

Abstract
KRAS mutations drive oncogenic alterations in numerous cancers, particularly in human pancreatic ductal adenocarcinoma (PDAC). About 93% of PDAC have KRAS mutations, with G12D (~42% of cases) and G12V (~32% of cases) being the most common. The recent approval of sotorasib (AMG510), a small-molecule, covalent, and selective KRASG12C inhibitor, for treating patients with non-small cell lung cancer represents a breakthrough in KRAS targeted therapy. However, there is a need to develop other much-needed KRAS mutant inhibitors for PDAC therapy. Notably, Mirati Therapeutics recently developed MRTX1133, a small-molecule, non-covalent, and selective KRASG12D inhibitor through extensive structure-based drug design. MRTX1133 has demonstrated potent in vitro and in vivo antitumor efficacy against KRASG12D-mutant cancer cells, especially in PDAC, leading to its recent initiation of a phase I/II clinical trial. Here, we provide a summary of the recent advancements related to the use of MRTX1133 for treating KRASG12D-mutant PDAC, focusing on its efficacy and underlying mechanistic actions. Additionally, we discuss potential challenges and future directions for MRTX1133 therapy for PDAC, including overcoming intrinsic and acquired drug resistance, developing effective combination therapies, and improving MRTX1133’s oral bioavailability and target spectrum. The promising results obtained from preclinical studies suggest that MRTX1133 could revolutionize the treatment of PDAC, bring about a paradigm shift in its management.
Keywords: Pancreatic Cancer, KRAS Targeted Therapy, MRTX1133
1. Introduction
Pancreatic cancer, mainly pancreatic ductal adenocarcinoma (PDAC), currently ranks as the third-leading cause of cancer-related death in the United States, and it is projected to be the second -leading cause of such death by 2030 (1). PDAC carries a bleak prognosis, with a median survival duration of 8 months following diagnosis and a 5-year survival rate of less than 12% across all stages (2). These discouraging statistics primarily stem from the late detection of the disease, its resistance to conventional chemoradiotherapy, and the lack of effective targeted therapy. Despite the success of immune therapy for numerous other malignancies, immune therapy for PDAC has yielded disappointing results. This outcome can be largely attributed to PDAC’s desmoplastic and immunosuppressive tumor microenvironment (TME), which consists of a dense extracellular matrix, cancer-associated fibroblasts (CAFs), and various immune cells, particularly cancer-associated macrophages (CAMs) and myeloid-derived suppressor cells (MDSCs). These components hinder the infiltration, activation, and elimination of tumor cells by effector T cells (3). Hence, there is an urgent need for novel therapeutic strategies that can significantly improve outcomes for patients with PDAC.
2. Oncogenic KRAS mutations drive PDAC development and progression
PDAC harbors a multitude of genetic alterations, including oncogenic KRAS mutations and inactivation of the CDKN2A, TP53, and SMAD4 tumor suppressors. These alterations pose significant challenges for molecular comprehension of PDAC and the development of targeted therapy for PDAC(4), as there are currently no effective inhibitors available for direct targeting of these driver alterations in clinical applications. Alternatively, researchers have been identifying and targeting downstream effectors of these driving oncogenic signaling pathways. For example, they have explored the use of MEK, EGFR, and PI3K inhibitors, but these approaches have shown limited clinical benefit in patients with PDAC (4–6). One promising PDAC targeted therapy utilizing small molecular inhibitors has been exemplified by olaparib. Olaparib, a small molecular inhibitor of poly (ADP-ribose) polymerase (PARP), has been approved as the first non-chemotherapy, targeted maintenance treatment for adult patients with BRCA-mutated metastatic pancreatic cancer based on the favorable outcomes in a phase III clinical trial (NCT02184195) (7).
KRAS mutations are considered a hallmark genetic alteration and represent one of the earliest events in pancreatic tumorigenesis (Fig. 1A). KRAS mutations are detected in 74% of human pancreatic intraepithelial neoplasia (PanIN) lesions and in 63% foci of acinar-ductal metaplasia associated with these PanINs (8), and approximately 93% of cases of human PDAC exhibit KRAS mutations (9). The mutations that promote cancer often occur at specific hotspots within the KRAS protein, such as codons 12, 13, and 61. The most common mutations are G12D (41.8%),G12V (31.6%), and G12R (16.1%), and G12C mutation is less prevalent, accounting for less than 2% of the mutations in PDAC (10,11) (Fig. 1B), which is in contrast with the much higher frequency of G12C observed in non-small cell lung cancer (NSCLC) (~13%) (12,13). Studies using genetically engineered mouse models revealed that activation of KrasG12D in pancreatic epithelial lineage of cells leads to the development of a spectrum of PanIN lesions, which progress spontaneously, albeit at a low frequency, to invasive and metastatic PDAC (14); and that KrasG12D is required for PDAC progression and maintenance (15–17). In comparison to G12V, G12C, and other KRAS mutations, the G12D mutation displays the highest oncogenic potential in in vitro and in vivo models of pancreatic, lung, and colorectal cancers (18,19). This heightened potential likely stems from its favorable structure conformation that impacts KRAS downstream signaling (19,20). Patients with PDAC carrying the KRASG12D mutation exhibit a shorter survival duration compared to those with KRAS wild-type, G12V, or G12R mutation (21).
Figure 1. Genetic mutations in PDAC initiation and development.
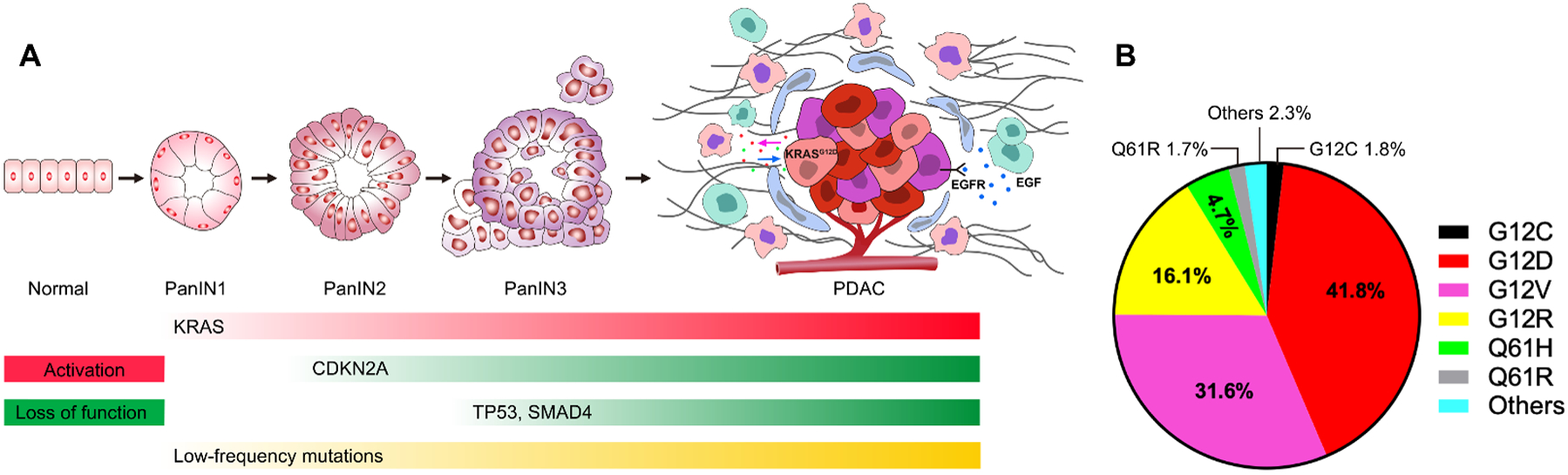
(A) Multi-stage initiation and progression model of PDAC. As cells acquire mutations in genes like KRAS, CDKN2A, TP53 and SMAD4, along with other less commonly mutated genes, the lesion progresses from low grade pancreatic intraepithelial neoplasia (PanIN1) through PanIN2 and high grade PanIN3, and ultimately to become invasive adenocarcinoma. In PDAC, intrinsic signals such as the KRASG12D mutation and extrinsic signals like EGFR activation contribute to an immune-suppressive and desmoplastic microenvironment through reciprocal interactions between tumor cells and diverse stroma cells. Examples of these stroma cells include cancer-associated fibroblasts (blue), cancer-associated macrophages (pink), myeloid-derived suppressor cells (green), among others. (B) Prevalence of KRAS mutant variants in human pancreatic cancer.
Normally, the KRAS protein functions as a molecular switch, transitioning between binding with guanosine-5′-triphosphate (GTP) in an active state and with guanosine-5′-diphosphate (GDP) in an inactive state. Mutations in KRAS disrupt the intrinsic GTPase activity of RAS, resulting in elevated levels of GTP-bound KRAS. This sustained RAS signaling activates pro-tumorigenic signaling through downstream effector pathways, including the Ras/Raf/MAPK (MEK)/ERK pathway and the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway (22) (Fig. 2). These pathways play critical roles in cellular processes, including promoting cell proliferation and survival, enhancing angiogenesis, facilitating metastasis, reprograming cellular metabolism to support the energy demands of rapidly dividing cancer cells (17,23–25), and inducing resistance to therapy (26). Furthermore, the activation of KRAS signaling in cancer cells can extend to and reprogram the surrounding microenvironment, thereby influencing cancer initiation, progression, and therapeutic response (27–30). These findings strongly indicate that directly targeting mutant KRAS could serve as an effective therapeutic strategy.
Figure 2. KRAS protein functioning switch and key downstream signaling pathways.
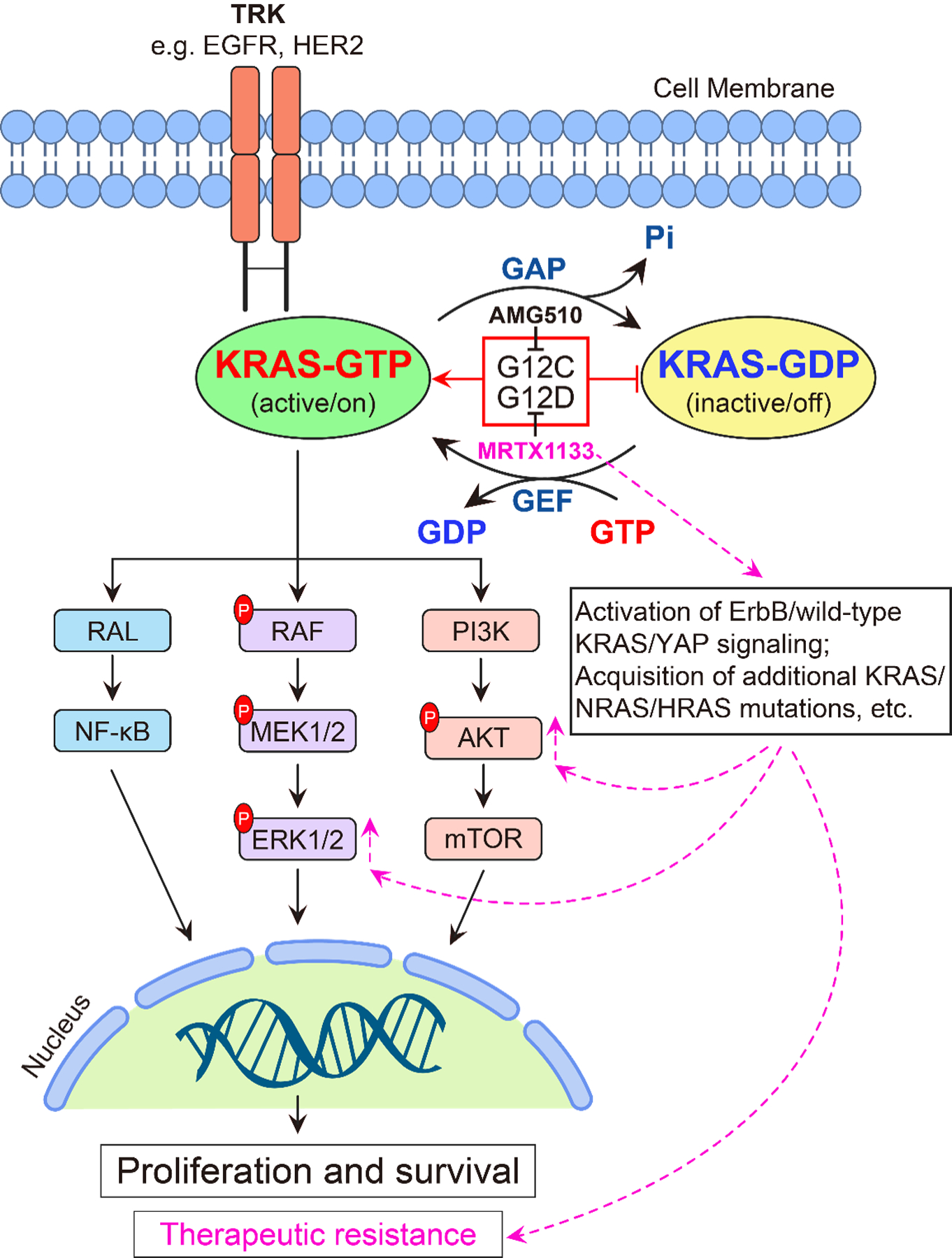
In normal conditions, when cells receive extracellular signals through transmembrane receptors, RAS-selective guanine nucleotide exchange factors (GEFs) and GTP-activating proteins (GAPs) regulate GDP- and GTP-bound KRAS cycling. In cancer cells, KRAS mutations disrupt its intrinsic GTPase activity, causing it to be locked in the GTP-bound and active form. This leads to the constitutive activation of downstream signaling pathways, such as the RAF/MEK/ERK pathway and the PI3K/AKT/mTOR pathway, promoting cancer cell proliferation and survival. The small molecular inhibitor AMG 510 specifically targets the KRASG12C mutation, while MRTX1133 specifically targets the KRASG12D mutation. However, cancer cells can potentially develop resistance to KRAS inhibitors through multiple mechanisms that may include compensation or feedback activation of the ErbB/wild-type KRAS and YAP signaling, acquisition of additional KRAS/NRAS/HRAS mutations, and etc.
3. Identification of MRTX1133, a first-in-class small-molecule, non-covalent, and selective KRASG12D inhibitor.
Given the high prevalence and functional significance of mutant KRAS in cancer, there has been considerable interest in developing therapeutic strategies to directly target mutant KRAS. However, three decades of effort have failed to identify clinically useful inhibitors of the KRAS proteins (31), because initial analyses of the KRAS proteins indicated that they lack a clear pocket for high-affinity binding of small molecules (32).
In 2013, Ostrem et al. reported a novel strategy to develop covalent inhibitors to target the reactive cysteins-12 (Cys-12) in the common KRASG12C mutant (33). Through structure-based design, they identified ARS-1620, a highly potent small molecular inhibitor that covalently binds to Cys-12 in the switch-II pocket of KRASG12C. This binding does not affect wild-type KRAS but subverts the native nucleotide preference to favor GDP over GTP and impairs binding to RAF, thus disrupting KRAS signaling (Fig. 2) (33). ARS-1620 demonstrated significant in vitro and in vivo activity against KRASG12C tumors (34). This discovery marked a milestone in targeting mutant KRAS, which was previously considered undruggable. Furthermore, using a structure-based design, Amgen developed a highly potent and selective KRASG12C inhibitor called AMG510, also known as sotorasib (Lumakras™), for a phase I/II clinical trial (NCT03600883) initiated in August 2018 (35). Based on the significant anti-tumor activity and manageable side effects in heavily pretreated patients with non-small cell lung cancer (NSCLC) with KRASG12C mutations in the clinical trial, the U.S. Food and Drug Administration (FDA) granted accelerated approval to sotorasib in May 2021 for the treatment of adult patients with locally advanced or metastatic NSCLC harboring the KRASG12C mutations, who have received at least one prior systemic therapy. This approval represents a breakthrough in KRAS targeted therapy, which is also demonstrated by recently updated clinical report showing that sotorasib achieved an overall response rate of 41%, a progression-free survival of 6.3 months, an overall survival (OS) of 12.5 months, and a 2-year OS rate of 33% (36). In parallel, Mirati developed Adagrasib (MRTX849) (37), an oral, small-molecule, covalent, and GDP-bound KRASG12C potent inhibitor with favorable pharmacological properties (38), for a phase I/II clinical trial in patients with advanced solid tumors with a KRASG12C mutation (NCT03785249). The trial was initiated in January 2019, and a recently updated report showed that among 57 patients with measurable solid tumors (excluding NSCLC and CRC patients), the objective response rate was 35.1% (20/57). This includes 7 out of 21 (33.3%) responses in pancreatic cancer and 5 out of 12 (41.7%) responses in biliary tract cancers (39). Notably, these developments have shown that the switch-II binding pocket, present in all KRAS proteins, is a feasible binding surface for the development of such selective KRASG12D inhibitors. The successful development of a KRASG12C-specific inhibitors provides valuable insights into and inspiration for the development of small molecular inhibitors targeting KRASG12D, which are desperately needed to treat PDAC.
Using a structure-based design and optimization, Mirati successfully developed a potent and selective non-covalent KRASG12D inhibitor called MRTX1133 (Fig. 3) in 2021 (40,41). Recent exciting results from preclinical studies indicate that the application of MRTX1133 has the potential to bring about a paradigm shift in the treatment of PDAC.
Figure 3. Chemical structure of MRTX1133 and the orally effective prodrug 9.
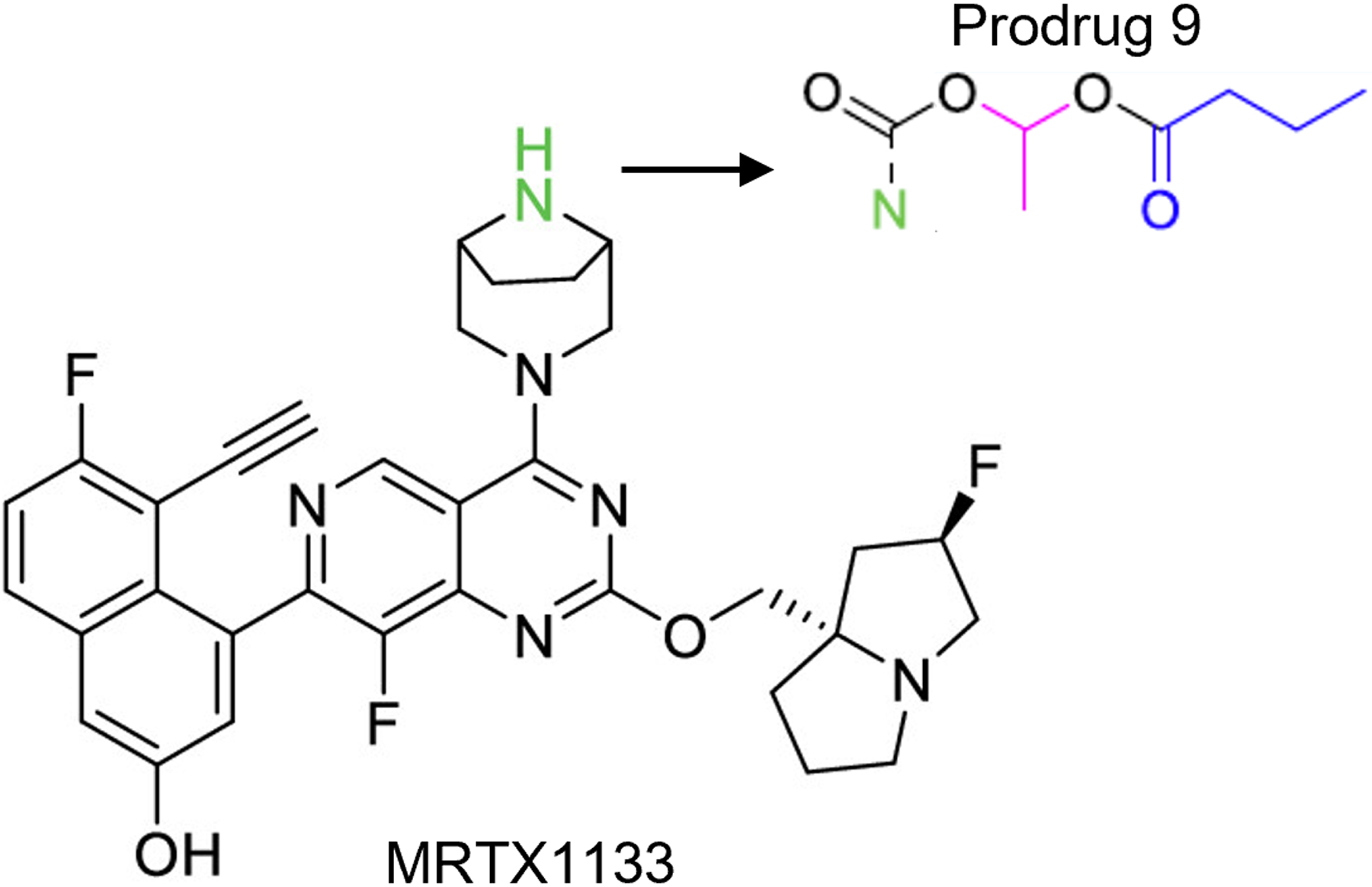
The amine moiety highlighted in green is believed to be a major contributor to its poor absorption in the gastrointestinal tract. Prodrug 9, which incorporates specific modifications to the amino group, demonstrated enhanced oral bioavailability compared to MRTX1133. Refer to ref. 62 for details.
4. Recent advances using MRTX1133 to target mutant KRASG12D in pancreatic cancer
MRTX1133 is a highly potent and selective non-covalent inhibitor of KRASG12D in vitro.
In a biochemical homogeneous time-resolved fluorescence assay, MRTX1133 exhibited binding to the GDP-bound, inactive form of KRASG12D with a half maximal inhibitory concentration (IC50) below 2nM, which was the lower limit of the detection of the assay (40). The selectivity of MRTX1133 for binding to the GDP-bound inactive KRASG12D was approximately 700-fold higher compared to that of MRTX1133 to the GDP-bound wild type KRAS (KRASWT). Additionally, MRTX1133 also inhibited the binding of a RAF-RAS binding domain peptide to the active form of KRASG12D.
Functionally, MRTX1133 effectively reduced cell viability and inhibited key signaling molecules in a dose-dependent manner in the KRAS pathway, such as phosphorylated extracellular signal-regulated kinase 1 and 2 (pERK1/2) and phosphorylated S6 (pS6), in KRASG12D-mutant pancreatic and other cancer cell lines, with a median IC50 value of approximately 5 nM (bioRxiv 2023.03.18.533261). But MRTX1133 treatment was found to upregulate the expression and phosphorylation of EGFR and HER2 in KRASG12D-mutant mouse and human pancreatic cancer cell lines (42). MRTX1133 exhibited selectivity over 1,000-fold larger for KRASG12D compared to that for KRASWT cell lines. However, certain established or patient-derived PDAC cell lines show intrinsic or acquired resistance to MRTX1133 treatment (42) (bioRxiv 2023.03.18.533261).
Notably, MRTX1133 demonstrated significantly higher efficacy in inhibiting cell proliferation in the three-dimensional (3D) organoid cell cultures compared to such two-dimensional (2D) cultures, even at concentrations that had no effect in the 2D setting (40) (bioRxiv 2023.03.18.533261). This finding suggests that anchorage-dependent cell growth may overcome KRAS dependency, which is consistent with the observed potent anti-tumor efficacy of MRTX1133 shown in in vivo studies (40)(bioRxiv 2023.03.18.533261).
MRTX1133 demonstrates KRASG12D-mutant tumor regression in xenograft mouse models of pancreatic cancer.
To evaluate the efficacy of MRTX1133 treatment on tumorigenesis, various preclinical models of pancreatic cancer have been used. In a xenograft model derived from the KRASG12D-mutant human HPAC cell line, MRTX1133 treatment via intraperitoneal injection demonstrated dose-dependently anti-tumor efficacy. The higher dose (30 mg/kg administered twice daily) resulted in a near-complete response, with an 85% regression rate without weight loss or overt signs of toxicity observed for up to 28 days. The antitumor effect was consistent with a dose-dependent reduction in a fraction of pERK- and pS6-positive cells, as well as a decrease in KRAS activity and increase in apoptosis in the tumor tissues (40).
In a panel of xenograft models derived from human KRASG12D-mutant cell-lines and patient-derived cells, MRTX1133 had excellent anti-tumor activity, especially in models of pancreatic cancer. Eight out of 11 (73%) models tested had at least showed 30% tumor regression, whereas only 2 out of 8 (25%) colorectal cancer models exhibited a similar response (40).
Interestingly, MRTX1133 displayed potent antitumor efficacy in xenograft tumors derived from patient-derived MRTX1133-resistant cells in 2D culture (EC50>1 μM) (bioRxiv 2023.03.18.533261). This finding highlights the sustained effectiveness of MRTX1133 in a 3D context and suggests that anchorage-dependent cell growth may contribute to the resistance observed in 2D cultures. Furthermore, MRTX1133 showed no significant anti-tumor efficacy in all four non-KRASG12D-mutant tumor models, further confirming the high-targeting specificity of MRTX1133 (40).
MRTX1133 is effective in immunocompetent models of pancreatic cancer.
Immune surveillance plays a crucial role in the development and progression of pancreatic cancer, and the presence of an immunosuppressive TME contributes to the limited efficacy of immune therapies in treating PDAC. To assess the efficacy of MRTX1133 in immunocompetent models of KRASG12D -mutated pancreatic cancer, both implantable and autochthonous PDAC models with an intact immune system were used. MRTX1133 demonstrated significant tumor regression in all tested models, leading to complete or near-complete remissions within 14 days. Evidence of tumor regression was observed as early as 2 days after MRTX1133 treatment initiation (43).
In addition to inducing tumor cell apoptosis and halting proliferation, MRTX1133 treatment induced notable changes in the TME. These changes included reprogramming CAFs and the extracellular matrix, an increase in CD8-positive effector T cells, and a decrease in neutrophils or myeloid cells (43) (44) (bioRxiv 2023.03.18.533261). The contribution of the immune-mediated antitumor effect elicited by MRTX1133 treatment was supported by the finding that MRTX1133 treatment resulted in potent tumor regression in syngeneic mouse models but not in immune-deficient mouse models (bioRxiv 2023.03.18.533261). The presence of T cells was essential for MRTX1133’s full antitumor effect, as T-cell depletion expedited tumor regrowth after therapy. Consistent with previous report (40), MRTX1133 treatment had no effect on tumor growth in immunocompetent mice carrying KRASG12C mutant pancreatic or lung tumors, or KRASWT colon tumors (43).
Furthermore, in autochthonous genetic mouse models with a KRASG12D mutation, MRTX1133 treatment resulted in the regression of both established pancreatic intraepithelial neoplasia (PanIN) and advanced PDAC. The regression of advanced PDAC relied on the presence of CD8-positive T cells and combining MRTX1133 with immune checkpoint blockade therapy (iCBT) targeting CTLA-4 and PD-1 (i.e., anti-CTLA-4 and anti-PD-1 antibodies) synergistically eradicated PDAC and extended overall survival compared to treatment with MRTX1133 alone (44). The ability of MRTX1133 to regress PanIN suggests that such inhibitors might be considered for use in a high-risk interception setting.
Mechanistically, the inhibition of KRASG12D by MRTX1133 in PDAC cell lines or tumor models resulted in the suppression of KRAS signaling and reduction of MAPK-dependent target gene expression, cell cycle regulator gene expression, and pro-survival gene expression, while pro-apoptotic and Fas genes were significantly upregulated in cancer cells (40) (44). MRTX1133 treatment also activated interferon-γ signaling and enhanced major histocompatibility complex (MHC) antigen presentation in the tumors, thus facilitating CD8-positive T cell-mediated cell death (44)(bioRxiv 2023.03.18.533261).
The promising results of MRTX1133 in preclinical studies have prompted exploration of its translation to human disease. MRTX1133 obtained clearance as an investigational new drug by the FDA in October 2021 for a phase I/II clinical trial (NCT05737706) that was initiated on March 20, 2023, targeting patients with advanced solid tumors (PDAC, NSCLC, colorectal cancer, and others) with KRASG12D mutations. This ongoing trial is expected to be completed on August 30, 2026.
5. Challenges and future directions
Despite the potent and selective antitumor effects observed in vitro and in vivo against KRASG12D-mutated pancreatic cancer cells, targeted therapy using MRTX1133 poses several challenges and necessitates further studies.
First, one of the major challenges in MRTX1133 treatment lies in overcoming intrinsic and acquired resistance to MRTX1133. Studies have clearly shown that not all PDAC cells with KRASG12D mutations are sensitive to MRTX1133 treatment (40) (bioRxiv 2023.03.18.533261), and cancer cells often develop resistance to the therapy over time (42,45–47). Understanding the underlying mechanisms of resistance and identifying strategies to overcome it will be crucial to achieving long-term success in MRTX1133 therapy.
In addition to a high frequency of oncogenic KRAS mutations, other alterations including inactivation of the CDKN2A, TP53, SMAD4, and PTEN tumor suppressors (4,48), activation of PIK3CA mutation (present in 3%−5% of PDAC) (49–52), contribute to the aggressiveness and therapeutic resistance of PDAC. In KRASG12C-targeted therapy for cancer patients, several mechanisms of primary and acquired resistance have been identified, including co-mutations in KEAP1, SMARCA4, CDKN2A, and STK11 (36,53,54), activation of YAP1 signaling (39,41,47,48), reactivation or feedback activation of the RAS/MAPK/PI3K pathway, acquired additional genetic alterations, such as in KRAS G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, and Y96C, a high level of amplification of the KRASG12C allele; activation mutations in NRAS, BRAF, MAP2K1, EGFR, and RET, and loss-of-function mutations in NF1 and PTEN (45). These findings may shed new light on our understanding of the resistance to MRTX1133 targeted therapy in PDAC and designing novel strategies to overcome such resistance.
Bioinformatics analyses have shown that cancer cells with lower PTEN and CDKN2A RNA expression are relatively insensitive to MRTX1133 treatment (40). SgRNAs-CRISPR/Cas9 mediated screening indicated that loss of tumor suppressors, including PTEN, KEAP1, NF1, and RB1, may confer partial resistance to MRTX1133 treatment in PDAC cells (40). In preclinical models, inhibition of mutant KRAS leads to feedback compensation to promote the expression of ERBB receptors or activation of EGFR/RASWT signaling was identified as a potential mechanism for acquired resistance of cancer cells to MRTX1133 treatment (46) (42). Therefore, elucidating the mechanisms for therapeutic resistance in MRTX1133 therapy would be facilitated by establishing MRTX1133-resistant cancer cell lines/clones or model systems, particularly the use of patient-derived samples, and exploring CRISPR/Cas9-based screening, single-cell RNA sequencing, and functional reverse-phase protein array (RPPA) approaches for comprehensive analysis, (40) (42).
Second, developing effective and mechanism-based combination therapy regimens, especially with immunotherapy, is essential to maximizing the antitumor efficacy of MRTX1133 and holds great promise for tumor elimination. For instance, MRTX1133 induces long-term regression of established tumors in immune-competent mice, which is not observed in immune-deficient mice. This indicates that, apart from tumor cell death induced by KRASG12D inhibition, an adaptive immune response is involved in the potent antitumor effect induced by MRTX1133 in immune-competent preclinical mouse model (43). These findings are supported by the observations that T-cell depletion accelerated tumor regrowth after monotherapy with MRTX1133 (43) and combination therapy with iCBT synergized with MRTX1133 to eradicate PDAC and prolong overall survival in a preclinical mouse model, whereas iCBT alone has been proven ineffective in treating PDAC (44). These results provide the rationale for future development of combination therapy of MRTX1133 with iCBT and other immunotherapies in clinical trials.
Since MRTX1133 treatment has been found to induce KRAS-dependent signaling pathway reactivation and/or feedback compensation (40,46) (42), combining MRTX1133 with inhibitors that target key KRAS-signaling components, both upstream and downstream effectors, such as the pan-ERBB inhibitor afatinib or EGFR inhibitor cetuximab, the PI3Kα inhibitor BYL719, CDK4/6 inhibitors, or YAP1 inhibitors, could be explored in future clinical trials (40). These combination therapies have the potential to enhance the efficacy of MRTX1133 treatment or overcome resistance based on the results of preclinical studies and the findings from KRASG12C targeted therapy (40,46,55–57) (42). Given that KRASG12D mutant PDAC cells can potentially develop the ability to bypass oncogenic KRAS dependency, utilizing mechanisms such as YAP activation, USP21 amplification, and HDAC5-mediated remodeling of TME (58–60), it is plausible to consider that a combination targeting of these altered KRAS bypass signaling pathways may enhance or sensitize the efficacy of MRTX1133 treatment in PDAC. Additionally, combining MRTX1133 with conventional chemotherapy and radiotherapy is also expected to enhance its therapeutic efficacy in treating patients with PDAC (61).
Third, there is additional need to develop novel, more potent, and broader KRAS mutant inhibitors for improved KRAS-targeted therapy. MRTX1133 has its limitations, including low oral bioavailability and high selectivity for G12D, as reported previously(40). To improve the low oral bioavailability of MRTX1133, researchers have designed and synthesized a series of prodrug compounds. These prodrugs aim to mask the amino group (refer to Fig. 3, highlighted in green) that is believed to be responsible for the molecule’s poor absorption in the gastrointestinal tract. Among these prodrugs, prodrug 9 has been discovered to have significantly better pharmacokinetic properties, oral bioavailability, and effective antitumor activity in a xenograft mouse model of human pancreatic cancer with KRASG12D mutation after oral administration(62). The high selectivity of MRTX1133 may restrict its broad use in treating patients with PDAC, such as for such patients whose tumors carry different variants of KRAS mutation due to heterogeneity or mutations acquired during treatment.
A recent study using isogenic “Ras-less” cell lines that express a single Ras allele demonstrated that, in addition to being highly sensitive to KRASG12D, MRTX1133 also exhibited significant activity against several other KRAS mutations (G12C, G12V, G13D) and wild-type KRAS but had no effect on HRAS (WT and G12D) and NRAS (WT and G12D) in the cells. Further studies indicated that while the 12D mutant residue is important, it is not essential to MRTX1133 activity. It is also important to note that although MRTX1133 exhibited activity against other KRAS mutations and wild-type KRAS, the concentrations of MRTX1133 needed to inhibit non-G12D variants were significantly higher and may not be pharmacologically achievable/relevant. The selectivity of MRTX1133 is associated with its binding to H95 on KRAS, a residue that is not conserved in HRAS and NRAS (63). This finding could be exploited for the development of the next generation of MRTX1133-like pan-KRAS inhibitors.
Moreover, the discovery of MRTX1133 has sparked a new wave of development of various KRASG12D inhibitors with alternative binding models, different allele specificities, selectivity for KRAS mutant variants, and distinct mechanistic actions (Table 1). Continuously characterizing, optimizing, and examining these inhibitors in cellular and animal models and translating them into clinical applications may usher in a new era of improved KRAS-targeted therapy for PDAC. It is reasonable to speculate, for instance, that combining MRTX1133 with ASP3080, a degrader of the KRASG12D protein, could lead to a synergistic or additive antitumor effect, helping overcome the therapeutic resistance caused by overactivation or feedback compensation of KRAS-dependent signaling.
Table 1.
New KRASG12D inhibitors under development and investigation
| Drug | Sponsor | Properties | Status | Reference |
|---|---|---|---|---|
| ASP3082 | Astellas | Binds to and targets KRASG12D for proteasome-mediated degradation | Phase I (NCT05382559) | Nagashima et al. (67) |
| HRS-4642 | Hengrui Medicine | KRASG12D inhibitor | Phase I (NCT05533463) | |
| RMC-9805 (RM-036) | Revolution Medicines | Covalent, GTP-bound and orally bioavailable KRASG12D(ON) inhibitor | IND-enabling | Jang et al. (68) |
| BI-2582 | Boehringer Ingelheim | Non-covalent KRASG12D inhibitor, with nanomolar binding affinity to GTP-bound KRASG12D. | Preclinical | Tran et al. (69) |
| ERAS-4 | Erasca | KRASG12D inhibitor | Preclinical | GlobeNewswire - 23 Aug 2022 |
| THZ835 | Tsinghua University | Inhibitor forms a salt bridge with Asp 12 of KRASG12D, induces switch-II fit pocket in KRASG12D and has off-target effect because inhibition is not fully dependent on KRAS mutation status. | Preclinical | Mao et al. (70) |
| QTX3046 | Quanta Therapeutics | Non-covalent and orally bioavailable KRASG12D inhibitor, picomolar-level binding affinity (0.01nM) to inactive KRASG12D | Preclinical | Vo et al. (71) |
| QTX3034 | Quanta Therapeutics | Selective, orally bioavailable, and picomolar-level binding affinity (0.6 nM) to GDP-bound KRASG12D | Preclinical | Zhang et al. (72) |
| VRTX153 | Selective KRASG12D inhibitor that binds to GDP-bound state, with a nanomolar-level IC50. | Preclinical | Bhavar et al. (73) | |
| Compound 3 | KRASG12D inhibitor with IC50 at nanomolar-level. | preclinical | Wang et al. (74) | |
| MRC-6236 | Revolution Medicines | Orally bioavailable, selective inhibitor for active RAS (ON) form of both wild and mutant variants of the canonical RAS isoforms (HRAS, NRAS, and KRAS) (pan-RAS/ RASMULTI inhibitor). | Phase I (NCT05379985) | Gustafson et al. (65) |
| JAB-23425 | Jacobio | Orally bioavailable, selective multiple KRAS mutant (G12A/C/D/R/V and Q61H) inhibitor, with sub-nanomolar-level binding affinity to GDP- and GTP-bound KRAS states. No inhibition of HRAS and NRAS (pan-KRAS/ RASMULTI inhibitor). | Preclinical | Wang et al. (75) |
| BI-2865 | Boehringer Ingelheim | Non-covalent, selective inactive form of KRAS (both wild and mutant) inhibitor, no effect on HRAS and NRAS (pan-KRAS or RASMULTI inhibitor) | Preclinical | Kim et al. (66) |
GTP, guanosine-5′-triphosphate; GDP, guanosine-5′-diphosphate; IND, investigative new drug.
RMC-6236 was identified as a first-in-class, orally bioavailable, selective KRAS (ON) inhibitor, exhibited high-antitumor efficacy in RAS-addictive cancer cells and RAS-driven cancer models, particularly KRASG12X (G12X includes G12A/C/D/R/S/V)(64). In pancreatic cancer, RMC-6236 treatment suppressed the phosphorylation of ERK kinases, leading to growth inhibition and apoptosis in several human cancer cell lines in vitro, and oral administration of RMC-6236 in cell line-derived and patient-derived xenograft models of KRAS-mutant PDAC resulted in profound and durable tumor regressions at doses that were well-tolerated (65). The promising preclinical data have led to a phase I clinical trial of RMC-6236, initiated on May 31, 2022, in adults with KRASG12X (exclude G12C) mutant advanced solid tumors (NCT05379985). BI-2865 is another example of non-covalent, selective pan-KRAS inhibitor, which blocks nucleotide exchange to impair the activation of KRASWT and a broad range of KRAS mutants, including G12A/C/D/F/V/S, G13C/D, V14I, L19F, Q22K, D33E, Q61H, K117N and A146V/T, while sparing NRAS and HRAS. BI-2865 treatment exhibited significant inhibition of KRAS signaling and cell proliferation that was restricted to KRAS-mutant cancer cells and led to KRAS-mutant tumor growth suppression in xenograft mouse models (66). If successfully translated into clinical applications, RMC-6236 and BI-2865 may be used to treat patients with PDAC carrying a variety of KRAS mutations with a lower frequency or delayed development of therapeutic resistance.
In summary, significant progress is being made rapidly in both preclinical and clinical studies regarding the use of MRTX1133, a non-covalent, selective, and potent KRASG12D inhibitor, for treating PDAC. However, it is crucial to elucidate and overcome treatment resistance, develop novel and more potent KRAS inhibitors, and explore combination approaches to optimize the efficacy of KRAS-targeted therapy and improve patient outcomes in PDAC. Furthermore, ongoing research and clinical trials are likely to reveal additional challenges, necessitating tremendous effort in the future.
Acknowledgements
The work was supported in part by the NIH-funded R01 grants (CA198090 to R Bresalier, CA220236 to A Maitra, and CA236905 to X Zuo) and Texas Medical Center Digestive Disease Center Research Core Center Program P30DK056338 (to R Bresalier and D Wei), and the grants from The University of Texas MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment, the MD Anderson Cancer Center Institutional Research Program (to D Wei). This work was also supported by the Sheikh Khalifa bin Zayed Foundation. We thank Ashli Nguyen-Villarreal, Associate Scientific Editor, and Bryan Tutt, Scientific Editor, in the Research Medical Library at MD Anderson Cancer Center, for editing this manuscript. The authors thank Wei Li for the graphic art used in the manuscript.
Footnotes
Potential Conflict of interest
AM is listed as an inventor on a patent that has been licensed by Johns Hopkins University to Thrive Earlier Detection and serves as a consultant for Freenome and Tezcat Biosciences. The other authors declare no conflicts of interest.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer research 2014;74(11):2913–21 doi 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17–48 doi 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 3.Schizas D, Charalampakis N, Kole C, Economopoulou P, Koustas E, Gkotsis E, et al. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat Rev 2020;86:102016 doi 10.1016/j.ctrv.2020.102016. [DOI] [PubMed] [Google Scholar]
- 4.Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: Advances and challenges. Cell 2023;186(8):1729–54 doi 10.1016/j.cell.2023.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aung KL, McWhirter E, Welch S, Wang L, Lovell S, Stayner LA, et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma. J Gastrointest Oncol 2022;13(6):3216–26 doi 10.21037/jgo-22-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neil BH, Scott AJ, Ma WW, Cohen SJ, Aisner DL, Menter AR, et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann Oncol 2015;26(9):1923–9 doi 10.1093/annonc/mdv264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. The New England journal of medicine 2019;381(4):317–27 doi 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi C, Hong SM, Lim P, Kamiyama H, Khan M, Anders RA, et al. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Molecular cancer research : MCR 2009;7(2):230–6 doi 10.1158/1541-7786.mcr-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer 2018;18(12):767–77 doi 10.1038/s41568-018-0076-6. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 2017;32(2):185–203 e13 doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer research 2012;72(10):2457–67 doi 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Judd J, Abdel Karim N, Khan H, Naqash AR, Baca Y, Xiu J, et al. Characterization of KRAS Mutation Subtypes in Non-small Cell Lung Cancer. Mol Cancer Ther 2021;20(12):2577–84 doi 10.1158/1535-7163.MCT-21-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRAS (G12C) Somatic Mutations across Race, Sex, and Cancer Type. The New England journal of medicine 2021;384(2):185–7 doi 10.1056/NEJMc2030638. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer cell 2003;4(6):437–50 doi 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 15.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122(2):639–53 doi 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell 2005;7(5):469–83 doi 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 17.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149(3):656–70 doi 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricciuti B, Son J, Okoro JJ, Mira A, Patrucco E, Eum Y, et al. Comparative Analysis and Isoform-Specific Therapeutic Vulnerabilities of KRAS Mutations in Non-Small Cell Lung Cancer. Clinical cancer research 2022;28(8):1640–50 doi 10.1158/1078-0432.ccr-21-2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zafra MP, Parsons MJ, Kim J, Alonso-Curbelo D, Goswami S, Schatoff EM, et al. An In Vivo Kras Allelic Series Reveals Distinct Phenotypes of Common Oncogenic Variants. Cancer discovery 2020;10(11):1654–71 doi 10.1158/2159-8290.cd-20-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Molecular cancer research : MCR 2015;13(9):1325–35 doi 10.1158/1541-7786.mcr-15-0203. [DOI] [PubMed] [Google Scholar]
- 21.Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P, et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clinical and translational gastroenterology 2016;7(3):e157 doi 10.1038/ctg.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 2018;8(9) doi 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497(7451):633–7 doi 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer discovery 2020;10(4):608–25 doi 10.1158/2159-8290.CD-19-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao W, Rose JL, Wang W, Seth S, Jiang H, Taguchi A, et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 2019;568(7752):410–4 doi 10.1038/s41586-019-1062-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nussinov R, Jang H, Tsai CJ, Liao TJ, Li S, Fushman D, et al. Intrinsic protein disorder in oncogenic KRAS signaling. Cell Mol Life Sci 2017;74(17):3245–61 doi 10.1007/s00018-017-2564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dias Carvalho P, Guimaraes CF, Cardoso AP, Mendonca S, Costa AM, Oliveira MJ, et al. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer research 2018;78(1):7–14 doi 10.1158/0008-5472.CAN-17-2084. [DOI] [PubMed] [Google Scholar]
- 28.Hafezi S, Saber-Ayad M, Abdel-Rahman WM. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. Int J Mol Sci 2021;22(19) doi 10.3390/ijms221910219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011;11(11):761–74 doi 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Velez-Delgado A, Donahue KL, Brown KL, Du W, Irizarry-Negron V, Menjivar RE, et al. Extrinsic KRAS Signaling Shapes the Pancreatic Microenvironment Through Fibroblast Reprogramming. Cell Mol Gastroenterol Hepatol 2022;13(6):1673–99 doi 10.1016/j.jcmgh.2022.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holderfield M Efforts to Develop KRAS Inhibitors. Cold Spring Harb Perspect Med 2018;8(7) doi 10.1101/cshperspect.a031864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014;13(11):828–51 doi 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503(7477):548–51 doi 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172(3):578–89 e17 doi 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. Journal of medicinal chemistry 2020;63(1):52–65 doi 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 36.Dy GK, Govindan R, Velcheti V, Falchook GS, Italiano A, Wolf J, et al. Long-Term Outcomes and Molecular Correlates of Sotorasib Efficacy in Patients With Pretreated KRAS G12C-Mutated Non-Small-Cell Lung Cancer: 2-Year Analysis of CodeBreaK 100. Journal of clinical oncology 2023;41(18):3311–7 doi 10.1200/jco.22.02524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. Journal of medicinal chemistry 2020;63(13):6679–93 doi 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- 38.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer discovery 2020;10(1):54–71 doi 10.1158/2159-8290.cd-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bekaii-Saab TS, Yaeger R, Spira AI, Pelster MS, Sabari JK, Hafez N, et al. Adagrasib in Advanced Solid Tumors Harboring a KRAS(G12C) Mutation. Journal of clinical oncology 2023:Jco2300434 doi 10.1200/jco.23.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nature medicine 2022;28(10):2171–82 doi 10.1038/s41591-022-02007-7. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. Journal of medicinal chemistry 2022;65(4):3123–33 doi 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- 42.Gulay KCM, Zhang X, Pantazopoulou V, Patel J, Esparza E, Pran Babu DS, et al. Dual inhibition of KRASG12D and pan-ERBB is synergistic in pancreatic ductal adenocarcinoma. Cancer research 2023. doi 10.1158/0008-5472.can-23-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J, et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer discovery 2023;13(2):298–311 doi 10.1158/2159-8290.CD-22-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahadevan KK, McAndrews KM, LeBleu VS, Yang S, Lyu H, Li B, et al. KRASG12D inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8+ T cells. Cancer cell 2023;41(9):1606–1620 DOI: 10.1016/j.ccell.2023.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. The New England journal of medicine 2021;384(25):2382–93 doi 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng J, Hu Z, Xia X, Liu X, Lian Z, Wang H, et al. Feedback activation of EGFR/wild-type RAS signaling axis limits KRAS(G12D) inhibitor efficacy in KRAS(G12D)-mutated colorectal cancer. Oncogene 2023;42(20):1620–33 doi 10.1038/s41388-023-02676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021;599(7886):679–83 doi 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park W, Chawla A, O’Reilly EM. Pancreatic Cancer: A Review. JAMA 2021;326(9):851–62 doi 10.1001/jama.2021.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Payne SN, Maher ME, Tran NH, Van De Hey DR, Foley TM, Yueh AE, et al. PIK3CA mutations can initiate pancreatic tumorigenesis and are targetable with PI3K inhibitors. Oncogenesis 2015;4(10):e169 doi 10.1038/oncsis.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tignanelli CJ, Herrera Loeza SG, Yeh JJ. KRAS and PIK3CA mutation frequencies in patient-derived xenograft models of pancreatic and colorectal cancer are reflective of patient tumors and stable across passages. Am Surg 2014;80(9):873–7. [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss GA, Rossi MR, Khushalani NI, Lo K, Gibbs JF, Bharthuar A, et al. Evaluation of phosphatidylinositol-3-kinase catalytic subunit (PIK3CA) and epidermal growth factor receptor (EGFR) gene mutations in pancreaticobiliary adenocarcinoma. J Gastrointest Oncol 2013;4(1):20–9 doi 10.3978/j.issn.2078-6891.2012.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu CY, Carpenter ES, Takeuchi KK, Halbrook CJ, Peverley LV, Bien H, et al. PI3K regulation of RAC1 is required for KRAS-induced pancreatic tumorigenesis in mice. Gastroenterology 2014;147(6):1405–16 e7 doi 10.1053/j.gastro.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Negrao MV, Araujo HA, Lamberti G, Cooper AJ, Akhave NS, Zhou T, et al. Comutations and KRASG12C Inhibitor Efficacy in Advanced NSCLC. Cancer discovery 2023;13(7):1556–71 doi 10.1158/2159-8290.cd-22-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. The New England journal of medicine 2021;384(25):2371–81 doi 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adachi Y, Kimura R, Hirade K, Yanase S, Nishioka Y, Kasuga N, et al. Scribble mis-localization induces adaptive resistance to KRAS G12C inhibitors through feedback activation of MAPK signaling mediated by YAP-induced MRAS. Nat Cancer 2023;4(6):829–43 doi 10.1038/s43018-023-00575-2. [DOI] [PubMed] [Google Scholar]
- 56.Goodwin CM, Waters AM, Klomp JE, Javaid S, Bryant KL, Stalnecker CA, et al. Combination Therapies with CDK4/6 Inhibitors to Treat KRAS-Mutant Pancreatic Cancer. Cancer research 2023;83(1):141–57 doi 10.1158/0008-5472.CAN-22-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hagenbeek TJ, Zbieg JR, Hafner M, Mroue R, Lacap JA, Sodir NM, et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat Cancer 2023;4(6):812–28 doi 10.1038/s43018-023-00577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer discovery 2020;10(7):1058–77 doi 10.1158/2159-8290.cd-19-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou P, Ma X, Yang Z, Zhang Q, Wu CJ, Li J, et al. USP21 deubiquitinase elevates macropinocytosis to enable oncogenic KRAS bypass in pancreatic cancer. Genes & development 2021;35(19–20):1327–32 doi 10.1101/gad.348787.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158(1):185–97 doi 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tajiknia V E-DW, Schwermann M, Huntington K, Zhou L, Srinivasan P. Combination of 5-FU plus KRAS G12D inhibitor MRTX1133 against human colorectal and pancreatic cancer cells and the affects on inhibition of pERK and immune-stimulatory cytokine patterns in in KRAS G12D and KRAS G12V tumor cells. Journal of Clinical Oncology 2023;41(16_suppl):e16301 doi 10.1200/JCO.2023.41.16_suppl.e16301. [DOI] [Google Scholar]
- 62.Ji X, Li Y, Kong X, Chen D, Lu J. Discovery of Prodrug of MRTX1133 as an Oral Therapy for Cancers with KRAS(G12D) Mutation. ACS Omega 2023;8(7):7211–21 doi 10.1021/acsomega.3c00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keats M, Han JJW, Lee YH, Lee CS, Luo J. A non-conserved histidine residue on KRAS drives paralog selectivity of the KRASG12D inhibitor MRTX1133. Cancer research 2023. doi 10.1158/0008-5472.CAN-23-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koltun ES, Rice MA, Gustafson WC, Wilds D, Jiang J, Lee BJ, et al. Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer research 2022;82(12_Supplement):3597. [Google Scholar]
- 65.Gustafson WC, Wildes D, Rice MA, Lee BJ, Jiang J, Wang Z, et al. Direct targeting of RAS in pancreatic ductal adenocarcinoma with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Journal of Clinical Oncology 2022;42(4_suppl):591. [Google Scholar]
- 66.Kim D, Herdeis L, Rudolph D, Zhao Y, Böttcher J, Vides A, et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023;619(7968):160–6 doi 10.1038/s41586-023-06123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagashima T, Inamura K, Nishizono Y, Suzuki A, Tanaka H, Yoshinari T, et al. ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. Eur J Cancer 2022;174S1:S30. [Google Scholar]
- 68.Jiang LMM, Weller C, Wang Z, Burnett L, Aronchik I, Steele S, et al. RMC-9805, a first-in-class, mutant-selective, covalent and oral KRASG12D(ON) inhibitor that induces apoptosis and drives tumor regression in preclinical models of KRASG12D cancers. Cancer research 2023;83(7_Supplement):526. [Google Scholar]
- 69.Tran TH, Alexander P, Dharmaiah S, Agamasu C, Nissley DV, McCormick F, et al. The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proceedings of the National Academy of Sciences of the United States of America 2020;117(7):3363–4 doi 10.1073/pnas.1918164117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mao Z, Xiao H, Shen P, Yang Y, Xue J, Yang Y, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell discovery 2022;8(1):5 doi 10.1038/s41421-021-00368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vo ED, Yang W, Zhang YW, Rominger D, Silva JM, Zhang YJ, et al. Discovery and characterization of QTX3046, a potent, selective, and orally bioavailable non-covalent KRASG12D inhibitor. Cancer research 2023;83(8_Supplement):LB321. [Google Scholar]
- 72.Zhang YW, Rominger D, Vo ED, Silva JM, Zhang YJ, Lee G, et al. Discovery and characterization of QTX3034, a potent, selective, and orally bioavailable allosteric KRAS inhibitor. Cancer research 2023;83(8_Supplement):LB320. [Google Scholar]
- 73.Bhavar PK, Surampudi UK, Sarma PP, Kshirsagar AR. Vrtx153, novel small molecule inhibitor of krasg12d. Cancer research 2023;83(7_Supplement):4481. [Google Scholar]
- 74.Wang Y, Zhang H, Li J, Niu MM, Zhou Y, Qu Y. Discovery of potent and noncovalent KRAS(G12D) inhibitors: Structure-based virtual screening and biological evaluation. Frontiers in pharmacology 2022;13:1094887 doi 10.3389/fphar.2022.1094887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang P, Wang Y, Sun X, Liu D, Liu X, Zhang W, et al. Preclinical investigation of orally bioavailable, potent KRASMulti inhibitor. Cancer research 2023;83(7_Supplement):1660. [Google Scholar]