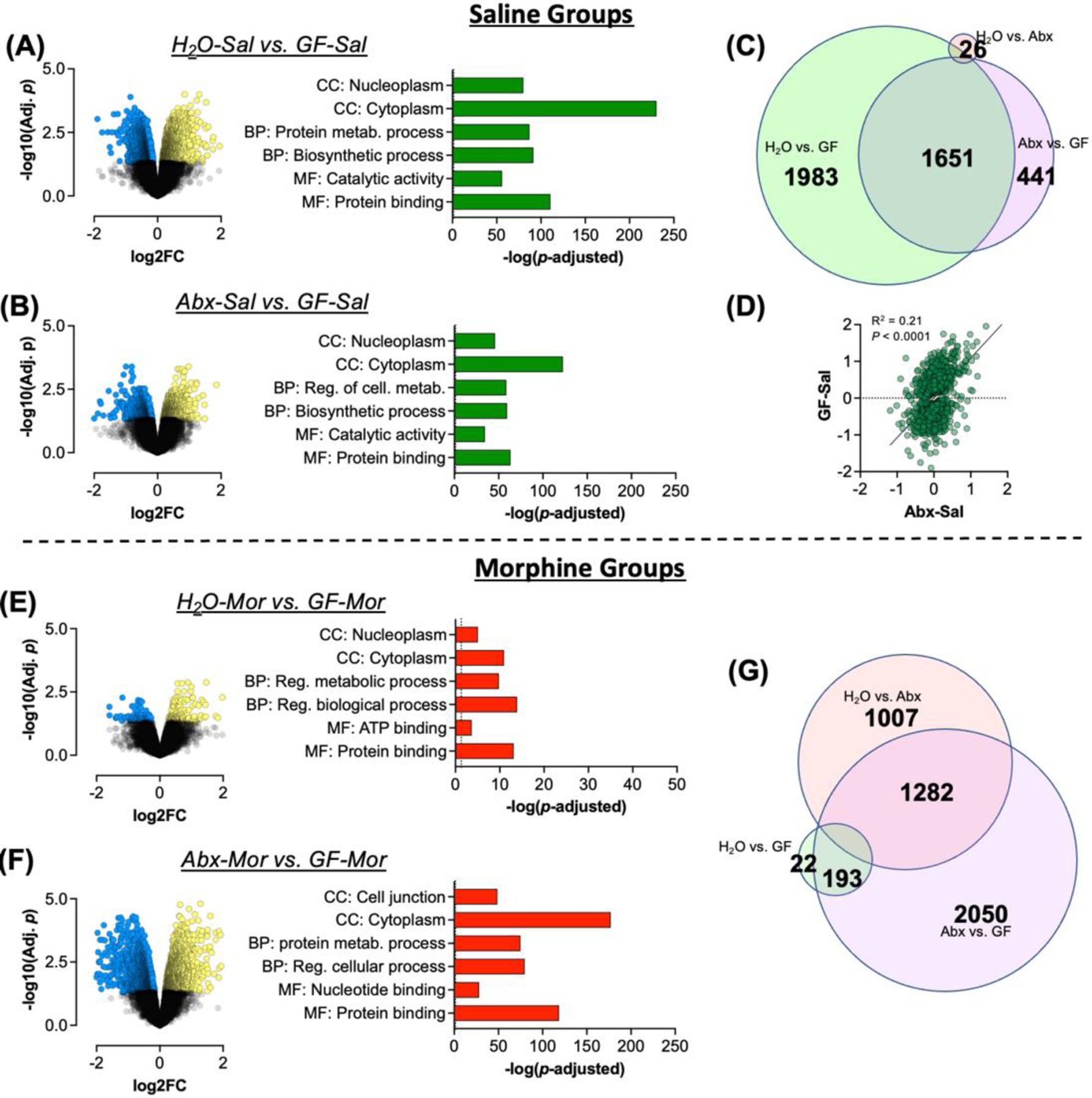
To ascertain effects of germ-free status on gene expression in the nucleus accumbens (NAc) we first compared saline treated groups. (A/B) Volcano plots and gene ontologies of interest for H2O-Sal and Abx-Sal compared to GF-Sal mice. (C) Venn diagram of significantly differentially expressed genes in all groups with proportions relative to the number of genes in each area. (D) Correlation analysis of all significant genes in the GF-Sal and Abx-Sal groups with Log2 fold change relative to H2O-Sal on each axis. (E/F) Volcano plots and gene ontologies of interest for H2O-Mor and Abx-Mor mice compared to GF-Mor mice. (G) Venn diagram of overlap between morphine treated groups.
We next compared gene expression patterns of Abx treated conventionally raised mice (Abx-Sal) with those of GF-Sal mice. As we showed in multiple previous studies, this Abx treatment regimen dramatically reduces both the amount and diversity of colonic bacteria (Kiraly et al., 2016; Hofford et al., 2021). This analysis thus allowed us to compare the effects of robust, though incomplete microbiome depletion in adult mice compared to the effects of lacking any microbiome throughout development. Interestingly, we find that NAc gene expression patterns of GF-Sal mice are also markedly different from Abx depleted mice with thousands of differentially expressed genes in this comparison as well (Fig. 2B – left). Pathway analysis demonstrated significant regulation of similar pathways in this comparison with robustly significant changes in genes in metabolic, nuclear, and cytosolic pathways (Fig. 2B – right & Full list in Table S2). Additionally, findings of specific genes of interest to OUD are presented as Fig. S1 which finds effects on genes related to glutamatergic and GABAergic signaling, transcription factors, and markers of multiple glial cell types.
Overlap of significantly altered genes between groups was then analyzed using BioVenn (Hulsen et al., 2008). While the largest subset of differentially expressed genes was unique to the H2O vs GF comparison, there was marked overlap with Abx vs GF differentially expressed genes (Fig. 2C – center). We then performed a correlation analysis with all genes that were regulated by depletion of the microbiome, using fold change in gene expression in Abx and GF mice compared to H2O controls. There was a significant positive correlation of fold change in gene expression between Abx and GF mice, with a larger average fold change seen in the GF mice (Fig. 2D – r2 =0.21, p < 0.0001). This suggests that while GF status was the largest driver of gene expression changes in this analysis, depletion of the microbiome in adulthood altered gene expression in a similar, though less pronounced, manner.
In our previous study, Abx and morphine treated mice (Abx-Mor) had the most robust changes in gene expression relative to H2O controls (Hofford et al., 2021). So here we compared gene expression patterns in GF morphine treated mice (GF-Mor) to the H2O and Abx morphine groups. Compared to H2O-Mor mice we saw fewer changes in GF-Mor mice than were noted with Abx-Mor previously, but still several hundred differentially regulated genes after false discovery rate (FDR) correction (Fig. 2E – left). Gene ontology analyses showed that these effects were in similar pathways as the other comparisons, but less robust (Fig. 2E – right, note altered x-axis scale compared to other panels; full list in Table S3). Comparison of Abx-Mor to GF-Mor mice showed more robust differential gene expression (Fig. 2F – left) and significant effects in numerous gene ontology pathways, with a marked effect in cell junction proteins and again in cellular metabolism (Fig. 2F – right & Table S4). BioVenn analysis to look at overlap of differentially expressed genes demonstrated greater transcriptomic differences between Abx and GF mice than H2O and Abx mice when the mice are treated with morphine (Fig. 2G). So GF mice treated with morphine show a different pattern of gene expression changes than morphine-treated mice with microbiome depletion in adulthood.
Given our specific interest in effects of the microbiome on transcriptional regulation following morphine treatment, we performed additional analysis looking at overlap of significantly altered gene ontologies among the morphine groups. As shown in Fig. 3, we found that each pairwise comparison resulted in multiple unique pathways, but there was significant overlap between the three comparisons, with the most robust overlap between the H2O vs Abx and Abx vs GF differentially expressed gene pathways (Fig. 3 – Center). In looking at the unique pathways in each comparison, we see that H2O vs GF mice had unique effects on mRNA processing and hormone secretion pathways (Fig. 3 – Left), H2O vs Abx had effects on protein transport, cell morphology, and sodium transport (Fig. 3 – Top). Abx vs GF yielded the most unique pathways with robust effects on cell metabolism, presynaptic, and transferase enzyme related pathways (Fig. 3 – Right).
Figure 3 – Overlap of gene ontology pathways between morphine groups.
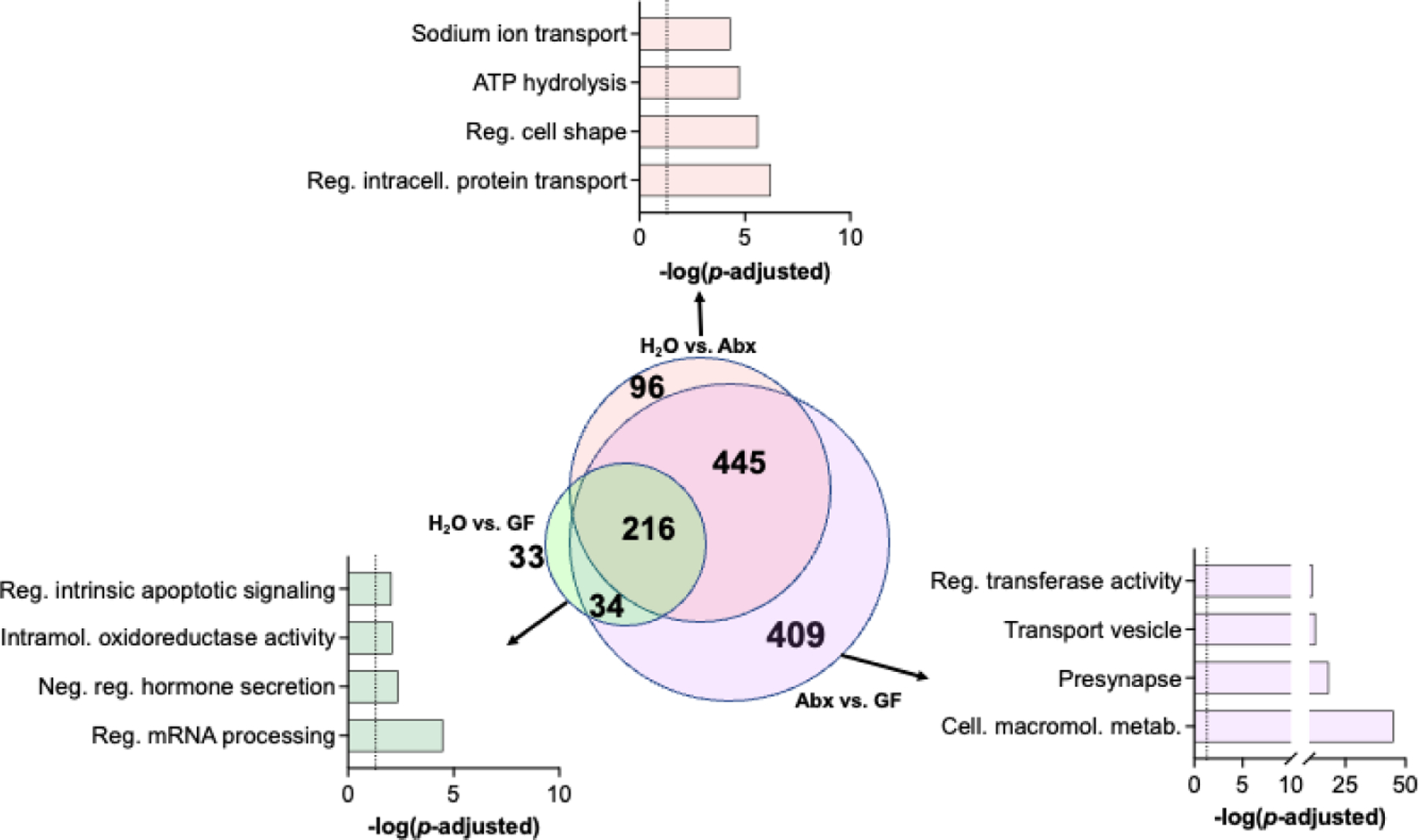
Venn diagram showing overlap of significant gene ontologies between all pairwise comparisons in morphine groups (center). Top significant pathways from H2O vs GF (left), H2O vs Abx (top), and Abx vs GF (right) groups are highlighted.
Effects of morphine in germ-free mice
As shown in Fig. 2, germ-free mice have a significantly altered transcriptome at baseline relative to conventionally raised animals. We next compared gene expression patterns of germ-free mice treated with saline or morphine. Interestingly, when the results were analyzed and FDR-corrected there were no significantly differentially expressed genes between the groups. There were approximately 300 genes that had a nominal p value < 0.05 which are presented in Fig. 4A. As can be seen from the patterns of expression, there are effects of morphine in GF mice, but the variability within groups is marked enough that the effects do not meet stricter criteria for statistical correction. This is in marked contrast to Abx treated mice, which exhibited marked differences compared to saline controls in our previously published work (Hofford et al., 2021). Given that the morphine solution for the germ-free mice had to be sterile filtered prior to injection, we wanted to ensure that the filtering process was not removing significant amounts of active compound. To test this, aliquots of the solutions used for injecting conventional mice and germ-free mice were analyzed via morphine ELISA. The sterile filtration did not result in any significant change in morphine concentration (Fig. 4B – t(6) = 0.38; p = 0.72). While we were not able to obtain serum samples from germ-free mice with acute morphine treatment for this study, we did previously find that Abx treated mice had no alterations in serum or brain distributions of morphine (Hofford et al., 2021).
Figure 4 – Effects of morphine treatment in germ-free mice.
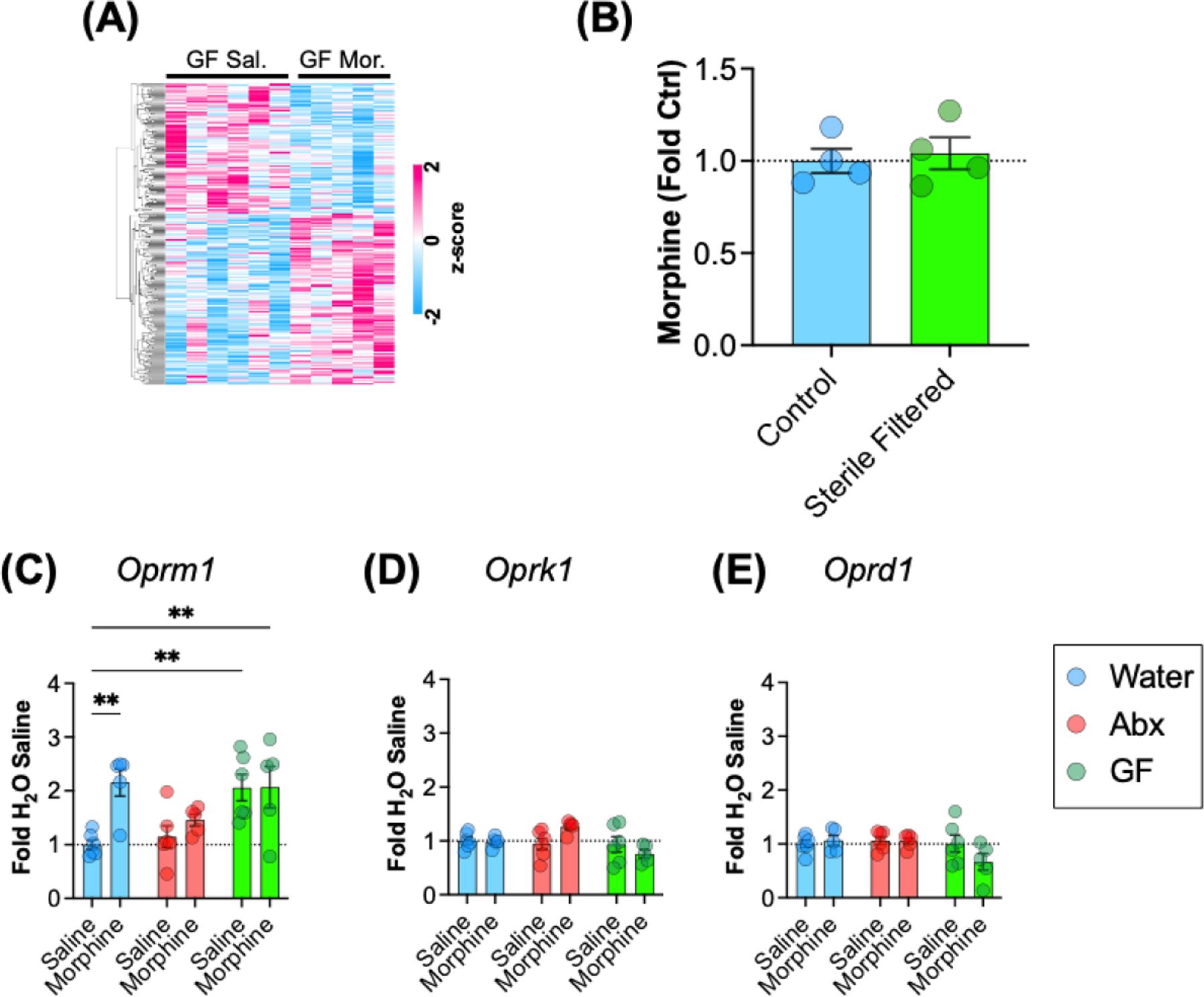
(A) Heatmap of relative expression of all genes with nominal p value < 0.05 between GF-Sal and GF-Mor groups. Scale is of z-scored FPKM values. (B) Concentration of morphine solutions used to inject conventionally raised mice (Control) and germ-free mice (Sterile filtered). (C-E) Between group comparisons of mu (Oprm1), kappa (Oprk1), and delta (Oprd1) receptor transcript levels for all groups. All data are presented as mean ± S.E.M.; ** p < 0.01 on Holm-Sidak post-hoc test.
Another potential explanation for the blunted transcriptional response to morphine in germ-free mice would be altered expression of opioid receptors in the NAc. Using FPKM values from our sequencing analyses, we performed targeted pairwise comparisons of opioid receptor subtypes. Interestingly, there was a main effect of microbiome group on mu opioid receptor (Oprm1) with germ-free mice expressing higher levels at baseline and without morphine response (Fig. 4C – main effect of microbiome group: F(2,27) = 5.65; p = 0.009). There was also a main effect of morphine (F(1,27) = 6.85; p=0.014) and a trend toward a morphine x microbiome interaction (F(2,27) = 3.33; p=0.051). Post-hoc testing comparing other groups to H2O-saline showed an increase in expression in morphine treated control animals, and increased expression in both GF groups. Analysis of the kappa opioid receptor gene (Oprk1) also showed a main effect of microbiome status with a decrease seen in the germ-free group (Fig. 4D – main effect of microbiome group: F(2,27) = 3.83; p =0.034), and while there was no main effect of morphine (p = 0.59), there was a significant group x morphine interaction (F(2,27) = 3.79; p = 0.035). There were no main effects or interactions on two-way analysis of the delta opioid receptor (Oprd1 – Fig. 4E; p > 0.1 for all).
Weighted gene coexpression network analysis (WGCNA)
Given the complexity of our experimental groups and the interrelated effects of microbiome status on gene expression patterns, we next performed WGCNA using data from all experimental groups as input to provide a threshold-free means of analyzing expression changes across multiple groups (Langfelder and Horvath, 2008). This method uses complex hierarchical clustering techniques to identify modules of highly correlated genes and allows for analysis of how these modules correspond with group variables. The first step in this process is identification of correlated modules of gene expression. Figure 5A shows our gene dendrogram obtained with the average linkage hierarchical clustering. The color row below each branch shows the module assignment as identified with dynamic tree cut. Figure 5B shows a topological outline of genes assigned to each of the modules. Only modules that had a weight of 0.99 were included in the graphic representation. Cytoscape ver 3.9.1 was used to generate network images. Modules and individual eigengenes are colored and labeled based on module assignment. Cytoscape software performed unsupervised clustering of individual eigengenes and nodes. All genes assigned to modules with this analysis can be found in Table S5.
Figure 5 – WGCNA module identification.
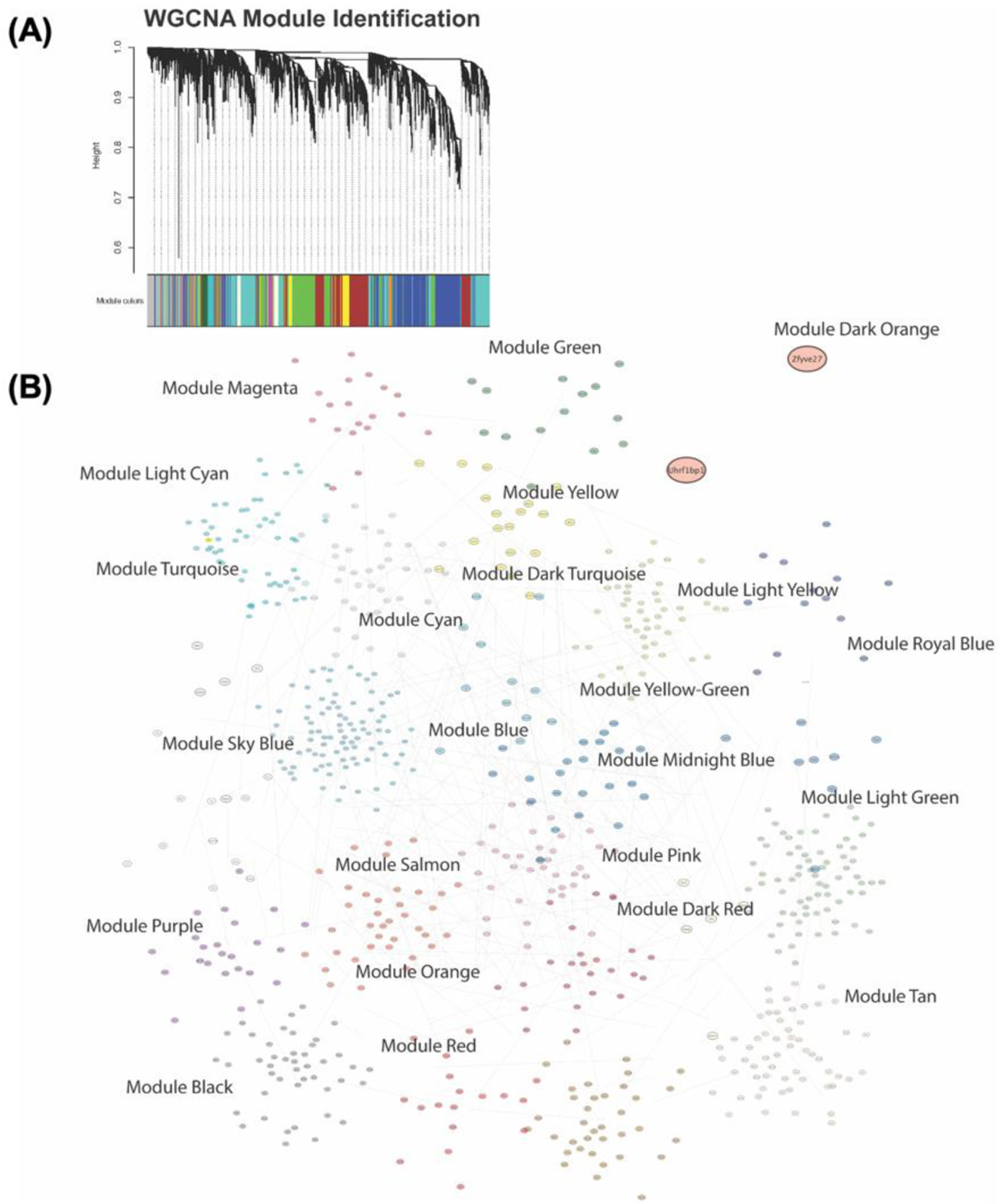
To identify modules of coexpressed genes in our sample groups WGCNA hierarchical clustering analyses were performed followed by branch cutting based on variance to separate samples into coexpressed modules. (A) Clustering dendrogram with the height represented as 1 – r for the correlation and the colored rows along the bottom representing the assigned modules. (B) Graphical representation of gene modules as topological network diagram of all modules and their contained genes and interactions.
To assess how individual microbiome groups and treatments related to gene expression patterns we then performed module trait correlation analysis with each individual treatment group used as a trait (Fig. 6A). Data in this plot represent positive or negative correlations of each trait with the module eigengene with the r correlation value on top of each significant module and the p value in parentheses below. As it is challenging to fit all these data in a single figure panel, the full table with r and p values is available as Table S6. As would be predicted from our previous pairwise comparison analyses, we see that patterns of gene expression are most significantly associated with germ-free status, and with Abx-Mor treatment – as these were the treatment groups with by far the most statistically significant gene effects in pairwise analyses (Fig. 2).
Figure 6 – Initial WGCNA analysis.
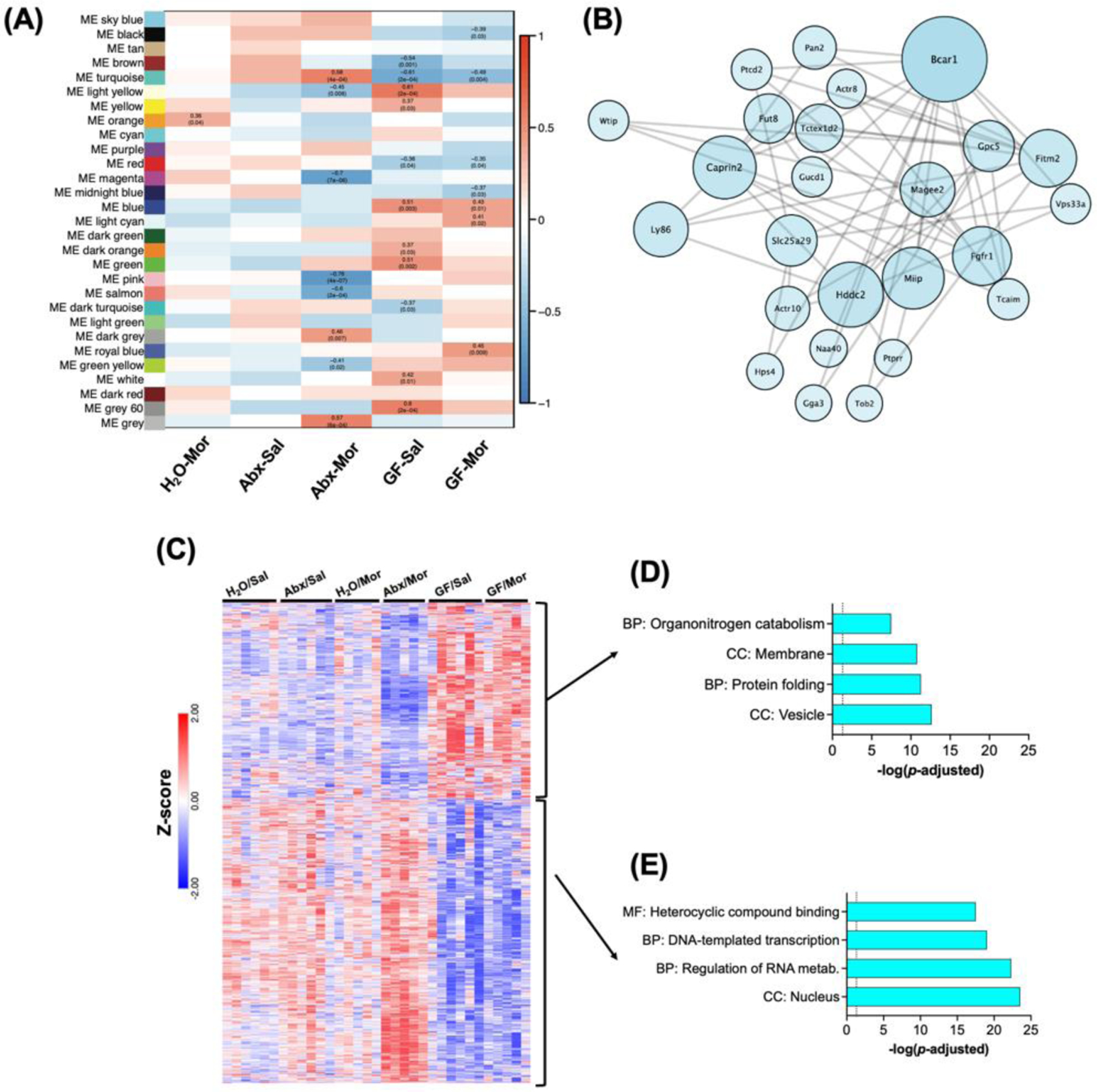
(A) Correlation matrix of module eigengene relationship with individual treatment groups. Significant cells are labeled with r value and p value. (B) Topological network diagram of the top 25 weighted genes in theTurquoise module, which is the largest gene module and the only one with significant effects in Abx-Mor and GF mice. (C) Heatmap of z-scored FPKM expression data for all genes in turquoise module followed by unsupervised hierarchical clustering. (D) Select significant gene ontologies from subset of Turquoise module genes decreased in Abx-Mor and increased in GF groups. (E) Select GO pathways for subset of Turquoise genes decreased in GF and increased in Abx-Mor.
To examine how these modules are associated with functional patterns of gene expression, we first performed detailed analysis of the turquoise module, which is both the largest module, and the only one in which significant correlations with both Abx-Mor and both germ-free groups were noted (Fig. 6A). Figure 6B is a topological network diagram of the interrelation between the top 25 heaviest weighted genes within this module. Graphical representation of networks was performed in Cytoscape. Individual eigengenes were colored based on module assignment and sized based on the weight of that eigengene within its assigned module. Eigengene layout was performed using Cytoscape’s default unsupervised clustering tool. We next mapped the relative expression of the genes assigned to this network using their FPKM values. Here, we see a clear pattern in which nearly half of the genes are decreased in Abx-Mor and up in the GF groups, and the remainder are increased in Abx-Mor, but decreased in GF groups (Fig. 6C). Given this disparate expression between the two largest drivers of gene expression, and the stark difference between microbiome depleted Abx mice and constitutively germ-free mice, we analyzed these two groups of genes in greater detail. The subset of genes that were down in Abx-Mor but increased in the GF groups were significantly enriched for genes coding for vesicles and membrane functions among others (Fig. 6D & Table S7). The larger subset, which were decreased in GF groups but increased in Abx-Mor mice, were robustly enriched for nuclear function, transcription, and regulation of RNA metabolism among others (Fig. 6E & Table S7).
Given that germ-free status was the factor with the strongest effect on gene expression, we also performed more detailed analysis on two modules with the strongest associations with germ-free groups. The first was the Blue module which had positive eigengene correlation with both germ-free groups (Fig. 6A). A topological network diagram showing relationships between the top 25 genes is presented as Figure 7A. Gene ontology analysis showed that this module was enriched for factors related to macromolecule metabolism, cellular localization, and intracellular transport among others (Fig. 7B & Table S8). The red module, which showed negative module correlation with germ free status, is shown in Fig. 7C, and gene ontology analysis in Fig. 7D & Table S9 which demonstrates alterations in gene pathways related to nervous system development, and metabolic processes. Taken together, our pairwise comparison analyses and coexpression network analyses demonstrate robust bi-directional effects of germ-free status on gene expression related to metabolism and transcription regulation in the nucleus accumbens.
Figure 7 – Analysis of modules associated with germ-free status.
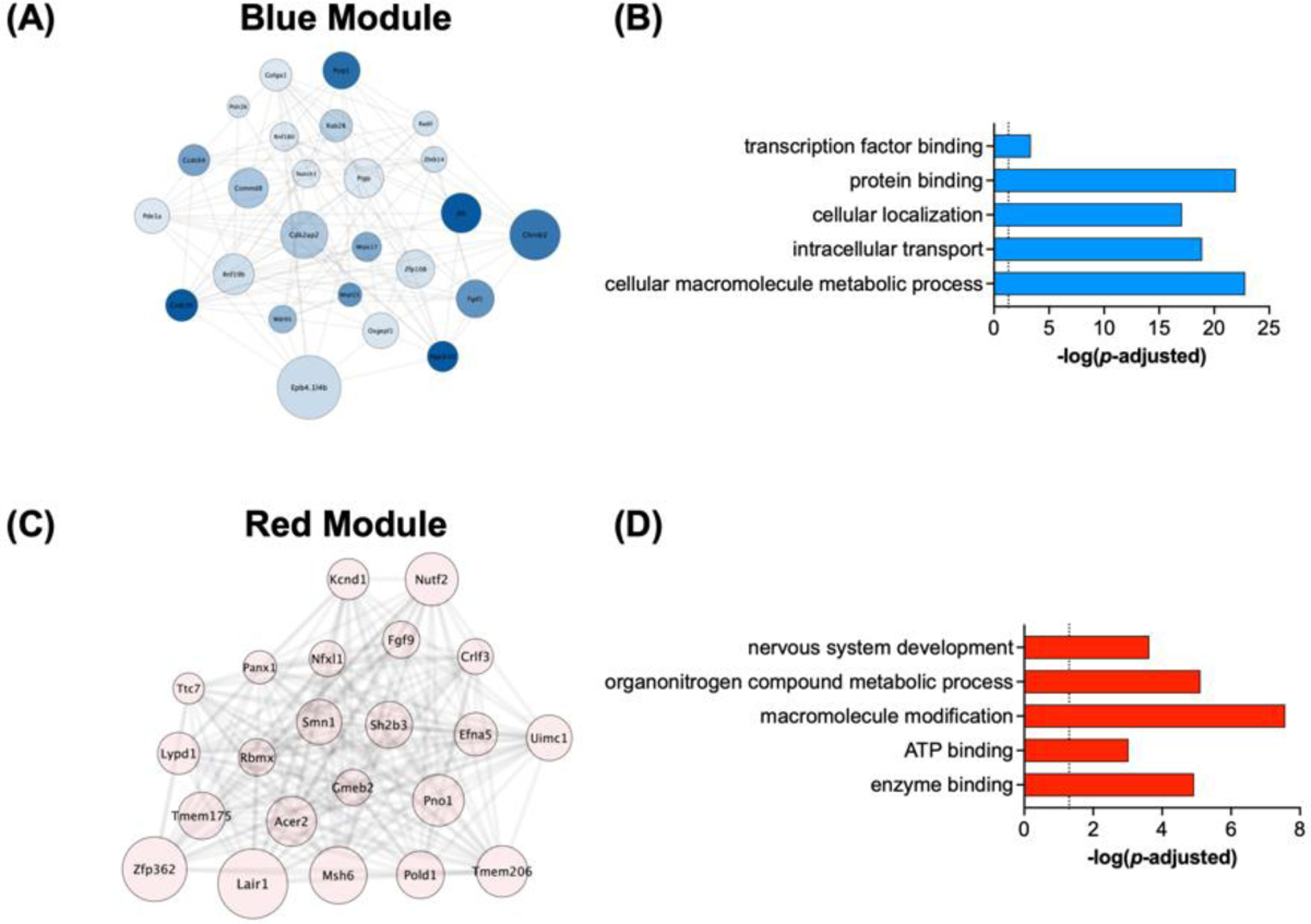
Detailed analysis of WGCNA modules with the strongest correlations with germ-free status. (A) Network diagram of the Blue module and interrelationships between genes in this module. (B) Significant gene ontologies of interest associated with genes in the Blue module. (C) Network diagram of genes expressed in the Red module. (D) Ontologies of interest associated with genes in the Red module.
Discussion
There are a growing number of studies examining the contribution of the gut microbiome to cellular and molecular responses to drugs of abuse (Kiraly et al., 2016; Lee et al., 2018; Zhang et al., 2019; Hofford et al., 2021, 2022; Simpson et al., 2022; García-Cabrerizo et al., 2023). Of the available tools used to alter the microbiome, two of the most common are administration of oral Abx and the use of GF animals. Both methods have advantages and appropriate applications in research. However, a full comparison of how the two manipulations alter the brain is not known and not often examined. In this study, we directly compared NAc gene expression of conventionally raised mice with intact or depleted with broad spectrum Abx microbiomes to GF animals with and without repeated morphine treatment. We found that mice that are GF from birth have markedly different transcriptomic profiles at baseline compared to conventionally raised microbiome intact and microbiome depleted mice. However, we also saw that these mice had a blunted transcriptional response to chronic morphine compared to conventionally raised Abx treated mice, which have an exaggerated transcriptional response to morphine (Hofford et al., 2021). These data provide important insight into potential developmental effects of microbiome content on brain signaling and how it affects plasticity in response to external stimuli.
In this study, we show that the complete lack of a microbiome found in GF mice significantly changes gene expression in the NAc at baseline when compared to both H2O and Abx treated conventionally raised mice (Fig. 2A–D). This contrasts with the effects of limited microbiome knockdown achieved in the Abx treated group — very few genes are differentially regulated in conventional mice on Abx compared to H2O (Hofford et al., 2021). Interestingly, morphine exposure significantly decreased the number of differentially regulated genes between GF and H2O groups; essentially, morphine made gene expression more similar between groups (Fig. 2E–G). This is unlike the results found in Hofford 2021, where there were more than 2,000 genes differentially regulated between H2O-Mor and Abx-Mor mice, demonstrating that disruption of an existing microbiome alters morphine-induced gene expression more profoundly than GF rearing, even though GF status has a large effect on gene expression in the absence of morphine. Similarly, morphine did not significantly alter gene expression in GF mice: direct comparison of the transcriptional profiles of GF-Sal and GF-Mor mice showed no significantly regulated genes after FDR correction (Fig. 3A). The blunted transcriptional response to morphine observed in GF mice could be a consequence of the marked alterations in gene expression at baseline. If gene expression is markedly disordered at baseline, the response to robust environmental stimuli, such as high dose morphine, may not appropriately induce transcriptional changes. Additionally, at least one study has identified changes in epigenetic regulation in the brain in GF mice (Thion et al., 2018). It is possible that baseline differences in epigenetics alters the propensity of genes to be expressed in response to an external stimulus. Similarly, alterations in blood-brain barrier integrity could explain the blunted transcriptional response in GF mice, but previous work has shown these mice have a more permeable blood-brain barrier (Braniste et al., 2014), which would likely not explain the effects seen here. While we did examine levels of morphine in the injectate in these experiments, we did not specifically measure levels of morphine in the brain of GF mice.
The blunted transcriptional response to morphine in GF mice could also be attributed to baseline opioid receptor transcript expression. Levels of Oprm are elevated at baseline in GF-Sal mice and remain high after morphine (Fig. 3C), and levels of Oprk decrease after morphine (Fig. 3D), although this didn’t reach significance. This is in contrast to mice in the H O group, where Oprm levels increase after morphine and levels of Oprk remain the same. These two receptors influence reward behavior in opposite directions, with mu agonism increasing reward and kappa agonism decreasing reward (Le Merrer et al., 2009; Fields and Margolis, 2015). There are other possible explanations for the reduced morphine response in GF mice, including attenuation of peripheral immune responses that could be unique to GF rearing, or altered morphine metabolism. Treatment with Abx does not produce changes in serum or brain levels of morphine (Hofford et al., 2021), but this might not be true for GF mice. More work is needed to determine why GF mice have a reduced transcriptional response to morphine.
There are multiple other published works that have evaluated the effects of germ-free rearing and microbiome status on the transcriptome and epigenome in the brain Previous work from the Cryan lab has identified effects of GF status on transcription in the amygdala (Hoban et al., 2018; Stilling et al., 2018). A number of groups have identified effects of microbiome composition in the prefrontal cortex (Stilling et al., 2015; Gacias et al., 2016; Hoban et al., 2016; Chu et al., 2019). In fact, Chu et al. performed single cell sequencing of the prefrontal cortex and found significant gene expression changes in all expressed neuronal and glial cell types (Chu et al., 2019). We and others have identified changes in striatal gene expression in response to antibiotics or other manipulations in conventionally raised mice(Jadhav et al., 2018; Hofford et al., 2021; Chivero et al., 2022; Cuesta et al., 2022; Lai et al., 2022), but the current study seems to be the first study examining effects of germ-free status on the striatal transcriptome. Another study by Simpson and colleagues found brain-wide patterns in expression of genes related to neuronal activation (Simpson et al., 2020). Additionally, a number of studies have examined isolated microglial populations from GF or Abx treated mice and found gene expression and chromatin accessibility to be particularly sensitive to microbiome status in these resident immune cells (Erny et al., 2015; Thion et al., 2018).
In the present study, pathway analysis from the list of differentially expressed genes and from threshold-free WGCNA identified terms associated with cellular metabolism, such as “protein metabolic process,” “regulation of metabolic process,” and “cellular macromolecule metabolic process,” as being highly regulated in GF mice compared to other groups both with and without morphine. Holistic analysis using WGCNA found the turquoise module to be of particular interest, as genes in this module showed significant relation to Abx-Mor, GF-Sal, and GF-Mor groups. Interestingly, genes within this module were altered in different directions between Abx-Mor and both GF groups (Fig. 5). Genes that were downregulated in Abx-Mor and upregulated in GF groups were associated with vesicles and the cell membrane, while genes that were upregulated in Abx-Mor and downregulated in GF groups were associated with the nucleus and gene transcription. Additional analysis of the blue and red modules, which were strongly correlated with GF status, identified terms related to metabolism in both modules and “nervous system development” within the red module. Given the known role of the microbiome in regulating the host metabolome, the findings that GF mice have markedly dysregulated genes related to multiple metabolic pathways suggests that this may be a key mechanism of gut-brain signaling.
Our previous work examining behavioral and transcriptional changes in conventionally raised mice determined that a reduction in SCFAs caused by Abx treatment likely contributed to the reduction in morphine conditioned place preference we observed, since SCFA replenishment was sufficient to reverse the behavior (Hofford et al., 2021). SCFAs are the byproducts of bacterial fermentation of soluble fiber, and are normally almost entirely derived from the microbiome (Koh et al., 2016). They are necessary for several cellular functions and are well-known histone deacetylase inhibitors (Waldecker et al., 2008; Koh et al., 2016; Dalile et al., 2019) with peripheral SCFAs shown to regulate histone acetylation and gene expression in the brain (Mews et al., 2019). Additionally, SCFAs can have direct effects on the activity of transcription factors (Rabelo et al., 2003; Mally et al., 2004; MacFabe et al., 2007; Nankova et al., 2014). As GF mice are well known to be deficient in SCFAs, as well as many other potential neuroactive metabolites, these metabolites may account for the robust baseline transcriptional differences seen in GF mice.
Importantly, the study as presented also has limitations that should be recognized. For these studies we utilized conventionally raised mice and GF mice, but did not have a group of mice born GF that were subsequently conventionalized. Thus, we cannot determine the critical phase of development for gene expression changes driven by GF status. In future studies, it would be informative to examine GF mice colonized shortly before opioid treatment to determine how this affects baseline and opioid induced transcriptional regulation. Additionally, we cannot directly account for the possibility that our GF mice have some altered level of morphine metabolism. While we know that the same amount of morphine was delivered to all mice, the logistics of getting timed serum samples in the gnotobiotic isolator precluded direct pharmacokinetic assessments. We do know that Abx treated mice have normal cocaine and opioid metabolism from our previous manuscripts (Kiraly et al., 2016; Hofford et al., 2021), but cannot definitively say this is the case for GF mice.
Supplementary Material
{kind=link}
Highlights.
Gene expression in the nucleus accumbens is markedly different in germ-free and conventionally raised mice.
Microbiome depletion in adulthood leads to altered patterns of gene expression in the nucleus accumbens compared to constitutively germ-free mice.
Germ-free mice have minimal transcriptional response in the nucleus accumbens in response to chronic morphine.
Weighted gene coexpression network analysis identifies unique signatures of gene coexpression amongst microbiome status groups and morphine treatment groups.
Germ-free mice have altered patterns of gene expression that suggest marked changes in cellular metabolism.
Acknowledgements
This work was supported by NIH grants DA051551 to D.D.K. and DA050906 to R.S.H. in addition to NARSAD Young Investigator Awards to both D.D.K. and R.S.H. Morphine was provided by the NIDA drug supply program. Dr. Kavya Devarakonda contributed key edits to the final draft of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare that they have no conflicts of interest with this work financial or otherwise.
Declaration of competing interests: The authors have no competing interests.
References Cited
- Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, Korecka A, Bakocevic N, Ng LG, Guan NL, Kundu P, Gulyás B, Halldin C, Hultenby K, Nilsson H, Hebert H, Volpe BT, Diamond B, Pettersson S (2014) The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 6:263ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne CJ, Godino A, Salery M, Nestler EJ (2020) Epigenetic Mechanisms of Opioid Addiction. Biological Psychiatry 87:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Systems Biology 8:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivero ET, Sil S, Singh S, Thangaraj A, Gordon L, Evah-Nzoughe GB, Ferguson N, Callen S, Buch S (2022) Protective Role of Lactobacillus rhamnosus Probiotic in Reversing Cocaine-Induced Oxidative Stress, Glial Activation and Locomotion in Mice. J Neuroimmune Pharmacol 17:62–75. [DOI] [PubMed] [Google Scholar]
- Chu C et al. (2019) The microbiota regulate neuronal function and fear extinction learning. Nature 574:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta S, Burdisso P, Segev A, Kourrich S, Sperandio V (2022) Gut colonization by Proteobacteria alters host metabolism and modulates cocaine neurobehavioral responses. Cell Host Microbe 30:1615–1629.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalile B, Van Oudenhove L, Vervliet B, Verbeke K (2019) The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol 16:461–478. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydyk AM, Jain NK, Gupta M (2022) Opioid Use Disorder StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK553166/ [Accessed April 18, 2023]. [PubMed] [Google Scholar]
- Egervari G, Landry J, Callens J, Fullard JF, Roussos P, Keller E, Hurd YL (2017) Striatal H3K27 Acetylation Linked to Glutamatergic Gene Dysregulation in Human Heroin Abusers Holds Promise as Therapeutic Target. Biol Psychiatry 81:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eipper-Mains JE, Kiraly DD, Duff MO, Horowitz MJ, McManus CJ, Eipper BA, Graveley BR, Mains RE (2013) Effects of cocaine and withdrawal on the mouse nucleus accumbens transcriptome. Genes Brain Behav 12:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny D et al. (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 18:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Margolis EB (2015) Understanding opioid reward. Trends in Neurosciences 38:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacias M, Gaspari S, Santos P-MG, Tamburini S, Andrade M, Zhang F, Shen N, Tolstikov V, Kiebish MA, Dupree JL, Zachariou V, Clemente JC, Casaccia P (2016) Microbiota-driven transcriptional changes in prefrontal cortex override genetic differences in social behavior. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cabrerizo R, Barros-Santos T, Campos D, Cryan JF (2023) The gut microbiota alone and in combination with a social stimulus regulates cocaine reward in the mouse. Brain Behav Immun 107:286–291. [DOI] [PubMed] [Google Scholar]
- Hedegaard H, Brigham Bastian, Trinidad J, Spencer M, Warner M (2018) Drugs Most Frequently Involved in Drug Overdose Deaths: United States, 2011–2016. National Vital Statistics Reports 67:14. [PubMed] [Google Scholar]
- Hoban AE, Stilling RM, Moloney G, Shanahan F, Dinan TG, Clarke G, Cryan JF (2018) The microbiome regulates amygdala-dependent fear recall. Mol Psychiatry 23:1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban AE, Stilling RM, Ryan FJ, Shanahan F, Dinan TG, Claesson MJ, Clarke G, Cryan JF (2016) Regulation of prefrontal cortex myelination by the microbiota. Transl Psychiatry 6:e774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofford RS, Meckel KR, Wang W, Kim M, Godino A, Lam TT, Kiraly DD (2022) Changes in gut microbiome composition drive fentanyl intake and striatal proteomic changes :2022.11.30.518531 Available at: https://www.biorxiv.org/content/10.1101/2022.11.30.518531v1 [Accessed December 27, 2022].
- Hofford RS, Mervosh NL, Euston TJ, Meckel KR, Orr AT, Kiraly DD (2021) Alterations in microbiome composition and metabolic byproducts drive behavioral and transcriptional responses to morphine. Neuropsychopharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W (2008) BioVenn – a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav KS, Peterson VL, Halfon O, Ahern G, Fouhy F, Stanton C, Dinan TG, Cryan JF, Boutrel B (2018) Gut microbiome correlates with altered striatal dopamine receptor expression in a model of compulsive alcohol seeking. Neuropharmacology 141:249–259. [DOI] [PubMed] [Google Scholar]
- Jalodia R, Abu YF, Oppenheimer MR, Herlihy B, Meng J, Chupikova I, Tao J, Ghosh N, Dutta RK, Kolli U, Yan Y, Valdes E, Sharma M, Sharma U, Moidunny S, Roy S (2022) Opioid Use, Gut Dysbiosis, Inflammation, and the Nervous System. J Neuroimmune Pharmacol 17:76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, Walker DM, Calipari ES, Labonte B, Issler O, Pena CJ, Ribeiro EA, Russo SJ, Nestler EJ (2016) Alterations of the Host Microbiome Affect Behavioral Responses to Cocaine. Sci Rep 6:35455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F (2016) From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 165:1332–1345. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Reed B, Butelman ER (2019) Current status of opioid addiction treatment and related preclinical research. Sci Adv 5:eaax9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai S, Wang J, Wang B, Wang R, Li G, Jia Y, Chen T, Chen Y (2022) Alterations in gut microbiota affect behavioral and inflammatory responses to methamphetamine in mice. Psychopharmacology (Berl) 239:1–16. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Becker JAJ, Befort K, Kieffer BL (2009) Reward Processing by the Opioid System in the Brain. Physiological Reviews 89:1379–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Vuong HE, Nusbaum DJ, Hsiao EY, Evans CJ, Taylor AMW (2018) The gut microbiota mediates reward and sensory responses associated with regimen-selective morphine dependence. Neuropsychopharmacology 43:2606–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F, Taylor AR, Kavaliers M, Ossenkopp K-P (2007) Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res 176:149–169. [DOI] [PubMed] [Google Scholar]
- Mally P, Mishra R, Gandhi S, Decastro MH, Nankova BB, Lagamma EF (2004) Stereospecific regulation of tyrosine hydroxylase and proenkephalin genes by short-chain fatty acids in rat PC12 cells. Pediatr Res 55:847–854. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ (2003) Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci 6:1208–1215. [DOI] [PubMed] [Google Scholar]
- Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, Garcia BA, Berger SL (2019) Alcohol metabolism contributes to brain histone acetylation. Nature 574:717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankova BB, Agarwal R, MacFabe DF, La Gamma EF (2014) Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells--possible relevance to autism spectrum disorders. PLoS One 9:e103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S (2016) Epigenetic Basis of Mental Illness. Neuroscientist 22:447–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessar SC, Boustead A, Ge Y, Smart R, Pacula RL (2021) Assessment of State and Federal Health Policies for Opioid Use Disorder Treatment During the COVID-19 Pandemic and Beyond. JAMA Health Forum 2:e213833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabelo FLA, Ropert C, Ramos MG, Bonjardim CA, Gazzinelli RT, Alvarez-Leite JI (2003) Inhibition of ERK1/2 and CREB phosphorylation by caspase-dependent mechanism enhances apoptosis in a fibrosarcoma cell line treated with butyrate. Biochem Biophys Res Commun 303:968–972. [DOI] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ (2009) Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ (2011) Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12:623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson S et al. (2022) Identification of pre-existing microbiome and metabolic vulnerabilities to escalation of oxycodone self-administration and identification of a causal role of short-chain fatty acids in addiction-like behaviors :2022.07.23.501268 Available at: https://www.biorxiv.org/content/10.1101/2022.07.23.501268v1 [Accessed December 27, 2022].
- Simpson S, Kimbrough A, Boomhower B, McLellan R, Hughes M, Shankar K, de Guglielmo G, George O (2020) Depletion of the Microbiome Alters the Recruitment of Neuronal Ensembles of Oxycodone Intoxication and Withdrawal. eNeuro 7:ENEURO.0312–19 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, Moloney GM, Ryan FJ, Hoban AE, Bastiaanssen TF, Shanahan F, Clarke G, Claesson MJ, Dinan TG, Cryan JF (2018) Social interaction-induced activation of RNA splicing in the amygdala of microbiome-deficient mice. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, Ryan FJ, Hoban AE, Shanahan F, Clarke G, Claesson MJ, Dinan TG, Cryan JF (2015) Microbes & neurodevelopment--Absence of microbiota during early life increases activity-related transcriptional pathways in the amygdala. Brain Behav Immun 50:209–220. [DOI] [PubMed] [Google Scholar]
- Thion MS et al. (2018) Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 172:500–516.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre D, Lachmann A, Ma’ayan A (2018) BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. cels 7:556–561.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 19:587–593. [DOI] [PubMed] [Google Scholar]
- Walker DM, Cates HM, Heller EA, Nestler EJ (2015) Regulation of chromatin states by drugs of abuse. Curr Opin Neurobiol 30:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Meng J, Zhang L, Johnson T, Chen C, Roy S (2018) Morphine induces changes in the gut microbiome and metabolome in a morphine dependence model. Sci Rep 8:3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Meng J, Ban Y, Jalodia R, Chupikova I, Fernandez I, Brito N, Sharma U, Abreu MT, Ramakrishnan S, Roy S (2019) Morphine tolerance is attenuated in germfree mice and reversed by probiotics, implicating the role of gut microbiome. Proc Natl Acad Sci U S A 116:13523–13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.