

Abstract
Repetitive mild traumatic brain injuries (r-mTBI) sustained in the military or contact sports have been associated with the accumulation of extracellular tau in the brain, which may contribute to the pathogenesis of neurodegenerative tauopathies. The expression of the apolipoprotein E4 (apoE4) isoform has been associated with higher levels of tau in the brain, and worse clinical outcomes after r-mTBI, though the influence of apoE genotype on extracellular tau dynamics in the brain is poorly understood. We recently demonstrated that extracellular tau can be eliminated across blood-brain barrier (BBB), which is progressively impaired following r-mTBI. The current studies investigated the influence of repetitive mild TBI (r-mTBI) and apoE genotype on the elimination of extracellular solutes from the brain. Following intracortical injection of biotin-labeled tau into humanized apoE-Tr mice, the levels of exogenous tau residing in the brain of apoE4 mice were elevated compared to other isoforms, indicating reduced tau elimination. Additionally, we found exposure to r-mTBI increased tau residence in apoE2 mice, similar to our observations in E2FAD animals. Each of these findings may be the result of diminished tau efflux via LRP1 at the BBB, as LRP1 inhibition significantly reduced tau uptake in endothelial cells and decreased tau transit across an in vitro model of the BBB (basolateral-to-apical). Notably, we showed that injury and apoE status, (particularly apoE4) resulted in chronic alterations in BBB integrity, pericyte coverage, and AQP4 polarization. These aberrations coincided with an atypical reactive astrocytic gene signature indicative of diminished CSF-ISF exchange. Our work found that CSF movement was reduced in the chronic phase following r-mTBI (>18 months post injury) across all apoE genotypes. In summary, we show that apoE genotype strongly influences cerebrovascular homeostasis, which can lead to age-dependent deficiencies in the elimination of toxic proteins from the brain, like tau, particularly in the aftermath of head trauma.
Keywords: ApoE, Traumatic brain injury, Glymphatic system, Blood brain barrier, Tau
1. Introduction
Repetitive exposure to head trauma is a risk factor for the development of chronic neurodegenerative diseases and dementia, most notably AD and CTE. Traumatic brain injury (TBI) causes the release of cerebral waste (e.g. tau, and/or Aβ), (Piantino et al., 2022) into the interstitial fluid (ISF) of the central nervous system (CNS). Elevated extracellular tau is highly linked to adverse clinical outcomes and cognitive decline (Magnoni et al., 2012). Chronically increased levels of extracellular tau following TBI (Marklund et al., 2009), can facilitate intraneuronal tau fibril formation (Yamada et al., 2011) and pathogenic tau seeding (Frost et al., 2009), resulting in neurodegenerative decline.
Despite the importance of extracellular tau in disease progression, the mechanisms governing tau removal from the brain are not well understood. However, recent studies have indicated a link between extracellular tau and the neurovascular unit. After neuronal waste is secreted into the ISF, it is carried by a network of perivascular channels that promote fluid movement into the CSF, which enables the elimination of waste products. This “glymphatic” pathway (Iliff et al., 2012) shuttles waste to the blood brain barrier (BBB) for elimination via transcytosis to the blood or CNS drainage along contiguous perivascular channels. When BBB transport capacity is overwhelmed by excessive local concentrations of protein and/or impaired transport machinery, perivascular fluid flow disperses the protein along a wider area of the BBB to enable efflux, eventually draining any remaining solutes along continuous perivascular pathways into the cervical lymph nodes. Forces driving these extracellular fluid flow dynamics are modulated by several aspects of the neurovascular health, including aquaporin 4 (AQP4) polarization (Iliff et al., 2012), cerebral arterial pulsation (Iliff et al., 2013), neurovascular coupling (Holstein-Rønsbo et al., 2023), vasomotion (van Veluw et al., 2020) and vessel wall compliance (Drieu et al., 2022).
Recent reports have indicated that these neurovascular elimination routes play a significant role in the clearance of extracellular tau (Iliff et al., 2014; Nimmo et al., 2020), reflecting what has been previously observed with Aβ. The cerebrovasculature can eliminate extracellular tau by a combination of both glymphatic clearance (Harrison et al., 2020; Ishida et al., 2022) and BBB transit (Banks et al., 2016; Eisenbaum et al., 2021). Recent work by our lab and others found that exposure to TBI can lead to progressive neurovascular decline, coinciding with impaired glymphatic (Iliff et al., 2014) function and decreased tau removal from the brain (Eisenbaum et al., 2021), culminating in prolonged tau retention in the brain(Eisenbaum et al., 2021; Ojo et al., 2021). Impaired perivascular clearance is highly associated with instances of neurovascular decline observed in aging (Kress et al., 2014), TBI (Iliff et al., 2014; Ren et al., 2013), cerebral small vessel disease (M. Wang et al., 2017), cerebral amyloid angiopathy (CAA) (Aldea et al., 2019), and AD (Peng et al., 2016; Zeppenfeld et al., 2017). However, it is not clear how BBB dysfunction influences tau elimination following r-mTBI, particularly in the presence of other genetic risk factors which may further disrupt these elimination pathways.
In humans, apoE exists in three common isoforms (apoE2, apoE3, and apoE4). The apoE4 allele is the strongest genetic risk factor for early and late onset-AD. ApoE4 carriers demonstrate impaired vasomotion (Bonnar et al., 2023), and a progressive decline in BBB integrity, and mural cell coverage (Barisano et al., 2022; R. D. Bell et al., 2012), which is further exacerbated by AD (Halliday et al., 2016; Montagne et al., 2021) or TBI (Main et al., 2018). Expression of apoE4 impairs BBB recovery following TBI (Cao et al., 2017; Main et al., 2018; Teng et al., 2017), which likely contributes to worse clinical outcomes in apoE4 carriers (Giarratana et al., 2020; McFadyen et al., 2021; Zhou et al., 2008). Findings from our lab (Muza et al., 2019) and others (Cao et al., 2017) indicate that pathogenic tau accumulation is increased in apoE4 carriers following TBI. Interestingly, pathological tau-associated neurodegeneration is accelerated by apoE4 (Y. Shi et al., 2017), and may contribute to recent reports demonstrating increased prevalence and severity of CTE in apoE4 carriers (Atherton et al., 2022). Finally, while computational models have predicted that apoE4 may impair glymphatic clearance (Kyrtsos and Baras, 2015), the influence of apoE isoforms on perivascular dynamics and solute elimination, particularly after TBI, remains poorly characterized. In this study we examined whether apoE isoforms influence mechanisms of extracellular tau elimination from the brain, and how TBI impacts those processes.
2. Materials and methods
2.1. Animals
Wild-type (C57BL/6) (JAX:000664) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). ApoE-targeted replacement (ApoE-TR) mice were purchased from Taconic Biosciences. The ApoE-TR mice were generated using a targeted replacement of the endogenous murine apoE gene with human apoE2, apoE3 or apoE4. The mice express, produce, lipidate and secrete human apoE at physiological levels, (Verghese et al., 2013). To generate EFAD mice, apoE2, apoE3 and apoE4 mice were crossed with 5xFAD mice (Tg6799 line, over-expressing human amyloid beta (A4) precursor protein 695 (APP), with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) Familial AD mutations, and human PS1 with two FAD mutations, M146L and L286V) (JAX:034848), producing the E2FAD, E3FAD and E4FAD mouse models respectively, as previously described (Youmans et al., 2012). All mice are on a C57BL/6 background, and additional details regarding these mice are described in the sources cited above. Studies used a mix of male and female mice. All studies used mice housed 3 per cage under standard laboratory conditions (23 ± 1 °C, 50 ± 5% humidity, and a 12-h light/dark cycle) with free access to food and water throughout the study. All procedures involving mice were performed in accordance with Office of Laboratory Animal Welfare and National Institutes of Health guidelines under a protocol approved by the Roskamp Institute Institutional Animal Care and Use Committee.
2.2. Tau aggregation
Tau aggregation was performed as described and characterized by our group previously (Eisenbaum et al., 2021). Briefly, 4 μM biotin-labeled human tau-441 (Cat. No: T08–54BN; SignalChem) was incubated with freshly prepared heparin (1 μM) (Cat. No: H3393–10KU; Millipore Sigma) for 6 h at 37 °C. Aggregation was monitored by Thioflavin S (Cat. No: T1892–25G; Millipore Sigma), as described previously (Eisenbaum et al., 2021). The aggregate-enriched solution of biotin-labeled tau (btau) was concentrated using a 100 kDa MWCO filter (Corning) by centrifugation at 14,000 g for 25 min at 4 °C to filter out monomeric tau species <100 kDa. The concentrated, aggregate enriched fraction (MW > 100 kDa) was collected and stored at −80 °C. Prior to use, the concentration was determined using a modified biotin-labeled human tau enzyme linked immunosorbent assay (ELISA) (Cat. No: KHB0041; ThermoFisher Scientific) as previously described (Eisenbaum et al., 2021).
2.3. Intraparenchymal tracer injections
Biotin-labeled tau species and Texas Red labeled 10 kDa dextran (Cat. No: D1863; ThermoFisher Scientific) (TxD) were used to evaluate solute elimination from the brain, as per our prior methods (Eisenbaum et al., 2021). Briefly, 12 month old mice (apoE2, apoE3, apoE4) were anesthetized via inhalation using a 3% isoflurane / oxygen mix and maintained at 37 °C using a homeothermic blanket. Mice were stereo-taxically injected (0.5 mm anterior to the bregma, 2 mm lateral to the midline, and 3 mm below the surface of the skull) into the caudate putamen with 3 μL of biotin-labeled tau (50 μg/mL) and TxD (80 mg/mL). Mice were euthanized 2 h after the intracranial injection, then the injected brain hemisphere was harvested and homogenized with probe sonication in 500 Ll of lysis buffer (Mammalian protein extraction reagent (Cat. N: 78505; ThermoFisher Scientific) supplemented with phenylmethanesulfonyl fluoride (1 mM) and Halt protease and phosphatase inhibitor cocktail (Cat. N: 78442; ThermoFisher Scientific)). The brain homogenate was evaluated for biotin-labeled tau using a modified hTau ELISA, and TxD fluorescence using a microplate spectrofluorometer, as previously described (Eisenbaum et al., 2021). The amount of exogenous biotin-labeled tau residing in the brain was normalized to TxD. In assessing TxD residence in the brain, TxD was normalized to the total protein content in brain homogenate as determined using the bicinchoninic acid (BCA) protein assay (Cat. No: 23225; ThermoFisher Scientific).
2.4. Isolation of cerebrovasculature
The cerebrovasculature was isolated from mouse tissue as characterized and described by our group previously (Eisenbaum et al., 2021). Briefly, after the removal of meninges, the freshly isolated mouse cortex (apoE-Tr mice, 12 months of age), was homogenized in a glass Dounce homogenizer using a Teflon pestle. The brain homogenate was suspended in a solution of 20% dextran, and immediately centrifuged at 6000 g for 15 min. The pellet at the bottom of the centrifuge tube, containing the cerebrovasculature from a single mouse, was collected and resuspended in lysis buffer. Cerebrovascular lysates were analyzed for caveolin-1 by ELISA (Cat. No: MBS721447; MyBioSource, Inc), and normalized to total protein content using the BCA protein assay.
2.5. In vitro BBB assay
2.5.1. In vitro model of the BBB
Monoculture and coculture in vitro BBB models were used to evaluate tau transcytosis, as described previously by our group (Bachmeier et al., 2010; Eisenbaum et al., 2021). Briefly, primary human brain microvascular endothelial cells (HBMEC, Cat. No: 1000; ScienCell Research Laboratories) were seeded at 50,000 cells/cm2 onto the interior membrane of the fibronectin-coated (Cat. No: F1141; ThermoFisher Scientific) 24-well 0.4 μm-pore membrane inserts to establish a polarized monolayer. The layer of cells separates this system into an apical (“blood” side) and basolateral (“brain” side) compartment. For the co-culture studies, one hour before HBMEC seeding, primary human brain vascular pericytes (HBVP, Cat. No: 1200; ScienCell Research Laboratories) were seeded at 25,000 cells/cm2 onto the exterior membrane of poly-l-lysine-coated (Cat. No: P4707; ThermoFisher Scientific) 24-well 0.4 μm-pore membrane inserts.
2.5.2. Transendothelial transport of tau
The basolateral compartment was exposed to monomeric or aggregated biotin-labeled tau (200 ng/mL) in the presence or absence of RAP (100 nM) (Cat. No: Ca553506-M; Millipore Sigma), while fresh media was placed in the apical compartment. The inserts containing media were exposed to the wells containing biotin-labeled tau and incubated at 37 °C. The basolateral compartment was sampled at time 0 to establish the initial concentration of biotin-labeled tau. Samples were collected from the apical compartment at 0, 30, and 60 min to assess the rate of biotin-labeled tau transcytosis across the cell monolayer (basolateral-to-apical) and analyzed for biotin-labeled tau using a modified hTau ELISA. Furthermore, each basolateral compartment was exposed to a known paracellular marker, 10 kDa lucifer yellow dextran (LyD 10 μM) (Cat. No: D1825; ThermoFisher Scientific), to monitor barrier integrity and/or nonspecific permeability, as we previously described (Bachmeier et al., 2010). The apparent permeability (Papp) was determined using the equation Papp = 1/AC0 * (dQ/dt), where A represents the surface area of the membrane, C0 is the initial concentration of biotin-labeled tau in the basolateral compartment, and dQ/dt is the amount of biotin-labeled tau appearing in the apical compartment in the given time period (Artursson, 1990).
2.6. Tau uptake in HBMEC
Fully confluent HBMECs were exposed to monomeric biotin-labeled tau (1 μg/mL) in the presence or absence of RAP (100 nM) for 1 h at 37 °C. Cells were washed with Hanks’ balanced salt solution (HBSS) (Cat. No: H8264; Millipore Sigma) and cell lysates were collected using lysis buffer. The cell lysates were analyzed for biotin labeled tau and normalized to total protein content using the BCA assay.
2.7. Repetitive mild traumatic brain injury protocol
Repetitive mild traumatic brain injury (r-mTBI) was administered using a mouse model of closed head injury as previously characterized by our group (Mouzon et al., 2012, 2014, 2018). Briefly, 12 week old mice (apoE2, apoE3, apoE4) were anesthetized with 1.5 L/min of oxygen and 3% isoflurane, then secured in a stereotaxic apparatus mounted with an electromagnetic controlled impact device. Prior to impact, a 5 mm blunt metal impactor tip was retracted and positioned midway in relation to the sagittal suture. Using the myNeuroLab controller, the injury was triggered at a strike velocity of 5 m/s, strike depth of 1.0 mm, and a dwell time of 200 ms. Randomly assigned three-month-old mice received two injuries per week, approximately 72 h apart, for 3 months (r-mTBI). In this model of closed head injury, there are no incisions and no craniotomy. As a control, sham animals did not receive the brain injury, but were exposed to anesthesia for the same length of time as the injured mice, and under the same paradigm. Mice were euthanized at 6 months after the final brain injury or anesthesia exposure (12 months of age) for tau residence, or euthanized at 18 months after the final injury (24 months of age) for immunohistochemical analysis or cisternal injections. For RNA sequencing of primary astrocytes, mice (apoE2, apoE3, apoE4) were subjected to an accelerated injury paradigm, previously characterized by our group (Pearson et al., 2023). Randomly assigned mice received five injuries per week for one month. Mice were euthanized at 3 months after the final brain injury (7 months of age) for astrocyte isolation.
2.8. Perfusion and vessel painting procedure
Vessel painting was carried out as described previously (Jullienne et al., 2018; Salehi et al., 2019). Briefly, apoE mice (r-sham and r-mTBI, 6 months post injury) were anesthetized with isoflurane, then mice were injected intracardially with 500 μL of 0.3 mg/mL DiI (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate (‘DiI’; DiIC18(3))) (Cat. No: D282; ThermoFisher Scientific). Following perfusion with Phosphate Buffered Saline (PBS) and 4% Paraformaldehyde (PFA), brains were extracted and coronal vibratome sections (100 μm) were mounted using Fluoroshield mounting media with DAPI. Brains were imaged on a confocal microscope using the 5× magnification, and whole slice montages were reconstructed. A threshold of vessel paint signal was established using a healthy control mouse as a baseline. Vessel leakage was quantified as the percent area of vessel paint signal above the healthy control threshold.
2.9. Immunohistochemistry
At 18 months post injury, apoE-Tr mice (r-mTBI and r-sham) were anesthetized with isofluorane and perfused transcardially with PBS followed by 4% PFA. The brains were post fixed in 4% PFA for 48 h and then blocked in paraffin using a Sakura Tissue-Tek (Leica Biosystems Inc), then cut with a Leica RM2235 microtome (Leica Biosystems Inc). For each group, sets of frontal cortex sections 6 μm thick were mounted on glass slides, and dried for 48 h at 37 °C. Slides were then deparaffinized in xylene and antigen retrieval was performed using Dako target retrieval citrate buffer solution (pH 6.0) under pressure for 8 min in the microwave. Tissue sections were blocked with 5% normal donkey serum (NDS) in 0.1% Triton X-100 for 45 min. Afterwards, slides were incubated with primary antibodies diluted in blocking solution at 4 °C overnight. Slides were washed three times with PBS, and incubated with fluorophore conjugated secondaries for 1 h in the dark at room temperature. Slides were washed three times in PBS, then immersed in 70% ethanol for 5 min. Lipofuscin was quenched with the Autofluorescence Eliminator Reagent (Cat. No: 2160; Millipore Sigma) for 5 min, then immersed in three changes of 70% ethanol for 1 min each, as per manufacturer’s instructions, prior to mounting using Fluoroshield mounting media with DAPI. To visualize brain microvessels, all sections were incubated with Dylight 488-conjugated Lycopersicon esculentum lectin (Cat. No: DL-1174–1; 1:200; Vector labs). In addition to the microvascular lectin stain, the primary and secondary antibody pairs used were: goat anti-mouse aminopeptidase N/ANPEP (CD13) (Cat. No: AF2335; 1:100; R&D Systems) primary with Alexa Fluor 568-conjugated donkey anti-goat (Cat. No: A-11057; 1:500; Invitrogen) secondary; rabbit anti-mouse collagen IV (Cat. No: AB756P; 1:100; Millipore Sigma) primary with Alexa Fluor 568-conjugated donkey anti-rabbit (Cat. No: A-10042; 1:500; Invitrogen) secondary; rabbit anti-human fibrinogen (Cat. No: A0080; 1:500; Dako) primary with Alexa Fluor 568-conjugated donkey anti-rabbit (Cat. No: A-10042; 1:500; Invitrogen) secondary; and rabbit anti-mouse AQP4 (Cat. No: HPA014784; 1:1000; Millipore Sigma) primary with Alexa Fluor 568-conjugated donkey anti-rabbit (Cat. No: A-10042; 1:500; Invitrogen) secondary.
2.10. Microscopy analysis
2.10.1. Quantification analysis
For the quantification of pericyte coverage, collagen IV coverage, extravascular fibrinogen leakage, and AQP4 polarization, sections were imaged using the confocal microscope LSM 800 (Carl Zeiss AG), the Zen Blue 2.3 software and a 20× objective, at 1-μm step intervals for z-stacks. Ten micron maximum projection z-stacks were reconstructed from images taken in 5 randomly selected fields (320 × 320 μm) per animal in the somatosensory cortex, in three to five nonadjacent sections (~ 100 μm apart). The number of animals used for each analysis was indicated in the respective figure legends.
2.10.2. Pericyte coverage
For pericyte coverage, CD13 and lectin signals from microvessels ≤6 μm in diameter were separately subjected to threshold processing. Lectin-positive microvessels ≤6 μm in diameter were selected and the area occupied by the selected vessels was quantified using the NIH Image J Area measurement tool, and used to establish a microvascular region of interest (ROI). Pericyte coverage of those vessels was determined by measuring percentage of the area within the previously selected, lectin-positive, microvascular ROI that was covered by CD13-positive pericyte surface area, using the NIH Image J Area measurement tool, as described previously (Barisano et al., 2022; R. D. Bell et al., 2012; Montagne et al., 2021). An average of 60 selected microvessels from an average of 5 randomly selected, non-adjacent (at least 100 μm apart) regions in the somatosensory cortex were used to determine the pericyte coverage per animal.
2.10.3. Collagen IV coverage
For collagen IV coverage, collagen IV and lectin signals from microvessels ≤6 μm in diameter were separately subjected to threshold processing, as described above for pericyte coverage. The area occupied by threshold adjusted collagen IV signal within the threshold adjusted lectin positive microvessel area was quantified using the NIH Image J Area measurement tool, as described previously (R. D. Bell et al., 2012; Lv et al., 2023).
2.10.4. Extravascular leakages
Blood-derived fibrin(ogen) extravascular pericapillary deposits were quantified using an antibody that detects both fibrin and fibrinogen-derived fibrin polymers. Lectin and fibrinogen positive signals were subjected to threshold processing. The fibrinogen signal was analyzed using the ImageJ software integrated density analysis measurement tool. The fibrinogen positive signal outside the blood vessels was quantified by subtracting the fibrinogen integrated density in the lectin positive capillary surface area, from the total fibrinogen integrated density per field (320 × 320 μm), as described previously (R. D. Bell et al., 2012; Montagne et al., 2018).
2.10.5. AQP4 polarity
For AQP4 polarization, AQP4 and lectin signals from microvessels ≤6 μm were subjected to threshold processing. The mean pixel intensity (MPI) of the AQP4 channel was taken using the ImageJ software mean gray value tool as measurement of parenchymal AQP4 fluorescence. To evaluate perivascular AQP4, 8–10 capillaries per image were identified by lectin immunoreactivity. The MPI of AQP4 signal within the lectin positive ROI was taken as a measurement of vascular AQP4. The average vascular to parenchymal MPI ratio was used as a measure of AQP4 polarization, as described previously (Kress et al., 2014; Munk et al., 2019). Diminished polarization reflects lower vascular AQP4 and/or higher parenchymal AQP4 (Kress et al., 2014).
2.11. Primary astrocyte isolation
Primary astrocytes (apoE r-mTBI and sham) were isolated from accelerated r-mTBI mice using the adult brain dissociation kit mouse and rat (Cat. No: 130-107-677; Miltenyi Biotec), as described previously (Eisenbaum et al., 2023). Samples were incubated with Fcγ receptor blocker for 15 min at 4 °C followed by incubation for 10 min at 4 °C with magnetic particles coated by an antibody raised against the panastrocyte marker, ACSA2 (Cat. No: 130-097-679; Miltenyi Biotec). Samples were then loaded onto a pre-conditioned LS separation column and rinsed three times to remove unlabelled cells. To elute ACSA2+ve cells the LS column was removed from the magnet and PB buffer was used to elute the sample. Enriched ACSA2+ve cells were briefly centrifuged and then stored in RNA-Ice (Cat. No: AM7030; ThermoFisher Scientific) at −80 °C for subsequent RNA isolation.
2.12. RNA isolation
RNA isolation and sequencing were performed as previously described (Eisenbaum et al., 2023). Briefly, isolated ACSA2+ve cells (N = 3/group) were lysed in Trizol, 100 μL of 1-bromo-3-chloropropane (BCP) was added to each sample, and samples were centrifuged to induce phase separation. The aqueous phase was carefully removed, and RNA was precipitated using 100% isopropanol. The precipitated RNA was pelleted by centrifugation and washed three times using 75% ethanol in DEPC water. Pellets were briefly air-dried and then resuspended in DEPC water, RNA concentrations (ng/μL) and RNA purity (260:280) were calculated using a Cytation 3 (Biotek). At least 500 ng/sample of RNA were sent to GENEWIZ LLC (South Plainfield, NJ, USA) for subsequent RNA library preparation and total RNA sequencing (20–30 million reads). RNA samples received by GENEWIZ LLC were quantified using Qubit 2.0 Fluorometer (Life Technologies) and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies).
2.13. Library preparation and RNA sequencing
Full details of RNA library preparation can be obtained from GENEWIZ LLC (South Plainfield, NJ, USA). Briefly, RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina using manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNAs were initially enriched with Oligo(dT) beads. Enriched mRNAs were fragmented for 15 min at 94 °C. First and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing library was validated on the Agilent TapeStation (Agilent Technologies), and quantified by using Qubit 2.0 Fluorometer (Invitrogen) as well as by quantitative PCR (KAPA Biosystems). The sequencing libraries were clustered on a single lane of a flow cell. After clustering, the flowcell was loaded on the Illumina HiSeq 4000 according to manufacturer’s instructions. The samples were sequenced using a 2×150bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
2.14. Bioinformatic analysis
The sequencing data were uploaded to the galaxy web platform, and the public server usegalaxy.org was used to analyse the data (Afgan et al., 2018). Briefly, raw data in the fastq format were first preprocessed using the sample quality control (QC) program FastQC (Andrews, 2010). QC data were then aggregated using MultiQC (Ewels et al., 2016). All data were then processed through Trimmomatic (Bolger et al., 2014), to remove adapter sequences, poly-N-containing reads, and low-quality reads (Phred score < 25). Paired end reads were then aligned to the mouse genome using HiSAT2 and the GRCm38 (mm10) mouse reference genome (Kim et al., 2015). Duplicate reads were removed, and gene expression in FPKM (Fragments Per Kilobase Million) was measured using FeatureCounts (Liao et al., 2014). Sample data were aggregated into a single data frame using column join, and gene IDs were annotated using the AnnotateMyIDs program (Dunning, 2017). Batch correction was performed using RUVSeq (Risso et al., 2014), and differential expression analysis was performed using the DESeq2 program (Love et al., 2014). Resulting p values were adjusted using the Benjamini and Hochberg approach to control for false discovery rate. Genes identified using DESeq2 that reported a log2FC of >1.0 (absolute value) false discovery rate corrected p value of <0.01 were regarded as differentially expressed genes (DEGs).
2.15. Pathway analysis
Gene ontology analysis was performed using Ingenuity pathway analysis (IPA, QIAGEN). Datasets were filtered for differentially expressed genes, and both significantly up and downregulated genes were analyzed together to identify enriched canonical pathways. The ingenuity knowledge base was used as a reference set to identify significantly enriched pathways following analysis by Fisher’s exact test and the activation or inhibition of enriched pathways was determined with the use of a Z-score.
2.16. Intracisternal tracer injections
The CSF tracer Ova-647 (Ovalbumin-647 conjugate, Cat. No: O34784; ThermoFisher Scientific) was reconstituted in artificial CSF at a concentration of 0.5%. Mice were anesthetized via inhalation using a 3% isoflurane / oxygen mix and fixed in a stereotaxic frame, whereupon the posterior atlanto-occipital membrane overlying the cisterna magna was surgically exposed. Viscous glycerol was applied to the exposed area to help prevent CSF backflow. The tracer was injected into the cisterna using a 33-gauge Hamilton syringe at a rate of 1 μL/min over 10 min with a syringe pump (Harvard Apparatus). The syringe was left in situ for 3 min to prevent reflux. Thirty minutes after the start of the injection, the still deeply anesthetized animals were perfused trans-cardially with 4% PFA. The 30 min time point was selected, as prior reports indicate that after the initial 30 min period of glymphatic tracer influx, the tracer signal begins to diminish, reflecting glymphatic efflux (Iliff et al., 2012). Brains were then removed and post-fixed in 4% PFA for 24 h. Coronal brain slices (100 μm) were acquired using a vibratome and mounted on slides using PROLONG anti-fade gold with DAPI. Pharmacological inhibition of AQP4 was conducted using TGN-020 (Cat. No: SML0136; Millipore Sigma) in 20% Captisol® (CeDex Pharmaceuticals) delivered via intraperitoneal injection, as described previously (Harrison et al., 2020). One hour before CSF tracer infusion, wild type mice (6 months of age) were treated with 250 mg TGN-020 per kg body weight, or vehicle (20% Captisol®).
2.17. Ex vivo imaging of fluorescent CSF tracer
Tracer recirculation along perivascular pathways and into the brain parenchyma was visualized using fluorescent microscopy. For each animal, high-magnification montage images of 5 coronal sections (500 μm apart) were imaged using a confocal microscope LSM 800 (Carl Zeiss AG), the Zen Blue 2.3 (Carl Zeiss AG) software and a 4× objective. The acquisition settings were kept the same for all genotypes within the same experiment. The parenchymal uptake of Ova-647 following cisternal injection was quantified using NIH image J software as described previously (Iliff et al., 2012). Briefly, CSF influx, reported as percent Ova coverage, was measured by quantifying the fractional Ova area of the total coronal slice area above an arbitrary threshold, uniformly applied across all images.
2.18. Statistical analysis
Randomization and blinding procedures were employed. Power analysis was performed on all studies. However, for certain groups, there was a lower number of mice than originally intended, as the very advanced age and the prolonged post-injury period resulted in attrition beyond what could be predicted. Quantitative data were plotted as mean + standard deviation. Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, Inc). The Shapiro-Wilk test was completed to assess normality. Statistical analysis was evaluated for significance as determined by students t-test, one-way, or two-way ANOVA and the Bonferroni’s post hoc test with multiple comparisons, as indicated in the figure legends. For all analyses, a p value of <0.05 was considered statistically significant.
3. Results
3.1. Tau residence in apoE sham mice
To examine the elimination profile of exogenous tau from the brain, we evaluated the time-course by which biotin-labeled monomeric tau is removed from the brain following intracranial injection in apoE3 mice. The half-life of exogenous biotin-labeled monomeric tau residing in the brain was 36.7 min, (Fig. 1a), while the half-life of 10 kDa TxD, which does not readily cross the BBB, was 164.5 min (Fig. 1a). Next, the influence of apoE isoforms on the elimination of exogenous tau species from the brain was evaluated after intracranial injection of biotin-labeled tau at 2 h. While we did observe an apoE-genotype dependent effect on monomeric tau retention in the brain (apoE2 < apoE3 < apoeE4), these results did not reach statistical significance (Fig. 1b). However, the brain residence of aggregate-enriched tau was 60% higher in r-sham apoE4 versus r-sham apoE3 animals, and 2-fold higher than r-sham apoE2 mice (Fig. 1c). Additionally, there was a significant increase in the amount of dextran residing in the brain in apoE4 mice, relative to other apoE isoforms (Fig. 1d). Our prior work demonstrated that endothelial caveolae contribute to tau transit across the BBB. Caveolin-1 levels were evaluated in isolated cerebrovessels from 12 month old apoE mice and were found to be two-fold higher in isolated apoE4 cerebrovessels relative to other apoE genotypes (Fig. 1e), despite diminished tau efflux.
Fig. 1.

Effect of apoE isoform on tau brain residence.
(a) The time course of tau elimination from the brain of apoE3 mice (12 months of age) was established by examining monomeric biotin-labeled tau (btau) levels (n = 6) and 10 kDa LyD (n = 5) levels in the brain at various time points following intracortical injection. Btau content was analyzed using ELISA while LyD was analyzed via fluorescence and normalized to total protein using the BCA protein assay. The half-life for both btau and LyD were determined using nonlinear regression and a one phase decay fit. (b-d) Following intracortical injection in apoE2, apoE3, or apoE4 mice (12 months of age), the amount of exogenous btau and co-injected LyD residing in the brain was determined at 2 h post injection. Monomeric (b) or aggregated (c) btau content was analyzed using an ELISA, while LyD was analyzed via fluorescence. Values represent mean + SEM (n = 5) and are expressed as pg of btau per μg of LyD. *P < 0.05 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (d) Dextran residence values represent mean + SEM (n = 10) and are expressed as μg of LyD per mg protein. *P < 0.05 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (e) Isolated cerebrovascular lysates from apoE2, apoE3 or apoE4 mice (12 months of age) were analyzed for caveolin-1 by ELISA and normalized to total protein using the BCA protein assay. Values represent mean + SEM (n = 5). *P < 0.05 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (f) Schematic diagram of in vitro BBB model used in (g, h). (g) Monomeric btau was added alongside the known paracellular marker 10 kDa LyD to the basolateral compartment of the monoculture or coculture in vitro BBB model. (h) Monomeric or aggregate enriched btau was added alongside LyD to the basolateral compartment of the coculture in vitro BBB model in the presence or absence of the LRP1 antagonist RAP (100 nM). (g, h) Samples were collected from the apical compartment at 0, 30, and 60 min to determine the permeability of btau and LyD across the BBB model. Values represent mean ± SEM (n = 3) and are expressed as the apparent permeability coefficient (Papp). *P < 0.05 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (i) HBMECs were exposed to 1 μg/mL of monomeric btau in the presence or absence of RAP (100 nM) for 1 h at 37 °C. The cell lysates were analyzed for btau content by ELISA and normalized to total protein using the BCA protein assay. Values represent mean + SEM (n = 3). * P < 0.05 as determined by unpaired two-tailed Student’s t-test.
3.2. Effect of pericytes, LRP1 inhibition on tau transcytosis across an in vitro model of the BBB
As BBB transcytosis via caveolin-1 is affected by vascular pericyte density (B. Bell et al., 2023) we examined the effect of pericyte coverage on tau transit and BBB integrity using an in vitro model of the BBB. In comparing the BBB transit of tau (basolateral-to-apical) between a monoculture (endothelia alone) (Bachmeier et al., 2010) and co-culture (endothelia and pericytes) (Eisenbaum et al., 2021) BBB model, there was significantly more tau transit in the absence of pericytes, coinciding with elevated dextran permeability (Fig. 1g). To examine whether noncaveolar transport mechanisms could contribute to tau transit, we evaluated the role of the apoE receptor LRP1, which has been shown to interact with tau in neurons (Rauch et al., 2020), astrocytes (Eisenbaum et al., 2023) and pericytes (Bosworth et al., 2023). We observed a significant reduction in tau transit across the BBB model (2-fold) in the presence of the LRP1 inhibitor, RAP. Pre-treatment of the co-culture model with RAP (100 nM) resulted in a significant decrease in the BBB transcytosis of both monomeric and aggregate enriched tau (Fig. 1h). Of note, neither RAP, nor either tau species had any effect on dextran BBB permeability, compared to control conditions. We also found pre-treatment with RAP significantly decreased tau uptake in HBMECs (Fig. 1i), suggesting an endothelial cell driven, LRP1-mediated tau transport mechanism.
3.3. Tracer efflux altered by apoE
To examine whether r-mTBI or FAD influenced tau residence in an apoE isoform dependent manner, tau residence in the brain was evaluated after intracortical injection of biotin-labeled tau (2 h). Monomeric biotin-labeled tau residence in the brain was increased by 60% in the apoE2 r-mTBI mice and two-fold in E2FAD mice, compared to apoE2 r-sham animals (Fig. 2b). Conversely, no significant difference was observed between r-mTBI or FAD when compared to r-sham apoE3 mice (Fig. 2c). Unexpectedly, in apoE4 mice, monomeric biotin-labeled tau residence in the brain decreased by 50% in r-mTBI mice, and 55% E4FAD mice, compared to apoE4 r-sham animals (Fig. 2d). With respect to the brain residence of aggregate enriched tau, we observed an apoE-dependent effect between the r-sham mice (apoE2 < apoE3 < <apoE4). Furthermore, as observed with the monomeric tau studies, the amount of aggregated enriched tau residing in the brain in r-mTBI apoE4 mice was significantly lower than that observed in r-sham apoE4 animals (Fig. 2e). As outlined in Fig. 1, dextran can be used as an indicator of paracellular permeability/BBB integrity. In our prior work, we observed that dextran residence in the brain was not altered by r-mTBI in WT animals, indicating a lack of BBB changes post-injury, at least at the time points examined (Eisenbaum et al., 2021). To evaluate any potential effects of apoE on BBB integrity, dextran residence in the brain was examined in apoE4 mice in the context of r-mTBI or FAD. Remarkably, dextran residence in the brain was diminished by 40% in both apoE4 r-mTBI and E4FAD animals, relative to apoE4 r-sham mice (Fig. 2f). No other significant differences in dextran brain residence were observed among the other groups, though there was a trend towards increased dextran residence in the brain following r-mTBI in apoE2 mice, relative to apoE2 r-sham animals (Fig. 2f). These results suggest a chronic loss of BBB integrity in response to insult that is unique to the apoE4 genotype. To further interrogate the state of the BBB in the apoE4 animals, we utilized a vessel paint technique as a complementary approach to our dextran studies above. A separate cohort of apoE r-mTBI mice received an intracardiac injection of aqueous DiI solution (VP), which is a tracer that incorporates within the cellular lipid membrane upon contact and becomes strongly fluorescent. When delivered through intracardiac injection, in the absence of BBB breakdown, VP uniformly stains the lumen of endothelial cells, without staining non-endothelial cells (Salehi et al., 2019). In age-matched r-sham and r-mTBI wild-type mice (i.e., murine apoE), there was minimal VP leakage detected (Fig. 2g). In contrast, quantification of VP coverage revealed substantial BBB permeability in apoE4 mice relative to other apoE genotypes, which was exacerbated by r-mTBI (Fig. 2h). Collectively, these results demonstrate that the cerebrovasculature is chronically impaired in apoE4 animals following cerebral insult, consistent with prior reports investigating apoE4 and TBI (Main et al., 2018) or AD (Montagne et al., 2021).
Fig. 2.
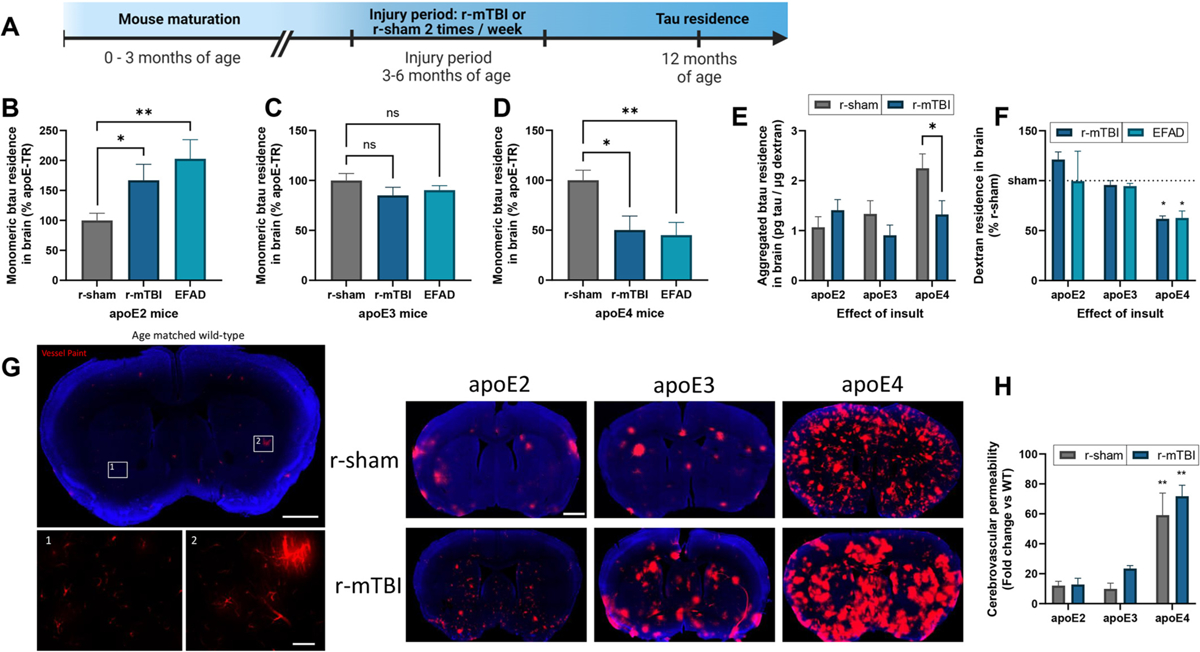
Influence of apoE and insult on tau brain residence.
(a) Timeline of closed head injury paradigm. (b-f) Following intracortical injection in 12 month old r-sham, r-mTBI and EFAD (apoE2, apoE3, and apoE4) mice, the amount of exogenous btau and co-injected LyD residing in the brain was determined at 2 h post injection. Monomeric (b-d) or aggregated (e) btau content was analyzed using an ELISA, while LyD (f) was analyzed via fluorescence. (b-d) Values represent mean + SEM (n = 5), and are expressed as the percentage tau residence normalized to each respective r-sham. *P < 0.05, **P < 0.01 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (e) Values represent mean + SEM (n = 5) and are expressed as pg of aggregated btau per μg of LyD. *P < 0.05 as determined by one-way ANOVA and Bonferroni’s multiple comparisons test. (f) Values represent mean + SEM (n = 10) and are expressed as the percentage dextran residence normalized to each respective r-sham. *P < 0.05 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test. (g, h) Vessel paint was applied to 12 month old r-sham and r-mTBI mice (apoE2, apoE3 and apoE4) via transcardiac injection. (g) Representative images of an age matched wild-type mouse showing vessel paint (red) leakage into the cortex with Dapi staining (blue). Insets 1 and 2 show boxed areas in (g). (h) Cerebrovascular permeability was quantified as the percent area of total cortex vessel paint signal normalized to age matched wild-type mice. Values represent mean + SEM (n = 3) and are expressed as the fold change normalized to age matched wild-type mice. *P < 0.05 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test.
3.4. TBI, apoE influence pericyte coverage
Pericytes maintain BBB permeability by regulating paravascular leakage and cellular transcytosis, and it has been shown that a loss of pericyte coverage can result in significant macromolecular tracer leakage (R. D. Bell et al., 2010). Furthermore, the altered cerebrovascular expression of endothelial caveolin-1 observed in r-sham apoE4 mice is also indicative of pericyte dysfunction, which is known to be progressively dysregulated following head trauma (Eisenbaum et al., 2021; Eser Ocak et al., 2020). Therefore, CD13 positive pericyte coverage was evaluated by immunofluorescence in a cohort of apoE r-sham and r-mTBI mice (Fig. 3a). Compared to other apoE isoforms, r-sham apoE4 mice demonstrated a substantial loss of pericyte coverage in the cortex (Fig. 3b). Following r-mTBI, the effect of r-mTBI on pericyte coverage was most predominant in apoE2 and apoE3 mice, whereas pericyte coverage in the apoE4 r-mTBI mice diminished to a lesser extent, relative to apoE4 controls (Fig. 3b).
Fig. 3.
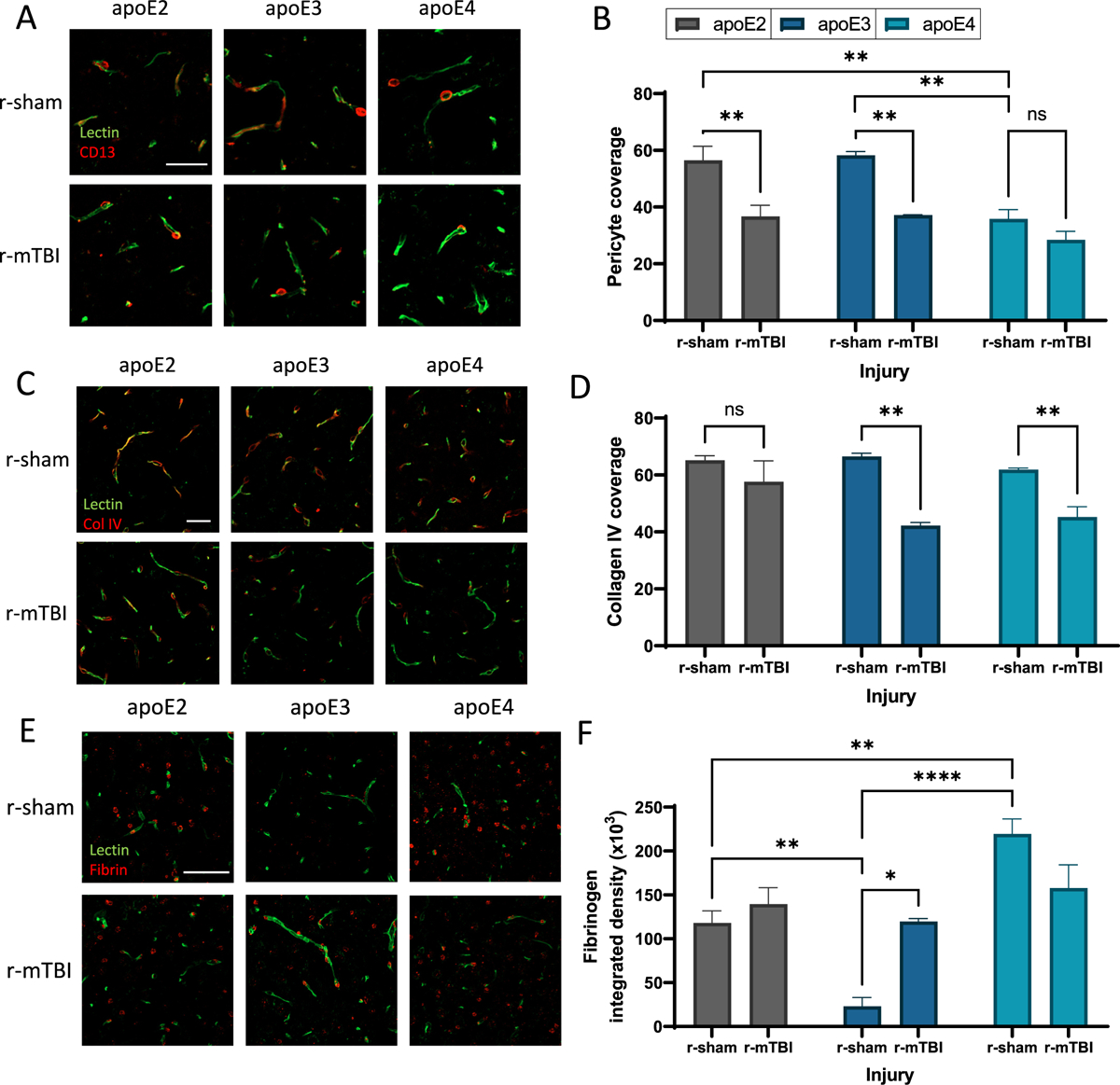
Influence of apoE and insult on pericyte coverage and BBB integrity.
(a) Confocal microscopy of CD13 immunodetection showing pericyte coverage (red) of lectin-positive brain capillaries (green) in the somatosensory cortex of 24 month old r-sham and r-mTBI mice (apoE2, apoE3 and apoE4). Scale bar, 25 μm. (b) Quantification of pericyte coverage on capillaries in r-sham and r-mTBI (apoE2, apoE3 and apoE4) mice. Values represent mean + SEM (n = 4). **P < 0.01 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test. (c) Confocal microscopy of Collagen IV (red) and lectin-positive brain capillaries (green) in the somatosensory cortex of 24 month old r-sham and r-mTBI mice (apoE2, apoE3 and apoE4). Scale bar, 25 μm. (d) Quantification of Collagen IV coverage of lectin-positive capillaries. Values represent mean + SEM (n = 4). **P < 0.01 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test. (e) Confocal microscopy of fibrin (red) and lectin-positive brain capillaries (green) in the somatosensory cortex of 24 month old r-sham and r-mTBI mice (apoE2, apoE3 and apoE4). Scale bar, 50 μm. (f) Quantification of extravascular pericapillary fibrinogen deposits. Values represent mean + SEM (n = 4). **P < 0.01 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test.
3.5. TBI, apoE influence extravascular leakage
Pericyte regulation of the BBB is partially modulated by the neurovascular basement membrane, which may be compromised following injury or mural cell dysfunction. Following r-mTBI, there were significant reductions in the microvascular coverage of collagen IV in apoE3 and apoE4 mice compared to their respective r-sham controls (Fig. 3c, d). Also, there was no significant difference in collagen IV coverage between r-sham apoE3 and apoE4 mice, consistent with prior reports of mice at this age (Oue et al., 2023). Loss of BBB integrity due to diminished pericyte coverage and a compromised basement membrane can enable the brain penetrance of macromolecules such as fibrin(ogen) (MW = 340 kDa) from the blood (R. D. Bell et al., 2010). A loss of pericyte coverage is frequently associated with increased regional accumulation of neurotoxic fibrinogen deposits (R. D. Bell et al., 2012; Montagne et al., 2018), and our studies observed a substantial increase in abluminal fibrinogen deposits in control apoE4 mice, relative to the other apoE genotypes (Fig. 3e, f). Notably, apoE2 control mice had a higher fibrinogen load (5-fold more) than apoE3 control mice (Fig. 3e, f). Fibrinogen deposition increased significantly in r-mTBI apoE3 mice compared to r-sham apoE3 animals, to level consistent with injured apoE2 mice (Fig. 3e, f). There was no significant injury effect on fibrinogen deposition in apoE2 or apoE4 mice compared to their respective shams.
3.6. Impaired neurovascular and glymphatic pathways by r-mTBI in apoE4 astrocytes
Our prior work found that astrocytes process and eliminate extracellular tau through an LRP1-mediated pathway, which was impaired by r-mTBI in an apoE isoform dependent manner (Eisenbaum et al., 2023). To examine how r-mTBI affects astrocytic function, we performed RNA sequencing on astrocytes isolated from apoE-Tr mice (7 months of age) in the context of r-mTBI (3 months post injury) (Fig. 4a). While there was no significant differences among apoE isoforms in the sham mice (Fig. 4b), RNA sequencing identified a total of 2297 differentially expressed genes (DEGs) and 507 DEGs in apoE4 and apoE2 r-mTBI vs r-sham astrocytes, respectfully (Fig. 4b). No significant injury effect on DEGs was observed in apoE3 astrocytes. Analysis of overlapping DEGs between r-mTBI apoE2 and apoE4 astrocytes revealed 415 shared DEGs. Notably, we observed that these common DEGs demonstrated a consistent directional shift across genotypes, with 135 upregulated DEGs and 280 downregulated DEGs (Fig. 4b–d). Using a hypothesis driven analysis of the overlapping DEGs, we observed the down-regulation of several genes such as Sox9, Slc1a2, Ptch1, AQP4, Dag1, Gja1, and Kcnj16. The diminished expression of this core astrocyte signature suggests the adoption of an atypical reactive astrocyte phenotype, reflecting a loss of homeostatic astrocytic functions. The adoption of this phenotypic signature is consistent with prior reports of mTBI-induced BBB leakage (George et al., 2022) (Fig. 4e), with the most pronounced effect observed in apoE4 astrocytes. The apoE4-associated vascular frailty demonstrated by injured apoE4 mice in Figs. 2 and 3, may contribute to the enriched atypical astrocyte gene signature in r-mTBI apoE4 astrocytes. To further assess how r-mTBI and apoE genotype impact key biological processes in astrocyte, we performed gene enrichment using Ingenuity pathway analysis (IPA). Enrichment analysis of DEGs between apoE4 r-mTBI vs r-sham astrocytes revealed that neurovascular coupling was significantly inhibited following injury (Fig. 4f, g). Furthermore, we identified several other dysregulated canonical pathways associated with altered neurovascular support (Fig. 4f, g) in apoE4 r-mTBI vs r-sham astrocytes, including those associated with WNT/β-catenin signaling, leukocyte extravasation, and inhibition of MMPs. Analysis of those dysregulated pathways predicted that r-mTBI astrocytes may impair processes critical to CSF-ISF influx and BBB integrity, and are exacerbated by apoE4.
Fig. 4.
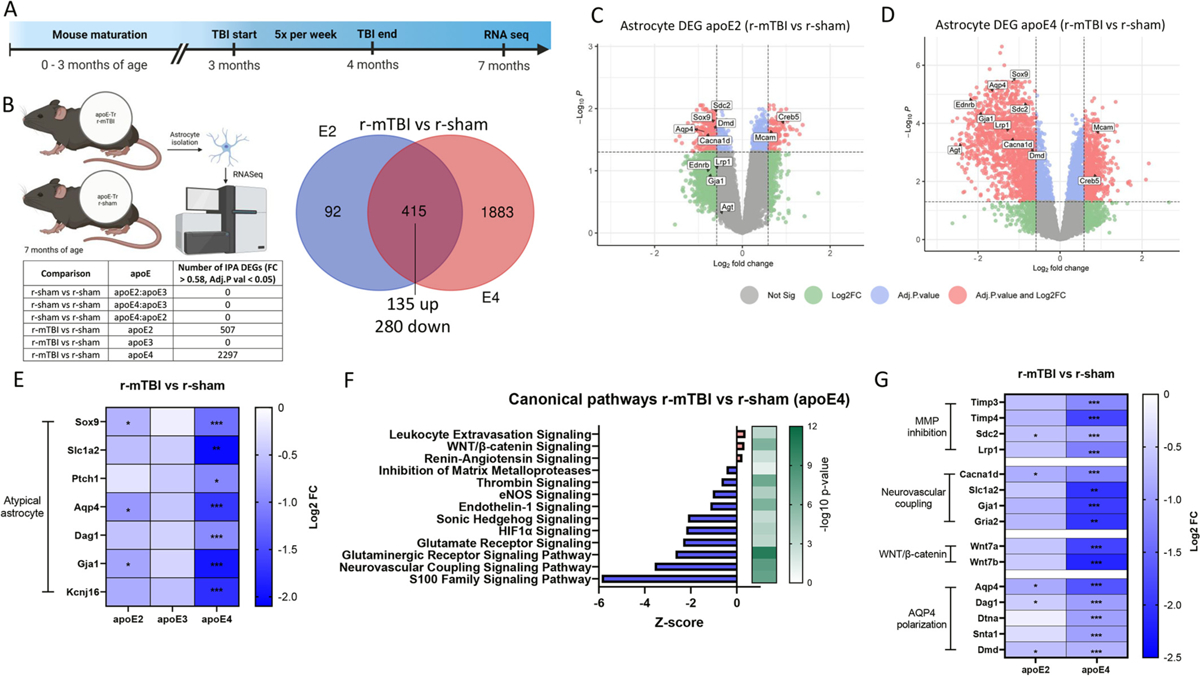
Enriched atypical reactive astrocyte profile by apoE4 in r-mTBI mouse model.
(a-f) RNA was extracted from astrocytes isolated from 7 month old r-sham and r-mTBI (apoE2, apoE3 and apoE4) mice, and gene ontology analysis of r-mTBI astrocytes (versus each respective r-sham) was performed to identify enriched canonical pathways. (a) Timeline of accelerated closed head injury paradigm. (b) Venn diagram of the significantly differentially expressed genes (DEGs) affected by injury for apoE2 and apoE4 (adjusted P < 0.05, |fold change| > 0.58). All common DEGs between apoE2 and apoE4 r-mTBI (compared to their respective shams) changed in a consistent direction. (c, d) Volcano plots of DEGs in r-mTBI versus sham astrocytes isolated from (c) apoE2 and (d) apoE4 mice (significant cut off set at adjusted P < 0.05, |fold change| > 0.58, red points indicate DEGs above both the adjusted p value and Log2FC cut off threshold). (e) Heatmap showing fold change (FC) values (compared to respective r-shams) of genes correlating with the atypical reactive astrocyte phenotype signatures across injury groups. (f) Results of enrichment analysis comparing the DEGs in astrocytes isolated from r-mTBI and r-sham apoE4 mice to the ingenuity knowledge base, showing top significantly dysregulated canonical pathways. Orange, upregulated; blue, downregulated genes/functions. (g) Heatmap showing downregulated FC values compared to values in respective r-shams of DEGs involved in Matrix metalloprotease inhibition (Timp3, Timp4, Sdc2, Lrp1), Neurovascular coupling (Cacna1d, Sl1a2, Gja1, Gria2), and WNT/β-catenin signaling (Wnt7a, Wnt7b), and glymphatic system-associated AQP4 polarization (Aqp4, Dag1, Dtna, Snta1, Dmd). Significance reflects adjusted P < 0.05, |FC| > 0.58.
3.7. TBI, apoE influence AQP4 polarization
AQP4 polarization is sensitive to both pericyte coverage (Munk et al., 2019) and fibrinogen deposition (Eide and Hansson, 2020). Pericytes express several key components including laminin α2 and agrin (Vanlandewijck et al., 2018) that bind to and anchor the astrocytic dystrophin-associated complex (DAC) for AQP4, enabling perivascular polarization of AQP4 in astrocyte endfeet (Munk et al., 2019). Diminished DAC function and AQP4 polarization can stagnate ISF flow and impair glymphatic clearance (Simon et al., 2022). Notably, our transcriptomic analysis demonstrated a significant decrease in AQP4 expression following r-mTBI, particularly in apoE4 animals (Fig. 4E), and characteristic of prior atypical astrocyte reports (George et al., 2022). The expression of Wnt7a and Wnt7b, which are required to maintain astrocyte endfoot integrity and AQP4 polarization (Guérit et al., 2021), were also repressed by r-mTBI in apoE4 astrocytes (Fig. 4g), predicting AQP4 delocalization. We next examined whether the diminished neurovascular health and astrocyte endfoot integrity predicted by our transcriptomic analysis of injured apoE4 astrocytes impacted other structural components of the DAC. We found that all endfoot-enriched genes affiliated with the DAC (e.g., AQP4, Snta1, Dmd, Dtna and Dag1), were significantly reduced by r-mTBI, particularly in apoE4 animals (Fig. 4h). Collectively, the transcriptomic analysis predicts that r-mTBI-induced BBB dysfunction may impair glymphatic clearance through multiple mechanisms (e.g., disrupted neurovascular coupling and depolarized AQP4). AQP4 becomes more depolarized with age (Kress et al., 2014), which does not significantly influence the r-mTBI mice evaluated by RNAseq (7 months of age), but would substantially contribute to the r-mTBI mice examined in Figs. 1–3. Therefore, we examined AQP4 polarization towards the vasculature and away from the neuropil in 24 month-old r-mTBI mice by immunofluorescence (Fig. 5a, b). Exposure to r-mTBI significantly reduced AQP4 polarization in apoE2 and apoE3 mice, relative to their respective controls (Fig. 5b). Notably, uninjured apoE4 carriers were demonstrated the same degree of AQP4 depolarization as injured mice (apoE2, apoE3 and apoE4), reflecting the chronic BBB instability described demonstrated in Figs. 2 and 3.
Fig. 5.
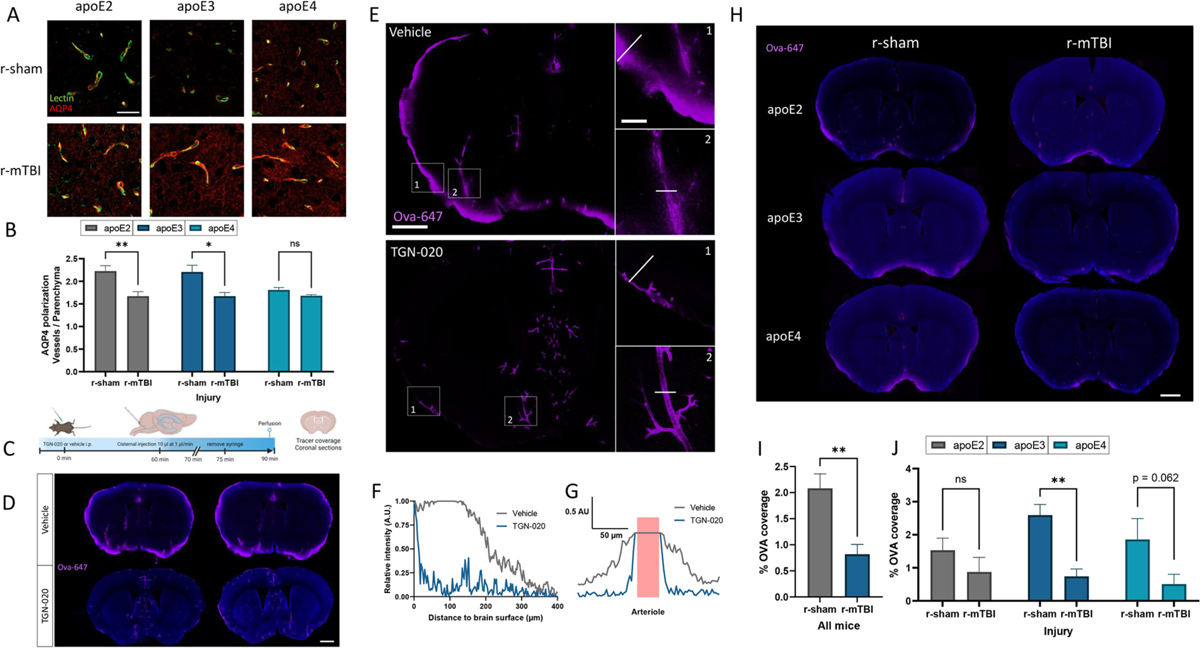
Influence of apoE and insult on glymphatic influx.
(a) Confocal microscopy of AQP4 (red) and lectin-positive brain capillaries (green) in the somatosensory cortex of 24 month old r-sham and r-mTBI mice (apoE2, apoE3 and apoE4). Scale bar, 25 μm. (b) Quantification of AQP4 polarization measured as the ratio of signal around the capillary divided by the signal in the surrounding parenchyma. Values represent mean + SEM (n = 4). *P < 0.05, **P < 0.01 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test. (c-g) The effect of pharmacological inhibition of AQP4 on the CSF-ISF exchange was examined in wild-type mice (6 months) by injecting TGN-020 (250 mg/kg) or vehicle i.p., followed by an intracisternal injection of the CSF tracer ovalbumin-647. 30 min after the intracisternal injection, mice were perfused with 4% PFA and the distribution of the CSF influx tracer was probed in brain slices. (d) Representative confocal microscopic images showing Ova (magenta) influx with Dapi staining (blue). Scale bar, 1.0 mm. (e) Representative images showing Ova (magenta) distribution and influx in coronal sections. Insets 1 and 2 show the boxed areas in (e). Scale bar full images, 1.0 mm. Scale bar insets, 250 μm. (f) Tracer penetration depth profile, normalized to the fluorescence at the pial surface of a coronal section. The line in insert 1 was placed orthogonal to the cortical surface at the most dorsal position in the TGN-020 treated mouse where tracer could be found at the pial surface, and depth was measured at the same position in the untreated mouse. (g) Tracer perivascular influx profile, normalized to the fluorescence at the pial surface of a cortical section. The line in insert 2 was placed across a penetrating artery, and distribution of Ova was assessed from the arterial lumen. Vessel lumen in pink. (h-j) The effect of r-mTBI on CSF-ISF exchange was examined in 24 month old r-sham and r-mTBI (apoE2, apoE3, and apoE4) mice by injecting the CSF tracer Ova into the cisterna magna. Brains were harvested 30 min following the injection, and the Ova influx was quantified. (h) Representative confocal microscopic images showing Ova (magenta) coverage with Dapi staining (blue). Scale bar, 1.0 mm. (i, j) Quantification of Ova coverage on brain coronal sections. (i) Values represent mean + SEM (n = 12). (j) Values represent mean + SEM (n = 4). **P < 0.01 as determined by two-way ANOVA and Bonferroni’s multiple comparisons test.
3.8. CSF influx diminished at 6 months post injury
Perivascular CSF influx depends, in part, on microvascular integrity and polarization of AQP4 (Munk et al., 2019). Prior reports have demonstrated an association between CSF inflow and tau elimination (Harrison et al., 2020; Ishida et al., 2022), which are both diminished in the early stages following neurotrauma (Iliff et al., 2014), though it is unclear whether this glymphatic impairment persists at a chronic stage following mild TBI. Thus, we evaluated the influence of r-mTBI and apoE on CSF influx at 18 months post injury. First, to validate our approach, we examined the influence of the AQP4 inhibitor TGN-020 on the influx and distribution of Ova-647 following cisterna magna infusion (Fig. 5c). Compared to vehicle, application of TGN-020 dramatically reduced Ova-647 influx (Fig. 5d) and penetration (Fig. 5e), both from the cortex surface and from arterioles (Fig. 5f, g). To determine whether glymphatic function was impaired at a chronic stage following r-mTBI, the CSF tracer Ova was delivered via cisterna magna infusion, and mice were sacrificed after 30 min. Using a common technique to assess CSF influx (Drieu et al., 2022; Iliff et al., 2012; Kress et al., 2014), Ova coverage was quantified in vibratome coronal sections. Compared to r-sham mice, CSF tracer penetration into the brain was significantly reduced at 12 months following r-mTBI (Fig. 5i). When stratified by apoE genotype, there was no significant effect of apoE genotype on CSF influx in uninjured animals (Fig. 5j). However, we did observe a significant reduction in CSF influx in apoE3 mice exposed to r-mTBI, followed by apoE4 and apoE2, when compared to their respective r-sham controls (Fig. 5j). It is important to note that age alone can significantly impair CSF dynamics in the brain, (Drieu et al., 2022; Kress et al., 2014), potentially minimizing any apoE-genotype or r-mTBI driven effects at this advanced age. Overall, the CSF tracer penetration profile in our studies was consistent with prior reporting (Drieu et al., 2022), and exposure to repetitive head trauma results in a persistent reduction in CSF dynamics, which may be a contributing factor in the development of pathogenic proteinopathies.
4. Discussion
Emerging reports have investigated the influence of apoE isoforms on tau propagation and pathology, with mixed results (Davies et al., 2023; Y. Shi et al., 2017; Williams et al., 2023; Zhao et al., 2018). Despite the accumulating observations into this topic, there is still very little understanding regarding the mechanisms by which extracellular tau is eliminated from the brain, and whether tau dynamics are modulated by apoE isoforms. To investigate whether apoE isoforms influence tau elimination from the brain, we evaluated the retention of monomeric or aggregated tau species in the brains of apoE-Tr mice following exogenous intracerebral administration, using a previously established method (Eisenbaum et al., 2021). We observed a greater retention of exogenous tau in the brains of apoE4 animals compared to mice expressing other apoE isoforms, indicating reduced tau elimination from the brain when apoE4 is expressed. These results are consistent with prior studies investigating Aβ retention in the brain, where both mice expressing apoE4 (Castellano et al., 2011), and mice with apoE4 infused into the ISF (Verghese et al., 2013), exhibited increased Aβ retention in the brain and elevated levels of Aβ in the ISF.
There are several potential mechanisms by which apoE may influence tau elimination from the brain. Like Aβ (Kanekiyo et al., 2014) neurovascular tau clearance is achieved through a combination of receptor-mediated efflux, caveolae-driven transcytosis, and/or perivascular drainage. We recently found that caveolin-1 contributes to tau transit across the BBB (Eisenbaum et al., 2021). Arterioles demonstrate significant caveolae vesicular transcytosis from the perivascular space to the blood (B. Bell et al., 2023), potentially enabling tau transit in larger vessels, however caveolae-mediated transit may be attenuated to some degree by the presence of pericytes in brain capillaries (Andreone et al., 2017). Prior reports have linked apoE4 with diminished pericyte health and coverage (Barisano et al., 2022), which would precipitate elevated caveolae-mediated transcytosis. Indeed, the current studies showed elevated cerebrovascular caveolin-1 expression in apoE4 vessels, relative to other apoE isoforms. Furthermore, using an in vitro BBB model, we found that the presence of pericytes increases BBB integrity overall, and significantly reduces tau transit, likely due in part to the suppression of caveolin-1 expression. Despite the regulatory effect of vascular pericytes on caveolin-1 activity at the BBB, additional pathways are present that also contribute to the movement of tau across the BBB.
As apoE4 mice demonstrated reduced tau clearance from the brain, despite increased cerebrovascular caveolin-1 levels, we examined other tau transport mechanisms and the influence of apoE genotype on these processes. LRP1 was recently identified as a molecular point of convergence for AD and CTE (Cooper et al., 2021), due to its critical role in tau trafficking alongside other disease-associated proteins including apoE and Aβ. Prior reports from our lab and others (Storck et al., 2016) have demonstrated that LRP1-mediated transit from the brain is influenced by apoE isoforms (Bachmeier et al., 2014). Interestingly, despite in vitro evidence for tau-apoE interactions (Strittmatter et al., 1994), direct binding between apoE and tau physiologically has been inconclusive. Rather, apoE may influence tau extracellular dynamics indirectly by interfering with LRP1-mediated transport. This is well characterized for Aβ, as apoE competitively inhibits LRP1 binding to Aβ in an isoform dependent manner (Verghese et al., 2013), and the apoE4 isoform in particular has a much stronger binding affinity to LRP1 than other apoE isoforms (Cooper et al., 2021). Recent reports from our lab and others have demonstrated that LRP1 contributes to tau endocytosis in both astrocytes (Eisenbaum et al., 2023) and pericytes (Bosworth et al., 2023). While cerebrovascular LRP1 is widely known to facilitate Aβ clearance at the BBB, its contribution to tau elimination from the brain has been under investigated. We found that LRP1 inhibition decreased tau uptake in brain endothelia and significantly diminished the BBB transit of both monomeric and aggregated tau species in a capillary co-culture model of the BBB. In terms of the manner by which apoE4 may impact tau transport via LRP1, the apoE4 isoform may competitively inhibit tau binding to LRP1 (Cooper et al., 2021), as has been demonstrated for Aβ (Montagne et al., 2021; Storck et al., 2016). ApoE4 can also indirectly modulate LRP1 activity through intracellular LRP1 sequestration (Prasad and Rao, 2018) which impairs the endosomal trafficking and processing of subcellular cargo (Narayan et al., 2020). Finally, we previously demonstrated that endothelial LRP1 ectodomain shedding is elevated in the presence of apoE4, which prevents the receptor from transporting extracellular ligands (Bachmeier et al., 2014). These findings, alongside our prior reporting on caveolintau efflux (Eisenbaum et al., 2021) suggests the cerebrovasculature uses several mechanisms for tau transport, each of which are impacted by apoE genotype.
Elevations in extracellular tau are commonly observed after head trauma (Marklund et al., 2009), (due in part to impaired tau elimination that results in prolonged tau residence in the brain (Eisenbaum et al., 2021). Our prior work found that cerebrovessels isolated from TBI or AD mice exhibited diminished cerebrovascular uptake of both tau and Aβ (Ojo et al., 2021). AD and TBI are associated with diminished endothelial LRP1 (Pop et al., 2013; H. Shi et al., 2020), and cerebrovascular cav-1 expression (Alsaqati et al., 2023; Ojo et al., 2021). In line with our prior findings examining the influence of r-mTBI on tau residence in the brain in wild-type animals (Eisenbaum et al., 2021), apoE2 mice exposed to r-mTBI (or FAD) displayed higher levels of tau residing in the brain compared to r-sham animals, indicating reduced tau elimination from the brain post-injury. Along these lines, it has been reported that apoE2 carriers have a higher incidence of primary tauopathies like PSP and CBD than other apoE genotypes (Zhao et al., 2018). Notably, the effect of r-mTBI or FAD on tau elimination from the brain was not observed in apoE3 mice. Interestingly, compared to murine apoE or other apoE isoforms, the presence of the apoE3 isoform substantially extended the lifespan in a mouse model of tauopathy (Williams et al., 2023). In line with our observations, the authors suggested that, relative to other isoforms, apoE3 confers some level of systemic resistance to the pathological effects of tau (Williams et al., 2023). With that being said, recent work from different groups have demonstrated tau associated decline to be worse for each apoE genotype: apoE2 (Zhao et al., 2018), apoE3 (Williams et al., 2022) and apoE4 (Y. Shi et al., 2017). These inconsistencies may be due in part to differences in tau models or measured outcomes, as apoE2 was worse for tau pathology (Zhao et al., 2018), apoE3 was worse for the spread of phosphorylated tau (ptau) (Williams et al., 2022), and apoE4 was worse for neurodegeneration in the presence of tau (Y. Shi et al., 2017). It may be the case that apoE2 can facilitate tau propagation to a greater degree than other apoE genotypes, while apoE4 is more vulnerable to the presence of neurotoxic tau species, but at this time, the mechanisms by which apoE isoforms influence pathological tau under different conditions are not currently understood.
As our apoE studies above indicated tau removal from the brain was significantly reduced with the apoE4 allele compared to other apoE genotypes, we anticipated head trauma would exacerbate the effect of apoE4 on tau retention in the brain. Unexpectedly, we found apoE4 mice under disease conditions (r-mTBI or FAD) showed an apparent increase in tau elimination from the brain. To further understand the influence of apoE4 on tau elimination from the brain, we interrogated the brain residence of a dextran tracer, which is a known paracellular marker that does not readily cross the BBB (Natarajan et al., 2017). We observed that apoE4 mice exposed to either r-mTBI or FAD both had significantly less tracer remaining in the brain, indicating substantial disruptions in BBB integrity and/or leakage from the brain to the periphery, compared to other apoE isoforms. Furthermore, a vessel paint marker detected substantial microvascular breakdown in both injured and sham apoE4 mice. Like cadaverine (Heithoff et al., 2021), both the size (< 1 kDa) and rapid cellular uptake of the vessel paint enables the detection of abnormal BBB integrity in vessels that otherwise appear intact (Munoz-Ballester et al., 2022), in contrast to larger molecules (≥ 10 kDa) which remain impermeable.
Observations of these subtle, apoE4-associated alterations in BBB integrity are consistent with prior reports (Halliday et al., 2016), and more recent work found accelerated BBB leakage of small molecules in apoE4 mice in the absence of larger BBB breakdown and fibrinogen deposition (Oue et al., 2023). However, these apoE4-associated alterations can enhance the microvascular susceptibility to rupture in the midst of insults such as r-mTBI or AD, leading to the extensive leakage of larger proteins like dextran (10 kDa) or tau (45 kDa). The lack of tau residing in the brain in the apoE4 animals with r-mTBI or AD in our studies is likely due to increased bi-directional cerebrovascular permeability as opposed to enhanced transport of tau out of the brain in these animals. Thus, our approach to evaluating tau elimination from the brain through exogenous intracerebral tau injection would not be amenable to this animal cohort as the aberrant vascular leakage would override the detection of any potential deficiencies in tau removal from the brain following r-mTBI and AD in the apoE4 animals. These results suggest that non-apoE3 isoforms are risk factors for injury-associated BBB dysfunction, though they may do so through different mechanisms. In AD for example, apoE2 carriers that develop AD have a different, nonamnestic, clinical presentation characterized by an increased severity of microbleeds and small vessel disease (Groot et al., 2018), with more fibrinogen deposition in the vessel wall (McCarron et al., 1999), whereas apoE4 carriers develop a more pronounced breakdown of the BBB at the capillary level. Our findings, and the reporting of others, suggest the apoE isoforms differentially promote neurovascular degeneration associated with head trauma and AD.
The BBB regulates the influx of nutrients like oxygen and glucose into the brain, while at the same time mitigating the influx of toxic blood-borne factors like fibrin, in addition to removing cerebral waste products and neurotoxic proteins like tau. BBB dysfunction precedes the clinical manifestations of AD and is characterized by pericyte loss alongside changes in paracellular and transcellular permeability. Individuals with apoE4 demonstrate progressive, life-long neurovascular degeneration, characterized by a loss of mural cell coverage and the extravasation of blood borne factors like fibrin (R. D. Bell et al., 2012). As apoE4 carriers are less capable of maintaining cerebrovascular homeostasis (Yamazaki et al., 2019), they are more susceptible to injury- or insult- induced neurovascular decline. As our prior work demonstrated a loss of pericyte health following r-mTBI (Ojo et al., 2021), we continued this line of investigation by examining pericyte coverage in the context of apoE genotype. We found that apoE4 microvessels had diminished mural cell coverage relative to other apoE isoforms, reflecting prior reports of substantial pericyte coverage loss in apoE4 mice, even in the absence of injury (Barisano et al., 2022; R. D. Bell et al., 2012; Montagne et al., 2021). Moreover, r-mTBI resulted in a loss of mural cell coverage for both apoE2 and apoE3 cerebrovessels, to a level similar to that observed in apoE4 r-sham mice.
Loss of pericyte functionality and/or coverage is associated with the parenchymal infiltration of immune cells, fibrinogen deposition, and dysregulated angiogenesis (Inoue et al., 2023). The BBB can follow a biphasic pattern after TBI, where it becomes more permeable following the initial injury, returns to homeostasis as pericytes and astrocytes restore BBB integrity (though this is impaired when apoE4 is present (Main et al., 2018)), but can potentially become more permeable at a more chronic phase post-injury (Gama Sosa et al., 2021), particularly when exposed to subsequent insults. Chronic, dysfunctional pericyteendothelial signaling can result in BBB disruption and basement membrane degeneration (Oue et al., 2023) via matrix metalloproteases (MMPs), even in the absence of altered pericyte coverage (Selhorst et al., 2022), which can be precipitated by TBI (Bhowmick et al., 2019). In fact, diminished functional collagen IV coverage is associated with micro-hemorrhage indicative of small vessel disease, particularly following TBI (Gould et al., 2006). Our prior work demonstrated an isoform-dependent effect of apoE on the regulation of MMP9 activity (apoE2 > apoE3> > apoE4) (Ringland et al., 2020). Consistent with these reports, we observed an injury-associated reduction of microvascular collagen IV coverage in apoE3 and apoE4 animals, while the apoE2 mice showed minimal changes in collagen IV post-injury.
The loss of pericyte coverage is associated with vascular frailty and pronounced BBB breakdown, which may facilitate the extravasation of larger blood-borne proteins including fibrinogen (R. D. Bell et al., 2010). In our studies, we observed that uninjured apoE4 mice at 24 months of age had extensive fibrinogen extravasation, which remained elevated following TBI. Compared to apoE4 animals, fibrinogen levels were not as high in apoE2 mice, and showed a non-significant increase following r-mTBI. Uninjured apoE3 mice had very low levels of fibrinogen, consistent with prior reporting (Montagne et al., 2021), but we did observe a significant increase following r-mTBI. These results are consistent with previous studies indicating that exposure to r-mTBI results in sustained, chronic neurovascular degeneration, even at a prolonged time point following head trauma (18 months post-injury). Notably, apoE2 mice had enhanced fibrinogen deposition at this advanced age, relative to age-matched apoE3 mice, consistent with clinical reports indicating that fibrinoid necrosis is most prevalent in vascularly compromised apoE2 carriers (McCarron et al., 1999). Following brain trauma or neurovascular injury, fibrin can leak into the brain through larger areas of BBB rupture and/or extravasate into the brain through non-ruptured microvessels via caveolar transcytosis (Sulimai et al., 2021). Fibrinogen can then deposit in the vascular-astrocyte endfeet interface (Muradashvili et al., 2017), where it can physically detach astrocytic endfeet from the BBB, and impair astrocytic homeostasis (Sulimai et al., 2021). Fibrinogen deposition can induce a wide range of neurotoxic insults, including white matter damage and demyelination (Montagne et al., 2018), as well as glial activation and AQP4 mislocalization (Eide and Hansson, 2020). Collectively, these results indicate that neurovascular degeneration is strongly influenced by apoE isoform expression, which could influence the susceptibility to traumatic microbleeds (TMB), and vascular dementia following brain injury.
Traumatic microbleeds, which persist for months or years after injury, are linked to worse clinical outcomes (Griffin et al., 2019) and are highly prevalent after mTBI in both mice (George et al., 2022) and human subjects (Griffin et al., 2019). While astrocytes play a dynamic role in cerebrovascular recovery following injury, sustained exposure to blood plasma proteins (≤ 10 kDa) due to microvascular leakage can induce a reactive astrocytic phenotype characterized by the loss of many proteins involved in brain homeostasis (George et al., 2022), which may be exacerbated by disease risk polymorphisms like apoE (Escartin et al., 2021). The observations in this study and others (Main et al., 2018) suggest that the BBB restoration that typically occurs following r-mTBI is attenuated in apoE4 carriers. Furthermore, we recently observed that apoE4 astrocytes from r-mTBI mice demonstrated chronically altered functionality (Eisenbaum et al., 2023), suggesting that apoE4 astrocytes may be more susceptible to this phenotype following brain injury. To expand upon this prior work, we performed RNA sequencing on isolated astrocytes from r-mTBI and r-sham apoE mice (7 months old) exposed to an accelerated injury model. This accelerated model was selected to specifically assess the impact of injury on apoE astrocyte DEGs, prior to the manifestation of any confounding age-associated DEGs among apoE sham astrocytes that may arise at the age of the mice used in the non-accelerated injury cohort. Consistent with previous reports (Lozupone and Panza, 2024), there was no genotype effect on astrocyte DEGs from sham mice at this age. However, we found that astrocytes from injured mice exhibited a DEG signature indicative of blood-borne protein extravasation, and subsequent atypical astrocyte reactivity (George et al., 2022), which was consistent among mice exposed to r-mTBI, but most significantly altered in apoE4 animals (apoE4 > apoE2 > apoE3).
Canonical pathway analysis of apoE4 r-mTBI vs r-sham predicted several mechanisms of injury induced alterations in astrocytic neurovascular support (Michinaga and Koyama, 2019), including dysregulated SHH, Wnt/β-catenin, Endothelin-1, and eNOS signaling, that are associated with a loss of BBB integrity (Yue and Hoi, 2023). The heightened susceptibility of apoE4 astrocytes to microvascular leakage-associated reactivity following injury is consistent with prior reports (Main et al., 2018), as apoE4 carriers have diminished endothelial junction restrictiveness (Nishitsuji et al., 2011), and other microvascular alterations (Oue et al., 2023) culminating in a diminished capacity to maintain homeostatic function (Steele et al., 2022), particularly after an additional insult (e.g., TBI). Critically, the cohort of mice used for transcriptomic analysis in the current study were 7 months of age, with a relatively short post injury time (3 months), reflecting an early degenerative progression following injury that was exacerbated by apoE4. Recent transcriptomic analysis of human CTE brains found that DEGs in apoE4 carriers were indicative of dysfunctional homeostasis processes that occurred prior to the onset of degenerative decline, and may predispose these individuals to the development of more severe pathology (Labadorf et al., 2023). Correspondingly, single nucleus RNA sequencing of astrocytes from human AD brains revealed a spectrum of reactivity largely characterized by a substantial loss of homeostatic genes (Dai et al., 2023). These findings suggest that initial differences in response to r-mTBI between apoE isoforms, particularly with respect to neurovascular homeostasis, may contribute to worse outcomes at later stages of the disease process (Atherton et al., 2022).
Waste removal relies heavily on net fluid transport, irrespective of the driving forces or pathways (Segeroth et al., 2023). Disruption of one mechanism of solute exchange, without adequate compensation by other solute exchange systems, will ultimately result in solute accumulation over time (Segeroth et al., 2023). Like Aβ (Wardlaw et al., 2020), extracellular tau is eliminated from the CNS though a combination of BBB transport and CFS-ISF exchange. Dynamic changes in arterial diameter, influenced by forces including vasomotion (van Veluw et al., 2020) and arterial pulsation (Iliff et al., 2013), drives CSF from the subarachnoid region into the brain along the perivascular space (Holstein-Rønsbo et al., 2023), where it disperses into the neuropil, and facilitates the movement of solutes towards the perivascular space for elimination. Moreover, AQP4 channels pump the ISF out of the neuropil, enabling the influx of new CSF from the perivascular space. The drainage of ISF along the perivascular space helps disperse solutes to BBB transporters for elimination, while at the same time preventing localized solute accumulation due to ISF stagnation. Our prior observations suggest that the apoE4 isoform impairs the capacity of LRP1 at the BBB to export pathogenic proteins like tau and Aβ (Bachmeier et al., 2014) out of the brain, potentiating a greater reliance on this drainage pathway to eliminate waste. Pathogenic proteins and waste that are not eliminated via BBB transporters as they travel along this pathway are ultimately removed from the brain to the peripheral lymphatic system.
Interestingly, several of the pathways that are known to facilitate extracellular solute clearance are diminished in atypical astrocytes (George et al., 2022) and were similarly impacted by injury in the current study, particularly in apoE4 mice. Neurovascular coupling, which drives perivascular CSF-ISF exchange and the elimination of tau and other neuronal waste products (Holstein-Rønsbo et al., 2023), was one of the most negatively enriched pathways after r-mTBI in apoE4 astrocytes. Consistent with this, astrocyte expression of the glymphatic pathway-linked water channel AQP4 (Iliff et al., 2012), along with most of the other genes encoding dystrophin-associated scaffold (DAC) proteins (i.e. Dmd, Dnta, Snta1, Dag1) that polarize and anchor AQP4 at the astrocytic endfoot-BBB interface (Simon et al., 2018) were diminished following r-mTBI, most prominently in apoE4 astrocytes. A loss of AQP4 and other DAC components is consistent with the atypical astrocyte phenotype signature, and several altered pathways predicted by RNAseq may exacerbate this endfoot dysfunction. Diminished astrocyte-derived Wnt7a/Wnt7b expression (Guérit et al., 2021) leads to BBB leakage of small molecules and impaired endfoot integrity, culminating in AQP4 delocalization. Canonical pathway analysis predicted that r-mTBI impaired apoE4 astrocytic inhibition of matrix metalloproteases, which is consistent with our prior work suggesting that apoE4 is least effective at binding and inactivating vascular MMP-9 activity (Ringland et al., 2020). A recent study found that secreted MMP-9 cleaves the scaffolding protein encoded by Dag1 (β-dystroglycan), which disturbs endfoot contact with the perivascular basement membrane, resulting in impaired AQP4 polarization and glymphatic dysfunction (Si et al., 2023). Critically, it is the distribution of AQP4 at the perivascular endfeet, and not total expression, that enables appropriate CSF-ISF exchange and solute elimination. Reduced AQP4 polarization is associated with pathogenic tau deposits (Simon et al., 2022), and impaired glymphatic tau clearance from the brain (Ishida et al., 2022). Diminished expression of AQP4 and/or other DAC proteins in previous AD mouse models are associated with diminished glymphatic influx, and an accumulation of pathogenic tau species (Harrison et al., 2020; Iliff et al., 2014; Simon et al., 2022).
Moderate TBI and vascular leakage of large blood-borne components fibrin (Eide and Hansson, 2020) may increase GFAP (Muñoz-Ballester and Robel, 2023) and AQP4 expression (Iliff et al., 2014), while mild TBI and size-restricted microvascular leakage (Munoz-Ballester et al., 2022) may diminish AQP4 expression (George et al., 2022). Vessel rupture initiates both types of astrocytes, ultimately resulting in AQP4 mislocalization. Recently, a report found that removal of astrocytic apoE4 expression in a mouse model of tauopathy was able to restore AQP4 polarization (C. Wang et al., 2021). As the transcriptomic profile of our r-mTBI astrocytes predicted dysregulated AQP4-DAC expression and impaired polarization, we evaluated the perivascular localization of AQP4 in aged apoE mice exposed to r-mTBI. We found that r-mTBI and the presence of apoE4 were both independently associated with diminished AQP4 polarization, consistent with our RNAseq data and other mTBI reports (George et al., 2022). Interestingly, while AQP4 was most prominently dysregulated by r-mTBI in apoE4 astrocytes at 7 months of age, the other apoE isoforms eventually demonstrated a similar magnitude of injury-induced AQP4 polarization by 18 months of age, suggesting that apoE4 accelerated the effect of injury on AQP4 delocalization. Diminished AQP4 polarization following r-mTBI (Cai et al., 2021) and in apoE4 mice in the current studies may also be influenced by a loss of pericyte coverage, as AQP4 is highly enriched at sites in direct contact with pericytes (Gundersen et al., 2014). These results indicate that the apoE4 allele exacerbates neurovascular degeneration at the capillary level following repetitive head trauma, resulting in a sustained loss of astrocytic homeostasis which impairs several mechanisms responsible for the elimination of tau and pathogenic waste from the brain.
Disruptions to AQP4 polarization, as observed in this study, can impair ISF/CSF drainage by reducing fluid export, which increases the resistance of the ISF chamber to CSF fluid influx from the periarterial space, ultimately suppressing the glymphatic system (Gomolka et al., 2023). To assess glymphatic activity (CSF-ISF exchange), we used a widely published protocol (Drieu et al., 2022; Iliff et al., 2012; Peng et al., 2016), and found that r-mTBI had a marked effect on glymphatic activity, consistent with our RNAseq and immunofluorescent results. Glymphatic impairment following moderate TBI has been widely reported (Iliff et al., 2014; Ren et al., 2013), though there has been limited preclinical investigation into the influence of mild TBI on glymphatic influx particularly at prolonged timepoints post-injury, which may be more clinically relevant (Piantino et al., 2022). To our knowledge, this is the first report of sustained glymphatic impairment at such a chronic time point following r-mTBI in mice, and the first investigation of glymphatic activity in apoE mice. Impaired AQP4 polarity and BBB breakdown following repetitive mTBI, as observed in this study, can result in chronically dysfunctional and swollen perivascular astrocytes (Gama Sosa et al., 2021) that limit solute permeability to the perivascular space via the narrowing of inter endfeet gaps (Ringstad et al., 2018). Several AQP4 independent mechanisms of apoE4 and/or TBI-associated neurovascular decline contribute to disrupted CSF influx, including the reduction in neurovascular coupling (Holstein-Rønsbo et al., 2023) as predicted by our RNAseq data. Both the diminished ability of apoE4 to inhibit MMP activity (Ringland et al., 2020), along with the influence of apoE4 on astrocyte-derived basement membrane components (Keable et al., 2020), would affect the mechanical and structural compliance of the vessel wall (Gama Sosa et al., 2021). Our transcriptomic analysis predicts that TBI exacerbates these processes, resulting in reduced vascular elasticity, thus attenuating the dynamic changes in arterial diameter required to drive glymphatic flow (Holstein-Rønsbo et al., 2023). Finally, a recently identified mechanism by which apoE4 can directly influence the glymphatic system is via border associated macrophage (BAM) dysregulation (Iadecola et al., 2023) which drives neurovascular dysfunction and arterial stiffness (Drieu et al., 2022). This BAM dysregulation can disrupt CSF influx (Drieu et al., 2022) and tau clearance (Drieu et al., 2023) independently of AQP4 polarization, and may become exacerbated by additional insults (e.g., TBI and/or AD). Interestingly, our studies also demonstrated that apoE4 mice exhibit diminished clearance of an inert tracer from the brain parenchyma, suggesting that apoE isoforms alone may modulate global CSF-ISF exchange.
It is important to note that glymphatic exchange is significantly reduced at the advanced age of the mice used in our studies (Da Mesquita et al., 2018; Kress et al., 2014). This age-dependent reduction in the glymphatic pathway (Kress et al., 2014) can be attributed to a multifaceted decline in neurovascular health. Several contributing factors include reduced CSF formation (Segeroth et al., 2023), diminished AQP4 polarization (Kress et al., 2014), attenuated vascular pulsatility (Kress et al., 2014), and greater prevalence of sleep disturbances (C. Wang et al., 2023). These factors result in a precipitous drop in tracer influx (Kress et al., 2014) and may mask the detection of more subtle genotype-specific alterations in glymphatic exchange via this method. Furthermore, at this advanced age, changes to AQP4 polarization may be less impactful on CSF influx (Kress et al., 2014), relative to other contributing factors like arterial wall compliance. Additionally, in humans, glymphatic impairment may have a more substantial impact on regional protein accumulation, as the complex architecture makes certain brain regions more reliant on CSF pulsations for the elimination of waste (Ringstad et al., 2018). Trauma-induced microbleeds, which disrupt localized protein clearance, persist for months to years (Griffin et al., 2019), resulting in regional pathogenic protein accumulation over the course of decades in humans (Ringstad et al., 2018), particularly if this impairment in fluid exchange does not resolve over time, as we observed in our current mouse studies. Lifelong apoE4 associated vascular impairments like arteriole blood flow reduction (Yamazaki et al., 2021), hypoperfusion (Dounavi et al., 2023) and vascular frailty (Steele et al., 2022) may predispose apoE4 carriers to an accelerated decline in glymphatic function, and earlier onset of pathogenic tauopathy. Collectively, these results suggest that apoE4 and/or injury-associated impairments in tau clearance could be the result of a multi-factorial homeostatic decline that includes AQP4 depolarization, neurovascular degeneration, and diminished elimination across the BBB.
Acknowledgments
This work was supported by the Department of Defense under award number W81XWH1910344-PRARP-CSRA. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. This work was also supported by Merit Review award number I01BX003709 from the Department of Veterans Affairs (VA) Biomedical Laboratory Research and Development Program. Dr. Bachmeier is a Research Scientist at the Bay Pines VA Healthcare System, Bay Pines, FL. Dr. Crawford is a Research Career Scientist at the James A. Haley Veterans Hospital, Tampa, FL. The contents do not represent the views of the Department of Veterans Affairs or the United States Government. Finally, we would like to thank the Roskamp Institute for their generosity in helping to make this work possible.
Footnotes
CRediT authorship contribution statement
Maxwell Eisenbaum: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. Andrew Pearson: Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. Camila Ortiz: Formal analysis, Investigation. Milica Koprivica: Investigation, Methodology. Arianna Cem-bran: Investigation, Methodology. Michael Mullan: Conceptualization. Fiona Crawford: Conceptualization. Joseph Ojo: Conceptualization, Writing – review & editing. Corbin Bachmeier: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Declaration of competing interest
The authors declare they have no conflict of interest.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Ech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D, 2018. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46 (W1), W537–W544. 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G, 2019. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front. Aging Neurosci 11 (JAN) 10.3389/fnagi.2019.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaqati M, Thomas RS, Kidd EJ, 2023. Upregulation of endocytic protein expression in the Alzheimer’s disease male human brain. Aging Brain 4, 100084. 10.1016/j.nbas.2023.100084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, Deik AA, Ginty DD, Clish CB, Gu C, 2017. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94 (3), 581–594.e5. 10.1016/j.neuron.2017.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S, 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available Online at. Http://Www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/.
- Artursson P, 1990. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorbtive (Caco-2) cells. J. Pharm. Sci 79 (6), 476–482. 10.1002/jps.2600790604. [DOI] [PubMed] [Google Scholar]
- Atherton K, Han X, Chung J, Cherry JD, Baucom Z, Saltiel N, Nair E, Abdolmohammadi B, Uretsky M, Khan MM, Shea C, Durape S, Martin BM, Palmisano JN, Farrell K, Nowinski CJ, Alvarez VE, Dwyer B, Daneshvar DH, Mez J, 2022. Association of APOE genotypes and chronic traumatic encephalopathy. JAMA Neurol 79 (8), 787–796. 10.1001/jamaneurol.2022.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmeier C, Mullan M, Paris D, 2010. Characterization and use of human brain microvascular endothelial cells to examine β-amyloid exchange in the blood-brain barrier. Cytotechnology 62 (6), 519–529. 10.1007/s10616-010-9313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmeier C, Shackleton B, Ojo J, Paris D, Mullan M, Crawford F, 2014. Apolipoprotein E isoform-specific effects on lipoprotein receptor processing. NeuroMolecular Med 16 (4), 686–696. 10.1007/s12017-014-8318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Kovac A, Majerova P, Bullock KM, Shi M, Zhang J, 2016. Tau proteins cross the blood-brain barrier. J. Alzheimers Dis 55 (1), 411–419. 10.3233/JAD-160542. [DOI] [PubMed] [Google Scholar]
- Barisano G, Kisler K, Wilkinson B, Nikolakopoulou AM, Sagare AP, Wang Y, Gilliam W, Huuskonen MT, Hung ST, Ichida JK, Gao F, Coba MP, Zlokovic BV, 2022. A “multi-omics” analysis of blood-brain barrier and synaptic dysfunction in APOE4 mice. J. Exp. Med 219 (11) 10.1084/jem.20221137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV, 2010. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68 (3), 409–427. 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV, 2012. Apolipoprotein e controls cerebrovascular integrity via cyclophilin A. Nature 485 (7399), 512–516. 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell B, Anzi S, Sasson E, Ben-Zvi A, 2023. Unique features of the arterial blood–brain barrier. Fluids Barriers CNS 20 (1). 10.1186/s12987-023-00450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick S, D’Mello V, Caruso D, Wallerstein A, Abdul-Muneer PM, 2019. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol 317, 260–270. 10.1016/j.expneurol.2019.03.014. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B, 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnar O, Shaw K, Anderle S, Grijseels DM, Clarke D, Bell L, King SL, Hall CN, 2023. APOE4 expression confers a mild, persistent reduction in neurovascular function in the visual cortex and hippocampus of awake mice. J. Cereb. Blood Flow Metab 10.1177/0271678X231172842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosworth A, Griffin C, Chakhoyan A, Sagare AP, Nelson AR, Wang Y, Kisler K, Montagne A, Clementel V, Tcw J, Rust R, Coba M, Zlokovic BV, 2023. Molecular Signature and Functional Properties of Human Pluripotent Stem Cell-Derived Brain Pericytes 10.1101/2023.06.26.546577. [DOI] [Google Scholar]
- Cai X, Harding IC, Sadaka AH, Colarusso B, Kulkarni P, Ebong E, Qiao J, O’Hare NR, Ferris CF, 2021. Mild repetitive head impacts alter perivascular flow in the midbrain dopaminergic system in awake rats. Brain Commun 3 (4) 10.1093/braincomms/fcab265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, El Gaamouch F, Meabon JS, Meeker KD, Zhu L, Zhong MB, Bendik J, Elder G, Jing P, Xia J, Luo W, Cook DG, Cai D, 2017. ApoE4-associated phospholipid dysregulation contributes to development of tau hyper-phosphorylation after traumatic brain injury. Sci. Rep 7 (1) 10.1038/s41598-017-11654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, Demattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM, 2011. Human apoE Isoforms Differentially Regulate Brain Amyloid-b Peptide Clearance 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JM, Lathuiliere A, Migliorini M, Arai AL, Wani MM, Dujardin S, Muratoglu SC, Hyman BT, Strickland DK, 2021. Regulation of tau internalization, degradation, and seeding by LRP1 reveals multiple pathways for tau catabolism. J. Biol. Chem 296 10.1016/j.jbc.2021.100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Mesquita S, Fu Z, Kipnis J, 2018. The meningeal lymphatic system: A new player in neurophysiology. In: Neuron (Vol. 100, Issue 2, pp. 375–388). Cell Press. 10.1016/j.neuron.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DL, Li M, Lee EB, 2023. Human Alzheimer’s disease reactive astrocytes exhibit a loss of homeostastic gene expression. Acta Neuropathol. Commun 11 (1), 127. 10.1186/s40478-023-01624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C, Tulloch J, Yip E, Currie L, Colom-Cadena M, Wegmann S, Hyman BT, Wilkins L, Hooley M, Tzioras M, Spires-Jones TL, 2023. Apolipoprotein E isoform does not influence trans-synaptic spread of tau pathology in a mouse model. Brain Neurosci. Adv 7 10.1177/23982128231191046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dounavi ME, Mak E, Swann P, Low A, Muniz-Terrera G, McKeever A, Pope M, Williams GB, Wells K, Lawlor B, Naci L, Malhotra P, Mackay C, Koychev I, Ritchie K, Su L, Ritchie CW, O’Brien JT, 2023. Differential association of cerebral blood flow and anisocytosis in APOE ε4 carriers at midlife. J. Cereb. Blood Flow Metab 10.1177/0271678X231173587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drieu A, Du S, Storck SE, Rustenhoven J, Papadopoulos Z, Dykstra T, Zhong F, Kim K, Blackburn S, Mamuladze T, Harari O, Karch CM, Bateman RJ, Perrin R, Farlow M, Chhatwal J, Brosch J, Buck J, Farlow M, Kipnis J, 2022. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 611 (7936), 585–593. 10.1038/s41586-022-05397-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drieu A, Du S, Kipnis M, Bosch ME, Herz J, Lee C, Jiang H, Manis M, Ulrich JD, Kipnis J, Holtzman DM, Gratuze M, 2023. Parenchymal border macrophages regulate tau pathology and tau-mediated neurodegeneration. Life Sci. Alliance 6 (11). 10.26508/lsa.202302087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning M, 2017. AnnotateMyIDs
- Eide PK, Hansson HA, 2020. Blood-brain barrier leakage of blood proteins in idiopathic normal pressure hydrocephalus. Brain Res 1727 10.1016/j.brainres.2019.146547. [DOI] [PubMed] [Google Scholar]
- Eisenbaum M, Pearson A, Gratkowski A, Mouzon B, Mullan M, Crawford F, Ojo J, Bachmeier C, 2021. Influence of traumatic brain injury on extracellular tau elimination at the blood–brain barrier. Fluids Barriers CNS 18 (1). 10.1186/s12987-021-00283-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbaum M, Pearson A, Ortiz C, Mullan M, Crawford F, Ojo J, Bachmeier C, 2023. ApoE4 expression disrupts tau uptake, trafficking, and clearance in astrocytes. GLIA 10.1002/glia.24469. [DOI] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Verkhratsky A, 2021. Reactive astrocyte nomenclature, definitions, and future directions. In: Nature Neuroscience (Vol. 24, Issue 3, pp. 312–325). Nature Research. 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser Ocak P, Ocak U, Sherchan P, Gamdzyk M, Tang J, Zhang JH, 2020. Overexpression of Mfsd2a attenuates blood brain barrier dysfunction via Cav-1/Keap-1/Nrf-2/HO-1 pathway in a rat model of surgical brain injury. Exp. Neurol 326 10.1016/j.expneurol.2020.113203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M, 2016. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI, 2009. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem 284 (19), 12845–12852. 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama Sosa MA, De Gasperi R, Pryor D, Perez Garcia GS, Perez GM, Abutarboush R, Kawoos U, Hogg S, Ache B, Janssen WG, Sowa A, Tetreault T, Cook DG, Tappan SJ, Gandy S, Hof PR, Ahlers ST, Elder GA, 2021. Low-level blast exposure induces chronic vascular remodeling, perivascular astrocytic degeneration and vascular-associated neuroinflammation. Acta Neuropathol. Commun 9 (1) 10.1186/s40478-021-01269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George KK, Heithoff BP, Shandra O, Robel S, 2022. Mild traumatic brain injury/concussion initiates an atypical astrocyte response caused by blood-brain barrier dysfunction. J. Neurotrauma 39 (1–2), 211–226. 10.1089/neu.2021.0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giarratana AO, Zheng C, Reddi S, Teng SL, Berger D, Adler D, Sullivan P, Thakker-Varia S, Alder J, 2020. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci. Rep 10 (1) 10.1038/s41598-020-76849-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomolka RS, Hablitz L, Mestre H, Giannetto M, Du T, Hauglund N, Xie L, Peng W, Martinez PM, Nedergaard M, Mori Y, 2023. Loss of aquaporin-4 results in glymphatic system dysfunction via brain-wide interstitial fluid stagnation. ELife 12. 10.7554/eLife.82232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould D, Phalan C, van Mil S, Sundberg J, Vahedi K, Massin P, Bousser M, Heutink P, Miner J, Tournier-Lasserve E, John S, 2006. Role of Col4a1 in small vessel disease and hemorrhagic stroke. N. Engl. J. Med 354 (14), 1489–1496. [DOI] [PubMed] [Google Scholar]
- Griffin AD, Turtzo LC, Parikh GY, Tolpygo A, Lodato Z, Moses AD, Nair G, Perl DP, Edwards NA, Dardzinski BJ, Armstrong RC, Ray-Chaudhury A, Mitra PP, Latour LL, 2019. Traumatic microbleeds suggest vascular injury and predict disability in traumatic brain injury. Brain 142 (11), 3550–3564. 10.1093/brain/awz290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot C, Sudre CH, Barkhof F, Teunissen CE, Van Berckel BNM, Seo SW, Ourselin S, Scheltens P, Cardoso MJ, Van Der Flier WM, Ossenkoppele R, 2018. Clinical phenotype, atrophy, and small vessel disease in APOE2 carriers with Alzheimer disease. Neurology 91 (20), E1851–E1859. 10.1212/WNL.0000000000006503. [DOI] [PubMed] [Google Scholar]
- Guérit S, Fidan E, Macas J, Czupalla CJ, Figueiredo R, Vijikumar A, Yalcin BH, Thom S, Winter P, Gerhardt H, Devraj K, Liebner S, 2021. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog. Neurobiol 199 10.1016/j.pneurobio.2020.101937. [DOI] [PubMed] [Google Scholar]
- Gundersen GA, Vindedal GF, Skare Ø, Nagelhus EA, 2014. Evidence that pericytes regulate aquaporin-4 polarization in mouse cortical astrocytes. Brain Struct. Funct 219 (6), 2181–2186. 10.1007/s00429-013-0629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV, 2016. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab 36 (1), 216–227. 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison IF, Ismail O, Machhada A, Colgan N, Ohene Y, Nahavandi P, Ahmed Z, Fisher A, Meftah S, Murray TK, Ottersen OP, Nagelhus EA, O’Neill MJ, Wells JA, Lythgoe MF, 2020. Impaired glymphatic function and clearance of tau in an Alzheimer’s disease model. Brain 143 (8), 2576–2593. 10.1093/brain/awaa179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S, 2021. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. GLIA 69 (2), 436–472. 10.1002/glia.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein-Rønsbo S, Gan Y, Giannetto MJ, Rasmussen MK, Sigurdsson B, Beinlich FRM, Rose L, Untiet V, Hablitz LM, Kelley DH, Nedergaard M, 2023. Glymphatic influx and clearance are accelerated by neurovascular coupling. Nat. Neurosci 26 (6), 1042–1053. 10.1038/s41593-023-01327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anfray A, Schaeffer S, Hattori Y, Santisteban M, Casey N, Wang G, Strickland M, Zhou P, Holtzman D, Anrather J, Park L, 2023. Cell autonomous role of border associated macrophages in ApoE4 neurovascular dysfunction and susceptibility to white matter injury. Res. Sq 10.21203/rs.3.rs-3222611/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M, 2012. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes. Includ. Amyloid B 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M, 2013. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci 33 (46), 18190–18199. 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M, 2014. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci 34 (49), 16180–16193. 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Shue F, Bu G, Kanekiyo T, 2023. Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease. In: Molecular Neurodegeneration (Vol. 18, Issue 1, p. 46). NLM (Medline). 10.1186/s13024-023-00640-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Yamada K, Nishiyama R, Hashimoto T, Nishida I, Abe Y, Yasui M, Iwatsubo T, 2022. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J. Exp. Med 219 (3) 10.1084/jem.20211275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullienne A, Salehi A, Affeldt B, Baghchechi M, Haddad E, Avitua A, Walsworth M, Enjalric I, Hamer M, Bhakta S, Tang J, Zhang JH, Pearce WJ, Obenaus A, 2018. Male and female mice exhibit divergent responses of the cortical vasculature to traumatic brain injury. J. Neurotrauma 35 (14), 1646–1658. 10.1089/neu.2017.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G, 2014. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners?. In: Neuron (Vol. 81, Issue 4, pp. 740–754) 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keable A, O’neill R, Macgregor Sharp M, Gatherer M, Yuen HM, Johnston DA, Weller RO, Carare RO, 2020. APOE4 astrocytes secrete basement membranes rich in fibronectin and poor in laminin compared to APOE3 astrocytes. Int. J. Mol. Sci 21 (12), 1–13. 10.3390/ijms21124371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL, 2015. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei Bs HS, Zeppenfeld D, Xie L, Hongyi Kang BS, Xu Q, Liew JA, Plog BA, Ding F, PhD RD, Nedergaard M, 2014. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol 76 (6), 845–861. 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrtsos CR, Baras JS, 2015. Modeling the role of the glymphatic pathway and cerebral blood vessel properties in Alzheimer’s disease pathogenesis. PLoS One 10 (10). 10.1371/journal.pone.0139574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadorf A, Agus F, Aytan N, Cherry J, Mez J, McKee A, Stein TD, 2023. Inflammation and neuronal gene expression changes differ in early versus late chronic traumatic encephalopathy brain. BMC Med. Genet 16 (1) 10.1186/s12920-023-01471-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W, 2014. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15 (12) 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone M, Panza F, 2024. Impact of apolipoprotein E isoforms on sporadic Alzheimer’s disease. Neural Regen. Res 19 (1), 80–83. 10.4103/1673-5374.375316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv C, Han S, Sha Z, Liu M, Dong S, Zhang C, Li Z, Zhang K, Lu S, Xu Z, Bie L, Jiang R, 2023. Cerebral glucagon-like peptide-1 receptor activation alleviates traumatic brain injury by glymphatic system regulation in mice. CNS Neurosci. Ther 10.1111/cns.14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, Zipfel GJ, Stocchetti N, Brody DL, 2012. Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 135 (4), 1268–1280. 10.1093/brain/awr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main BS, Villapol S, Sloley SS, Barton DJ, Parsadanian M, Agbaegbu C, Stefos K, McCann MS, Washington PM, Rodriguez OC, Burns MP, 2018. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol. Neurodegener 13 (1) 10.1186/s13024-018-0249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Blennow K, Zetterberg H, Ronne-Engström E, Enblad P, Hillered L, 2009. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury: clinical article. J. Neurosurg 110 (6), 1227–1237. 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- McCarron M, Nicoll J, Stewart J, Ironside J, Mann D, Love S, Graham D, Dewar D, 1999. The apolipoprotein E E2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J. Neuropathol. Exp. Neurol 58 (7), 711–718. [DOI] [PubMed] [Google Scholar]
- McFadyen CA, Zeiler FA, Newcombe V, Synnot A, Steyerberg E, Gruen RL, Rosand J, Palotie A, Maas AIR, Menon DK, 2021. Apolipoprotein E4 polymorphism and outcomes from traumatic brain injury: A living systematic review and meta-analysis. In: Journal of Neurotrauma (Vol. 38, Issue 8, pp. 1124–1136). Mary Ann Liebert Inc. 10.1089/neu.2018.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michinaga S, Koyama Y, 2019. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. In: International Journal of Molecular Sciences (Vol. 20, Issue 3). MDPI AG. 10.3390/ijms20030571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, Barnes SR, Daianu M, Ramanathan A, Go A, Lawson EJ, Wang Y, Mack WJ, Thompson PM, Schneider JA, Varkey J, Langen R, Mullins E, Jacobs RE, Zlokovic BV, 2018. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med 24 (3), 326–337. 10.1038/nm.4482. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D, Rege SV, Grond A, Zuniga E, Barnes SR, Prince J, Sagare M, Hsu CJ, LaDu MJ, Jacobs RE, Zlokovic BV, 2021. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 1 (6), 506–520. 10.1038/s43587-021-00073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon BC, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F, 2012. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J. Neurotrauma 29 (18), 2761–2773. 10.1089/neu.2012.2498. [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F, 2014. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann. Neurol 75 (2), 241–254. 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ojo JO, Acker CM, Ferguson S, Paris D, Ait-Ghezala G, Crynen G, Davies P, Mullan M, Stewart W, Crawford F, 2018. Lifelong behavioral and neuropathological consequences of repetitive mild traumatic brain injury. Ann. Clin. Transl. Neurol 5 (1), 64–80. 10.1002/acn3.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk AS, Wang W, Bèchet NB, Eltanahy AM, Cheng AX, Sigurdsson B, Benraiss A, Mäe MA, Kress BT, Kelley DH, Betsholtz C, Møllgård K, Meissner A, Nedergaard M, Lundgaard I, 2019. PDGF-B is required for development of the glymphatic system. Cell Rep 26 (11), 2955–2969.e3. 10.1016/j.celrep.2019.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munñoz-Ballester C, Robel S, 2023. Astrocyte-mediated mechanisms contribute to traumatic brain injury pathology. In: WIREs Mechanisms of Disease John Wiley and Sons Inc. 10.1002/wsbm.1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Ballester C, Mahmutovic D, Rafiqzad Y, Korot A, Robel S, 2022. Mild traumatic brain injury-induced disruption of the blood-brain barrier triggers an atypical neuronal response. Front. Cell. Neurosci 16 10.3389/fncel.2022.821885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi SC, Lominadze D, 2017. Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sci 7 (7) 10.3390/brainsci7070077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza P, Bachmeier C, Mouzon B, Algamal M, Rafi NG, Lungmus C, Abdullah L, Evans JE, Ferguson S, Mullan M, Crawford F, Ojo JO, 2019. APOE genotype specific effects on the early neurodegenerative sequelae following chronic repeated mild traumatic brain injury. Neuroscience 404, 297–313. 10.1016/j.neuroscience.2019.01.049. [DOI] [PubMed] [Google Scholar]
- Narayan P, Sienski G, Bonner JM, Lin YT, Seo J, Baru V, Haque A, Milo B, Akay LA, Graziosi A, Freyzon Y, Landgraf D, Hesse WR, Valastyan J, Barrasa MI, Tsai LH, Lindquist S, 2020. PICALM rescues endocytic defects caused by the Alzheimer’s disease risk factor APOE4. Cell Rep 33 (1) 10.1016/j.celrep.2020.108224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan R, Northrop N, Yamamoto B, 2017. Fluorescein isothiocyanate (FITC)-dextran extravasation as a measure of blood-brain barrier permeability. Curr. Protoc. Neurosci 2017, 9.58.1–9.58.15. 10.1002/cpns.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo J, Johnston DA, Dodart JC, MacGregor-Sharp MT, Weller RO, Nicoll JAR, Verma A, Carare RO, 2020. Peri-arterial pathways for clearance of α-Synuclein and tau from the brain: implications for the pathogenesis of dementias and for immunotherapy. Alzheimer’s Dementia 12 (1). 10.1002/dad2.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M, 2011. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem 286 (20), 17536–17542. 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo J, Eisenbaum M, Shackleton B, Lynch C, Joshi U, Saltiel N, Pearson A, Ringland C, Paris D, Mouzon B, Mullan M, Crawford F, Bachmeier C, 2021. Mural cell dysfunction leads to altered cerebrovascular tau uptake following repetitive head trauma. Neurobiol. Dis 150 10.1016/j.nbd.2020.105237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oue H, Yamazaki Y, Qiao W, Yuanxin C, Ren Y, Kurti A, Shue F, Parsons TM, Perkerson RB, Kawatani K, Wang N, Starling SC, Roy B, Mosneag IE, Aikawa T, Holm ML, Liu CC, Inoue Y, Sullivan PM, Kanekiyo T, 2023. LRP1 in vascular mural cells modulates cerebrovascular integrity and function in the presence of APOE4. JCI Insight 8 (7). 10.1172/jci.insight.163822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A, Ortiz C, Eisenbaum M, Arrate C, Browning M, Mullan M, Bachmeier C, Crawford F, Ojo JO, 2023. Deletion of PTEN in microglia ameliorates chronic neuroinflammation following repetitive mTBI. Mol. Cell. Neurosci 125 10.1016/j.mcn.2023.103855. [DOI] [PubMed] [Google Scholar]
- Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, Regan S, Kasper T, Peng S, Ding F, Benveniste H, Nedergaard M, Deane R, 2016. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis 93, 215–225. 10.1016/j.nbd.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantino JA, Iliff JJ, Lim MM, 2022. The bidirectional link between sleep disturbances and traumatic brain injury symptoms: A role for glymphatic dysfunction?. In: Biological Psychiatry (Vol. 91, Issue 5, pp. 478–487). Elsevier Inc. 10.1016/j.biopsych.2021.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Sorensen DW, Kamper JE, Ajao DO, Paul Murphy M, Head E, Hartman RE, Badaut J, 2013. Early brain injury alters the blood-brain barrier phenotype in parallel with β-amyloid and cognitive changes in adulthood. J. Cereb. Blood Flow Metab 33 (2), 205–214. 10.1038/jcbfm.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad H, Rao R, 2018. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. U. S. A 115 (28), E6640–E6649. 10.1073/pnas.1801612115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, Leshuk C, Hernandez I, Wegmann S, Hyman BT, Gradinaru V, Kampmann M, Kosik KS, 2020. LRP1 is a master regulator of tau uptake and spread. Nature 580 (7803), 381–385. 10.1038/s41586-020-2156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M, 2013. “Hit & Run” model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab 33 (6), 834–845. 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringland C, Schweig JE, Paris D, Shackleton B, Lynch CE, Eisenbaum M, Mullan M, Crawford F, Abdullah L, Bachmeier C, 2020. Apolipoprotein E isoforms differentially regulate matrix metallopeptidase 9 function in Alzheimer’s disease. Neurobiol. Aging 95, 56–68. 10.1016/j.neurobiolaging.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad G, Valnes LM, Dale AM, Pripp AH, Vatnehol SAS, Emblem KE, Mardal KA, Eide PK, 2018. Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight 3 (13). 10.1172/jci.insight.121537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S, 2014. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32 (9), 896–902. 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Jullienne A, Wendel KM, Hamer M, Tang J, Zhang JH, Pearce WJ, DeFazio RA, Vexler ZS, Obenaus A, 2019. A novel technique for visualizing and analyzing the cerebral vasculature in rodents. Transl. Stroke Res 10 (2), 216–230. 10.1007/s12975-018-0632-0. [DOI] [PubMed] [Google Scholar]
- Segeroth M, Wachsmuth L, Gagel M, Albers F, Hess A, Faber C, 2023. Disentangling the impact of cerebrospinal fluid formation and neuronal activity on solute clearance from the brain. Fluids Barriers CNS 20 (1). 10.1186/s12987-023-00443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selhorst S, Nakisli S, Kandalai S, Adhicary S, Nielsen CM, 2022. Pathological pericyte expansion and impaired endothelial cell-pericyte communication in endothelial Rbpj deficient brain arteriovenous malformation. Front. Hum. Neurosci 16 10.3389/fnhum.2022.974033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Holtzman DM, 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549 (7673), 523–527. 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs DT, Kramerov AA, Ljubimov AV, Dumitrascu OM, Rodriguez AR, Barron E, Hinton DR, Black KL, Miller CA, Mirzaei N, Koronyo-Hamaoui M, 2020. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol 139 (5), 813–836. 10.1007/s00401-020-02134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si X, Dai S, Fang Y, Tang J, Wang Z, Li Y, Song Z, Chen Y, Liu Y, Zhao G, Zhang B, Pu J, 2023. Matrix metalloproteinase-9 inhibition prevents aquaporin-4 depolarization-mediated glymphatic dysfunction in Parkinson’s disease. J. Adv. Res 10.1016/j.jare.2023.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Murchison C, Iliff JJ, 2018. A transcriptome-based assessment of the astrocytic dystrophin-associated complex in the developing human brain. J. Neurosci. Res 96 (2), 180–193. 10.1002/jnr.24082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Wang MX, Ismail O, Braun M, Schindler AG, Reemmer J, Wang Z, Haveliwala MA, O’Boyle RP, Han WY, Roese N, Grafe M, Woltjer R, Boison D, Iliff JJ, 2022. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimer’s Res. Therapy 14 (1). 10.1186/s13195-022-00999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele OG, Stuart AC, Minkley L, Shaw K, Bonnar O, Anderle S, Penn AC, Rusted J, Serpell L, Hall C, King S, 2022. A multi-hit hypothesis for an APOE4-dependent pathophysiological state. Eur. J. Neurosci 56 (9), 5476–5515. 10.1111/ejn.15685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storck SE, Meister S, Nahrath J, Meißner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU, 2016. Endothelial LRP1 transports amyloid-β1–42 across the blood-brain barrier. J. Clin. Investig 126 (1), 123–136. 10.1172/JCI81108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong L, Jakes R, Huangt DY, Pericak-vance M, Schmechel D, Roses AD, 1994. Isoform-Specific Interactions of Apolipoprotein E with Microtubule-Associated Protein Tau: implications for Alzheimer Disease, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulimai N, Brown J, Lominadze D, 2021. Fibrinogen interaction with astrocyte icam-1 and prpc results in the generation of ros and neuronal death. Int. J. Mol. Sci 22 (5), 1–16. 10.3390/ijms22052391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Z, Guo Z, Zhong J, Cheng C, Huang Z, Wu Y, Tang S, Luo C, Peng X, Wu H, Sun X, Jiang L, 2017. ApoE influences the blood-brain barrier through the NF-κB/MMP-9 pathway after traumatic brain injury. Sci. Rep 7 (1) 10.1038/s41598-017-06932-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veluw SJ, Hou SS, Calvo-Rodriguez M, Arbel-Ornath M, Snyder AC, Frosch MP, Greenberg SM, Bacskai BJ, 2020. Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron 105 (3), 549–561.e5. 10.1016/j.neuron.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C, 2018. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554 (7693), 475–480. 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM, 2013. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U. S. A 110 (19) 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ding F, Deng SY, Guo X, Wang W, Iliff JJ, Nedergaard M, 2017. Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J. Neurosci 37 (11), 2870–2877. 10.1523/JNEUROSCI.2112-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, Manis M, Schroeder C, Yin Z, Madore C, Butovsky O, Artyomov M, Ulrich JD, Holtzman DM, 2021. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109 (10), 1657–1674.e7. 10.1016/j.neuron.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Nambiar A, Strickland MR, Lee C, Parhizkar S, Moore AC, Musiek ES, Ulrich JD, Holtzman DM, 2023. APOE-ε4 synergizes with sleep disruption to accelerate Aβ deposition and Aβ-associated tau seeding and spreading. J. Clin. Investig 10.1172/jci169131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ, Tannenbaum A, Ballerini L, Rungta RL, Boido D, Sweeney M, Montagne A, Charpak S, Joutel A, Smith KJ, Black SE, 2020. Perivascular spaces in the brain: anatomy, physiology and pathology. In: Nature Reviews Neurology (Vol. 16, Issue 3, pp. 137–153). Nature Research. 10.1038/s41582-020-0312-z. [DOI] [PubMed] [Google Scholar]
- Williams T, Ruiz AJ, Ruiz AM, Vo Q, Tsering W, Xu G, McFarland K, Giasson BI, Sullivan P, Borchelt DR, Chakrabarty P, 2022. Impact of APOE genotype on prion-type propagation of tauopathy. Acta Neuropathol. Commun 10 (1) 10.1186/s40478-022-01359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Bathe T, Vo Q, Sacilotto P, McFarland K, Ruiz AJ, Hery GP, Sullivan P, Borchelt DR, Prokop S, Chakrabarty P, 2023. Humanized APOE genotypes influence lifespan independently of tau aggregation in the P301S mouse model of tauopathy. Acta Neuropathol. Commun 11 (1) 10.1186/s40478-023-01581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VMY, Holtzman DM, 2011. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci 31 (37), 13110–13117. 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G, 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. In: Nature Reviews Neurology (Vol. 15, Issue 9, pp. 501–518). Nature Publishing Group. 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Liu CC, Yamazaki A, Shue F, Martens YA, Chen Y, Qiao W, Kurti A, Oue H, Ren Y, Li Y, Aikawa T, Cherukuri Y, Fryer JD, Asmann YW, Kim BYS, Kanekiyo T, Bu G, 2021. Vascular ApoE4 impairs behavior by modulating Gliovascular function. Neuron 109 (3), 438–447.e6. 10.1016/j.neuron.2020.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C, LaDu MJ, 2012. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of alzheimer disease. J. Biol. Chem 287 (50), 41774–41786. 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Q, Hoi MPM, 2023. Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease. In: Neural Regeneration Research (Vol. 18, Issue 9, pp. 1890–1902). Wolters Kluwer Medknow Publications. 10.4103/1673-5374.367832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppenfeld DM, Simon M, Haswell JD, D’Abreo D, Murchison C, Quinn JF, Grafe MR, Woltjer RL, Kaye J, Iliff JJ, 2017. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol 74 (1), 91–99. 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, Attrebi ON, Petrucelli L, Fryer JD, Wszolek ZK, Graff-Radford NR, Caselli RJ, Sanchez-Contreras MY, Rademakers R, Murray ME, Bu G, 2018. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat. Commun 9 (1) 10.1038/s41467-018-06783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA, 2008. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J. Neurotrauma 25 (4), 279–290. 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.