

Abstract
In both excitable and non-excitable cells, diverse physiological processes are linked to different calcium microdomains within nanoscale junctions that form between the plasma membrane and endo-sarcoplasmic reticula. It is now appreciated that the junctophilin protein family is responsible for establishing, maintaining, and modulating the structure and function of these junctions. We review foundational findings from more than two decades of research that have uncovered how junctophilin-organized ultrastructural domains regulate evolutionarily conserved biological processes. We discuss what is known about of junctophilin family of proteins. Our goal is to summarize the current knowledge of junctophilin domain structure, function and regulation, and to highlight emerging avenues of research that helps our understanding of the transcriptional, translational, and post-translational regulation of this gene family and their role in health and during disease.
Keywords: Junctophilin, junctional membrane complex, transverse tubule, endo-sarcoplasmic reticulum, calcium, Calpain
INTRODUCTION
The plasma membrane bidirectionally communicates with endo-sarcoplasmic reticula to mediate essential physiological processes such as muscle contraction and brain activity. Such events are typically initiated by the conversion of plasma membrane (PM) depolarization into Ca2+ release from nearby endoplasmic (ER) or sarcoplasmic reticula (SR) Ca2+ stores (1–3). Early microscopy studies revealed the PM of many cell types is frequently aligned within 10–30 nm of SR/ER membrane patches (4–9). The physical nature of these junctional membrane contacts varies by tissue and are called ER/PM junctions in non-excitable cells, subsurface cisternae in neurons, and peripheral couplings in smooth and striated muscle (7, 10) (Figure 1a–e). Striated myocytes have additional junctional structures known as dyads and triads that associate with specialized interiorly projecting extensions of PM, i.e., transverse (T-) tubules. Dyads are the major junction of cardiomyocytes in which single SR cisternae make contacts with a T-tubule. Skeletal myocytes have both dyad and triad junctions along relatively thinner T-tubules. Triads are named by the appearance of SR terminal cisternae making extensive contacts on opposite sides of a given T-tubule. Each myocyte possesses thousands of T-tubules arranged in a highly organized network (11, 12). Dyad and triad junctions within this network are responsible for synchronous SR Ca2+ release and uniform myofilament contraction in response to PM depolarization (13–15).
Figure 1.
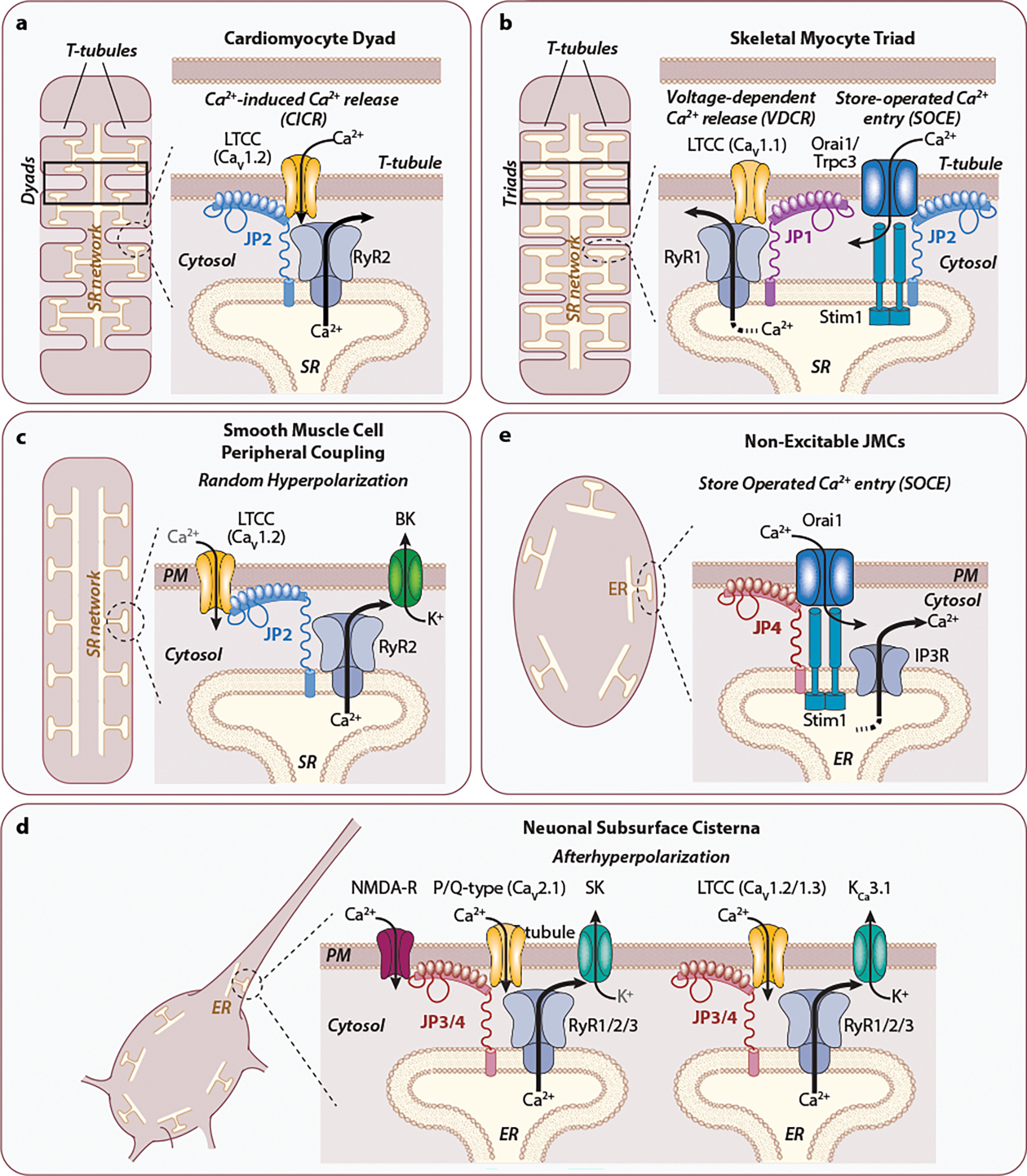
Junctophilin-mediated junctional membrane complexes (JMCs). Cellular JMCs (dashed circles) have different names in different cell types to elicit different phenomena. JMCs are commonly stabilized by Junctophilin proteins for localizing ion channels on the plasma membrane with Ca2+ permeable channels and sensors on the SR/ER membrane. (a) In cardiomyocytes, the dyad formed by T-tubular PM—SR network contacts facilitates Ca2+-induced Ca2+ release (CICR). (b) Skeletal muscle triads (two junctional SR cisternae flanking one T-tubule) are responsible for voltage-dependent Ca2+ release in parallel with store-operated Ca2+ entry (SOCE). (c) PM—SR Peripheral couplings in smooth muscle cells determine random hyperpolarization. (d) SOCE is the primary function of non-excitable cell JMCs and is likely present in most excitable cells. (e) Neurons have subsurface cisternae for mediating afterdepolarization phenomena. Evidence in support of the junctophilin homolog(s), ion channels, and Ca2+ sensors that constitute each JMC type are given in the text.
[**Note to Annual Reviews: We created this figure for this article; it is not based on any previously published image.**]
Electron dense material was revealed by original studies to concentrate within junctional PM – ER/SR clefts (4–6) and was later recognized to be made of proteins that enable PM – ER/SR communication (7–9). The junctions and their constituent proteins are broadly referred to as junctional membrane complexes, or JMCs (10). JMCs incorporate surface Ca2+ (and other) ion channels that partner with ER/SR specific Ca2+ channels and sensors. Although the precise combination of JMC proteins vary by cell type to mediate specific physiological outcomes, all JMCs require at least one member of a fascinating family of proteins known as junctophilins. Junctophilins have a unique multivalent capacity to bind to PM phospholipids, PM ion channels, the ER/SR membrane, ER/SR ion channels, and their associated regulatory molecules. It has therefore been proposed that junctophilins provide a physiological “tether” or “glue” in stabilizing the JMC (16). Furthermore, junctophilins can functionally modify the biophysical properties of the ion channels with which they interact.
This review aims to provide the reader with a greater appreciation of junctophilins in health and disease. We survey the physiological functions of junctophilin family members at JMCs in different cell types and discuss how their domain structure, post-translational modifications, disease associated mutations, and transcriptional regulation impact their overall function. Throughout, we pose current questions the field is attempting to address including possible junctophilin-focused therapies in treating diseases associated with JMC dysfunction.
JUNCTOPHILINS STABILIZE PLASMA MEMBRANE—ENDO-SARCOPLASMIC RETICULUM COMMUNICATION
The four mammalian junctophilin paralogs were identified as JP1-JP4 by Dr. Takeshima and colleagues over twenty years ago (17, 18). Current nomenclature, however, now designate junctophilins as with JPH or Jph prefixes (see sidebar “Junctophilin diversification”). As both naming conventions are still widely used, we will attribute “JPH/Jph” to junctophilin genes and RNAs and “JP” to their protein counterparts. Mammalian Jph1–4 genes are transcribed in spatially unique patterns for performing cell type specific functions. Jph1 is most abundantly expressed in skeletal muscle (19–21). Jph2 is also expressed in skeletal muscle and is the primary junctophilin of the heart and smooth muscle (17, 22–26). Jph3 and Jph4 are produced throughout the brain (18, 27) and in endo/exocrine glands (28). Jph3 is also reported throughout the gastrointestinal tract and reproductive organs (29). Vertebrate JP proteins typically range in size from 600–800 amino acids in length, but shorter alternatively spliced isoforms are predicted in human. The function of these shorter proteins remains to be defined. The four junctophilin paralogs have distinct functions in the tissues in which they are expressed, but they are homologous to each other, and what we have learned about one may be generally applied to the others. The most investigated is JP2 and studies related its structure and function have led to robust insights into how junctophilins maintain JMC integrity and mediate PM – ER/SR communication.
SIDEBARS.
Junctophilin diversification
Genome databases have increasingly indicated Jph genes are conserved across phyla including invertebrates and sponges (75, 142). While a single junctophilin gene is present in most lower organisms, repeated duplication events in the vertebrate lineage suggest multiple paralogs have arisen to perform more nuanced functions. Most vertebrates have three Jph loci (Jph1–3), while Jph4 is found primarily in mammals. Instead of a distinct Jph4 gene, some ray-finned fish have two homologous Jph1 genes. An elegant study in Drosophila melanogaster demonstrates that cardiac, muscle, and neuronal specific knockout of its lone junctophilin gene (jp) generates phenotypes that largely parallel the phenotypes of mammalian Jph1, Jph2, and Jph3+Jph4 knockout models, respectively (57).
CICR, VDCR, and E-C coupling
CICR is the process in cardiomyocytes by which modest Ca2+ influx through T-tubule localized L-type Ca2+ channels (LTCCs, specifically CaV1.2) induce efficient and massive Ca2+ efflux from nearby SR terminal cisterna expressing type 2 ryanodine receptor (RyR2) channels. E-C coupling converts membrane depolarizations into CICR-induced spikes of cytoplasmic Ca2+ that trigger myofilament contraction. Cytosolic Ca2+ levels then return to baseline through Ca2+ uptake into the SR via SR Ca2+-ATPase (SERCA2a) and Ca2+ extrusion by PM Na+/Ca2+ exchangers allowing for myofilament relaxation. Skeletal myocytes depend less on CICR and instead rely on a similar phenomenon known as voltage dependent Ca2+ release (VDCR) in which T-tubule localized CaV1.1 LTCCs activate skeletal myocyte expressing RyR1 channels on the SR membrane via a direct voltage-dependent confirmational change.
Junctophilin-2 is essential for cardiomyocyte dyad function
The primary role of JP2 in cardiomyocytes is to establish, maintain, and regulate dyad junctions. The importance of an intact T-tubule/SR dyadic system was inferred from observations that cardiomyocytes from failing hearts of patients (30, 31) and in animal models of heart failure exhibit disrupted T-tubule and dyad ultrastructure that correlate with defects in Ca2+ handling (31–40). Mice with global knockout (KO) of Jph2 have a weak and irregular heart rhythm in utero with half of homozygous animals dying by embryonic day 10.5 from cardiac arrest (17). Lethality was proposed to be due to a drastic reduction in the frequency of immature dyad junctions and abnormal Ca2+ transients. This notion was validated a decade later in studies showing that cardiomyocyte expression of a Jph2 shRNA hairpin causes cardiac dilatation, acute heart failure, pulmonary congestion, and mortality within one week in the absence of heart failure-related hypertrophic remodeling (41). Similar studies in isolated adult rat cardiomyocytes showed Jph2 knockdown reduces the frequency and length of dyad junctions correlating with lower cytosolic Ca2+ transient amplitudes and slower rise times (37). These results demonstrated JP2 plays an essential role in dyad stability by mediating phenomena in cardiomyocytes called Ca2+-induced Ca2+ release (CICR) and excitation-contraction (E-C) coupling (Figure 1a–b and sidebar “CICR, VDCR, and E-C coupling”).
JP2 protein expression in the mouse heart is detectable at embryonic day 9.5 (17) and gradually increases throughout cardiac development (42, 43), particularly in ventricles (44, 45). Cardiomyocyte PM structure in newborn rodents is rather unremarkable (46) but begins to display invaginating T-tubules within 10 days of post-natal life (42, 43). Dyad structure and function become fully mature around three weeks of age (42, 46, 47). JP2 is first documented at the PM of non-tubulated postnatal mouse myocytes (48) before concentrating along early invaginating T-tubules (42, 43, 46). A correlation then develops between JP2 colocalization with RyR2 Ca2+ channels already present on the SR membranes and a significant increase in E-C coupling (46). Proper JP2 trafficking to the dyad requires microtubule dynamics. Increasing microtubule filament density, as occurs during heart failure, re-localizes JP2 away from T-tubules (49–51). Conversely, depolymerizing microtubules in failing hearts promotes JP2 re-distribution back into dyads, attenuates T-tubule degeneration, and normalizes contractile dysfunction (49).
Jph2 overexpression in the mammalian heart, in contrast to Jph2 loss of function, is well tolerated having no effect on cardiac function or viability (52). Postnatal JP2 overexpression accelerates the development of T-tubules (43) and increases SR/T-tubule contact length and number (52). Approximately 10% of dyads in these hearts exhibit extended and complex T-tubules with multiple SR contacts consistent with the role of JP2 in initiating and stabilizing JMCs. Despite such changes, overexpressing hearts do not have changes to cardiac ejection fraction, left ventricular mass, or Ca2+ handling function at baseline but do confer protection against pathological insult. Overexpressing mice have increased survival, attenuated cardiac hypertrophy, and sustained T-tubule integrity after pressure overload stress relative to wildtype mice (52).
Studies using super-resolution microscopy techniques, primarily from Soeller and colleagues, have shed light on how JP2 and RyR2 channels are arranged within cardiomyocyte dyads and at peripheral coupling sites located within and outside of T-tubules, respectively. JP2 molecules intersperse among RyR2 channels in both junction types with dyads having more than four times as many RyR2 channels on average than peripheral couplings (53–55). RyR2 clustering is induced by Jph2 overexpression but is not affected by Jph2 knockdown despite a loss of T-tubules and E-C coupling efficiency (56). Both Jph2 overexpression and knockdown approaches increase the proportion of longitudinal tubules that interconnect T-tubules increases, but why this occurs is not understood.
Taken together, the loss of JP2 is recognized as a major factor in stress-induced T-tubule disorganization and degeneration, inefficient CICR and maladaptive cardiac remodeling. Studies too numerous to list here have monitored JP2 protein levels as an early and reliable indicator of heart failure from most etiologies, including inherited and acquired cardiomyopathies, ischemia/reperfusion injury, arrhythmias, and pressure overload. Similarly, studies testing the efficacy of heart failure interventions now commonly assess JP2 levels, its localization, and its correlation with improved T-tubule structure, E-C coupling efficiency and cardiac function. The precise function of peripheral couplings and longitudinal tubules, however, is not nearly as well understood as T-tubular associated dyads. Novel studies designed to differentiate the role of JP2 at different junctional sites are likely to unveil the unique contribution and composition of specific JMCs in cardiac pathophysiology.
Skeletal myocyte junctions incorporate Junctophilins-1 and -2
Mouse JP2 is first expressed in embryonic skeletal myocytes and correlates with the formation of dyad structures. JP1 is then induced prior to birth when triad structures are observed (see Figure 1a and 1b). Although mice with specific depletion of Jph2 in mammalian skeletal myocytes have yet to be developed, global Jph1 KO mice have muscle weakness and die perinatally likely from an inability to suckle (21). Skeletal myocytes in Jph1 KO mice consist of swollen, irregular, and partially vacuolated SR features similar to jp KO in D. melanogaster (57) and exhibit a specific reduction in muscle triad junctions. These observations suggest JP1 and JP2 support different JMC structures and/or functions (20). Both JP1 and JP2 are capable of interacting with the C-terminus of skeletal myocyte expressed CaV1.1 LTCCs (58), but only JP1 copurifies with skeletal myocyte expressed RyR1 (59). Similar voltage-dependent SR Ca2+ release phenomena can be replicated in heterologous cells when RyR1, CaV1.1, and Stac3 are co-expressed with either JP1 or JP2 as well as with neuronal JP3 (60) (61).
Early studies examining junctophilin function in adult mouse skeletal muscle involved inducible and simultaneous knockdown of both Jph1 and Jph2 (26, 62). Dual silencing results in the formation of irregular triad and JMC structures with misaligned or missing SR – T-tubule contacts. Furthermore, voltage-induced Ca2+ release is attenuated and store-operated Ca2+ entry (SOCE)-dependent mechanisms, activated by SR Ca2+ depletion, becomes uncoupled. Both studies show reversal of Jph1 and Jph2 shRNA expression restored myocyte ultrastructure, SR Ca2+ release and SOCE coupling (26, 62). Specific loss of JP1 appears to be sufficient to explain many of the phenotypes caused by double Jph1 and Jph2 silencing as Jph1 KO in mouse myotubes impairs SOCE through a reduction and/or displacement of its associated Orai1, STIM1, and Trpc1/3 channels despite no change in JP2 expression (63). The full function of JP2 in skeletal myocytes has yet to be defined but a differential role for JP1 and JP2 in anterograde versus retrograde PM-SR mechanisms likely exists (64, 65).
While the consequences of increased expression of JP1 in mammalian skeletal muscle has not been reported, transgenic overexpression of Jph1 in the murine heart results in increased dyad contacts, but not triads. Similar to Jph2 overexpression (52), cardiac Jph1 expression facilitates extended, thin and “convoluted” SR membrane structures that are able to surround T-tubules (66). These findings, combined with the developmental JP1 versus JP2 studies described above, suggest skeletal myocytes possess a yet to be characterized factor that assists JP1-dependent triad formation that is not present in cardiomyocytes.
Junctophilin function in other cell types
Jph2 is the primary junctophilin in smooth muscle peripheral coupling junctions (Figure 1c) as validated for cerebral and mesenteric arteries (24, 25) and bladder (23). Jph2 silencing in smooth muscle cells does not affect spontaneous SR Ca2+ release events (also known as Ca2+ sparks), total SR Ca2+ uptake, or SR Ca2+ load as it does in cardiomyocytes but uncouples RyR2 from PM Ca2+ activated big conductance K+ (BK) channels (24). BK channels are localized near RyR2 channels and within caveolar microdomains that contain JP2. Physiologically, BK channels are responsible for membrane hyperpolarization that controls the magnitude and duration of smooth muscle constriction in response to elevated hemodynamic pressure (25).
The mammalian brain primarily expresses Jph3 and Jph4 for regulating subsurface cisternae junctions (Figure 1d). In D. melanogaster, neuronal-specific jp overexpression or silencing reduces survival with overexpressing flies showing a more dramatic phenotype (57). Adult Jph4 KO mice are phenotypically normal (67) while Jph3 disruption leads to modest age- and sex-dependent coordination and balance deficits (27, 68). Double Jph3 / Jph4 knockout (DKO) animals have a more dramatic phenotype in displaying severe neuromuscular defects, growth retardation, short- and long-term memory deficits, and lethality by one month of age (67, 69). Hippocampal NMDA receptors become uncoupled from both RyR-dependent ER Ca2+ release and Ca2+-dependent activation of hyperpolarizing small conductance K+ (SK) channels (69). RyR-SK channel crosstalk is also affected in DKO Purkinje neurons in response to CaV2.1 P/Q-type Ca2+ channel activation. Furthermore, there is a requirement for JP3 and JP4 in the hippocampus to stabilize a tripartite CaV1.3 (or CaV1.2) LTCC - RyR2 - KCa3.1 channel complex responsible for slow afterhyperpolarization (70). JP3 and JP4 are not entirely functionally redundant as their regulation of neuronal voltage dependent Ca2+ channels and their coupling with different RyR isoforms are different (61, 71).
Emerging evidence points to a greater expression pattern for Junctophilins in tissues beyond muscle and brain. For example, Jph3 knockdown in pancreatic beta islet cells attenuates glucose stimulated insulin secretion (28). JP4 expression is enriched in dorsal root ganglia and injection of Jph4 siRNAs into the lumbar spine of rats blunts bradykinin-induced hind limb pain responses (72). In non-excitable T-cells, SOCE depends on STIM1 channel recruitment to PM-ER junctions via a direct interaction with the JP4 C-terminus (Figure 1e). Silencing of JP4 blocks STIM1 recruitment and prevents T-cell activation (73).
JUNCTOPHILIN STRUCTURE AND FUNCTION
Junctophilin domain structure
Genetic gain- and loss-of-function approaches clearly establish an essential role for junctophilins in JMC organization, but how these proteins stabilize PM-ER/SR contacts for efficient Ca2+ handling processes cannot be understood without further molecular dissection. A tandem array of glycine-rich Membrane Organization and Recognition Nexus (MORN) motifs was first identified in the junctophilin family (17) and is the consensus feature of all junctophilin paralogs and predicted spliced isoforms. Full length isoforms have eight N-terminal MORN motifs split into a 6 + 2 arrangement separated by a serine-rich “joining” or “linker” region that are followed by pseudo-MORN, alanine-rich α-helical, proline-rich, and hydrophobic transmembrane (TM) helix sequences (Figure 2a and 2b). Human JP paralogs share 42–52% identity with the JP4 sequence, which is more divergent than the other three (Figure 2c). MORN motifs are the most conserved of the domains while the proline-rich region has the lowest degree of homology and is aptly referred to as the “divergent region.” There is generally a high degree of species-to-species conservation for each paralog except within the proline-rich divergent region.
Figure 2.

Sequence and structural similarity among Junctophilin family members. (a) Schematic representing our current understanding of junctophilin structure within a typical junctional membrane complex. For JP2, the N-Terminal MORN motifs are reported to bind plasma membrane phospholipids (81) and interact with the α-helical region (78). The C-terminal transmembrane domain (TM) tethers junctophilins to the ER/SR membrane. (b) Protein alignments of human JP1–4 isoforms highlighting their functional domains and regions enriched for specific amino acids. (c) Heatmap representations of percent human JP1–4 protein sequence identity across full-length (overall) and individual domain regions as indicated in (b). (d) Summary of reported intra- and interprotein junctophilin interactions. MORN, membrane and occupation recognition nexus domain; P, pseudo-MORN; TM, transmembrane domain; NLS, nuclear localization signal; bNLS, bipartite NLS; JP3-as, antisense JP3 protein implicated in Huntington’s Disease-like 2 syndrome.
[**Note to Annual Reviews: We created this figure for this article; it is not based on any previously published image.**]
Function of Junctophilin domains
The MORN, joining, α-helical, divergent, and transmembrane domains serve specialized roles within the larger junctional membrane complex and beyond. Here, we summarize how specific domains contribute to junctophilin structure and function.
Membrane Organization and Recognition Nexus (MORN) motifs mediate plasma membrane ion channel and phospholipid interactions.
Beyond junctophilins, MORN motifs have been identified in a handful of other protein classes in plants and animals (74). MORN motifs are typically found as tandem arrays in proteins known to be involved in membrane junctions and fission events. Each motif has a core fourteen residue consensus sequence (Y-X-G-X-W/F-X2-G-X3-G-X-G) (17, 75) that may stretch to 23 residues considering other conserved glycine and hydrophobic residues (74). Structural studies show these motifs fold into a repetitive antiparallel beta hairpin configuration that collectively form a concave binding pocket for peptide-substrate interactions (76, 77). LTCCs and SK channels are now reported to interact with the MORN motifs of JP1 and JP2 (78, 79). The van Petegem group recently solved the crystal structures of N-terminal JP1 and JP2 fragments showing that all eight MORN motifs fold into a single twisting concave configuration (78). Interestingly, a “pseudo-MORN” segment just after MORN8 was found to continue the beta hairpin fold despite lacking a MORN consensus sequence. This study went on to show a highly conserved LTCC C-terminal peptide derived from CaV1.1 binds within the concave groove formed by the first three MORN motifs of JP2.
Although absent in crystal structures, prior evidence suggests the junctophilins and their MORN motifs have affinity for phospholipids, particularly phosphatidylinositol phosphates (PIPs). Overlay of an array of spotted lipids with recombinant JP1 lacking its C-terminal TM domain (80) or a JP21–452 fragment (81) show interaction with PIP, PIP2 and PIP3. The N-terminal JP2 fragment also simultaneously binds phosphatidyl serine (PS) for regulating JP2 flexibility. It is noteworthy that full-length JP2 has increased affinity for phospholipids relative to a truncated JP21–452 fragment suggesting that C-terminal regions also contribute (81). An elegant study by Rossi et al. demonstrated the PM localization of GFP-tagged JP1 and JP2 in HeLa cells is sensitive to PIP2 levels. Redistribution of GFP fluorescence away from the PM was triggered by phospholipase-C activation and readily reversed by inhibition with atropine (82). This phenomenon likely occurs in cardiomyocytes as cardiac knockout of phosphoinositide-3-kinase p100α and p110β catalytic subunits promotes JP2 translocation away from dyad sites and induces a lethal heart failure phenotype reminiscent of Jph2 shRNA expressing hearts (36). Similarly, transfection of skeletal muscle fibers with mutant JP1 devoid of its MORN1–6 or MORN7–8 motifs along with the joining region increases JP1 mobility away from triads (82). The possible requirement for junctophilin MORN motif interactions with specific phospholipids at dyad and triad junctions is intriguing and in need of further investigation. It would be important for future studies to resolve the ongoing issue whether MORN motifs directly bind phospholipids (e.g., (78)) and whether phospholipids alter junctophilin interactions with their ion channel substrates. Determining the physiological role of predicted JP2 and JP3 short isoforms (Figure 2b) may be helpful in this respect.
The Serine-rich joining region supports multiple protein interactions
Emerging literature supports the notion that the serine-rich joining domain between MORNs 1–6 and MORNs 7–8 is responsible for self-dimerization (82) and mediating multivalent protein-protein interactions within the JMC (Figure 2d). In addition to MORN motif binding to the proximal C-terminus of striated muscle LTCCs (58, 78), LTCCs also interact with JP1230–369 and JP2216–399 fragments that encompass the second half of the joining region through most of the Ala-rich region (83). The Houser group pinpointed the functional interaction responsible for stabilizing cardiac T-tubules and dyad assembly to seven conserved residues in the human JP2 joining region (84). Viral infection of feline cardiomyocytes with JP2 mutated at these sites increases T-tubule degeneration, reduces LTCC association and leads to arrhythmogenic Ca2+ handling dysfunction in response to the catecholamine isoproterenol. The interaction between JP2 and Trpc3 also maps to the analogous sequence in mouse and specifically to E227 (E234 in human) (65). JP1 lacks homology with this segment and fails to co-immunoprecipitate with Trpc3 thus providing an explanation for how JP2 functions apart from JP1 - RyR1 complexes in skeletal myocytes.
In cardiomyocytes, Jph2 knockdown is known to increase RyR2 open probability in SR lipid bilayer assays and increase RyR2 activities, e.g., more frequent spontaneous Ca2+ sparks, compared to wildtype samples (85). The stabilizing effect of JP2 on RyR2 gating is thought to be mediated by the proximal joining region of JP2. The JP2 E169K mutation associated with cardiac hypertrophy and arrhythmia reduces the co-immunoprecipitation efficiency between JP2 and RyR2. Addition of a 25-residue wildtype peptide spanning E169 attenuates RyR2 opening when added to SR lipid bilayers and reduces Ca2+ spark frequency when incubated with atrial myocytes (86). RyR2 stabilization by JP2, however, remains controversial as JP2 overexpression was unable to suppress RyR2 gating properties in transgenic hearts (52) and JP2 silencing did not alter RyR2-mediated Ca2+ activities in pressurized cerebral arteries (24). Finally, the joining region may be important for assembling JP2 and JMCs into lipid rafts (83, 87). Jph2 overexpression promotes LTCC and caveolin-3 recruitment to cardiomyocyte T-tubules in a cholesterol dependent manner (88). JP2 also interacts with caveolin-1 in mouse mesenteric artery smooth muscle cells with joining residues T286TTET290 lying just upstream of MORN7 (25). This interaction may explain why knockdown of Jph2 uncouples caveolar BK channels from RyRs and results in suppressed spontaneous BK channel transient outward currents and vascular hypercontractility (24, 89).
The Alanine-rich α-helical and Proline-rich divergent domains
Experimental evidence shows that the central alanine-rich region of JP1 and JP2 forms a single ~45 Å α-helix which folds back onto the similarly sized antiparallel β-sheet array formed by the MORN motifs and adjacent sequences (78) (see Figure 2a). The intermolecular interactions between the MORN and α-helical domains as revealed by the crystal structure is potentially plastic as will be discussed below with respect to the cytosolic versus nuclear function of JP2 (see Transcriptional regulation by JP2). The divergent region, as highlighted above, also contributes to JP3- and JP4- mediated regulation of SOCE and differential RyR isoform binding (71, 73). It is postulated that the divergent region may be involved in other unknown functions to mediate tissue-specific physiological responses and warrants further investigation.
The C-terminal transmembrane domain anchors junctophilin to the endo-sarcoplasmic reticulum membrane
Each of the four full-length junctophilin paralogs end in a hydrophobic α-helix that tethers it to the ER/SR membrane (Figure 2). Except for a terminal threonine residue in JP1, JP2, and JP4, no amino acids are thought to project into the SR lumen. Injection of newt embryos with wildtype Jph1 cRNA promotes JMC formation, while cRNAs lacking TM codons do not (17). Without a TM segment, JP1 localization becomes diffusely distributed throughout the PM instead of being restricted to nearby ER. Reciprocally, electroporation of muscle fibers with GFP fused JP1 or JP2 TM domain constructs show a clear sub-sarcolemmal RyR2-like localization pattern (82). One final intriguing finding from Rossi et al. is that JP1 and JP2 can form homo- and heterodimers in non-muscle cells either through their TM or joining region domains (82). Parallel experiments with JP4 were unsuccessful and underscore that inherent functional differences exist between muscle and non-muscle junctophilin TM motifs.
Other interactions
JP2 forms stable interactions with several proteins involved in intracellular Ca2+ handling. For example, JP2 immune complexes isolated from canine hearts were found to be enriched with SR transmembrane triadin and calnexin proteins and PM CaV1.2 LTCCs and KCNQ1 voltage gated potassium channels (90). JP2 binding appears to have opposite effects on CaV1.2 and KCNQ1. Heterologous expression of Jph2 reduces the surface biotinylation of CaV1.2 channels but increases LTCC current amplitude which is somewhat different from cardiomyocytes where JP2 overexpression increases both CaV1.2 expression and current density at the membrane (88). Conversely, JP2 increases KCNQ1 surface expression but attenuates K+ current amplitude in COS7 cells by shifting its voltage-dependent activation in the positive direction (90). These findings could explain how JP2 promotes CICR by enhancing Ca2+ currents through LTCCs while suppressing inward K+ currents responsible for action potential repolarization.
Mass spectrometry analyses of affinity complexes have found JP2 interacts with a wide spectrum of cardiac proteins (91–95). SPEG (striated muscle preferentially expressed protein kinase), a Ser/Thr directed kinase, was separately identified in two studies using co-immunoprecipitation and proximity biotinylation purification methods (94, 95). As with Jph2 silencing, conditional cardiac knockout of SPEG results in T-tubule degeneration, abnormal SR Ca2+ release, and cardiac failure (94). SPEG appears to phosphorylate JP2 (94), but the consequences of this event remain unclear. There are many other JP2 interacting proteins that are awaiting functional characterization and it is our hope that similar studies will be designed to identify and characterize novel partners of JP1, JP3, and JP4.
POST-TRANSLATIONAL MODIFICATIONS
Full length junctophilins have predicted molecular masses between 65 kDa (JP4) and 82 kDa (JP3). Published immunoblot results, however, are variable and frequently report apparent masses 15–25 kDa larger than expected (27, 59, 72, 81, 96–100). The reason for this discrepancy remains incompletely resolved. Although the rod-shaped nature of JP2 could slow migration, purified bacterially expressed JP2 migrates at its expected size of 75kDa (81). Alternative transcripts are also insufficient to account for these differences. Here, we discuss the events and factors that may contribute to differences in apparent mass and highlight those that are likely to generate important insight into junctophilin function.
Calpain proteolysis
Pathological increases in cytosolic Ca2+ in cardiac and skeletal myocytes disrupt E-C coupling in part through proteolytic cleavage of JP1 and JP2 (100, 101). Evidence supports cleavage primarily occurs through Ca2+-activated calpain proteases (102, 103) but matrix metalloproteinase-2 (MMP-2) may also contribute (104). Calpain catalytic subunits μ-Calpain (Capn1) and m-Calpain (Capn2) are ubiquitously expressed and activated by autolysis in response to μM and mM Ca2+, respectively. Proteolysis of JP1 occurs at sub-micromolar Ca2+ levels in skeletal muscle coinciding with μ-Calpain autocleavage at two possible sites (100, 105). Likewise, JP2 cleavage correlates with increased calpain activity, cardiac functional decline, T-tubule degeneration and Ca2+ handling dysfunction (101) and is largely prevented with in vivo calpain inhibition (103). Conversely, Jph2 overexpression partially overcomes the dysfunction in cardiac contractility, Ca2+ handling, and T-tubule integrity caused by Capn1 transgenic overexpression (103) supporting JP2 as a major calpain substrate.
Understanding the mechanisms by which calpains recognize and cleave junctophilins in disease could have far reaching therapeutic implications but characterization of cleavage events, defining cleavage site(s), and identifying calpain isoform(s) involved in JP1 and JP2 proteolysis has produced disparate results. In skeletal muscle, for example, JP1 has been reported to be expressed as a 90 kDa protein that is cleaved into N-terminal cytosolic 75 kDa and C-terminal SR-bound 15 kDa species (100), or as a 72 kDa protein that yields a 44 kDa C-terminal nuclear fragment (105) (Figure 3a). JP2 is typically observed as a ~100 kDa protein and is readily cleaved into 75 and 25 kDa fragments in stressed or failing hearts (96, 97, 100, 101, 106). Our group was the first to map a primary Capn1 cleavage event responsive to mM Ca2+ to a conserved site in the divergent region (R565/T566 in mouse JP2) (96). Wehrens and colleagues, however, reasoned that JP2 remains largely intact in unstressed hearts during normal Ca2+ cycling conditions sufficient for Capn1 activation but becomes preferentially cleaved by Capn2 during pathological stress when cytosolic Ca2+ reaches mM levels (98). Based on this reasoning, the authors reported Capn2 cleavage of JP2 at G482/T483 produces similar 75 kDa N-terminal and the 25 kDa C-terminal cleavage products (98) as those from R565/T566 cleavage (96). But how similar fragments arise from cleavage sites 83 amino acids apart was not resolved at the time. Two independent studies have since systematically compared the two sites in human and mouse JP2 that demonstrate a clear requirement for R565/T566, but not G482/T483, in mediating JP2 cleavage by both Capn1 and Capn2 in generating these proteolytic products (106, 107). Furthermore, immunoblots indicate in vivo produced N- and C-terminal JP2 fragments migrate at the same molecular mass as heterologously expressed JP2 1–565 and 566–696 peptides, respectively, but not those of 1–482 and 483–696 (106), indicating that cleavage of JP2 in vivo does not involve the G482/T483 site.
Figure 3.
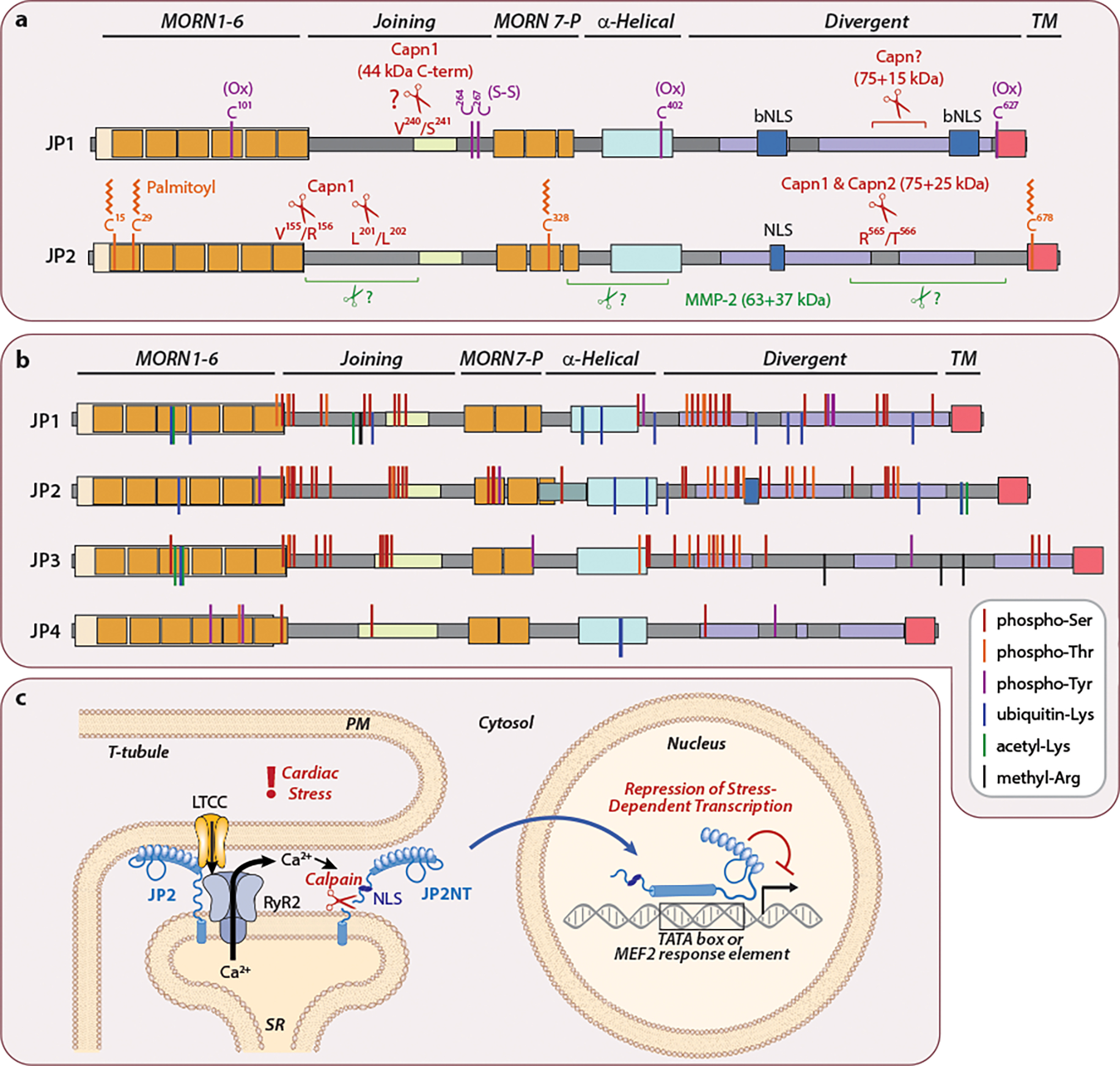
Junctophilin post-translational modifications. (a) Residues in JP1 and JP2 implicated in oxidation, palmitoylation and proteolysis. Cysteine residues in JP1 are oxidized (Ox) with a disulfide bond between C264 and C267 (S-S) (59). JP1 and JP2 are proteolytically cleaved by calpains (96, 100, 105, 106) and MMP-2 (104). The precise calpain cleavage site(s) of JP1 and MMP-2 site(s) of JP2 are not known. JP2 is also palmitoylated on Cys residues (99). (b) JP1–4 proteins are remarkably phosphorylated, ubiquitinated, acetylated, and methylated. Curated proteomics data for human, rat, and mouse JP1–4 were downloaded from phosphosite.org. Modifications are shown relative to human protein sequences. Modifications identified in rat or mouse are included if the amino acid is conserved in human. The physiological relevance for the vast majority of these modifications is unknown. (c) Stress events and promote the generation of a cardioprotective N-terminal fragment of JP2 (JP2NT) (97). Excessive SR Ca2+ release and cytosolic Ca2+ overload activates calpain which proteolyzes JP2 downstream of its NLS. JP2NT translocates into the nucleus to attenuate maladaptive gene expression by binding to promoter TATA-boxes and MEF2 response elements.
[**Note to Annual Reviews: We created this figure for this article; it is not based on any previously published image.**]
Cysteine palmitoylation and oxidation
JP2 lipidation on conserved cysteine residues tethers JP2 to JMCs and facilitates its incorporation into caveolae as ER-localized puncta (99) (Figure 3a). The majority of non-palmitoylated JP2 remains ER-localized but more distant from the PM. Palmitoylated JP2 is detected only at 100 kDa and a quadruple mCherry-JP2 C15/C29/C328/C678 mutant migrates faster than its WT counterpart (99). Whether dynamic JP2 palmitoylation occurs in cardiomyocytes is less clear as it is inefficiently palmitoylated and only at the PM outside of T-tubules (99) and JP2 crystal structures suggests C15 and C328 are inaccessible and C29 is buried when bound to LTCCs (78). Three of these Cys residues are conserved in other junctophilins suggesting their JMC localization may also be regulated through lipidation. Protein palmitoylation is specifically regulated by zinc finger and Asp-His-His-Cys (zDHHC) S-acyltransferases. The Brody group validated JP2 is palmitoylated in the heart but not further palmitoylated in zDHHC9 transgenic mice (108), implicating these modifications are probably meditated by other S-acyltransferase family members.
Other cysteines in JP1 act as redox sensors for modulating Ca2+ dynamics during oxidative stress conditions. RyR1 open conditions promote disulfide bond formation between JP1 C264 and C267, which increases its association with RyR1 (59). Similarly, the reactivity of JP1 C101, C402 and C627 are reduced under conditions favoring RyR1 open versus closed states. To what extent modification of junctophilins by palmitoylation or oxidation contributes to their function remains to be investigated and could be helped by knock-in in vivo models.
Phosphorylation and other modifications
Several kinases are predicted to phosphorylate junctophilins (75) but only PKC has been shown to mediate a specific phosphorylation event. The JP2 Ser165Phe mutation is associated with hypertrophic cardiomyopathy (HCM) (109) and its expression in skeletal myotubes reduces PKC-dependent JP2 phospho-Ser levels by ~60% (110). The mutation promotes skeletal myocyte hypertrophy, inhibits RyR1 channel activity, and interferes with JP2 - Trpc3 interactions. Knockout of the SPEG kinase reduces JP2 Ser phosphorylation by half and leads to the disorganization of T-tubules and E-C uncoupling (94) but which JP2 residue is responsive to SPEG remains unknown. Curated proteomics datasets (e.g., phosphosite.org) reveal JP1–4 proteins are endogenously and extensively phosphorylated on Ser, Thr, and Tyr residues, especially within their joining and divergent regions (Figure 3b), revealing we know very little about the post-translational regulation of junctophilins. Furthermore, several ubiquitinated and acetylated lysine and methylated arginine residues are present. The field is therefore ready for determining how these modifications contribute to junctophilin function.
TRANSCRIPTIONAL REGULATION BY JUNCTOPHILIN PROTEOLYTIC FRAGMENTS
Investigation of JP2 proteolysis by calpain led to the unexpected discovery that the 75 kDa JP2 N-terminal cleavage product (JP2NT) acts as a cardioprotective transcription factor that reports E-C coupling dysfunction through a direct excitation-transcription coupling mechanism (97, 111). JP2NT localizes exclusively to the nucleus due to a nuclear localization signal (NLS) within the divergent region lying upstream of the R565/T566 calpain cleavage site (Figure 3a and 3c). Hearts expressing JP2NT via transgene or viral gene therapy approaches are protected against pathological cardiac remodeling while those with an NLS deletion knock-in have exacerbated pressure overload-induced cardiac dysfunction (97, 112). JP2NT directly binds TATA-box like DNA sequences (including MEF2 response elements), enriches at transcriptional start sites, and represses stress-inducible transcripts involved in cardiac hypertrophy, fibrosis, and inflammation.
Interestingly, JP2NT DNA binding activity (97) maps to the central pseudo-MORN plus Ala-rich “backbone” α-helical sequences found to interact with the larger MORN array in the crystal structure (78). How such a structure allows for DNA binding is not readily predicted and will require additional structural studies. It is possible that the central JP2 region adopts one conformation at junctional membranes and another in nuclei. Similarly, JP1 proteolysis promotes nuclear enrichment of a 44kDa C-terminal fragment that includes two potential bipartite NLS’s (bNLS) (105). While this fragment requires more thorough investigation, its heterologous expression represses genes involved PI3K-Akt signaling and glycogenolysis. Whether other junctophilins (JP3, JP4) have a role in regulating gene transcriptions after pathological cleavage is worth further investigation.
PATHOLOGAL GENE MUTATIONS AND ALLELES
JPH2 mutations and cardiac disease
JP2 mutations were already hypothesized to be a risk factor for heart disease at the time of its identification based on the similar calcium handling defects observed between embryonic Jph2 knockout mouse myocytes and hypertrophic and failing hearts (17). Rare JPH2 mutations have since been identified in patients with hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), atrial fibrillation (AF) among other cardiac conditions (Figure 4a, see also (113–115). Dozens of other unvalidated JPH2 mutations are annotated as likely or possibly pathogenic in the ClinVar database as are infrequent mutations in JPH1 and JPH3 associated with Charcot-Marie-Tooth disease (116) and Huntington’s Disease-Like 2 (68, 117), respectively. Mutations in JPH4 have yet to be reported. Computational algorithms can predict how damaging a given mutation is reveal there are likely numerous deleterious alleles for JP1, JP2, and JP3 (Figure 4b–d) which, if pursued, could provide novel mechanistic insights.
Figure 4.

Pathogenic probability of junctophilin missense mutations. (a) Junctophilin mutations that are clinically associated with disease as annotated in the ClinVar database and/or have been published as is the case for JP1 R213P (116); JP1 T520M (125); JP2 E85K (126); JP2 S101R, Y141H, and S165F (81, 109); JP2 T161K (127); JP2 E169K (86); JP2 A189T (128); JP2 L204R, R363L, E402K, and R522W (129); JP2 T237A and I414L (130); JP2 E338G (131); JP2 A405S (132); JP2 A405T (133); JP2 R436C and G505S (134); JP2 E641* (135); and JP3 CAG/CTG repeat expansion (117, 136–138). *, nonsense mutation; CMT2K, Charcot-Marie-Tooth Disease Type 2K; HCM, Hypertrophic Cardiomyopathy; CA, cardiac amyloidosis; DCM, Dilated Cardiomyopathy; LVNC, Left Ventricular Non-Compaction; AF, Atrial Fibrillation; VT, ventricular tachycardia; SD, sudden death; HDL2, Huntington’s Disease-Like 2; NA, not applicable. (b and c) Predicted consequences of missense mutations on junctophilin function. Mutations predicted to be deleterious by both algorithms in each plot are shaded by gray boxes. HumDiv probability calculated by PolyPhen2 is based on the degree of all human disease variants relative to sequence divergence (139). Probably damaging missense mutations have values >0.95. PROVEAN score is derived from alignment-based correlations and are considered deleterious when scores are < −2.50 (140). SIFT predicts whether a mutation affects protein function based on sequence homology and the physical properties of the amino acid using a score cutoff < 0.05 (141). (d) List of missense mutations predicted by all three algorithms to be deleterious. Specific mutations in (b) and (c) are those listed in (a) and indicated in bold in (d).
[**Note to Annual Reviews: We created this figure for this article; it is not based on any previously published image.**]
Future efforts will be important for validating the pathogenicity of mutations identified in patients. Approaches for functionally characterizing JPH2 mutations include introduction into human embryonic stem cells (118), Jph2 genetic editing in the adult mouse heart through cardiotropic adeno-associated virus (AAV2/9) delivery of CRISPR/Cas9 and Jph2 gRNAs (119) and viral delivery of Jph2 transgenes (112, 120). For example, injection of mice after pressure overload surgery with AAV2/9 particles expressing intact JP2 or nuclear localizing JP2NT blunts T-tubule disorganization, Ca2+ handling dysfunction, cardiac remodeling processes, and hypertrophic gene expression (112, 120).
TRANSCRIPTIONAL REGULATION OF JUNCTOPHILINS
The mechanisms responsible underlying junctophilin gene transcription remain more enigmatic than their post-translational modifications. Only two transcription factors have been identified ot directly regulate Jph2 expression. Cardiac contraction in hibernating ground squirrels was found to be stronger than in non-hibernating animals due to a compensatory reduction in LTCC current density and Ca2+ influx combined with a near doubling in EC-coupling gain. This is achieved through increased dyad contact surface area and decreased junctional gap distance. Not surprisingly, JP2 expression was increased along with the cardiac transcription factor myocardin (121). Jph2 expression was found to require promoter CArG motifs known to bind SRF/myocardin dimers and depend on the synergistic actions of SRF and myocardin.
Non-coding RNAs help shape the transcriptome during growth, differentiation, and adaptation processes. Of these, a handful of mircoRNAs (miRNAs) have been experimentally shown to regulate junctophilin mRNA levels. MiR-24, miR-34, and miR-331 are induced in different forms of heart disease and increasing their expression or abundance in cardiomyocytes leads to Jph2 downregulation, E-C coupling dysfunction, and the initiation of heart failure (38, 122, 123). Mutation of two conserved miR-24 binding sites within the Jph2 3’ untranslated region prevents regulation by miR-24 (38). Injection of either miR-24 or miR-34 antagomirs in preclinical mouse models restores Jph2 levels and attenuates cardiac dysfunction following cardiac stress (122, 124). These encouraging pre-clinical studies demonstrate that targeting the miRs that downregulate Jph2 expression is theoretically possible and of translational value.
CONCLUDING REMARKS
After extensive research over the last 20 years junctophilins are now widely acknowledged to have an essential role in mediating cellular Ca2+ homeostasis within the narrow interface that lies between the PM and ER/SR terminal cisternae. Junctophilins have gone from an obscure family of conserved molecules into a fascinating quartet of multipurpose proteins with physiologically relevant functions. It will be interesting to see how our knowledge of junctophilin function expands over the next ten to twenty years particularly if the questions listed in Future Issues are effectively addressed.
SUMMARY POINTS.
Junctophilins organize plasma membrane-ER/SR junctional contacts involved in physiologically important processes in excitable and non-excitable cells.
Junctophilins bind to plasma membrane phospholipids, membrane ion channels, ER/SR localized ion channels, the ER/SR membrane and nuclear DNA through its MORN, joining, divergent, transmembrane and α-helical domains.
The essential requirement of JPH2 in the heart has provided great insights into the overall function of the junctophilin gene family.
JP2 is a dual-function protein for maintaining JMC integrity and E-C coupling at baseline and for regulating gene transcription and protecting against pathological remodeling under stress conditions.
Identification of disease-associated mutations have provided important insights into the tissue-specific functions of individual junctophilin genes.
Many unknowns remain regarding junctophilins including fine details underlying their transcription and the purpose of their post-translational modifications.
FUTURE ISSUES.
What is the function of the smaller, alternatively spliced junctophilin isoforms containing only the N-terminal MORN domains and do they act separate from or interfere with the function of full-length JP proteins?
Why do JPH2 gene mutations associate with cardiac disease but not with skeletal muscle defects?
How do MORN domains exactly bind to or associate with the plasma membrane? Do membranous phospholipids compete with and/or modulate junctophilin interactions with other proteins?
Do specific domain(s) of junctophilins determine the 12–15 nm physical gap of junctophilins-organized PM - ER/SR junction?
What is the function of the C-terminal divergent region in the intact and cleaved junctophilin proteins?
Do JP3, or JP4 possess nuclear translocating and DNA binding activity similar to JP1 and JP2 proteolytic fragments, and if so, can such fragments be leveraged as gene therapy agents?
How do the many endogenous post-translational modifications and interacting partners regulate junctophilin function?
What are the mechanisms underlying JPH gene transcription beyond the few that have been reported thus far?
ACKNOWLEDGEMENTS
This work was supported by National Heart, Lung and Blood Institute, Department of Veterans Affairs of the United States, and American Heart Association.
Footnotes
CONFLICT OF INTEREST
LSS is an inventor on a patent regarding the use of Junctophilin-2 fragments for the treatment of heart failure and other disease (WO2017214296A1; US 11,351,270 B2).
LITERATURE CITED
- 1.Dai S, Hall DD, Hell JW. 2009. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89: 411–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bravo-Sagua R, Parra V, Munoz-Cordova F, Sanchez-Aguilera P, Garrido V, et al. 2020. Sarcoplasmic reticulum and calcium signaling in muscle cells: Homeostasis and disease. Int Rev Cell Mol Biol 350: 197–264 [DOI] [PubMed] [Google Scholar]
- 3.Chen YJ, Quintanilla CG, Liou J. 2019. Recent insights into mammalian ER-PM junctions. Curr Opin Cell Biol 57: 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter KR, Palade GE. 1957. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol 3: 269–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbluth J 1962. Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol 13: 405–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henkart M, Landis DM, Reese TS. 1976. Similarity of junctions between plasma membranes and endoplasmic reticulum in muscle and neurons. J Cell Biol 70: 338–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang CL, Chen YJ, Liou J. 2017. ER-plasma membrane junctions: Why and how do we study them? Biochim Biophys Acta Mol Cell Res 1864: 1494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzini-Armstrong C, Protasi F, Ramesh V. 1999. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J 77: 1528–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi T, Martone ME, Yu Z, Thor A, Doi M, et al. 2009. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci 122: 1005–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeshima H, Hoshijima M, Song LS. 2015. Ca(2)(+) microdomains organized by junctophilins. Cell Calcium 58: 349–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang SQ, Wei C, Zhao G, Brochet DX, Shen J, et al. 2004. Imaging microdomain Ca2+ in muscle cells. Circ Res 94: 1011–22 [DOI] [PubMed] [Google Scholar]
- 12.Williams GS, Chikando AC, Tuan HT, Sobie EA, Lederer WJ, Jafri MS. 2011. Dynamics of calcium sparks and calcium leak in the heart. Biophys J 101: 1287–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rios E 2018. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J Gen Physiol 150: 521–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. 1998. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol 512 ( Pt 3): 677–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. 2001. beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ Res 88: 794–801 [DOI] [PubMed] [Google Scholar]
- 16.Zhao X, Yamazaki D, Kakizawa S, Pan Z, Takeshima H, Ma J. 2011. Molecular architecture of Ca2+ signaling control in muscle and heart cells. Channels (Austin) 5: 391–6 [DOI] [PubMed] [Google Scholar]
- 17.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. 2000. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6: 11–22 [DOI] [PubMed] [Google Scholar]
- 18.Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H. 2003. Coexpression of junctophilin type 3 and type 4 in brain. Brain Res Mol Brain Res 118: 102–10 [DOI] [PubMed] [Google Scholar]
- 19.Nishi M, Mizushima A, Nakagawara K, Takeshima H. 2000. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun 273: 920–7 [DOI] [PubMed] [Google Scholar]
- 20.Komazaki S, Ito K, Takeshima H, Nakamura H. 2002. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett 524: 225–9 [DOI] [PubMed] [Google Scholar]
- 21.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, et al. 2001. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 154: 1059–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, et al. 2011. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail 4: 214–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotta S, Morimura K, Ohya S, Muraki K, Takeshima H, Imaizumi Y. 2007. Ryanodine receptor type 2 deficiency changes excitation-contraction coupling and membrane potential in urinary bladder smooth muscle. J Physiol 582: 489–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pritchard HAT, Griffin CS, Yamasaki E, Thakore P, Lane C, et al. 2019. Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci U S A 116: 21874–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saeki T, Suzuki Y, Yamamura H, Takeshima H, Imaizumi Y. 2019. A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and BKCa channels in the Ca(2+) microdomain of vascular smooth muscle. J Biol Chem 294: 13093–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko JK, Choi KH, Zhao X, Komazaki S, Pan Z, et al. 2011. A versatile single-plasmid system for tissue-specific and inducible control of gene expression in transgenic mice. FASEB J 25: 2638–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishi M, Hashimoto K, Kuriyama K, Komazaki S, Kano M, et al. 2002. Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292: 318–24 [DOI] [PubMed] [Google Scholar]
- 28.Li L, Pan ZF, Huang X, Wu BW, Li T, et al. 2016. Junctophilin 3 expresses in pancreatic beta cells and is required for glucose-stimulated insulin secretion. Cell Death Dis 7: e2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu X, Kuang Y, Li L, Tang H, Shi Q, et al. 2017. Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics 7: 2150–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. 2011. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One 6: e17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, et al. 2009. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A 106: 6854–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen B, Li Y, Jiang S, Xie YP, Guo A, et al. 2012. beta-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J 26: 2531–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, et al. 2008. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res 102: 338–46 [DOI] [PubMed] [Google Scholar]
- 34.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, et al. 2006. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 574: 519–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. 2006. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103: 4305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu CY, Jia Z, Wang W, Ballou LM, Jiang YP, et al. 2011. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS One 6: e24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu HD, Xu M, Li RC, Guo L, Lai YS, et al. 2012. Ultrastructural remodelling of Ca(2+) signalling apparatus in failing heart cells. Cardiovasc Res 95: 430–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu M, Wu HD, Li RC, Zhang HB, Wang M, et al. 2012. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res 111: 837–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang HB, Li RC, Xu M, Xu SM, Lai YS, et al. 2013. Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res 98: 269–76 [DOI] [PubMed] [Google Scholar]
- 40.Wei S, Guo A, Chen B, Kutschke W, Xie YP, et al. 2010. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107: 520–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, et al. 2011. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123: 979–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen B, Guo A, Zhang C, Chen R, Zhu Y, et al. 2013. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc Res 100: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, et al. 2013. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res 100: 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, et al. 2014. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ Res 115: 986–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandenburg S, Pawlowitz J, Eikenbusch B, Peper J, Kohl T, et al. 2019. Junctophilin-2 expression rescues atrial dysfunction through polyadic junctional membrane complex biogenesis. JCI Insight 4: e127116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziman AP, Gomez-Viquez NL, Bloch RJ, Lederer WJ. 2010. Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol 48: 379–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han J, Wu H, Wang Q, Wang S. 2013. Morphogenesis of T-tubules in heart cells: the role of junctophilin-2. Sci China Life Sci 56: 647–52 [DOI] [PubMed] [Google Scholar]
- 48.Liu C, Spinozzi S, Chen JY, Fang X, Feng W, et al. 2019. Nexilin Is a New Component of Junctional Membrane Complexes Required for Cardiac T-Tubule Formation. Circulation 140: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C, Chen B, Guo A, Zhu Y, Miller JD, et al. 2014. Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129: 1742–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prins KW, Asp ML, Zhang H, Wang W, Metzger JM. 2016. Microtubule-Mediated Misregulation of Junctophilin-2 Underlies T-Tubule Disruptions and Calcium Mishandling in mdx Mice. JACC Basic Transl Sci 1: 122–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prins KW, Tian L, Wu D, Thenappan T, Metzger JM, Archer SL. 2017. Colchicine Depolymerizes Microtubules, Increases Junctophilin-2, and Improves Right Ventricular Function in Experimental Pulmonary Arterial Hypertension. J Am Heart Assoc 6: e006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo A, Zhang X, Iyer VR, Chen B, Zhang C, et al. 2014. Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc Natl Acad Sci U S A 111: 12240–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jayasinghe ID, Baddeley D, Kong CH, Wehrens XH, Cannell MB, Soeller C. 2012. Nanoscale organization of junctophilin-2 and ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophys J 102: L19–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hou Y, Jayasinghe I, Crossman DJ, Baddeley D, Soeller C. 2015. Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes. J Mol Cell Cardiol 80: 45–55 [DOI] [PubMed] [Google Scholar]
- 55.Jayasinghe I, Clowsley AH, Lin R, Lutz T, Harrison C, et al. 2018. True Molecular Scale Visualization of Variable Clustering Properties of Ryanodine Receptors. Cell Rep 22: 557–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munro ML, Jayasinghe ID, Wang Q, Quick A, Wang W, et al. 2016. Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J Cell Sci 129: 4388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calpena E, Lopez Del Amo V, Chakraborty M, Llamusi B, Artero R, et al. 2018. The Drosophila junctophilin gene is functionally equivalent to its four mammalian counterparts and is a modifier of a Huntingtin poly-Q expansion and the Notch pathway. Dis Model Mech 11: dmm029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakada T, Kashihara T, Komatsu M, Kojima K, Takeshita T, Yamada M. 2018. Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc Natl Acad Sci U S A 115: 4507–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phimister AJ, Lango J, Lee EH, Ernst-Russell MA, Takeshima H, et al. 2007. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J Biol Chem 282: 8667–77 [DOI] [PubMed] [Google Scholar]
- 60.Perni S, Lavorato M, Beam KG. 2017. De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca(2+) release. Proc Natl Acad Sci U S A 114: 13822–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perni S, Beam K. 2022. Junctophilins 1, 2, and 3 all support voltage-induced Ca2+ release despite considerable divergence. J Gen Physiol 154: e202113024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, et al. 2006. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J 90: 4418–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Ding X, Lopez JR, Takeshima H, Ma J, et al. 2010. Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J Biol Chem 285: 39171–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee EH, Cherednichenko G, Pessah IN, Allen PD. 2006. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J Biol Chem 281: 10042–8 [DOI] [PubMed] [Google Scholar]
- 65.Woo JS, Hwang JH, Ko JK, Kim DH, Ma J, Lee EH. 2009. Glutamate at position 227 of junctophilin-2 is involved in binding to TRPC3. Mol Cell Biochem 328: 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Komazaki S, Nishi M, Takeshima H. 2003. Abnormal junctional membrane structures in cardiac myocytes expressing ectopic junctophilin type 1. FEBS Lett 542: 69–73 [DOI] [PubMed] [Google Scholar]
- 67.Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, et al. 2006. Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci U S A 103: 10811–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, et al. 2012. Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol 71: 245–57 [DOI] [PubMed] [Google Scholar]
- 69.Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, et al. 2007. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J 26: 1924–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sahu G, Wazen RM, Colarusso P, Chen SRW, Zamponi GW, Turner RW. 2019. Junctophilin Proteins Tether a Cav1-RyR2-KCa3.1 Tripartite Complex to Regulate Neuronal Excitability. Cell Rep 28: 2427–42 e6 [DOI] [PubMed] [Google Scholar]
- 71.Perni S, Beam K. 2021. Neuronal junctophilins recruit specific CaV and RyR isoforms to ER-PM junctions and functionally alter CaV2.1 and CaV2.2. Elife 10: e64249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hogea A, Shah S, Jones F, Carver CM, Hao H, et al. 2021. Junctophilin-4 facilitates inflammatory signaling at plasma membrane-endoplasmic reticulum junctions in sensory neurons. J Physiol 599: 2103–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Woo JS, Srikanth S, Nishi M, Ping P, Takeshima H, Gwack Y. 2016. Junctophilin-4, a component of the endoplasmic reticulum-plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc Natl Acad Sci U S A 113: 2762–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou J, Liu H, Lin Y, Zhao J. 2022. Membrane occupation and recognition nexus (MORN) motif controls protein localization and function. FEBS Lett 596: 1839–50 [DOI] [PubMed] [Google Scholar]
- 75.Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. 2009. Molecular evolution of the junctophilin gene family. Physiol Genomics 37: 175–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Liu H, Raval MH, Wan J, Yengo CM, et al. 2019. Structure of the MORN4/Myo3a Tail Complex Reveals MORN Repeats as Protein Binding Modules. Structure 27: 1366–74 e3 [DOI] [PubMed] [Google Scholar]
- 77.Liu H, Li Z, Yang Q, Liu W, Wan J, et al. 2019. Substrate docking-mediated specific and efficient lysine methylation by the SET domain-containing histone methyltransferase SETD7. J Biol Chem 294: 13355–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang ZF, Panwar P, McFarlane CR, Tuinte WE, Campiglio M, Van Petegem F. 2022. Structures of the junctophilin/voltage-gated calcium channel interface reveal hot spot for cardiomyopathy mutations. Proc Natl Acad Sci U S A 119: e2120416119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fan HK, Luo TX, Zhao WD, Mu YH, Yang Y, et al. 2018. Functional interaction of Junctophilin 2 with small- conductance Ca(2+) -activated potassium channel subtype 2(SK2) in mouse cardiac myocytes. Acta Physiol (Oxf) 222: e12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kakizawa S, Moriguchi S, Ikeda A, Iino M, Takeshima H. 2008. Functional crosstalk between cell-surface and intracellular channels mediated by junctophilins essential for neuronal functions. Cerebellum 7: 385–91 [DOI] [PubMed] [Google Scholar]
- 81.Bennett HJ, Davenport JB, Collins RF, Trafford AW, Pinali C, Kitmitto A. 2013. Human junctophilin-2 undergoes a structural rearrangement upon binding PtdIns(3,4,5)P3 and the S101R mutation identified in hypertrophic cardiomyopathy obviates this response. Biochem J 456: 205–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rossi D, Scarcella AM, Liguori E, Lorenzini S, Pierantozzi E, et al. 2019. Molecular determinants of homo- and heteromeric interactions of Junctophilin-1 at triads in adult skeletal muscle fibers. Proc Natl Acad Sci U S A 116: 15716–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Golini L, Chouabe C, Berthier C, Cusimano V, Fornaro M, et al. 2011. Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J Biol Chem 286: 43717–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gross P, Johnson J, Romero CM, Eaton DM, Poulet C, et al. 2020. Interaction of the Joining Region in Junctophilin-2 with the L-type Ca(2+) Channel Is Pivotal for Cardiac Dyad Assembly and Intracellular Ca(2+) Dynamics. Circ Res 128: 92–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang W, Landstrom AP, Wang Q, Munro ML, Beavers D, et al. 2014. Reduced junctional Na+/Ca2+-exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin-2-deficient mice. Am J Physiol Heart Circ Physiol 307: H1317–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, et al. 2013. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol 62: 2010–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, et al. 2004. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun 325: 852–6 [DOI] [PubMed] [Google Scholar]
- 88.Poulet C, Sanchez-Alonso J, Swiatlowska P, Mouy F, Lucarelli C, et al. 2020. Junctophilin-2 tethers T-tubules and recruits functional L-type calcium channels to lipid rafts in adult cardiomyocytes. Cardiovasc Res 117: 149–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bergdahl A, Sward K. 2004. Caveolae-associated signalling in smooth muscle. Can J Physiol Pharmacol 82: 289–99 [DOI] [PubMed] [Google Scholar]
- 90.Jiang M, Zhang M, Howren M, Wang Y, Tan A, et al. 2016. JPH-2 interacts with Cai-handling proteins and ion channels in dyads: Contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 13: 743–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mathiesen SB, Lunde M, Stensland M, Martinsen M, Nyman TA, et al. 2020. The Cardiac Syndecan-2 Interactome. Front Cell Dev Biol 8: 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hennessey JA, Wei EQ, Pitt GS. 2013. Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ Res 113: 381–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, et al. 2013. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 10: 1886–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, et al. 2017. SPEG (Striated Muscle Preferentially Expressed Protein Kinase) Is Essential for Cardiac Function by Regulating Junctional Membrane Complex Activity. Circ Res 120: 110–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feng W, Liu C, Spinozzi S, Wang L, Evans SM, Chen J. 2020. Identifying the Cardiac Dyad Proteome In Vivo by a BioID2 Knock-In Strategy. Circulation 141: 940–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guo A, Hall D, Zhang C, Peng T, Miller JD, et al. 2015. Molecular Determinants of Calpain-dependent Cleavage of Junctophilin-2 Protein in Cardiomyocytes. J Biol Chem 290: 17946–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guo A, Wang Y, Chen B, Wang Y, Yuan J, et al. 2018. E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 362: eaan3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lahiri SK, Quick AP, Samson-Couterie B, Hulsurkar M, Elzenaar I, et al. 2020. Nuclear localization of a novel calpain-2 mediated junctophilin-2 C-terminal cleavage peptide promotes cardiomyocyte remodeling. Basic Res Cardiol 115: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang M, Hu J, White FKH, Williamson J, Klymchenko AS, et al. 2019. S-Palmitoylation of junctophilin-2 is critical for its role in tethering the sarcoplasmic reticulum to the plasma membrane. J Biol Chem 294: 13487–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Murphy RM, Dutka TL, Horvath D, Bell JR, Delbridge LM, Lamb GD. 2013. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J Physiol 591: 719–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu CY, Chen B, Jiang YP, Jia Z, Martin DW, et al. 2014. Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J Am Heart Assoc 3: e000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanzaki K, Watanabe D, Kuratani M, Yamada T, Matsunaga S, Wada M. 2017. Role of calpain in eccentric contraction-induced proteolysis of Ca(2+)-regulatory proteins and force depression in rat fast-twitch skeletal muscle. J Appl Physiol (1985) 122: 396–405 [DOI] [PubMed] [Google Scholar]
- 103.Wang Y, Chen B, Huang CK, Guo A, Wu J, et al. 2018. Targeting Calpain for Heart Failure Therapy: Implications From Multiple Murine Models. JACC Basic Transl Sci 3: 503–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chan BYH, Roczkowsky A, Cho WJ, Poirier M, Lee TYT, et al. 2019. Junctophilin-2 is a target of matrix metalloproteinase-2 in myocardial ischemia-reperfusion injury. Basic Res Cardiol 114: 42. [DOI] [PubMed] [Google Scholar]
- 105.Tammineni ER, Figueroa L, Manno C, Varma D, Kraeva N, et al. 2023. Muscle calcium stress cleaves junctophilin1, unleashing a gene regulatory program predicted to correct glucose dysregulation. Elife 12: e78874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J, Ciampa G, Zheng D, Shi Q, Chen B, et al. 2021. Calpain-2 specifically cleaves Junctophilin-2 at the same site as Calpain-1 but with less efficacy. Biochem J 478: 3539–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weninger G, Pochechueva T, El Chami D, Luo X, Kohl T, et al. 2022. Calpain cleavage of Junctophilin-2 generates a spectrum of calcium-dependent cleavage products and DNA-rich NT1-fragment domains in cardiomyocytes. Sci Rep 12: 10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Essandoh K, Subramani A, Ferro OA, Teuber JP, Koripella S, Brody MJ. 2023. zDHHC9 Regulates Cardiomyocyte Rab3a Activity and Atrial Natriuretic Peptide Secretion Through Palmitoylation of Rab3gap1. JACC Basic Transl Sci 8: 518–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, et al. 2007. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol 42: 1026–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Woo JS, Hwang JH, Ko JK, Weisleder N, Kim DH, et al. 2010. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem J 427: 125–34 [DOI] [PubMed] [Google Scholar]
- 111.Padmanabhan A, Haldar SM. 2018. Unusual transcription factor protects against heart failure. Science 362: 1359–60 [DOI] [PubMed] [Google Scholar]
- 112.Wang J, Shi Q, Wang Y, Dawson LW, Ciampa G, et al. 2022. Gene Therapy With the N-Terminus of Junctophilin-2 Improves Heart Failure in Mice. Circ Res 130: 1306–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, et al. 2019. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ Genom Precis Med 12: e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, et al. 2021. An Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 144: 7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parker LE, Kramer RJ, Kaplan S, Landstrom AP. 2021. One gene, two modes of inheritance, four diseases: A systematic review of the cardiac manifestation of pathogenic variants in JPH2-encoded junctophilin-2. Trends Cardiovasc Med 33: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pla-Martin D, Calpena E, Lupo V, Marquez C, Rivas E, et al. 2015. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet 24: 213–29 [DOI] [PubMed] [Google Scholar]
- 117.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, et al. 2001. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29: 377–8 [DOI] [PubMed] [Google Scholar]
- 118.Wu F, Li X, Bai R, Li Y, Gao J, Lan F. 2020. Generation of a Junctophilin-2 homozygous knockout human embryonic stem cell line (WAe009-A-36) by an episomal vector-based CRISPR/Cas9 system. Stem Cell Res 48: 101930. [DOI] [PubMed] [Google Scholar]
- 119.Guo Y, VanDusen NJ, Zhang L, Gu W, Sethi I, et al. 2017. Analysis of Cardiac Myocyte Maturation Using CASAAV, a Platform for Rapid Dissection of Cardiac Myocyte Gene Function In Vivo. Circ Res 120: 1874–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, et al. 2016. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca(2+) release. Int J Cardiol 225: 371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang L, Li RC, Xiang B, Li YC, Wang LP, et al. 2021. Transcriptional regulation of intermolecular Ca(2+) signaling in hibernating ground squirrel cardiomyocytes: The myocardin-junctophilin axis. Proc Natl Acad Sci U S A 118: e2025333118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu J, Gao C, Wei C, Xue Y, Shao C, et al. 2019. RBFox2-miR-34a-Jph2 axis contributes to cardiac decompensation during heart failure. Proc Natl Acad Sci U S A 116: 6172–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang J-J, Wang L-P, Li R-C, Wang M, Huang Z-H, et al. 2019. Abnormal expression of miR-331 leads to impaired heart function. Science Bulletin 64: 1011–17 [DOI] [PubMed] [Google Scholar]
- 124.Li RC, Tao J, Guo YB, Wu HD, Liu RF, et al. 2013. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res 112: 601–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kanwal S, Perveen S. 2019. Association of SNP in JPH1 gene with severity of disease in Charcot Marie Tooth 2K patients. J Pak Med Assoc 69: 241–43 [PubMed] [Google Scholar]
- 126.Sabater-Molina M, Navarro M, Garcia-Molina Saez E, Garrido I, Pascual-Figal D, et al. 2016. Mutation in JPH2 cause dilated cardiomyopathy. Clin Genet 90: 468–69 [DOI] [PubMed] [Google Scholar]
- 127.Vanninen SUM, Leivo K, Seppala EH, Aalto-Setala K, Pitkanen O, et al. 2018. Heterozygous junctophilin-2 (JPH2) p.(Thr161Lys) is a monogenic cause for HCM with heart failure. PLoS One 13: e0203422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Narula N, Tester DJ, Paulmichl A, Maleszewski JJ, Ackerman MJ. 2015. Post-mortem Whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol 36: 768–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sahlin E, Green A, Gustavsson P, Lieden A, Nordenskjold M, et al. 2019. Identification of putative pathogenic single nucleotide variants (SNVs) in genes associated with heart disease in 290 cases of stillbirth. PLoS One 14: e0210017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Miura A, Kondo H, Yamamoto T, Okumura Y, Nishio H. 2020. Sudden Unexpected Death of Infantile Dilated Cardiomyopathy with JPH2 and PKD1 Gene Variants. Int Heart J 61: 1079–83 [DOI] [PubMed] [Google Scholar]
- 131.Neubauer J, Haas C, Bartsch C, Medeiros-Domingo A, Berger W. 2016. Post-mortem whole-exome sequencing (WES) with a focus on cardiac disease-associated genes in five young sudden unexplained death (SUD) cases. Int J Legal Med 130: 1011–21 [DOI] [PubMed] [Google Scholar]
- 132.Quick AP, Landstrom AP, Wang Q, Beavers DL, Reynolds JO, et al. 2017. Novel junctophilin-2 mutation A405S is associated with basal septal hypertrophy and diastolic dysfunction. JACC Basic Transl Sci 2: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Abou Ziki MD, et al. 2017. Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults. Circ Cardiovasc Genet 10: e001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, et al. 2007. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet 52: 543–48 [DOI] [PubMed] [Google Scholar]
- 135.Jones EG, Mazaheri N, Maroofian R, Zamani M, Seifi T, et al. 2019. Analysis of enriched rare variants in JPH2-encoded junctophilin-2 among Greater Middle Eastern individuals reveals a novel homozygous variant associated with neonatal dilated cardiomyopathy. Sci Rep 9: 9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stevanin G, Camuzat A, Holmes SE, Julien C, Sahloul R, et al. 2002. CAG/CTG repeat expansions at the Huntington’s disease-like 2 locus are rare in Huntington’s disease patients. Neurology 58: 965–7 [DOI] [PubMed] [Google Scholar]
- 137.Krause A, Mitchell C, Essop F, Tager S, Temlett J, et al. 2015. Junctophilin 3 (JPH3) expansion mutations causing Huntington disease like 2 (HDL2) are common in South African patients with African ancestry and a Huntington disease phenotype. Am J Med Genet B Neuropsychiatr Genet 168: 573–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, et al. 2003. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 126: 1599–603 [DOI] [PubMed] [Google Scholar]
- 139.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Choi Y, Chan AP. 2015. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31: 2745–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ng PC, Henikoff S. 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yoshida M, Sugimoto A, Ohshima Y, Takeshima H. 2001. Important role of junctophilin in nematode motor function. Biochem Biophys Res Commun 289: 234–9 [DOI] [PubMed] [Google Scholar]