

Abstract
Autoantibodies are an immunological resource ripe for exploitation in cancer detection and treatment. Key to this translation is a better understanding of the self-epitopes autoantibodies target in tumor tissue, but do not bind to in normal tissue. Posttranslational modifications on self-proteins are known to break tolerance in many autoimmune diseases and have also recently been described in cancer. This scope of possible autoantigens is quite broad and new high-dimensional and -throughput technologies to probe this repertoire will be necessary to fully exploit their potential. Here, we discuss the strengths and weaknesses of existing high-throughput platforms to detect autoantibodies, review the current methods for characterizing immunogenic posttranslational modifications, describe the main challenges to identifying disease-relevant antigens and suggest the properties of future technologies that may be able to address these challenges. We conclude that exploiting the evolutionary power of the immune system to distinguish between self and non-self has great potential to be translated into antibody-based clinical applications.
Graphical Abstract:

Introduction
Antibodies have revolutionized the field of cancer therapy and diagnostics[1]. The presence of B lymphocytes, their more differentiated progeny antibody-secreting cells, and autoantibodies are frequently associated with positive prognosis in many cancer types[•2]. Autoantibodies are antibodies that target ‘self’ antigens, and their ability to target cancer-specific antigens without causing overt autoimmunity indicates an ability to distinguish self from tumor. Identifying these tumor-specific autoantibodies would allow us to harness these autoantibodies as powerful tools to detect, image, and treat cancer with high specificity.
For these reasons, there has been a spike in interest in autoantibodies and their relationship to cancer, particularly in the last few years (Figure 1). Part of this recent research has focused on the discovery that posttranslational modifications (PTMs) can be a critical part of the neoantigens responsible for inducing autoantibody-mediated autoimmune diseases[3,4]. Autoimmunity-inducing PTMs, like citrullination or isoaspartylation, often occur as a result of acute or chronic inflammation. Cellular stress is also a consequence of oncologic transformation[5], and very recently, it has become clear that autoantibodies to the same types of PTMs can also occur in cancer[6]. Normally, maturing B cells in the bone marrow are eliminated or edited if their receptor-binding domains react too strongly to self-peptides, a process known as central tolerance[7]. However, the PTMs that occur on self-peptides in stressed or aging cells may not be represented during this selection process. As a result, autoreactive B cells to PTM-modified antigens can escape central tolerance, undergo activation, and eventually lead to pathology in ‘self’-antigen expressing tissues[8]. Autoantibodies to PTM-modified self-antigens have been characterized as pathogenic in several autoimmune diseases including rheumatoid arthritis, system lupus erythematosus, and celiac disease [9]. In the context of cancer, PTM-targeting autoantibodies potentially target highly specific neoantigens that differentiate cancer from normal tissues and reshape the adaptive immune response[10,11]. Here, we will focus on the PTMs with the strongest experimental and clinical evidence for immunogenicity and the technological workflows or platforms that have recently been developed to discover PTM-targeting autoantibodies.
Figure 1.

PubMed citations of autoantibodies (AAb) described in autoimmunity or cancer have steadily risen since 1963. In the last decade, AAb citations have almost doubled for a total of 2565 autoimmunity and 908 cancer publications in 2022.
Posttranslational modifications inducing autoantibodies
A well-described group of autoantibodies targeting PTMs are anti-citrullinated protein/peptide autoantibodies (ACPA) in rheumatoid arthritis[12]. Some ACPA are very disease specific (98% specificity)[13], targeting specific citrullinated epitopes that can be used for both diagnostic and prognostic assessments[14]. Other ACPAs react with a range of citrullinated peptides and do not have a clear pathogenic function [15,16]. Citrulline is a noncoding amino acid generated from deimination of peptidyl-arginine, a reaction catalyzed by a peptidyl-arginine deiminase (PAD) enzyme. An enzyme to reverse this reaction has not been identified. The conversion of positively charged arginine into electrically neutral citrulline influences protein folding, function, and stability[17]. Of the five known PAD isoforms, PAD2 and PAD4 have been shown to be upregulated in numerous solid tumors compared to normal tissues[18]. Cigarette smoking has also been shown to upregulate PAD2 expression in human lungs and increase citrullinated proteins in cells from bronchoalveolar lavage fluid[19]. In breast cancer, PAD2 was highly expressed and positively correlated with tumor-infiltrating B cells[20]. Interestingly, ACPA-specific B cells isolated from rheumatoid arthritis patients demonstrate continual activation without reaching quiescence, indicating enhanced effector function against PTM antigens[21]. Autoantibodies that target citrulline modifications have also been shown to serve as circulating tumor biomarkers in breast cancer[20] and small cell lung cancer[•22].
Another PTM associated with autoantibody production is the spontaneous deamination of asparagine or isomerization of aspartic acid to form isoaspartyl (isoAsp) residues in stressed or aging cells. IsoAsp modifications can cause conformational bends in protein structure that compromise protein function, but activity can be restored by the repair enzyme protein L-isoaspartate O-methyltransferase (PIMT)[23]. The importance of this repair enzyme is reflected in mice with homozygous knockout of PIMT, which results in increased isoAsp-modified proteins, growth retardation and fatal epileptic seizures 30-60 days after birth[24]. PIMT knockout mice do not have abnormal lymphoid development, but do have an enhanced T cell effector phenotype[25]. PIMT knockout CD4+ T cells were hyperresponsive to multiple types of activation stimuli. Over time, the hyper-effective function of PIMT knockout lymphocytes produced anti-DNA autoantibodies and autoimmune-mediated kidney pathology in irradiated wild type mice reconstituted with PIMT knockout bone marrow. Higher expression of PIMT (i.e., lower expression of isoAsp) is a predictor of poor prognosis in human lung adenocarcinoma[26], cervical cancer[27], bladder cancer[28] and breast cancer[29]. In mouse models of systemic lupus erythematosus, immunization with isoAsp-modified model antigens, but not unmodified antigens, stimulated lymphocytes[30]. Immunization with isoAsp-modified peptides generated autoantibodies that were cross-reactive to both isoAsp-modified and unmodified peptides, even though T cells did not respond to the unmodified peptides. In two cancer models, isoAsp-modified, but not unmodified, antigens were highly immunogenic and elicited strong T and B cell responses[31]. Similar to the autoimmunity models, isoAsp-containing antigens caused B cells to produce autoantibodies to both isoAsp-modified and unmodified antigens. Immunization of rabbits with an isoAsp peptide of ELAVL4, a known autoantigen widely expressed in SCLC, induced antibodies to both isoAsp-ELAVL4 and unmodified ELAVL4, but, after careful screening, antibodies with specific binding to isoAsp-ELAVL4 were isolated[32]. Cumulatively, these findings support the hypothesis that isoAsp modifications are potent lymphocytic immunogens that can induce the generation of PTM-specific autoantibodies.
Newly discovered PTM epitopes and their associated autoantibodies represent an area of active investigation. Recently, the PTM carboxyethylation was identified on ITGA2B as the epitope inducing the autoimmune disease ankylosing spondylitis (AS)[33]. In this study, the authors examined possible amino acid derivatives across the proteome in peripheral blood mononuclear cells from patients with AS and healthy donors. They then measured the mass differences between the coded amino acids and the actual residues by mass spectrometry and found a specific cysteine in ITGA2B had a higher mass, indicating carboxyethylation. This PTM of ITGA2B was significantly enriched in AS patients. In a mouse model of AS, immunization with a carboxyethylated ITGA2B peptide induced a significant pathogenic immune response (colitis, spondylitis), whereas immunization with the unmodified ITGA2B peptide had minimal effect. In another example, pancreatic islet proteins from pre-diabetic mice were separated by 2D-gel electrophoresis, derivatized with 2,4-dinitrophenylhydrazine (DNPH), probed with an anti-dinitrophenyl (DNP) antibody to detect carbonyl modifications, and IgG-bound protein spots were examined by mass spectrometry to identify carbonyl-modified P4Hb as an early autoantigen in type 1 diabetes[34]. Autoantibodies targeting carbonyl-P4Hb were upregulated in the serum from both a pre-diabetic mouse model and early type 1 diabetes patients. These powerful studies reveal important pathology, but also reflect the laborious process to discover PTM-autoantigens and PTM-targeted autoantibodies. Alternative high-dimensional approaches would be useful to increase throughput for autoantibody and autoantigen discovery.
High-throughput capture approaches
Biotechnological advancements have allowed the production of binding or capture reagent libraries, several of which cover almost the entire human proteome. The largest proteomic coverage appears to be the Human Proteome (HuProt) Microarrays, which contain >21,000 unique human proteins, isoforms and protein fragments produced in yeast and spotted on a nitrocellulose slide[35]. After incubation with plasma or serum, the amount of immunoglobulin (e.g., autoantibodies) bound to each protein on the array can be assayed via a fluorescent detection antibody (Figure 2a). Nucleic Acid Programmable Protein Arrays (NAPPA) provide an alternative approach to producing protein using a cell-free expression system, simplifying the need to purify recombinant proteins. Using DNA templates printed on the surface of an array, proteins are produced and captured locally by affinity reagents[36]. After incubation with sera, affinity tag captured proteins with bound autoantibodies are detected by dye-labelled anti-human immunoglobulin secondary antibody. Protein identity is known during library construction and each protein is mapped to a specific location on the array surface. The serological identification of antigens by recombinant expression cloning (SEREX) method was published over 20 years ago and involves constructing cDNA libraries directly from tumor tissue and cloning them into lambda phage expression vectors[37]. These recombinant phages are then used to produce proteins in bacteria that are transferred to nitrocellulose membranes. After addition of patient sera, immunoglobulins bound to the protein spots are determined and sequencing of the expression vector insert is used to determine the identity of the antigen. More recently, phage immunoprecipitation sequencing (PhIP-Seq) was developed using an oligonucleotide-encoded library representing overlapping peptide antigens up to 90 amino acids long that are presented via phage display (Figure 2b)[38]. After incubation with serum, IgGs bound to peptides are immunoprecipitated using protein A/G-coated magnetic beads. The enrichment of each encoded peptide is quantified via next generation sequencing (NGS) and normalized to IgG input via ELISA. The advantage of phage display over synthetic peptides is the increase in the usable peptide length to ~90 amino acids, compared to ~20 amino acids typical for synthetic peptide arrays. Alternatively, the rapid extracellular antigen profiling (REAP) platform contains over 2,600 barcoded extracellular proteins displayed by yeast[•39]. Similar to the PhIP-Seq platform, after incubation with serum, IgG is separated using a protein G magnetic column and barcoded yeast proteins are quantified via NGS. In both PhIP-Seq and REAP, the capture antigens are known and barcoded, enabling easy identification of the antigens targeted by the bound autoantibodies. A key limitation for all these high-throughput approaches is that the antigens produced are not subject to human or disease-specific PTMs and consequently may not react with relevant autoantibodies. However, if a particular PTM is of known interest, high-throughput capture antigens produced in non-human cells can be post-translationally modified with the inclusion of PTM-associated enzymes. For example, PhIP-Seq libraries treated with PAD2 and PAD4 to induce citrulline PTMs on capture peptides bound rheumatoid arthritis-associated autoantibodies[•40]. Similarly, the Contra Capture Protein Array platform uses a modified version of NAPPA to synthesize an array of proteins from a library of cDNA templates[41]. The protein array is then treated with recombinant PAD2 to yield citrullinated residues. This platform has been used to confirm known citrullinated autoantigen-autoantibody pairs as well as to discover novel citrullinated autoantigen-autoantibody pairs in sera samples from rheumatoid arthritis patients.
Figure 2. Two approaches to high-throughput autoantibody identification.

(A) Platforms like SEREX, NAPPA, and HuProt use recombinant proteins bound to a coated slide or other surface in a spatially specific manner in order to map protein identification. Human serum or plasma are then added, unbound autoantibodies are washed away, and bound autoantibodies are often detected by a fluorescent anti-human immunoglobulin secondary antibody. (B) Platforms like PhIP-Seq or REAP produce barcoded proteins and after incubation with human serum or plasma, autoantibodies are immunoprecipitated by antibody-binding magnetic beads or columns. The identify of autoantibody-bound proteins are then identified by barcode sequencing. Image created with BioRender.com.
Technologies to link disease-specific autoantibody discovery and PTM-containing epitope identification
In order to be most useful clinically, autoantibodies must be relatively disease specific. To identify these autoantibodies, the capture antigen should be derived from the diseased tissue of interest (e.g., tumor cells). Serological proteome analysis (SERPA) separates antigens from tumor tissue, cell lines or other inputs of interest by 2D electrophoresis and after transfer, these immunoblots are probed with patient serum and quantified by secondary detection reagents (Figure 3a)[42]. The identity of disease-specific proteins that bind autoantibody is then determined by excising the specific spot followed by mass spectrometry. A key limitation of this approach is the relatively low throughput (so determining whether the tumor antigen is shared across many patients is more difficult). Our group has developed a more high-throughput approach in a large-format antibody array that captures circulating autoantibody-antigen complexes from human plasma using commercially available antibodies (Figure 3b)[43,44]. These capture antibodies enable straightforward identification of antigen bound to autoantibody. By isolating autoantibody-antigen complexes, we are able to detect truly disease-specific autoantibodies bound to their native tumor-derived epitopes. However, we are limited by the number and type of capture antibodies.
Figure 3. Approaches for PTM-targeting autoantibody identification.
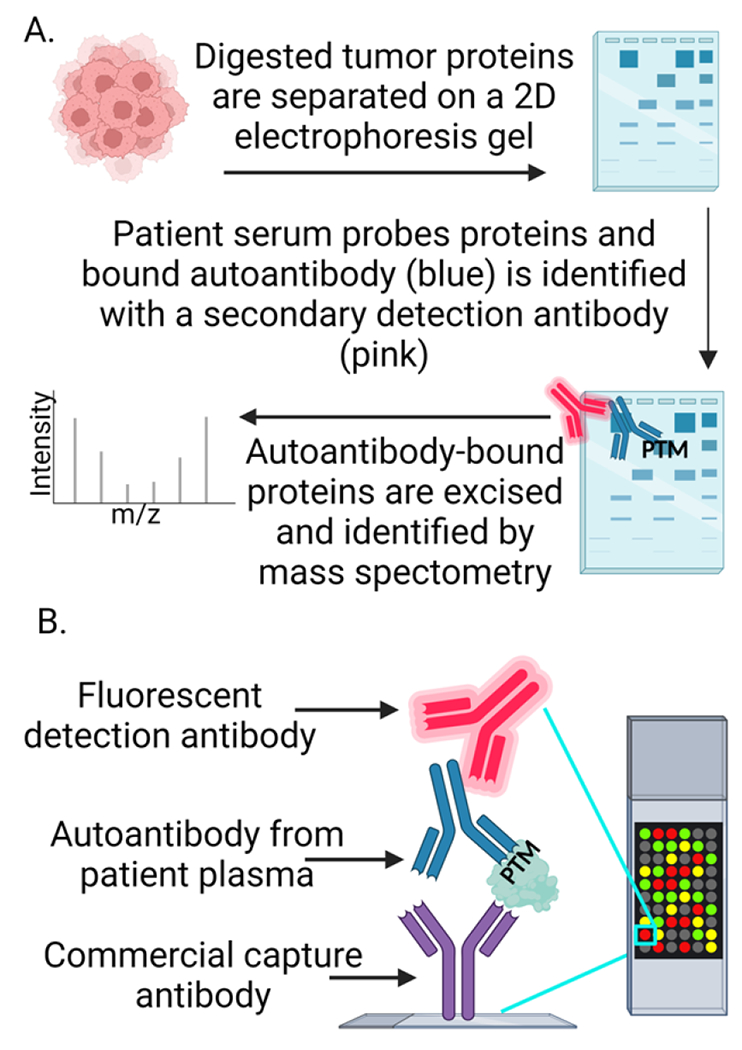
(A) Methods like SERPA use tumor or cancer cell lysates that are separated on 2D electrophoresis gels and probed with serum from diseased or healthy patients. Bound autoantibodies are detected with a fluorescent or chemiluminescent secondary antibody. Proteins with autoantibody binding in diseased but not healthy patients are excised and sent for identification via mass spectrometry. (B) Commercially available antibodies are spotted on coated microarray slides in a specific spatial orientation and used to capture autoantibody-antigen complexes upon incubation with patient serum or plasma. Bound autoantibody-antigen complexes are detected by a fluorescent secondary antibody. Image created with BioRender.com
A limitation for all of these approaches is the need to further define the immunogenic epitopes that autoantibodies target (e.g., PTM and surrounding sequence). This step is necessary for the creation of specific assay reagents with clinical utility. To enable the identification of immunogenic epitopes, we’ve used model peptides with or without PTMs in the extracellular domains of antigens identified with our autoantibody-antigen complex array. These model peptides are used as capture reagents, and after incubation with plasma, assayed for autoantibody binding with a luminescent anti-human immunoglobulin detection antibody. With this approach, we recently described epitopes containing multiple types of PTMs, including citrullination, isoaspartylation and glycosylation, that are targeted by autoantibodies in small cell lung cancer patients without autoimmune disease[20]. The therapeutic potential of antibodies targeting tumor-specific PTMs is illustrated by the PankoMab antibody[45,46]. This antibody targets tumor-specific glycan PTM motifs on MUC1, can distinguish tumor MUC1 from MUC1 expressed in normal tissue, and has shown preliminary clinical utility as a glycoengineered antibody promoting Fc-mediated anti-tumor immunity. Chimeric antigen receptor (CAR) T regulatory cells targeting citrullinated vimentin are undergoing evaluation with encouraging preclinical results[47]. Thus, methods to capture disease-specific autoantibodies and identify the targeted autoantigen epitopes, which may include PTMs, are key to clinical translation.
Conclusions and outlook
Existing methods for autoantibody discovery have begun to illustrate the importance of autoantibodies as potential clinical tools. After the epitope for a clinically relevant autoantibody is defined, converting autoantibody assays into a clinically acceptable platform is relatively straightforward[48]. However, current autoantibody discovery methods lack the ability to both screen for and readily identify disease-specific, human PTMs that might comprise the epitope. We believe one path forward is to use platforms such as our autoantibody-autoantigen array to identify the antigen, and then use platforms such as the Contra Capture Protein Array to discover if specific PTM epitopes of the antigen are recognized by the disease-specific autoantibodies. A crucial requirement for the therapeutic application of autoantibodies is the ability to target tumor antigens but avoid targeting the same proteins in healthy tissues, which would cause harmful autoimmunity. Autoantibodies have been shown to be upregulated prior to clinical detection of cancer and can be found at high concentrations even when autoantigens are at low abundance due to an amplified immune response[49]. Tumor-reactive autoantibodies can undergo antigen-driven maturation[•50], and we believe leveraging of the immune system’s intrinsic ability to distinguish self from foreign antigens could open the door to highly specific autoantibody-derived therapeutic agents. After the epitope for a clinically relevant autoantibody is defined, one can use model antigens (e.g., peptides containing the PTM that caused autoimmunity) to “pan” for and isolate antigen-specific human B cells from patients with the disease of interest. After single-cell sequencing of the antibody variable regions, tumor-specific reagents can be used to produce CAR T-cell therapies or antibody drug conjugates. Such approaches utilize the immune system to select for disease specificity – a strategy that has already been fruitful in other disease settings[51]. Antibody-based therapeutics have already been clinical success stories for the treatment of a variety of different cancers, and we believe that progress will continue with the novel antibody-based technologies being utilized and newly developed.
Highlights:
Autoantibodies can be used for cancer early detection, imaging, and therapeutics.
Posttranslational modifications can form neoantigens, stimulating autoantibody production.
New high-dimensional techniques to identify autoantigens can aid in epitope analysis.
Acknowledgements
This work is supported by National Institutes of Health (P50CA228944, R01CA243328) to PDL, (KL2TR002317, U01CA258066) to KJL, and (P30CA015704) to Fred Hutchinson Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflicts of interests.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:• of special interest
- [1].Jin S, Sun Y, Liang X, Gu X, Ning J, Xu Y, et al. Emerging new therapeutic antibody derivatives for cancer treatment. Sig Transduct Target Ther 2022;7:1–28. 10.1038/s41392-021-00868-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer 2022;22:414–30. 10.1038/s41568-022-00466-l. [DOI] [PMC free article] [PubMed] [Google Scholar]; A thorough review describing the clinical associations and translational implications of tumor-infiltrating B cells.
- [3].Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Current Opinion in Immunology 2012;24:112–8. 10.1016/j.coi.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Doyle HA, Mamula MJ. Posttranslational protein modifications: new flavors in the menu of autoantigens. Curr Opin Rheumatol 2002;14:244–9. 10.1097/00002281-200205000-00009. [DOI] [PubMed] [Google Scholar]
- [5].Li W, Li F, Zhang X, Lin H-K, Xu C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Sig Transduct Target Ther 2021;6:1–30. 10.1038/s41392-021-00825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Srivastava AK, Guadagnin G, Cappello P, Novelli F. Post-Translational Modifications in Tumor-Associated Antigens as a Platform for Novel Immuno-Oncology Therapies. Cancers (Basel) 2022;15:138. 10.3390/cancersl5010138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol 2017;17:281–94. 10.1038/nri.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Scherer HU, van der Woude D, Toes REM. From risk to chronicity: evolution of autoreactive B cell and antibody responses in rheumatoid arthritis. Nat Rev Rheumatol 2022;18:371–83. 10.1038/s41584-022-00786-4. [DOI] [PubMed] [Google Scholar]
- [9].Doyle HA, Mamula MJ. Post-translational protein modifications in antigen recognition and autoimmunity. Trends in Immunology 2001;22:443–9. 10.1016/S1471-4906(01)01976-7. [DOI] [PubMed] [Google Scholar]
- [10].Kacen A, Javitt A, Kramer MP, Morgenstern D, Tsaban T, Shmueli MD, et al. Post-translational modifications reshape the antigenic landscape of the MHC I immunopeptidome in tumors. Nat Biotechnol 2023;41:239–51. 10.1038/s41587-022-01464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brentville VA, Vankemmelbeke M, Metheringham RL, Durrant LG. Post-translational modifications such as citrullination are excellent targets for cancer therapy. Seminars in Immunology 2020;47:101393. 10.1016/j.smim.2020.101393. [DOI] [PubMed] [Google Scholar]
- [12].Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y. Citrullination and autoimmunity. Autoimmun Rev 2015;14:490–7. 10.1016/j.autrev.2015.01.013. [DOI] [PubMed] [Google Scholar]
- [13].de Brito Rocha S, Baldo DC, Andrade LEC. Clinical and pathophysiologic relevance of autoantibodies in rheumatoid arthritis. Advances in Rheumatology 2019;59:2. 10.1186/s42358-018-0042-8. [DOI] [PubMed] [Google Scholar]
- [14].van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. Journal of Autoimmunity 2020;110:102392. 10.1016/j.jaut.2019.102392. [DOI] [PubMed] [Google Scholar]
- [15].Ge C, Holmdahl R. The structure, specificity and function of anti-citrullinated protein antibodies. Nat Rev Rheumatol 2019;15:503–8. 10.1038/s41584-019-0244-4. [DOI] [PubMed] [Google Scholar]
- [16].He Y, Ge C, Moreno-Giró À, Xu B, Beusch CM, Sandor K, et al. A subset of antibodies targeting citrullinated proteins confers protection from rheumatoid arthritis. Nat Commun 2023;14:691. 10.1038/s41467-023-36257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuzhalin AE. Citrullination in Cancer. Cancer Res 2019;79:1274–84. 10.1158/0008-5472.CAN-18-2797. [DOI] [PubMed] [Google Scholar]
- [18].Chang X, Han J, Pang L, Zhao Y, Yang Y, Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009;9:40. 10.1186/1471-2407-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Makrygiannakis D, Hermansson M, Ulfgren A-K, Nicholas AP, Zendman AJW, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis 2008;67:1488–92. 10.1136/ard.2007.075192. [DOI] [PubMed] [Google Scholar]
- [20].Katayama H, Kobayashi M, Irajizad E, Sevillano AM, Patel N, Mao X, et al. Protein citrullination as a source of cancer neoantigens. J Immunother Cancer 2021;9:e002549. 10.1136/jitc-2021-002549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kristyanto H, Blomberg NJ, Slot LM, van der Voort EIH, Kerkman PF, Bakker A, et al. Persistently activated, proliferative memory autoreactive B cells promote inflammation in rheumatoid arthritis. Science Translational Medicine 2020;12:eaaz5327. 10.1126/scitranslmed.aaz5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lastwika KJ, Kunihiro A, Solan JL, Zhang Y, Taverne LR, Shelley D, et al. Posttranslational modifications induce autoantibodies with risk prediction capability in patients with small cell lung cancer. Sci Transl Med 2023;15:eadd8469. 10.1126/scitranslmed.add8469. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes an antibody-based microarray platform that captures autoantibodies complexed to disease-specific epitopes ex vivo.
- [23].Mishra PKK, Mahawar M. PIMT-Mediated Protein Repair: Mechanism and Implications. Biochemistry (Mosc) 2019;84:453–63. 10.1134/S0006297919050018. [DOI] [PubMed] [Google Scholar]
- [24].Kim E, Lowenson JD, MacLaren DC, Clarke S, Young SG. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc Natl Acad Sci U S A 1997;94:6132–7. 10.1073/pnas.94.12.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Doyle HA, Gee RJ, Mamula MJ. A failure to repair self-proteins leads to T cell hyperproliferation and autoantibody production. J Immunol 2003;171:2840–7. 10.4049/jimmunol.171.6.2840. [DOI] [PubMed] [Google Scholar]
- [26].Saito H, Yamashita M, Ogasawara M, Yamada N, Niisato M, Tomoyasu M, et al. Chaperone protein L-isoaspartate (D-aspartyl) O-methyltransferase as a novel predictor of poor prognosis in lung adenocarcinoma. Hum Pathol 2016;50:1–10. 10.1016/j.humpath.2015.ll.006. [DOI] [PubMed] [Google Scholar]
- [27].Shan L, Wang X, Li Y, Li L, Wu S, Xi X, et al. Elevated expression of protein-L-isoaspartate O-methyltransferase-1 (PCMT1) in cervical cancer. Transl Cancer Res 2022;11:2582–90. 10.21037/tcr-21-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dong L, Li Y, Xue D, Liu Y. PCMT1 is an unfavorable predictor and functions as an oncogene in bladder cancer. IUBMB Life 2018;70:291–9. 10.1002/iub.1717. [DOI] [PubMed] [Google Scholar]
- [29].Guo J, Du X, Li C. PCMT1 Is a Potential Prognostic Biomarker and Is Correlated with Immune Infiltrates in Breast Cancer. Biomed Res Int 2022;2022:4434887. 10.1155/2022/4434887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones P-J, et al. Isoaspartyl Post-translational Modification Triggers Autoimmune Responses to Self-proteins. Journal of Biological Chemistry 1999;274:22321–7. 10.1074/jbc.274.32.22321. [DOI] [PubMed] [Google Scholar]
- [31].Doyle HA, Zhou J, Wolff MJ, Harvey BP, Roman RM, Gee RJ, et al. Isoaspartyl Post-translational Modification Triggers Anti-tumor T and B Lymphocyte Immunity. Journal of Biological Chemistry 2006;281:32676–83. 10.1074/jbc.M604847200. [DOI] [PubMed] [Google Scholar]
- [32].Pulido MA, DerHartunian MK, Qin Z, Chung EM, Kang DS, Woodham AW, et al. Isoaspartylation Appears to Trigger Small Cell Lung Cancer-Associated Autoimmunity against Neuronal Protein ELAVL4. J Neuroimmunol 2016;299:70–8. 10.1016/j.jneuroim.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhai Y, Chen L, Zhao Q, Zheng Z-H, Chen Z-N, Bian H, et al. Cysteine carboxyethylation generates neoantigens to induce HLA-restricted autoimmunity. Science 2023;379:eabg2482. 10.1126/science.abg2482. [DOI] [PubMed] [Google Scholar]
- [34].Yang M-L, Connolly SE, Gee RJ, Lam TT, Kanyo J, Peng J, et al. Carbonyl Posttranslational Modification Associated With Early-Onset Type 1 Diabetes Autoimmunity. Diabetes 2022;71:1979–93. 10.2337/db21-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Li J, Zhang X, Liu M, Ji L, Yang T, et al. Autoantibody signatures discovered by HuProt protein microarray to enhance the diagnosis of lung cancer. Clinical Immunology 2023;246:109206. 10.1016/j.clim.2022.109206. [DOI] [PubMed] [Google Scholar]
- [36].Díez P, González-González M, Lourido L, Dégano RM, Ibarrola N, Casado-Vela J, et al. NAPPA as a Real New Method for Protein Microarray Generation. Microarrays (Basel) 2015;4:214–27. 10.3390/microarrays4020214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sahin U, Tüeci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci USA 1995;92:11810–3. 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, et al. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat Protoc 2018;13:1958–78. 10.1038/s41596-018-0025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang EY, Dai Y, Rosen CE, Schmitt MM, Dong MX, Ferré EMN, et al. High-throughput identification of autoantibodies that target the human exoproteome. Cell Rep Methods 2022;2:100172. 10.1016/j.crmeth.2022.100172. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a high-throughput method to identify autoantibodies to known extracellular antigens.
- [40].Román-Meléndez GD, Monaco DR, Montagne JM, Quizon RS, Konig MF, Astatke M, et al. Citrullination of a phage-displayed human peptidome library reveals the fine specificities of rheumatoid arthritis-associated autoantibodies. eBioMedicine 2021;71. 10.1016/j.ebiom.2021.103506. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper adapts a high-throughput phage-display library to identify autoantibodies targeting citrullinated antigens.
- [41].Karthikeyan K, Barker K, Tang Y, Kahn P, Wiktor P, Brunner A, et al. A Contra Capture Protein Array Platform for Studying Post-translationally Modified (PTM) Auto-antigenomes. Mol Cell Proteomics 2016;15:2324–37. 10.1074/mcp.M115.057661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Massa O, Alessio M, Russo L, Nardo G, Bonetto V, Bertuzzi F, et al. Serological Proteome Analysis (SERPA) as a tool for the identification of new candidate autoantigens in type 1 diabetes. Journal of Proteomics 2013;82:263–73. 10.1016/j.jprot.2013.02.030. [DOI] [PubMed] [Google Scholar]
- [43].Rho J, Lampe PD. High-throughput screening for native autoantigen-autoantibody complexes using antibody microarrays. J Proteome Res 2013;12:2311–20. 10.1021/pr4001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lastwika KJ, Kargl J, Zhang Y, Zhu X, Lo E, Shelley D, et al. Tumor-derived Autoantibodies Identify Malignant Pulmonary Nodules. Am J Respir Crit Care Med 2019;199:1257–66. 10.1164/rccm.201804-06280C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Danielczyk A, Stahn R, Faulstich D, Löffler A, Märten A, Karsten U, et al. PankoMab: a potent new generation anti-tumour MUC1 antibody. Cancer Immunol Immunother 2006;55:1337–47. 10.1007/s00262-006-0135-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fiedler W, DeDosso S, Cresta S, Weidmann J, Tessari A, Salzberg M, et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur J Cancer 2016;63:55–63. 10.1016/j.ejca.2016.05.003. [DOI] [PubMed] [Google Scholar]
- [47].Raffin C, Muller Y, Barragan J, Zhou Y, Piccoli L, Lanzavecchia A, et al. Development of citrullinated-vimentin-specific CAR for targeting Tregs to treat autoimmune rheumatoid arthritis. J Immunol 2019;202:133.2–133.2. 10.4049/jimmunol.202.Supp.133.2. [DOI] [Google Scholar]
- [48].Ma H, Murphy C, Loscher CE, O’Kennedy R. Autoantibodies - enemies, and/or potential allies? Front Immunol 2022;13:953726. 10.3389/fimmu.2022.953726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kobayashi M, Katayama H, Fahrmann JF, Hanash SM. Development of autoantibody signatures for common cancers. Seminars in Immunology 2020;47:101388. 10.1016/jsmim.2020.101388. [DOI] [PubMed] [Google Scholar]
- [50].Mazor RD, Nathan N, Gilboa A, Stoler-Barak L, Moss L, Solomonov I, et al. Tumor-reactive antibodies evolve from non-binding and autoreactive precursors. Cell 2022;185:1208–1222.e21. 10.1016/j.cell.2022.02.012. [DOI] [PubMed] [Google Scholar]; This article describes the evolution of autoantibodies arising from both pre-existing autoantibodies and a tumor-reactive autoantibody response.
- [51].Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. Journal of Biomedical Science 2020;27:1. 10.1186/sl2929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]