

Abstract
Initiating and regulating humoral immunity, Fc gamma receptors (FcγRs) have been identified both as therapeutics and as drug targets, and thus production of biologically active FcγRs is highly demanded for biopharmaceutical development. Focusing on low-affinity FcγRs IIA (131H/R allotypes), IIB, and IIIA (176F/V), this study used human 293-F cells to achieve correct post-translational modifications (PTMs) including biotinylation, N-glycosylation, and disulfides. Approaches involving co-expression of FcγR-AviTag and E. coli biotin ligase BirA, endoplasmic reticulum retention, stable and transient transfections, and optimization of transgene ratio were investigated. Protein electrophoresis under reducing and non-reducing conditions, enzymatic deglycosylation, streptavidin pulldown assays, and binding kinetic analysis collectively indicated that the produced FcγR ectodomains were fully biotinylated, N-glycosylated, disulfide bond formed, and exhibited expected binding affinities towards IgG1 trastuzumab and its Fc mutants. A clear trade-off between production yield and PTM quality was also observed. Achieving complete PTMs of multiple types by one-step cell culture should have applications on production of a variety of complex proteins of biomedical importance.
Keywords: FcγR, glycosylation, biotinylation, PTM
1. INTRODUCTION
By covalently incorporating chemical moieties, post-translational modifications (PTMs) influence the structure, stability, and function of proteins1. Accordingly, achieving correct and complete PTMs is a critical aspect to produce biologic substances2. As major forms of PTMs, N-glycosylation and disulfide bonds are prevalent especially in membrane and secreted proteins. In fact, it is estimated that more than 50% of human proteins are glycosylated3, and proteomic analysis suggested that the content of proteins with intra-chain disulfide(s) increases during evolution4—78% of secretory proteins in Eukarya contain disulfide bonds5. Though a rare PTM in nature, thanks to the strong interaction between streptavidin and biotin, biotinylation has been extensively used in biotechnology ranging from detection and purification to imaging and drug delivery6. In this study, we developed mammalian cell expression processes, aiming to synthesize recombinant proteins carrying multiple PTMs including N-glycosylation, disulfides, and biotinylation.
Presenting on surface of immune cells, Fc gamma receptors (FcγRs) recognize the Fc portion of IgG and thus bridge the humoral and the cellular branches of immunity7,8. The FcγR family can be divided into activating (such as Ia, IIa and IIIa) or inhibitory (IIb) receptors for positive or negative regulations of immune responses respectively. FcγR-triggered regulations / effector functions include B cell proliferation, mast cell degranulation, macrophage-mediated phagocytosis, and NK cell-mediated cytolysis. In addition, FcγRs are responsible for internalization of captured immune complexes (ICs), which leads to pathogen degradation and antigenic peptide presentation9. Besides these protective functions, FcγRs are also associated with initiation of inflammatory cascades, pathogenesis of infections, and autoimmune diseases. For example, FcγR polymorphism such as FcγRIIIa-176F phenotype has been suggested as a susceptibility factor for systemic lupus erythematosus10. Furthermore, evidence indicates that FcγR-mediated viral entry provokes antibody-dependent enhancement in dengue virus and SARS-CoV infections11. Due to these important and paradoxical roles, FcγRs have been proposed both as drug targets (e.g., engineering Fc for optimal interactions with specific FcγRs) and as therapeutics themselves (e.g., potential uses of FcγR ectodomains as decoy receptors in anti-inflammatory treatments). Therefore, production of biologically active FcγRs is highly demanded for biopharmaceutical development.
FcγRs are single-pass transmembrane glycoproteins carrying intra-domain disulfides and phosphorylation sites in their C-terminal cytoplasmic domains. Notably, proper glycosylation and domain folding of FcγRs are critical for their interactions with IgG12. This study focuses on the low affinity FcγRs IIa, IIb, and IIIa, which bear two, three, and five N-glycan sites respectively, and two disulfide bonds each13. As clinically important allotypes, FcγRIIa variants -131H/R and FcγRIIIa variants -176F/V are included in this work. Binding with ICs initiates the intracellular biochemical signals of FcγRs via tyrosine phosphorylation. However, this PTM is not subjected for investigation in this study because FcγR ectodomains are the targeted products. Traditionally, biotinylation has been done by chemical conjugations. However, this method yields inadequate control over labeling degree and location. To achieve site-specific modification, enzymatic biotinylation has been developed. Particularly, biotin-protein ligases (EC 6.3.4.15, E. coli BirA, for example) catalyze the formation of an amide bond between the biotin moiety and the target lysine residue14. Pioneering work has identified a 15-residue highly efficient substrate of BirA known as AviTag15,16, appliable for both in vivo and in vitro biotinylation17,18. In this study, we use BirA and AviTag mediated in vivo biotinylation to achieve one-step production of functional FcγR ectodomains with desired PTMs.
2. MATERIALS AND METHODS
2.1. Plasmid construction.
Gene fragments encoding extracellular domains (ECDs, or N-terminal topological domains) of human FcγRIIa-131H (34–217aa), FcγRIIb (43–217aa) and FcγRIIIa-176F (17–208aa) with an AZU1 signal peptide fused at their N-termini and a His6-tag fused at their C-termini were chemically synthesized (IDT). Corresponding fragments encoding allotypes FcγRIIa-131R and FcγRIIIa-176V were generated by overlapping PCR with the site-directed mutations introduced by primers. These fragments were digested with HindIII/EcoRI and cloned into the same sites on pcDNA-Intron-WPRE-SP-HC-IRES-Zeo19 to obtain expression plasmids pFcγR for non-biotinylated FcγRs. The DNA fragment encoding a GSGSS linker, an AviTag (GLNDIFEAQKIEWHE), and a His6-tag was assembled by synthesized oligonucleotides and cloned into NsiI/EcoRI sites to replace the His6-tag on FcγR expression plasmids, resulting in FcγR-AviTag expression plasmids pFcγR-Avi. The gene of E. coli biotin ligase (BirA) was PCR-amplified from pET21a-BirA (Addgene) and fused at its C-terminus by extension PCR with an endoplasmic reticulum (ER) retention signal KDEL. The obtained fragment was cloned via XhoI/HpaI into pcDNA-Intron-WPRE-SP-LH-IRES-Hygro19 to construct BirA expression plasmid pBirA. Restriction enzymes, polymerases and DNA ligase were purchased from NEB. All cloned plasmids were confirmed by sequencing.
2.2. Mammalian cell culture, transfection, and selection.
Expi293F (Gibco) was cultured in Expi293 medium supplemented with 40 U/ml penicillin and 40 μg/ml streptomycin at 37 °C, in 8% CO2 humidified atmosphere with a shaking speed of 130 rpm. Typically, Expi293F cells were transfected at a cell density of 2–2.5×106/ml with 1 μg/ml of each pFcγR-Avi, 1 μg/ml of pBirA and 7 μg/ml of PEI MAX 40K (Polysciences). After 1 day of incubation, cells were cultured in fresh media and selected with 700 μg/ml of zeocin and 250 μg/ml of hygromycin for 5 days, then maintained with 100 μg/ml of zeocin and 50 μg/ml of hygromycin to generate polyclonal stable cells expressing each biotinylated FcγRs. Similarly, cells were transfected with 2 μg of each pFcγR expression plasmid with 7 μg of PEI, selected with 700 μg/ml of zeocin and maintained with 100 μg/ml of zeocin to generate the polyclonal stable cells expressing each non-biotinylated FcγRs.
2.3. FcγR production and purification.
Obtained polyclonal stable cells were cultured in Expi293 medium without antibiotics supplements for 5 days for FcγR production. 100 μM D-biotin was supplemented to culture media for in vivo biotinylation. In transient expressions, cells were co-transfected with 30 μg pFcγR-AviTag and pBirA DNA at a mass ratio of 1:2 or 1:1 with 105 μg PEI, and cells were cultured in Expi293 media for 5 days. After expression, supernatants were collected and the produced FcγRs were purified by using HisPur Ni-NTA resin (Thermo Scientific) following manufacturer’s manual. Eluted proteins were dialyzed against PBS at 4°C overnight and concentrated using Amicon 3kDa MWCO filter units (Millipore) by centrifugation at 4000 rpm 4°C. Concentrations of purified pFcγRs were measured by spectrometry at 280 nm using an Epoch plate reader (BioTek), and the purity was verified by SDS-PAGE. Similarly, trastuzumab IgGs carrying WT or Fc variants were produced from associated Expi293F stable cell lines and purified using protein A resin (Genscript).
2.4. SDS-PAGE, streptavidin pulldown assay, and deglycosylation.
15 μl of purified biotinylated FcγRs at 8 μM were mixed with 10 μl of 4 × Laemmli sample buffer (Bio-Rad) for non-reducing condition or sample buffer containing 5% β-mercaptoethanol (β-ME, Sigma-Aldrich) for reducing condition. Mixtures were incubated at 95 °C for 5 minutes by using a thermocycler (Bio-Rad) with its lid heated to 105 °C. After cooled down to room temperature, 15 μl of streptavidin (NEB) at its monomer concentrations ranging from 0 to 24 μM was added, and samples were incubated for 10 min then analyzed by 12% SDS-PAGE. Deglycosylation of FcγRs was performed by using PNGase F (NEB) in denaturing reaction condition following manufacturer’s manual and analyzed by reducing SDS-PAGE. Gels were stained with Coomassie Blue, destained with Milli-Q water and imaged using GelDoc (Bio-Rad).
2.5. Bio-layer interferometry.
Binding kinetics were measured in PBS at 25 °C by using BLItz (FortéBio). 5 ng/μl of biotinylated FcγRs were loaded on streptavidin biosensors for 2 min. Association was monitored for 1 min with 125 nM - 1 μM purified IgGs, and dissociation was monitored with PBS for 2 min. Kinetic analysis and curve fitting were performed with BLItz Pro.
3. RESULTS
3.1. Experiment design.
To achieve site-specific in vivo biotinylation, AviTag was fused to the C-termini of FcγR ectodomains and FcγRs-AviTag were co-expressed with E. coli biotin-protein ligase BirA (Fig. 1). As PTMs mainly take place in endoplasmic reticulum (ER), we reasoned that, with free biotin added in culture media, BirA retained in ER can metabolically biotinylate the secretory FcγRs when processed through ER. Therefore, an ER-retention signal sequence KDEL20 was fused to the C-terminus of BirA to facilitate its catalytic action on the newly synthesized FcγRs at ER. Notably, this strategy has been successfully exploited for diabody biotinylation and outperformed the co-secretion approach17. To achieve high production yields, stable polyclonal cells were generated, in which both BirA and FcγR transgenes were genomically integrated (Fig. 1, route 1). As it is not uncommon that high expression levels could compromise the quality of recombinant products, a second approach via transient transfection was also implemented to intentionally reduce the synthesis rate, likely resulting in improved product quality with a trade-off on yields (Fig. 1, route 2). Cells without BirA transgene were also prepared to produce non-biotinylated FcγRs.
Figure 1. In vivo biotinylation and glycosylation of FcγRs.

Genes encoding FcγRs-AviTag and biotin ligase (BirA) are either stably integrated in genome (route 1) or transiently transfected to cells (route 2). BirA is tagged with endoplasmic reticulum (ER) retention signal peptide KDEL. Synthesized FcγRs are further processed in Golgi apparatus for their secretion to culture media
3.2. Production of biotinylated FcγRIIIa glycoproteins.
From stable polyclonal cells carrying FcγRIIIa and BirA transgenes, extracellular domains of FcγRIIIa-176F and -176V were produced and purified with yields of 15 and 9.1 μg per mL culture media respectively (Table 1). Non-reducing SDS-PAGE revealed broad bands—a signature of glycoproteins—with apparent molecular weights (MWs) ranging from ~40 to ~52 kDa (Fig. 2A), which were consistent with FcγRIIIa glycoproteins reported in literature12 and clearly larger than unglycosylated FcγRIIIa ectodomains with a calculated MW of 22 kDa. Under reducing condition, these bands migrated to slightly larger MWs at 43–55 kDa, presumably reflecting intra-domain disulfide breakdowns and release of fully linearized polypeptide chains. Produced FcγRIIIa-176F and -176V were further tested for deglycosylation with peptide:N-glycosidase F (PNGase F) in denature condition (Fig. 2B). Reducing SDS-PAGE showed that PNGase F treatment completely shifted the FcγRIIIa ECD bands to lower MWs of 36–40 kDa while larger than that of unglycosylated receptors. This incomplete deglycosylation may attribute to the fact that a combination of several endoglycosidases is usually needed to achieve effective deglycosylation12. Nevertheless, above results collectively suggested the formation of fully glycosylated FcγRIIIa ECDs containing disulfides.
Table 1.
Yields of purified FcγRs at μg per mL of cell culture
| Yield (μg/mL) | Stable cells | Transient transfection | ||
|---|---|---|---|---|
| Not Biotinylated | Biotinylated | Not biotinylated | Biotinylated | |
| FcγRIIa-131H | 46 | 20 | 7.0 | 5.0 |
| FcγRIIa-131R | 30 | 17 | 3.6 | 2.8 |
| FcγRIIb | 58 | 26 | n.d. | n.d. |
| FcγRIIIa-176F | 25 | 15 | n.d. | n.d. |
| FcγRIIIa-176V | 27 | 9.1 | n.d. | n.d. |
Figure 2. Production of fully glycosylated and biotinylated FcγRIIIa in stable cell lines.
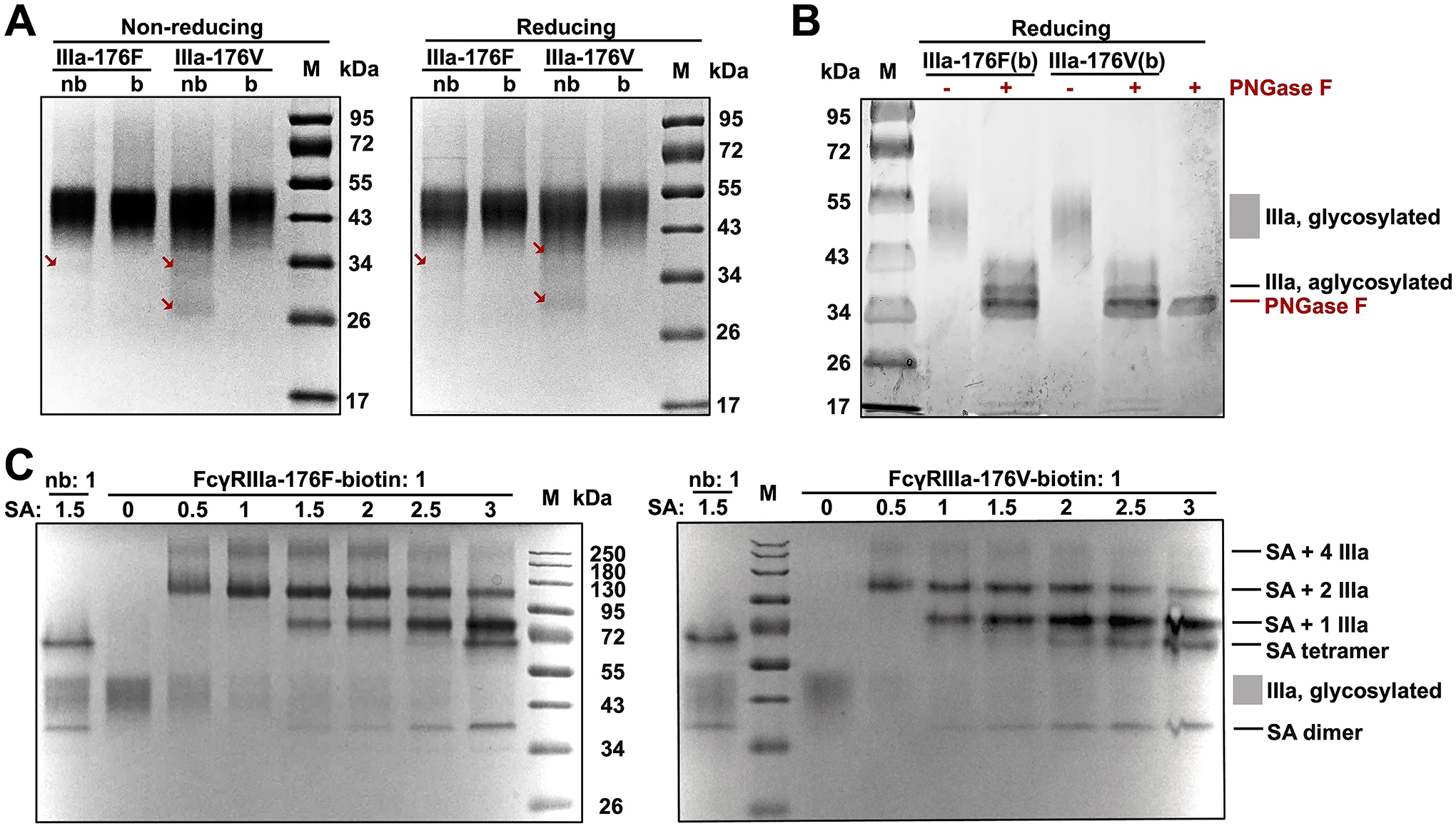
(A) FcγRIIIa variants produced with in vivo biotinylation (b) were analyzed at non-reducing and reducing conditions. FcγRIII variants without in vivo biotinylation (nb) were prepared as controls. Allotypes 176F and 176V were studies. Arrows indicate partially glycosylated FcγRIIIa. (B) Enzymatic deglycosylation of biotinylated FcγRIIIa with PNGase F. (C) Pulldown assays at increasing streptavidin / FcγRIIIa molar ratios.
Degree of biotinylation of produced FcγRIIIa was measured by streptavidin (SA) pulldown assays, in which SA was added at its molar ratio to FcγRIIIa ranging from 0:1 to 3:1 (Fig. 2C). SDS-PAGE results revealed that responding to the increasing amounts of SA, bands steadily evolved from high orders of complexes such as SA with four copies of FcγRIIIa (~260 kDa) and SA with two copies of FcγRIIIa (~110 kDa) to SA with one copy of FcγRIIIa (~80 kDa) and free SA tetramer (~70 kDa) and dimer (~38 kDa). Importantly, the 40–52 kDa broad bands associated with FcγRIIIa glycoproteins gradually diminished and finally disappeared at high SA/FcγRIIIa ratios, strongly suggesting that the produced FcγRIIIa were fully biotinylated. Non-biotinylated FcγRIIIa-176F and -176V produced from the stable cells without BirA transfection were also purified and examined. SDS-PAGE results showed that non-biotinylated FcγRIIIa behaved similarly to biotinylated FcγRIIIa but exhibited trace amounts of lower MW bands (arrowed in Fig. 2A). Considering FcγRIIIa have five N-glycan sites, these bands were likely partially glycosylated FcγRIIIa. Notably, the yields of non-biotinylated FcγRIIIa were 1.6–2.9 folds higher than biotinylated ones (Table 1), suggesting that the compromised quality on glycosylation could be associated with their high expression levels.
3.3. Production of biotinylated FcγRIIb glycoprotein.
FcγRIIb ectodomain was produced similarly from the stable cells co-transfected with BirA and FcγRIIb genes. 26 μg purified protein per mL of culture was yielded. Analysis by non-reducing SDS-PAGE indicated a thick band at 34 kDa, which migrated slightly slower in reducing gel with an apparent MW of 36 kDa (Fig. 3A). Notably, FcγRIIb glycoprotein showed a smaller MW than FcγRIIIa glycoproteins, largely because IIb has three N-glycan sites whereas IIIa has five. Strikingly, non-biotinylated FcγRIIB showed additional and sharper doublet bands (24 and 28 kDa under non-reducing and reducing conditions respectively), representing aglycosylated FcγRIIb presumably caused by its high expression level (Table 1). Treatment of biotinylated FcγRIIb with PNGase F resulted in deglycosylation and reduced MWs (Fig. 3B). Streptavidin titration experiments further confirmed that produced FcγRIIb was fully biotinlylated (Fig. 3C).
Figure 3. Production of fully glycosylated and biotinylated FcγRIIb in stable cell lines.
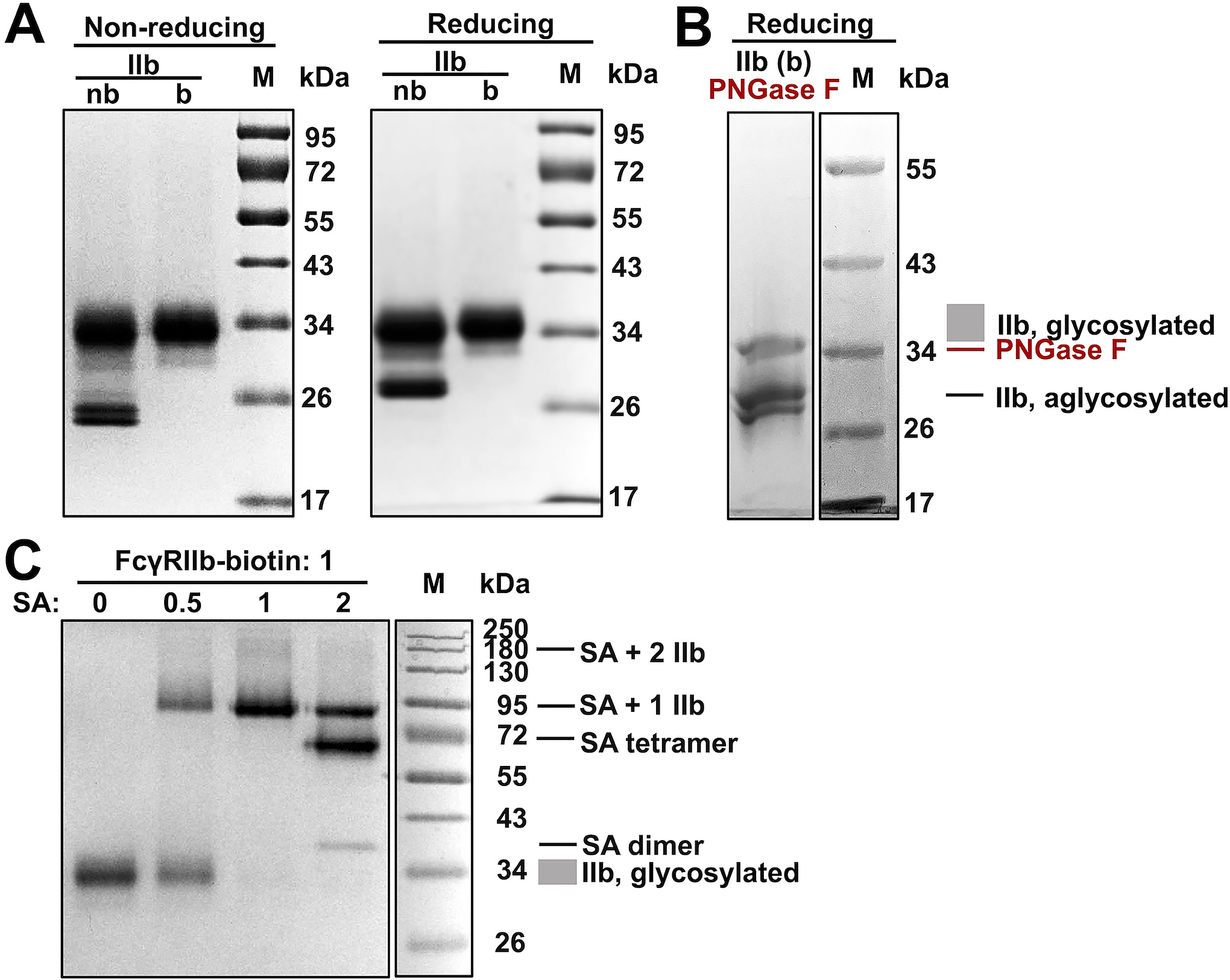
(A) FcγRIIb produced with in vivo biotinylation (b) was analyzed at non-reducing and reducing conditions. FcγRIIb without in vivo biotinylation (nb) was prepared as controls. (B) Enzymatic de-glycosylation of biotinylated FcγRIIb with PNGase F. (C) Pulldown assay at increasing streptavidin / FcγRIIb molar ratios
3.4. Production of biotinylated FcγRIIa glycoproteins by transient transfection.
When the same strategy of using stably transfected cells was applied to FcγRIIa-131H and -131R, significant amounts of aglycosylated bands were seen on SDS-PAGE (Fig. 4A). In addition, the aglycosylated bands of biotinylated FcγRIIa-131R (arrowed in Fig. 4A) did not respond to reducing agent, suggesting abnormal / incomplete disulfide bond formation. Pulldown assays further showed that even with streptavidin at 2-fold excessive molar amount, ~20% of FcγRIIa glycoproteins remained at ~32 kDa without migration to high MWs, indicating the in vivo biotinylation was not efficient in stable cells (Fig. 4B).
Figure 4. Incomplete glycosylation and incomplete biotinylation of FcγRIIa with stable cell lines.

(A) FcγRIIa variants produced with in vivo biotinylation (b) were analyzed at non-reducing and reducing conditions. FcγRIIa variants without in vivo biotinylation (nb) were prepared as controls. FcγRIIa allotypes 131H and 131R were studies. Arrows indicate abnormal disulfide formation. (B) Pulldown assays at increasing streptavidin / FcγRIIa molar ratio
As FcγRIIa has two N-glycosylation sites (less than IIIa and IIb) and relatively high expression levels were obtained (17–20 μg per mL of culture, Table 1), we reasoned that the quality can be improved at a slower production rate. Therefore, cells were transiently co-transfected with FcγRIIa and BirA expression plasmids at their mass ratios of 1:1 or 1:2. As expected, the yields of purified proteins were reduced to 2.8–5.0 μg per mL of culture (Table 1). And SDS-PAGE results indicated the quality of produced FcγRIIa was dramatically improved with >90% FcγRIIa-131H and >95% FcγRIIa-131R were glycosylated (Fig. 5A). In addition, all produced FcγRIIa correctly responded to the reducing agent indicating disulfides were fully formed. Deglycosylation with PNGase F showed complete shifts of bands, confirming glycosylation was efficient (Fig. 5B). Titrations with streptavidin also indicated full biotinylation, with evidence showing that all FcγRIIa glycoproteins formed complexes with streptavidin at the SA / receptor molar ratio of 2:1 (Fig. 5C). Notably, comparing 1:1 and 1:2 of FcγRIIa / BirA plasmid mass ratios, there is no significant difference in term of the quality of produced biotinylated FcγRIIa glycoproteins, suggesting that at the tested conditions, the amount of BirA was not the limiting factor. Therefore, the 1:1 ratio was used for FcγRIIa productions as this condition yielded higher amounts of products.
Figure 5. Production of fully glycosylated and biotinylated FcγRIIa with transient transfection.

(A) Cells were co-transfected with FcγRIIa and BirA expression plasmids at DNA mass ratio of 1:1 or 1:2. Purified FcγRIIa variants were analyzed at non-reducing and reducing conditions. FcγRIIa allotypes 131H and 131R were studies. (B) Enzymatic deglycosylation of biotinylated FcγRIIa variants with PNGase F. (C) Pulldown assays at increasing streptavidin / FcγRIIa molar ratios
3.6. Binding kinetics of produced FcγRs.
Biolayer interferometry experiments were conducted to validate whether the produced FcγRs had expected binding kinetics / affinities towards IgG molecules. In this study, human IgG1 trastuzumab and its Fc mutants G236A (clone GA, for FcγRIIa)21, P238D (clone PD, for FcγRIIb)22, and F243L/R292P/Y300L (clone LPL, for FcγRIIIa)23 were used to test their bindings with associated FcγRs. On streptavidin biosensors, biotinylated FcγRs were immobilized, and IgGs were loaded then washed to measure association and dissociation rate constants respectively. As results shown in Table 2, consistence was largely achieved between our measurements and literature reported values. For example, FcγRIIa-131H:GA showed an equilibrium dissociation constant KD of 120±35 nM (Fig. 6), in a good agreement with reported 130±10 nM21; and FcγRIIIa-V167:WT showed a KD of 608±39 nM, within the reported range of 554–850 nM24,25.
Table 2.
Binding affinities of produced FcγRs and comparison with reported values (nM)
| IgG wt | IgG carrying Fc mutation | ||||
|---|---|---|---|---|---|
| This study | Literature (Ref) | Mutant | This study | Literature (Ref) | |
| FcγRIIa-131H | 290 ± 67 | 292[24]; 800[25] | GA | 120 ± 35 | 130 ± 10[26] |
| FcγRIIa-131R | 610 ± 260 | 203[24]; 800[25] | GA | 210 ± 109 | 161 ± 3[26] |
| FcγRIIb | 360 ± 53 | 1274[24]; 3100[25] | PD | 310 ± 42 | N/A |
| FcγRIIIa-176F | 47 | 502[24]; 850[25] | LPL | 110 | N/A |
| FcγRIIIa-176V | 610 ± 39 | 554[24]; 850[25] | LPL | 250 ± 57 | N/A |
Figure 6.

Biolayer Interferometry of representative tests
4. DISCUSSION
Mammalian expression systems hold the key to recombinant production of monoclonal antibodies (mAbs) and complex proteins for preclinical and clinical applications. Over decades, Chinese hamster ovary (CHO) cell lines have been established as the industry-standard platform for therapeutic protein expression26. To date, ~70% of biopharmaceutic drugs on market are produced in CHO cells27, demonstrating a strong regulatory track record. However, due to its rodent origin, CHO cells lack some types of human glycosylation such as α2,6-sialylation28, and produce non-human glycoforms including galactose-α1,3-galactose (α-gal) and N-glycolylneuraminic acid (Neu5Gc)29,30. These features have raised immunogenicity and in vivo stability concerns – for example, it was reported that antibodies to Neu5Gc were prevalent in humans31; and severe hypersensitivity reactions associated with pre-existing IgE antibodies against α-gal were observed in clinical trials of cetuximab32. Therefore, to provide fully human PTMs, human cell lines have emerged as a valuable system for biopharmaceutic production. Notably, several therapeutic agents produced in human cell lines have been approved by FDA or European Medicines Agency recently27, indicating the potential of human cells on biologic production. Particularly, suspension HEK293-F cells have advantages including fast growth, high cell density, use of serum-free media, efficient transfection, and high production yields. By using this cell line as the expression host, we produced fully biotinylated and glycosylated FcγRs with yields up to 58 μg per milliliter of culture.
Large-scale production of therapeutic proteins often relies on stable cell lines which have the transgenes gnomically integrated. Upon selection, high-yield stably transfected clones can usually be obtained. Alternatively, transient transfection, an approach avoiding the laborious and time-consuming process of cell line establishment, is also commonly used for the rapid production. As expected, our results validated that stable cells produced larger amounts of FcγRIIa compared to using transiently transfected cells (Table 1). However, there was a remarkable trade-off for quality – the PTMs of FcγRIIa produced in stable cells were generally suboptimal (Fig. 4). It has been well understood that following transcription, a series of cellular procedures is involved in the synthesis and secretion of functional glycoproteins. These post-transcriptional steps include translation, polypeptide translocation to ER, protein folding and assembly, addition of PTMs, and secretion. Although a high level of transcription is beneficial to achieve high productivity, excessive and rapid synthesis can be detrimental for the quality of produced proteins when any of above steps becomes the bottleneck. By applying transient transfection, intentionally reduced synthesis rates allow better coordination of post-transcriptional machinery, and thus improve the overall quality (e.g., FcγRIIa shown in Fig. 5). Therefore, for successful production of bioactive proteins, the balance between yield and quality needs to be carefully adjusted.
Polyclonal stable cells with random genomic integration of FcγR transgenes were established and applied in this study. To improve the yields, monoclonal cell lines can be selected, and site-specific integration into transcriptional hotspots (e.g., via CRISPR, recombinases, or TALEN) can also be exploited33. Other optimizations can focus on engineering expression elements such as promoter, 5’-UTR, polyA tail, etc, and engineering culture media and bioprocess such as using fed-batch34. In addition, incorporating an ER retention signal to the GOIs themselves can be an attractive approach to further improve the PTM quality of produced proteins. Conclusively, in this work, human FcγRIIa (-131H and -131R), FcγRIIb, and FcγRIIIa (-176F and -176V) carrying complete N-glycosylation, disulfides, and biotinylation were produced via co-expression of FcγRs-AviTag and ER retained BirA. Produced FcγRs showed expected binding kinetics towards IgG1 and Fc mutated trastuzumab. Achieving PTMs of multiple types by one-step cell culture and the revealed trade-off between yield and quality can be valuable knowledge on guiding the production of a variety of complex biopharmaceutic drugs of biomedical importance.
ACKNOWLEDGMENTS
This work is supported by NIGMS grant R35GM141089 to X.G.
Footnotes
AUTHOR CONTRIBUTION
Minhyo Kang: Data curation (Lead); Formal analysis (Equal); Investigation (Equal); Methodology (Supporting); Writing – original draft (Supporting); Writing – review & editing (Supporting). Zening Wang: Conceptualization (Equal); Data curation (Supporting); Formal analysis (Equal); Investigation (Equal); Methodology (Lead); Writing – original draft (Supporting); Writing – review & editing (Supporting). Xin Ge: Conceptualization (Equal); Formal analysis (Equal); Funding acquisition (Lead); Investigation (Equal); Supervision (Lead); Writing – original draft (Lead); Writing – review & editing (Lead).
CONFLICTS OF INTEREST
No conflicts of interest
DATA AVAILABILITY STATEMENT
The datasets supporting the conclusions of this article are included within the article.
REFERENCES
- 1.Ramazi S, Zahiri J. Posttranslational modifications in proteins: resources, tools and prediction methods. Database (Oxford). 2021:baab012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann T, Schmieder V, Faustrup Kildegaard H, Borth N, Andersen MR. Genetic engineering approaches to improve posttranslational modification of biopharmaceuticals in different production platforms. Biotechnol Bioeng. 116:2778–2796 (2019). [DOI] [PubMed] [Google Scholar]
- 3.An HJ, Froehlich JW, Lebrilla CB. Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol. 13:421–426 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiedemann C, Kumar A, Lang A, Ohlenschläger O. Cysteines and Disulfide Bonds as Structure-Forming Units: Insights From Different Domains of Life and the Potential for Characterization by NMR. Front Chem. 8:280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bošnjak I, Bojović V, Šegvić-Bubić T, Bielen A. Occurrence of protein disulfide bonds in different domains of life: a comparison of proteins from the Protein Data Bank. Protein Eng Des Sel. 27:65–72 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Dundas CM, Demonte D, Park S. Streptavidin-biotin technology: improvements and innovations in chemical and biological applications. Appl Microbiol Biotechnol. 97:9343–9353 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Takai T Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2:580–592 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 8:34–47 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Junker F, Gordon J, Qureshi O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front Immunol. 11:1393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillis C, Gouel-Chéron A, Jönsson F, Bruhns P. Contribution of Human FcγRs to Disease with Evidence from Human Polymorphisms and Transgenic Animal Studies. Front Immunol. 5:254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 20:633–643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes JM, Frostell A, Cosgrave EF, Struwe WB, Potter O, Davey GP, Karlsson R, Anneren C, Rudd PM. Fc gamma receptor glycosylation modulates the binding of IgG glycoforms: a requirement for stable antibody interactions. J Proteome Res. 13:5471–5485 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Hayes JM, Wormald MR, Rudd PM, Davey GP. Fc gamma receptors: glycobiology and therapeutic prospects. J Inflamm Res. 9:209–219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman-Smith A, Cronan JE Jr. Molecular biology of biotin attachment to proteins. J Nutr. 129: 477S–484S (1999). [DOI] [PubMed] [Google Scholar]
- 15.Schatz PJ. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (NY). 11:1138–1143 (1993). [DOI] [PubMed] [Google Scholar]
- 16.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8:921–929 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barat B, Wu AM. Metabolic biotinylation of recombinant antibody by biotin ligase retained in the endoplasmic reticulum. Biomol Eng. 24:283–291 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fairhead M, Howarth M. Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol. 1266:171–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen KE, Chen C, Lopez T, Radecki KC, Bustamante K, Lorenson MY, Ge X, Walker AM. Use of a novel camelid-inspired human antibody demonstrates the importance of MMP-14 to cancer stem cell function in the metastatic process. Oncotarget 9:29431–29444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelham HR. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem Sci. 15:483–486 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. 7:2517–2527 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Mimoto F, Katada H, Kadono S, Igawa T, Kuramochi T, Muraoka M, Wada Y, Haraya K, Miyazaki T, Hattori K. Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIa(131R) and FcγRIIa(131H). Protein Eng Des Sel. 26:589–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, Vijh S, Johnson S, Bonvini E, Koenig S. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 67:8882–8890 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Zhang N, Liu L, Dumitru CD, Cummings NR, Cukan M, Jiang Y, Li Y, Li F, Mitchell T, Mallem MR, Ou Y, Patel RN, Vo K, Wang H, Burnina I, Choi BK, Huber HE, Stadheim TA, Zha D. Glycoengineered Pichia produced anti-HER2 is comparable to trastuzumab in preclinical study. mAbs 3:289–298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel R, Johnson K, Andrien B, Tamburini P. IgG subclass variation of a monoclonal antibody binding to human Fc-gamma receptors. Am J Biochem Biotechnol. 9:206–218 (2013). [Google Scholar]
- 26.Dumont J, Euwart D, Mei B, Estes S, Kshirsagar R. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol. 36:1110–1122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Hatton D. New Mammalian Expression Systems. Adv Biochem Eng Biotechnol. 165:9–50 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Raymond C, Robotham A, Spearman M, Butler M, Kelly J, Durocher Y. Production of α2,6-sialylated IgG1 in CHO cells. mAbs. 7:571–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosques CJ, Collins BE, Meador JW 3rd, Sarvaiya H, Murphy JL, Dellorusso G, Bulik DA, Hsu IH, Washburn N, Sipsey SF, Myette JR, Raman R, Shriver Z, Sasisekharan R, Venkataraman G. Chinese hamster ovary cells can produce galactose-α-1,3-galactose antigens on proteins. Nat Biotechnol. 28:1153–1156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 28:147–175 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 28:863–867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, Slebos RJ, Zhou Q, Gold D, Hatley T, Hicklin DJ, Platts-Mills TA. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 358:1109–1117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng JK, Lewis AM, Kim DS, Dyess T, Alper HS. Identifying and retargeting transcriptional hot spots in the human genome. Biotechnol J. 11:1100–1109 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Lai T, Yang Y, Ng SK. Advances in Mammalian cell line development technologies for recombinant protein production. Pharmaceuticals (Basel). 6:579–603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article.