

Abstract
Exposure to inorganic arsenic through drinking water is widespread and has been linked to many chronic diseases, including cardiovascular disease. Arsenic exposure has been shown to alter hypertrophic signaling in the adult heart, as well as in utero offspring development. However, the effect of arsenic on maternal cardiac remodeling during pregnancy has not been studied. As such, there is a need to understand how environmental exposure contributes to adverse pregnancy-related cardiovascular events. This study seeks to understand the impact of trivalent inorganic arsenic exposure during gestation on maternal cardiac remodeling in late pregnancy, as well as offspring outcomes. C57BL/6J mice were exposed to 0 (control), 100 or 1000 μg/L sodium arsenite (NaAsO2) beginning at embryonic day (E) 2.5 and continuing through E17.5. Maternal heart function and size were assessed via transthoracic echocardiography, gravimetric measurement, and histology. Transcript levels of hypertrophic markers were probed via qRT-PCR and confirmed by western blot. Offspring outcomes were assessed through echocardiography and gravimetric measurement. We found that maternal heart size was smaller and transcript levels of Esr1 (estrogen receptor alpha), Pgrmc1 (progesterone receptor membrane component 1) and Pgrmc2 (progesterone receptor membrane component 2) reduced during late pregnancy with exposure to 1000 μg/L iAs vs. non-exposed pregnant controls. Both 100 and 1000 μg/L iAs also reduced transcription of Nppa (atrial natriuretic peptide). Akt protein expression was also significantly reduced after 1000 μg/L iAs exposure in the maternal heart with no change in activating phosphorylation. This significant abrogation of maternal cardiac hypertrophy suggests that arsenic exposure during pregnancy can potentially contribute to cardiovascular disease. Taken together, our findings further underscore the importance of reducing arsenic exposure during pregnancy and indicate that more research is needed to assess the impact of arsenic and other environmental exposures on the maternal heart and adverse pregnancy events.
Keywords: Arsenic, Maternal exposure, Cardiotoxicity, Cardiovascular Disease, Pregnancy
INTRODUCTION
Inorganic arsenic (iAs) is a widespread drinking water contaminant which ranks as the top chemical on the Agency for Toxic Substances and Disease Registry’s (ATSDR) Substance Priority List1. iAs is naturally found in the Earth’s crust, and activities such as mineral extraction, waste processing, pesticide application, and additives to poultry and swine feed have mobilized iAs into food and drinking water supplies2. While also found in air, the greatest public health threat stems from the ingestion of iAs-contaminated groundwater, and food prepared with such groundwater2,3. There are two forms of iAs, trivalent and pentavalent arsenic, with trivalent arsenic being the most toxic form4. iAs has been linked to the etiology of many chronic diseases, including cancer, chronic respiratory disease, and many forms of cardiovascular disease (CVD)5–13, including hypertension, atherosclerosis, coronary heart disease, and stroke14. Research from our group demonstrated that chronic iAs exposure induced sex-specific pathological remodeling of the adult heart and altered susceptibility to ischemic injury15,16. However, studies have yet to examine the impact of iAs on the cardiovascular system during physiological stressors, such as pregnancy.
During pregnancy, the heart undergoes hypertrophic remodeling, increasing chamber size and wall thickness in response to the increase in blood volume that occurs during pregnancy17,18. Blood volume in the pregnant mother increases significantly starting at 6 weeks of gestation, resulting in a 45% increase in blood volume compared to non-pregnant women19–21. Cardiac output also increases during pregnancy and remains constant after the end of the second trimester22,23. Pregnancy-induced hypertrophy is reversible24,25 and regulated by various phases of hormonal signaling during pregnancy. Both progesterone and estradiol increase throughout pregnancy and have been shown to modulate cardiac hypertrophy26,27. Progesterone is known to induce cardiomyocyte hypertrophy, while estradiol has a biphasic effect, promoting hypertrophy at low concentrations and inhibiting hypertrophy at higher concentrations28,29.
Emerging evidence suggests that the environment plays a role in cardiovascular-related pregnancy complications. Exposure to air pollution and metals is associated with reduced conception rates, infertility, and increased pregnancy loss30–32, suggesting interference with hormone signaling. However, it is unclear how exposure to environmental factors is associated with peripartum cardiomyopathy and other cardiovascular complications during pregnancy, as the mechanism through which the environment impacts the cardiovascular system during pregnancy has yet to be explored. Metals, such as iAs, have negative effects on the cardiovascular system, which undergoes physiological alterations during pregnancy8,33–36. However, the pathways and physiologic processes that are altered with iAs exposure during pregnancy have not been established. In the US, 23.8 maternal deaths occurred for every 100,000 live births in 2020 and CVD is the leading cause of pregnancy-related mortality37,38. Women who develop complications during pregnancy, such as preeclampsia, hypertension, or gestational diabetes, are also at higher risk for adverse pregnancy outcomes and CVD postpartum39.
Our previous studies demonstrated that iAs exposure altered cardiac hypertrophy in adult male mice, and we next sought to examine the impact of iAs exposure on maternal cardiac remodeling during pregnancy15,16. Pregnant C57BL6/J mice were exposed to either 0, 100 or 1000 μg/L iAs in drinking water during gestation, and a late pregnancy (E17.5) endpoint was examined. We found that pregnant female mice exposed to 1000 μg/L iAs showed blunted maternal cardiac growth and decreased transcriptional levels of estrogen receptor alpha (ERα), progesterone receptor membrane component 1 and 2 (Pgrmc1/2), and atrial natriuretic peptide (ANP), and decreased protein expression of Akt in the maternal heart during late pregnancy.
METHODS
Animals and Arsenic Exposure
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH; Publication No. 85–23, Revised 2011) and was approved by the Institutional Animal Care and Use Committee of Johns Hopkins University. Timed pregnant female C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice (2 per cage) were housed under specific pathogen-free conditions and maintained on AIN-93G chow (Research Diets, New Brunswick, NJ) and Pure Life water (Nestle Waters North America, Stamford, CT). Research Diets has reported levels of iAs in AIN-93G chow to be below the limit of detection, and our lab has previously confirmed levels of iAs in Pure Life water to be below the limit of detection using inductively coupled plasma mass spectrometry measurements15,16. Pregnant and non-pregnant female mice (6–12 weeks of age) were given Pure Life water containing 0 (control), 100 or 1000 μg/L sodium arsenite (NaAsO2) beginning at embryonic (E) 2.5 and continuing until harvest at E17.5 or parturition (Fig. 1). Fresh water bottles containing control or NaAsO2 drinking water were replenished every 2–3 days to maintain concentrations of iAs and minimize oxidation. Body weight, and food and water consumption did not significantly differ between treatment groups throughout the exposure (Supplemental Figure 1).
Figure 1: Diagram of Experimental Timeline and Endpoints.

Experimental timeline of iAs exposure and experimental endpoints for dams and offspring. Created with Biorender.
Tissue Harvest and Collection
Mice were anesthetized with a mixture of ketamine (90 mg/kg; Hofspira) and xylazine (10 mg/kg; Sigma-Aldrich) via intraperitoneal injection and anticoagulated with heparin (Fresenvis Kabi, Lake Zurich, IL) prior to tissue collection. Hearts were excised, cannulated on a Langendorff apparatus, and perfused in a retrograde manner with Krebs-Henseleit buffer (95% O2, 5% CO2; pH 7.4) under constant pressure (100 cmH2O) and temperature (37°C) as previously described for 5 minutes to washout blood16. Buffer consisted of (in mmol/L): NaCl (120), KCl (4.7), KH2PO4 (1.2), NaHCO3 (25), MgSO4 (1.2), d-Glucose (14), and CaCl2 (1.75). Following perfusion, hearts were weighed and either sectioned for histology, or equally halved, and snap-frozen in liquid nitrogen and stored at −8°C for molecular studies. Tissue was collected for prepartum endpoints at E17.5, and postpartum endpoints at P12.5. Cross-fostering of offspring was not performed in this study, and dams were kept with their respective litters until P12.5.
Echocardiography
All echocardiography procedures were performed by an investigator blinded to experimental conditions using a preclinical ultrasound imaging system (Vevo 2100; FUJIFILM VisualSonics, Toronto, ON, Canada) with a 40-MHz linear transducer, as previously described16. Echocardiography was performed on pregnant mice and embryonic pups under anesthesia using isoflurane. Mice were induced with 3–4% isoflurane and maintained at 1–2% thereafter. Postnatal pups underwent conscious transthoracic echocardiography. The M-mode echocardiogram was acquired from the short-axis view of the left ventricle at the level of the midpapillary muscles (200 m/s sweep speed). Semiautomated continuous tracing of the left ventricular walls from this axis view measured, calculated, or extrapolated the following cardiac parameters using 3–5 cardiac cycles: left ventricular posterior wall thickness at end diastole (LVPWd), left ventricular posterior wall thickness at end systole (LVPWs), left ventricular internal diameter at end diastole (LVIDd), left ventricular internal diameter at end systole (LVIDs), percent fractional shortening (FS), percent ejection fraction (EF), heart rate (H), and cardiac output (CO). Parameters were calculated using the LVTool function in Vevo LAB (VisualSonics, Fujifilm). Embryonic and maternal echocardiography was performed at E16.5 and E17.5, and at days P4 and P5 for pups.
Histology and Cardiomyocyte Cross Sectional Area Measurement
After perfusion and gravimetric measurement, a transverse cross-section of the heart was taken, including both the left and right ventricle. Sections were taken from the middle third of the heart. Tissues were fixed in 4% formalin, embedded in paraffin, sectioned (4μM) and mounted on slides. Heart sections were stained using Hematoxylin and Eosin (H&E) to visualize cardiomyocyte integrity (2 sections) or Masson’s Trichrome to determine the presence of fibrosis (4 sections) and delineate cell boundaries to assess cardiomyocyte cross-sectional area as detailed in prior studies40–43. Sections were then imaged at 20x using Aperio ScanScope CS (Leica Biosystems, Deer Park, IL). Transverse sections were analyzed for fibrosis expressed as a percentage of total area using an Aperio ImageScope macro (Leica Biosystems, Deer Park, IL). Cross-sectional area of cardiomyocytes was determined from Masson’s Trichrome stained tissue, with 25 measurements taken from one region per transverse section across 4 sections, totaling 100 different measurements per heart. Each cell was outlined by a blinded investigator using Concentric by Proscia, allowing the area of the section to be calculated.
RNA Isolation, Extraction, and cDNA Conversion
Hearts were homogenized with TRIzol (1 mL, Ambion) using a bead-mill tissue homogenizer (2 × 30 s cycles, 0°C, 7,200 rpm; Precellys Evolution 24). Lysates were mixed with chloroform (200 μL, Thermo Fisher), incubated (5 min, 25°C), and centrifuged (12,000 g, 15 min, 4°C) for phase separation. RNA collected from the upper phase was precipitated by incubation (10 min, 25°C) with isopropyl alcohol (500 μL) and centrifugation (12,000 g, 8 min, 4°C). RNA pellets were washed with 75% ethanol, centrifuged (12,000 g, 10 min, 4°C), and air-dried under a laminar flow hood (30 min). Following RNA solubilization (100 μL, DEPC-treated water), RNA concentration and purity (A260/A280 range 1.98 to 2.06) were measured via spectrophotometry (1 mL sample, NanoDrop 100, Thermo Fisher). RNA was subsequently converted to cDNA per manufacturer’s instructions (High-Capacity cDNA Reverse Transcription Kit, 4368814, Thermo Fisher). Briefly, the reverse transcription reaction mix was prepared on ice, RNA (2 μg) was added, and the samples were run on a thermocycler (60 min, 37°C; 5 min, 95°C; held, 4°C; Applied Biosystems); cDNA was stored at −20°C until use.
Quantitative RT-PCR
Expression levels of mRNA transcripts were measured using generated cDNA, a PCR (Polymerase Chain Reaction) master mix (TaqMan Fast Advanced Master Mix, Applied Biosystems) and the following validated primers (TaqMan, Applied Biosystems) on a thermocycler (2 min, 50°C; 2 min, 95°C; (1 s, 95°C; 20 s, 60°C) × 40), (QuantStudio 3, Applied Biosystems, Thermo Fisher) in a 96-well plate (USA Scientific, Beltsville MD): Acta1 (Mm00808218_g1), Akt1 (Mm01331626_m1), Esr1 (Mm00433149_m1), Esr2 (Mm00599821_m1), Gapdh (Mm99999915_g1), Kncd3 (Mm01302126_m1), Nos3 (Mm00435217_m1), Myh6 (Mm00440359_m1), Myh7 (Mm00600555_m1), Nppa (Mm01255747_g1), Nppb (Mm01255770_g1), Pgrmc1 (Mm00443985_m1), Pgrmc2 (Mm01283154_m1), Prlr (Mm04336676_m1), and Vegfa (Mm00437304_m1). Expression was determined using the ΔΔCT method and normalized to Gapdh, which did not change in cycle time with iAs treatment.
Heart Homogenization and Protein Isolation
Hearts were homogenized in cell lysis buffer (1 mL; Cell Signaling Technology, Danvers, MA) supplemented with a protease and phosphatase-inhibitor cocktail (Cell Signaling Technology) using a hard tissue lysing kit (Precellys CK28 Lysing Kit, Bertin Instruments) with a bead-mill tissue homogenizer (2 × 30s cycles, 0°C, 7,2000 rpm; Precellys Evolution 24, Bertin Instruments). Supernatant was collected as total crude homogenate; protein concentration was determined via Bradford assay and aliquots of homogenate were stored at −80°C.
Western Blot
Protein homogenates (30 μg) were separated (20 min, 75 V; 100 min, 175 V) on a gradient Bis-Tris SDS-PAGE gel (4–12%, NuPAGE; Thermo Fisher, Carlsbad, CA) and transferred (90 min, 220 mA) to a PVDF membrane (Thermo Fisher). Every gel included two molecular-weight markers for separate regions of interest (High Range Color-Coded Prestained Protein Marker; Cell Signaling Technology; and Novex Prestained Protein Standard, Thermo Fisher) on opposite ends of the gel. Total protein served as the loading control by covalently labeling lysine residues (No-Stain Protein Labeling Reagent, Thermo Fisher) following the manufacturer’s protocol and visualized by fluorescence imaging (488 nm). Membranes were blocked (1h) with bovine serum albumin (5% wt/vol, Sigma-Aldrich) in Tris-buffered saline with Tween-20 (0.1%), and subsequently incubated (overnight, 4°C) with primary antibodies for p-Akt (S473) (1:1000, Rabbit mAb, Cell Signaling, 4060S), Akt (pan) (1:1000, Rabbit mAb, Cell Signaling, 4691S), p44/42 MAPK (ERK1/2) (1:1000, Rabbit mAb, Cell Signaling, 9102S), and p-p44/42 MAPK (T202/Y204)(1:1000, Rabbit Ab, Cell Signaling, 9101S). Membranes were then incubated with secondary antibodies (Anti-rabbit IgG, HRP-linked Ab, Thermo Fisher) and visualized using chemiluminescence substrate (SuperSignal West Pico PLUS, Thermo Fisher) and an iBright imager (Invitrogen, Thermo Fisher). Membranes were stripped with Reblot Plus Mild Solution (Millipore) when necessary to re-probe total protein following examination of phosphorylation status. No-Stain total protein and full western blot scans are included in Supplemental Figure 2.
Statistical Analysis
Sample sizes of mice for each experiment were estimated a priori via power analysis (power = 0.80, effect size = 0.25, alpha = 0.05) based on data generated in previous studies from our group15,16. Mice were randomized during collection to minimize batch effects. All offspring data were averaged by litter to minimize litter effect. All data are expressed as the mean ± SEM and were analyzed using GraphPad Prism (La Jolla, CA). Statistical outliers were identified using the ROUT method (Q=1%). Statistical comparisons between groups were determined using an ordinary one-way ANOVA with Sidak’s multiple comparisons test, a two-way ANOVA with Tukey’s multiple comparisons test or a two-tailed Mann-Whitney test as appropriate. Significance was set at p < 0.05.
RESULTS
iAs exposure alters cardiac function in late pregnancy
To determine whether iAs exposure alters maternal cardiac structure or function during pregnancy, transthoracic echocardiography was performed at E16.5 and E17.5 during the late pregnancy period (Fig. 2). We found that pregnant dams exposed to 1000μg/L iAs had an increase in ejection fraction trending towards significance compared to non-exposed pregnant control and 100 μg/L iAs exposed pregnant dams, but no changes were observed between non-pregnant exposed groups (Fig. 2A).Pregnant dams exposed to 1000 μg/L iAs also exhibited a significant increase in fractional shortening compared to control and 100 μg/L iAs exposed dams with no change between non-pregnant exposure groups (Fig. 2B). Additionally, pregnant mice exposed to 1000 μg/L iAs showed a significant increase in cardiac output compared to control and 100 μg/L exposed pregnant dams, as well as 100 and 1000 μg/L iAs exposed non-pregnant females (Fig. 2C). Since cardiac output is the product of heart rate and stroke volume, we examined these two parameters. We found no change in heart rate across pregnant or non-pregnant females (Fig. 2D), but stroke volume was significantly increased in 1000 μg/L iAs exposed pregnant dams compared to non-pregnant females exposed to 1000 μg/L iAs (Fig. 2E). We also found no change in end systolic or diastolic volumes, or in average wall thickness across all pregnant and non-pregnant exposure groups (Figs. 2F–H).
Figure 2: iAs exposure increases maternal cardiac function during pregnancy.

Cardiac parameters as measured by transthoracic echocardiography in both pregnant and non-pregnant mice including: cardiac output (A), stroke volume (B), heart rate (C), ejection fraction (D), fractional shortening (E), end diastolic volume (EDV; F), end systolic volume (ESV; G), average wall thickness (H), left ventricular internal diameter in diastole (LVID;d; I), and left ventricular internal diameter in systole (LVID;s; J) (*= P < 0.05, ** = P< 0.01 vs. control; n=7–9 mice/group). Significance was determined by two-way ANOVA with Tukey’s multiple comparisons test.
iAs exposure alters prepartum hypertrophic cardiac remodeling
Reversible cardiac hypertrophy is known to occur during pregnancy24,25, so we investigated heart size across all groups to potentially account for our observed changes in cardiac output and ejection fraction. As expected, there was a significant increase in heart size in control pregnant mice compared to control non-pregnant females (Figs. 3A, 3B). Surprisingly, during late pregnancy, we found that maternal hearts were significantly smaller in dams exposed to 1000 μg/L iAs compared to non-exposed pregnant controls and were not significantly different from non-pregnant females exposed to 1000 μg/L iAs (Fig. 3A). Exposure to 100 μg/L iAs, on the other hand, was not sufficient to reduce heart growth relative to non-exposed pregnant controls as heart size was increased as expected compared to non-pregnant mice with the same exposure (Fig. 3B). Next, we examined cardiomyocyte cross-sectional area in histological sections, and consistent with our observed differences in heart size, we found that cardiomyocyte cross-sectional area was significantly less in 1000 μg/L iAs-exposed hearts compared to controls (Figs. 3E, 3F). Masson’s trichrome staining was also performed on transverse heart sections to examine changes in collagen deposition, but no differences were seen in collagen deposition or fibrosis (Figs. 3C, 3D).
Figure 3: iAs exposure blunts maternal cardiac hypertrophy.

Heart weight divided by tibia length in pregnant and non-pregnant females with 1000 μg/L (A) and 100 μg/L (B) iAs exposure compared to non-exposed controls (n=11–18 mice/group; *= P < 0.05, ** = P< 0.01 vs. control). Quantification of fibrosis (C) and representative images (D, scale bar 2mm) in pregnant mice exposed to 1000 μg/L iAs vs. Non-exposed pregnant controls (Ctrl). Quantification of cardiomyocyte cross-sectional area (E) and representative images of cardiomyocyte cross-section (F, scale bar 100 μm) taken from 4 sections per heart with 25 cardiomyocytes per section (n=5 hearts/group, 100 cardiomyocytes/heart; **= P<0.01 vs. control). Significance was determined by two-way ANOVA with Sidak’s multiple comparisons test or Mann-Whitney test.
iAs exposure during pregnancy alters the transcriptional profile of hormone receptors
Hormone signaling is important for cardiovascular adaptation and heart growth during pregnancy, so we examined the expression of various hormone receptors in the maternal heart during late pregnancy. Estrogen impacts hypertrophic remodeling in the pregnant heart29,44 and we found that exposure to iAs induced a dose-dependent decrease in Esr1 (Estrogen Receptor α, ERα) mRNA levels in the whole heart (Fig. 4A). Esr2 (Estrogen receptor β) was also probed, but the cycle time (CT) values were greater than 38, indicating low levels of expression in whole myocardium. Progesterone is also known to regulate pregnancy-induced hypertrophy26–28, and we found that exposure to both 100 and 1000 μg/L iAs during gestation downregulated cardiac transcription of the progesterone membrane receptors Pgrmc1 and Pgrmc2 (Figs. 4B, 4C). Cytosolic progesterone receptors were also probed, but levels were too low in whole cardiac tissue to be detected. We also examined Prlr (prolactin receptor) expression in the heart, as prolactin is another hormone that is important during pregnancy, but we found that the levels were not significantly altered (Fig. 4D).
Figure 4: iAs downregulates estrogen receptor alpha transcripts during late pregnancy.

Myocardial mRNA transcript levels of estrogen receptor alpha (ESR1; A), progesterone receptor membrane component 1 (Pgrmc1; B), progesterone receptor membrane component 2 (Pgrmc2; C), and prolactin receptor (Prlr; D) in pregnant females at E17.5 as measured via RT-qPCR (**= P<0.01 vs. control). Transcript levels were normalized to Gapdh mRNA expression, which did not change with iAs exposure. Outliers were determined by the ROUT method (Q=1%), and significance was determined by one-way ANOVA with Sidak’s multiple comparisons test or Mann-Whitney test (n=7–9 mice/group).
iAs exposure and transcriptional markers of hypertrophy
Next, we examined the expression of gene transcripts typically associated with physiologic and pathologic hypertrophy, as well as genes which have been implicated in pregnancy-induced hypertrophy45. In hearts exposed to both 100 and 1000 μg/L iAs during pregnancy, we found a decrease in Akt1 (Akt), a major regulator of physiologic hypertrophy46, which was trending towards significance (Fig. 5A). We also found a reduction in Nppa (ANP) at both doses (Fig. 5B). Interestingly, we found a significant reduction in Vegfa transcripts at 100 μg/L iAs only (Fig. 5C). Other markers of physiologic and pathologic hypertrophy were not altered (Fig. 5D–I). Transcriptional results were confirmed via western blot and protein levels of Akt were significantly reduced in 1000 μg/L iAs exposed hearts, while Akt phosphorylation (Ser473) relative to total Akt showed a trending decrease (Fig. 6A and 6B). Extracellular-regulated kinase 1 and 2 (ERK1/2) is also a major regulator of pregnancy-induced hypertrophy46. However, we found that levels of ERK1/2, as well as phosphorylation of the activation site, were unchanged with iAs exposure (Fig. 6C and 6D).
Figure 5: iAs exposure reduces Akt and ANP transcripts in the prepartum heart.

Myocardial mRNA transcript levels of protein kinase B (Akt; A), natriuretic peptide A (ANP; B), vascular endothelial growth factor (Vegfa; C), potassium voltage-gated channel 4.3 (Kv4.3; D), endothelial nitric oxide synthase (Nos3; E), natriuretic peptide B (Nppb; F), β-myosin heavy chain (Myh7; G), α-myosin heavy chain (Myh6; H), skeletal muscle α-actin (Acta1; I), (*=P<0.05, n=7–9 hearts/group). Outliers were determined by the ROUT method (Q=1%), and significance was determined by a one-way ANOVA with Sidak’s multiple comparisons test.
Figure 6: iAs exposure reduces protein levels of Akt in the prepartum heart.

Myocardial protein expression of phosphorylated Akt at the activating site, S437, normalized to total Akt (A) in dam hearts (P= 0.0776 vs. control, n= 5 hearts/group). Protein expression of total Akt (B) in dam hearts (***P=0.0005 vs. control). Protein expression of phosphorylated ERK1/2 at the activating sites, T202/Y204 (C), and of total ERK1/2 (D). Protein levels of each target were normalized to total transferred protein levels. Outliers were determined by the ROUT method (Q=1%), and significance was determined by a Mann-Whitney test.
In utero exposure to iAs and offspring outcomes
Finally, we examined the impact of gestational iAs on offspring outcomes with a focus on cardiac development. We found that in utero exposure to 100 or 1000 μg/L iAs during gestation resulted in no change to litter size (Fig. 7A) or embryonic body weight (Fig. 7B). However, heart weight relative to body weight was increased at E17.5 after exposure to 1000 μg/L iAs (Fig. 7C). To further delineate changes in cardiac structure and function, echocardiography was performed in utero at E17.5, but we found no change in average wall thickness (Fig. 7D), LVPWd (Fig. 7E), IVSd (Fig. 7F), EF (Fig. 7H), or FS (Fig. 7I). However, we did find an increase in the left ventricular internal diameter during diastole at E17.5 with exposure to 1000 μg/L IAs (LVIDd, Fig. 7G).
Figure 7: Offspring from iAs exposed dams show increased heart weight and left ventricular internal diameter at E17.5.
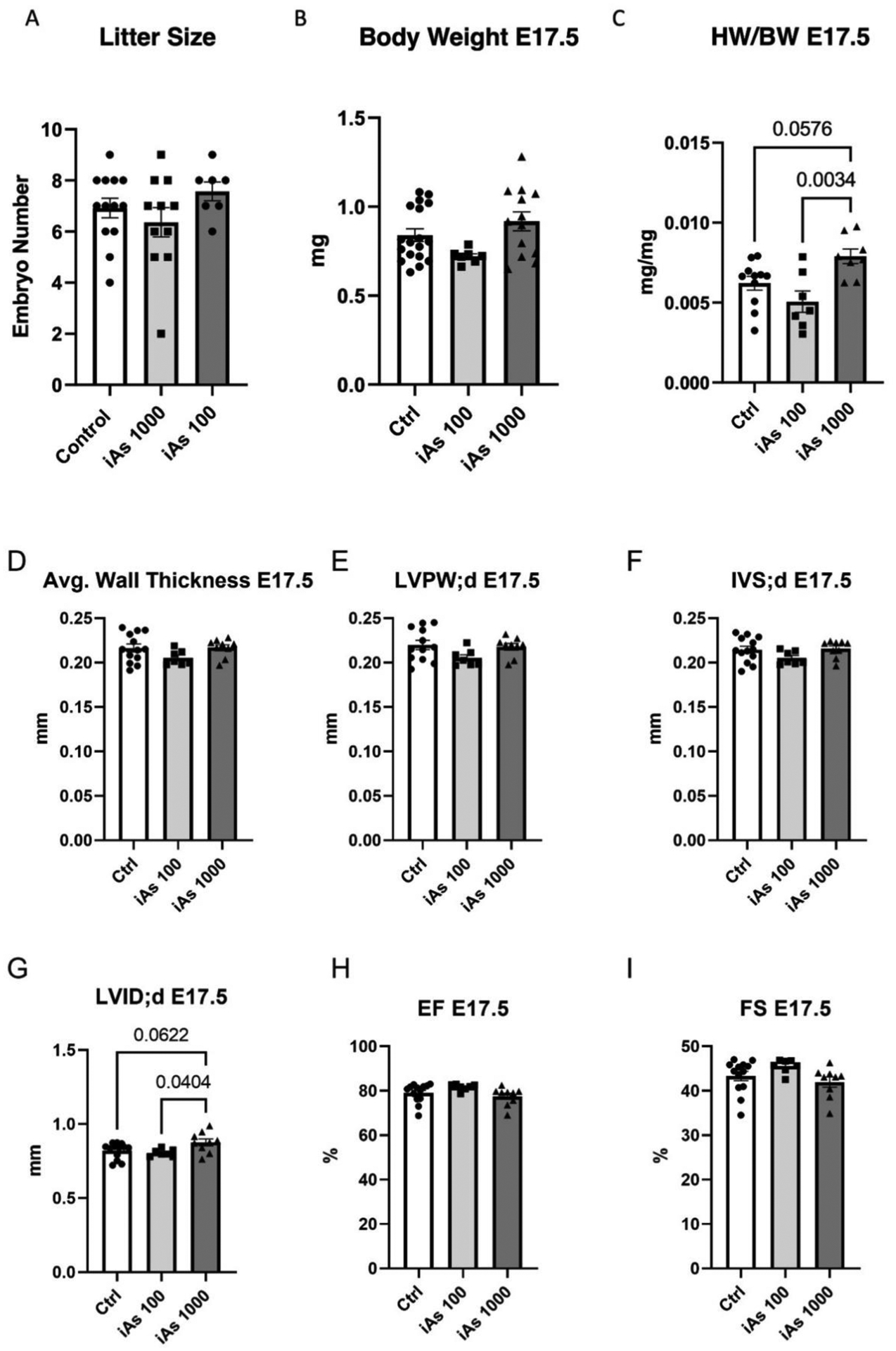
Litter size (A), body weight (B), and embryonic heart weight normalized to body weight (C), in offspring exposed to 100 and 1000 μg/L iAs during gestation (n= 16–18 litters/group). Cardiac parameters measured in utero using echocardiography including average wall thickness (D), left ventricular posterior wall thickness (LVPW; E), interventricular septum thickness (IVS; F), left ventricular internal diameter (LVID; G), ejection fraction (EF; H), and fractional shortening (FS; I). Outliers were determined by the ROUT method (Q=1%), and significance was determined by one-way ANOVA with Sidak’s multiple comparisons test.
These same parameters were measured again postnatally at P12, and we found that exposure to 100 μg/L iAs in utero reduced body weight at P12 (Fig. 8A). When stratified by sex, both sexes showed a similar significant reduction body weight (Fig. 8B). We also saw no difference in distribution of sex by exposure (Supplemental Table 1). However, while an increase in heart weight was noted at E17.5, we found no change compared to controls at P12, with or without sex stratification (Figs. 8C, 8D).Echocardiography also revealed no change in average wall thickness (Fig. 8E), IVS (Fig. 8F), LVPW (Fig. 8G), LVIDd (Fig. 8H), EF (Fig. 8I), or FS (Fig. 8J).
Figure 8: Offspring exposed to iAs in utero have reduced body weight at P12.

Body weight at P12 (A), body weight stratified by sex (B), heart weight relative to body weight (C), and heart weight relative to body weight stratified by sex (D) measured gravimetrically. Cardiac functional parameters measured via echocardiography including average wall thickness (E), interventricular septum thickness (IVS; F), left ventricular posterior wall thickness (LVPW; G), left ventricular internal diameter (LVID; H), ejection fraction (EF; I), and fractional shortening (FS; J). Significance was determined by one-way ANOVA with Sidak’s multiple comparisons test.
DISCUSSION
We report here for the first time that environmentally relevant iAs exposure during gestation blunts pregnancy-induced growth of the maternal heart during late pregnancy and downregulates hormone receptors ERα and PGRMC1/2, as well as ANP and Akt expression. Pregnancy-induced cardiac hypertrophy is critical for meeting the circulatory demands of the mother and the developing fetus during gestation, and we find that iAs exposure disrupts this important hypertrophic process17,18. We observed a compensatory change in cardiac function in iAs-exposed dams (Fig. 2), despite the lack of proper heart enlargement (Fig. 3), along with downregulation of ERα, PGRMC1/2, (Fig. 4), ANP and Akt (Figs. 5 and 6). Additionally, we found an increase in heart weight relative to body weight in offspring (Fig. 7) and decreased growth at P12 (Fig. 8) suggesting that either direct iAs exposure or improper maternal cardiac remodeling may contribute to adverse outcomes in offspring.
While our previous study demonstrated that iAs induced sex-dependent changes in cardiac hypertrophy, these changes were exclusive to adult males and adult females remained unaffected after an 8-week exposure16. However, in the current study, we find that pregnant females are particularly vulnerable to gestational iAs exposure. This study is the first to examine the impact of iAs exposure on maternal cardiac remodeling during pregnancy and our findings identify a critical disruption of pregnancy-induced maternal cardiac growth.
iAs exposure and maternal cardiac function
Heart growth or hypertrophy is a critical process that occurs during pregnancy. Prior work demonstrates that heart weight significantly increases in late pregnancy and post-partum; a non-significant increase in cardiomyocyte cross-sectional area during late pregnancy was also noted in this study, and this increase in cardiomyocyte cross-sectional area became significant postpartum47. Consistent with this prior work, our findings demonstrated a significant increase in heart weight in control and 100 μg/L iAs-exposed dams compared to iAs-exposed and non-exposed non-pregnant females (Fig. 3). However, cardiac growth was blunted in maternal hearts exposed to 1000 μg/L iAs during pregnancy, as we noted significantly smaller heart weight with 1000 μg/L iAs exposure compared to control and 100 μg/L iAs exposed dams (Fig. 3). Cross-sectional area was also significantly lower in cardiomyocytes from 1000 μg/L iAs exposed dams compared to non-exposed pregnant controls (Fig. 3), providing further confirmation that pregnant dams exposed to 1000 μg/L iAs during gestation have smaller hearts during late pregnancy. Left ventricular hypertrophy during pregnancy results from volume overload, and a lack of hypertrophy vs. pregnant controls may indicate a reduction in blood volume expansion17,48. As such, plasma volume will be examined with iAs exposure in future studies at different timepoints during pregnancy.
Furthermore, we found that cardiac function (i.e., fractional shortening) was significantly enhanced in pregnant dams exposed to 1000 μg/L iAs compared to non-exposed pregnant controls or dams exposed to 100 μg/L iAs (Fig. 2), likely as a compensatory mechanism to increase blood flow to the mother and developing fetus. However, it is important to note that we did not observe the expected increase in cardiac output in non-exposed pregnant dams vs. non-pregnant control mice that has been observed previously47. Nonetheless, the increase in cardiac function that we did observe with 1000 μg/L iAs-exposed dams may result from several mechanisms that include enhanced cardiomyocyte contractility or increased β-adrenergic stimulation, and future studies will examine these potential compensatory mechanisms. Importantly, sustained alterations to either of these pathways could result in detrimental changes to the heart, which could include a transition from pregnancy-induced hypertrophy to pathological hypertrophy and/or an increased risk for prolonged cardiovascular disease postpartum49. As such, postpartum remodeling and specifically the impact of iAs exposure during pregnancy on the postpartum heart, is a critical area that will be addressed in future mechanistic studies. Importantly, we find no alterations in cardiac size or function with iAs exposure alone in non-pregnant females at E17.5, or 15 days of exposure (Figs. 2 and 3), which is consistent with our prior studies where we found no change to cardiac structure and function in adult female mice with iAs exposure at 4 and 8 weeks15,16. Taken together, our findings suggest that the unique combination of iAs exposure and pregnancy are necessary for increasing cardiac function (Fig. 2) and blunting pregnancy-induced cardiac growth (Fig. 3).
iAs exposure decreases myocardial hormone receptor expression
Pregnancy-induced hypertrophy is governed largely by sex-hormones, such as progesterone and estradiol, and previous research has shown that iAs can act as an endocrine disruptor50–53. In the current study we found that exposure to 1000 μg/L iAs downregulated cardiac Esr1, Pgrmc1, and Pgrmc2 during late pregnancy (Fig. 4). Prior work has shown that iAs exposure downregulates various steroid hormone receptors and decreases ERα expression in the uterus, leading to alterations in uterine hypertrophy50,51. Estradiol has been shown to be anti-hypertrophic in mice at high levels and pro-hypertrophic at low levels in cultured cells, mediating the activation of certain downstream signaling pathways that are crucial for cardiac hypertrophy25,45,46. Exposure to iAs has also been shown to impact progesterone signaling, decreasing serum progesterone in rats, as well as progesterone receptors in culture54–56. Consistent with these findings, our data showed decreased transcripts of receptors for progesterone (Pgrmc1, Pgrmc2) and estradiol (Esr1) in the heart (Figs. 4A–4C). Interestingly, we observed that doses of 100 and 1000 μg/L iAs downregulated Pgrmc1 and Pgrmc2 at equal levels, while a dose-dependent reduction in Esr1 was noted with 100 and 1000 μg/L iAs.
Dampened estradiol signaling during pregnancy can impact blood volume expansion and the ability of the heart to grow, as pregnancy-induced cardiac hypertrophy is reliant upon major signaling pathways activated by a combination of mechanotransduction due to increased blood volume and hormone signaling25,46,57. The increase in plasma volume during pregnancy is also a consequence of sodium resorption and water retention, signaled via placental estrogen production58. As such, altered Esr1 and Pgrmc1/2 expression in the maternal heart during pregnancy may contribute to our observed changes in cardiac function and heart size in pregnant mice exposed to 1000 μg/L iAs, but further studies are needed to further characterize the impact of iAs on hormonal changes at different time points throughout pregnancy and establish causality with a specific focus on hormone levels.
Akt and ANP reduction in maternal hearts exposed to iAs
Transcriptionally, pregnancy-induced hypertrophy resembles exercise-induced hypertrophy, but there are notable differences24. For example, Acta1, ANP, and BNP are found to be upregulated in late pregnancy24,59,60. When probing for genes associated with pregnancy-induced cardiac hypertrophy17,45, we found that dams exposed to either 100 or 1000 μg/L iAs showed reduced transcript levels of Akt1, a primary mediator of pregnancy-induced hypertrophy46, in the maternal heart but this change was not significant (Fig. 5A). However, we did find a significant reduction in the protein expression of Akt in the maternal heart, and phosphorylation of the active site showed a trending decrease with iAs exposure (Figs. 6A, 6B). Our findings are consistent with a prior study, which demonstrated that exposure to iAs in adult rats reduced Akt expression in cardiac tissue irrespective of pregnancy61. While iAs exposure reduced myocardial protein expression of Akt in our model, additional research is needed to determine if this reduction in Akt expression specifically contributes to changes heart function and the blunted hypertrophy observed in 1000 μg/L iAs-exposed pregnant mice.
Exposure to 100 or 1000 μg/L iAs induced a ~50% reduction in transcript levels of Nppa (ANP) in the maternal heart (Fig. 5B). ANP is secreted by the heart in response to atrial distension and is found to be increased consistently in blood plasma throughout pregnancy59,62,63. ANP is a vasodilator and transcription is induced by atrial stretching due to volume overload64. These findings further suggest that iAs exposure could alter plasma volume expansion during pregnancy. In humans, exposure to iAs increased hemoglobin concentration and decreased blood protein concentration, indicating that iAs exposure may affect plasma volume regulation65,66. However, this has not been explored in the context of pregnancy. While a reduction in both ANP and Akt are associated with cardiovascular alterations during pregnancy, the reduction at both doses and the resulting pathophysiologic changes only at the highest dose may have several implications. While it is possible that ANP and Akt are acting in a synergistic manner with our observed changes in hormone receptor signaling, it is also possible that altered ANP and Akt levels are not associated with the changes in hypertrophic signaling and heart size at this timepoint. However, the full molecular pathway has yet to be elucidated, and thus it will be important to determine how the molecular profile of the maternal heart changes with iAs exposure during early- and mid-pregnancy to further our understanding of iAs exposure on the maternal heart.
Effects of gestational iAs exposure on offspring
In offspring, we found that heart weight was increased with 1000μ/L iAs exposure, but not with 100 μg/L iAs (Fig. 7C). Left ventricular diameter was also increased at E17.5 with 1000 μg/L iAs exposure (Fig. 7G). In mammals, heart cells proliferate from conception until late gestation and then switch to cell hypertrophy shortly after birth67. Pregnancies with metabolic dysfunction, such as obesity and diabetes, result in increased fetal cardiac hypertrophy due to increased cardiomyocyte cell size68–70. In adult hearts, enlarged left ventricular diameter in combination with decreased ejection fraction culminates in heart failure71. However, we do not see changes to ejection fraction with either iAs exposure dose (Fig. 7H), which suggests that there are only structural changes to the heart at E17.5. Several epidemiologic studies have associated in utero exposure to iAs with congenital heart defect72–74, but this has yet to be replicated in a murine model with a relevant exposure dose or route of exposure. We also examined postnatal cardiac function at P12 but found no alterations to cardiac function or structure across doses (Fig. 8). These findings suggest that changes observed in the fetal heart at E17.5 may potentially prime the heart for disease as an adult and many epidemiologic studies have found that in utero exposure to iAs induces cardiovascular disease later in life75–78.
Furthermore, we did not see alterations to litter size, embryonic body weight, or differences in sex distribution of litters at E17.5 with exposure to either 100 or 1000 μg/L iAs beginning at E2.5 (Fig. 7A, 7C). In humans, in utero exposure to iAs has been shown to increase spontaneous abortion and decrease birthweight79–81. While it is still unclear if this phenotype is seen in mice, it is possible that disrupted embryonic growth is due to exposure occurring before E2.5. We characterized offspring body and organ growth postnatally at P12 and found that exposure to 100 μg/L iAs in utero impaired growth as measured by body weight at P12 (Fig. 8A). Similar findings were observed in a prior study using doses as low as 10 μg/L iAs82. However, that study showed that cross-fostering the pups reduced the growth deficit, suggesting that the reduction in growth was due to breast milk nutrition rather than altered in utero programming82. Since our model halted iAs exposure at birth, and we did not cross-foster, it is possible that iAs may have lasting effects on breast milk nutrition. Additionally, iAs has been shown to alter epigenetic re-programming in offspring after in utero exposure83,84. In human studies, iAs has been shown to regulate birthweight via placental-derived miRNAs85,86. While we see evidence of some phenotype in offspring exposed to iAs in utero, more experiments are needed to define exposure windows and understand how these changes persist into adulthood.
LIMITATIONS
While this study provides important insights into the effects of iAs on maternal health, there are limitations that require acknowledgement. Firstly, the findings of this study are largely descriptive and although we do report molecular changes, additional work is required to establish mechanism and causality. Furthermore, this study used only a single prepartum timepoint, so future studies will be needed to characterize the physiologic growth of the maternal heart during early and mid-pregnancy timepoints throughout the 21-day window of murine pregnancy. We also cannot rule out arsenic-induced atrophy since atrophic signaling was not examined. The exposure was also started at E2.5 during gestation, while a real-world exposure would encompass a lifetime. Even though this is a limitation of our study, this exposure scenario is important for defining a critical window of maternal exposure. Mice were also housed in pairs to reduce stress during pregnancy, and water and food consumption was measured per cage, so individual variability is not accounted for and is a limitation. Additionally, experiments were conducted using a mouse model, and while mice are well characterized and commonly utilized for studies examining pregnancy and iAs exposure, it is important to note that iAs metabolism in mice is different compared to humans87–89. The mouse metabolizes iAs more quickly than humans, making higher doses in mice equivalent to lower doses in humans. There are also key differences in murine pregnancy compared to that of humans. Many rodent studies have not been able to replicate some of the findings associated with in utero exposure of iAs on development in humans, indicating that there are differences between the two species, and this should be taken into consideration when interpreting these findings. iAs has also been shown to target mitochondria and affect cellular metabolism90–95. Shifts in cardiac metabolism during pregnancy have been shown to be essential for cardiac hypertrophy47,96–100. While we did not examine iAs-dependent effects on myocardial metabolism in the current study, these pathways need to be further investigated. Additionally, we found that cardiac function was dampened after the administration of isoflurane, which is consistent with previous studies101,102. Since isoflurane reduced baseline cardiac function, it is important to note that isoflurane could affect our experimental findings, but all groups were exposed to the same amount of isoflurane during echocardiography. Indeed, the lack of increased cardiac output in non-exposed dams vs. non-pregnant control mice may very well result from the masking of changes in cardiac function by isoflurane, and as such is a limitation of this study that will be further investigated in future studies. Our use of M-mode echocardiography (vs. B-mode), may also affect our ability to resolve structural and functional differences in the heart with pregnancy. Finally, we only examined two iAs doses in our study, 100 μg/L and 1000 μg/L iAs. Lower doses should be used in future studies to understand if these effects, specifically postpartum, persist at lower doses, such as the recommended limit of 10 μg/L provided by the Environmental Protection Agency.
CONCLUSION
iAs exposure during gestation blunted pregnancy-cardiac growth and increased cardiac function during late pregnancy, and led to the downregulation of ERα, PRMGC1/2, ANP and Akt. As such, these changes have the potential to contribute to adverse pregnancy-related cardiovascular events, impact developing offspring, as noted, and may lead to increased risk for maternal CVD postpartum. Taken together, our findings suggest that exposure to iAs during pregnancy causes adverse effects on both the pregnant dam and the offspring. Our findings also add to the growing body of evidence implicating iAs as a driver of cardiovascular disease and a potential risk factor for maternal and fetal health during pregnancy, underscoring the importance of removing iAs from human consumption.
Supplementary Material
HIGHLIGHTS.
Arsenic exposure blunts maternal cardiac hypertrophy during late pregnancy
Arsenic exposure alters myocardial hormone receptor and hypertrophic gene expression in late pregnancy
Offspring from this exposure model showed enlarged hearts at embryonic day 17.5 and reduced body weight at P12
ACKNOWLEDGEMENTS
We acknowledge the technical assistance of the Oncology Tissue Services Core and the Small Animal Cardiovascular Phenotyping and Model Core at the Johns Hopkins University School of Medicine.
FUNDING
This work was supported by the National Institutes of Health [T32 ES007141 (NT), F31 HL165820 (HG), R21 HL157800 (MK) and R01 HL136496 (MK)].
Declaration of interests
Mark Kohr reports financial support was provided by National Heart Lung and Blood Institute. Nicole Taube reports financial support was provided by National Institute of Environmental Health Sciences. Haley Garbus reports financial support was provided by National Heart Lung and Blood Institute. Mark Kohr reports a relationship with National Heart Lung and Blood Institute that includes: funding grants. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare they have nothing to disclose.
REFERENCES
- 1.Substance Priority List | ATSDR. https://www.atsdr.cdc.gov/spl/index.html.
- 2.Nordstrom DK PUBLIC HEALTH: Enhanced: Worldwide Occurrences of Arsenic in Ground Water. Science (1979) 296, 2143–2145 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Abernathy CO, Thomas DJ & Calderon RL Toxicity and Risk Assessment of Trace Elements Health Effects and Risk Assessment of Arsenic 1,2.
- 4.Vahter M Mechanisms of arsenic biotransformation. Toxicology 181–182, 211–217 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Ferreccio C et al. Lung cancer and arsenic concentrations in drinking water in Chile. Epidemiology 11, 673–679 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Mabuchi K, Lilienfeld AM & Snell LM Cancer and occupational exposure to arsenic: A study of pesticide workers. Prev Med (Baltim) 9, 51–77 (1980). [DOI] [PubMed] [Google Scholar]
- 7.Yuan Y et al. Kidney cancer mortality: fifty-year latency patterns related to arsenic exposure. Epidemiology 21, 103–108 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Domingo-Relloso A et al. Arsenic Exposure, Blood DNA Methylation, and Cardiovascular Disease. Circ Res 131, E51–E69 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y et al. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study. BMJ 342, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moon KA et al. Association between Low to Moderate Arsenic Exposure and Incident Cardiovascular Disease. A Prospective Cohort Study NIH Public Access. Ann Intern Med 159, 649–659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zierold KM, Knobeloch L & Anderson H Prevalence of Chronic Diseases in Adults Exposed to Arsenic-Contaminated Drinking Water. 10.2105/AJPH.94.11.1936 94, 1936–1937 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Fu J, Wang H, Hou Y & Pi J Arsenic Exposure and Lifestyle-Related Diseases. Environ Health Prev Med 83–118 (2019) doi: 10.1007/978-981-13-2565-6_6.31888460 [DOI] [Google Scholar]
- 13.Farzan SF, Karagas MR & Chen Y In utero and early life arsenic exposure in relation to long-term health and disease. Toxicol Appl Pharmacol 272, 384–390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.States JC, Srivastava S, Chen Y & Barchowsky A Arsenic and Cardiovascular Disease. Toxicological Sciences 107, 312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veenema R et al. Inorganic arsenic exposure induces sex-disparate effects and exacerbates ischemia-reperfusion injury in the female heart. Am J Physiol Heart Circ Physiol 316, H1053–H1064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabir R et al. Inorganic arsenic induces sex-dependent pathological hypertrophy in the heart. Am J Physiol Heart Circ Physiol 320, H1321–H1336 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eghbali M, Wang Y, Toro L & Stefani E Heart hypertrophy during pregnancy: a better functioning heart? Trends Cardiovasc Med 16, 285–291 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Chung E & Leinwand LA Pregnancy as a cardiac stress model. Cardiovasc Res 101, 561–570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berlin NI, Goetsch C, Hyde GM & Parsons RJ The blood volume in pregnancy as determined by P32 labeled red blood cells. Surg Gynecol Obstet 97, 173–176 (1953). [PubMed] [Google Scholar]
- 20.Dieckmann WJ & Wegner CR THE BLOOD IN NORMAL PREGNANCY: I. BLOOD AND PLASMA VOLUMES. Arch Intern Med 53, 71–86 (1934). [Google Scholar]
- 21.Hytten FE & Paintin DB Increase in plasma volume during normal pregnancy. Journal of Obstetrics and Gynaecology of the British Commonwealth 70, 402–407 (1963). [DOI] [PubMed] [Google Scholar]
- 22.Robson SC, Hunter S, Boys RJ & Dunlop W Serial study of factors influencing changes in cardiac output during human pregnancy. 10.1152/ajpheart.1989.256.4.H1060 256, (1989). [DOI] [PubMed] [Google Scholar]
- 23.Mabie WC, DiSessa TG, Crocker LG, Sibai BM & Arheart KL A longitudinal study of cardiac output in normal human pregnancy. Am J Obstet Gynecol 170, 849–856 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Chung E, Heimiller J & Leinwand LA Distinct Cardiac Transcriptional Profiles Defining Pregnancy and Exercise. PLoS One 7, e42297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung E, Yeung F & Leinwand LA Calcineurin activity is required for cardiac remodelling in pregnancy. Cardiovasc Res 100, 402–410 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murr SM, Stabenfeldt GH, Bradford GE & Geschwind II Plasma progesterone during pregnancy in the mouse. Endocrinology 94, 1209–1211 (1974). [DOI] [PubMed] [Google Scholar]
- 27.McCormack JT & Greenwald GS PROGESTERONE AND OESTRADIOL-17β CONCENTRATIONS IN THE PERIPHERAL PLASMA DURING PREGNANCY IN THE MOUSE. Journal of Endocrinology 62, 101–107 (1974). [DOI] [PubMed] [Google Scholar]
- 28.Goldstein J, Sites CK & Toth MJ Progesterone stimulates cardiac muscle protein synthesis via receptor-dependent pathway. Fertil Steril 82, 430–436 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Pedram A et al. Estrogen Inhibits Cardiac Hypertrophy: Role of Estrogen Receptor-β to Inhibit Calcineurin. Endocrinology 149, 3361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conforti A et al. Air pollution and female fertility: a systematic review of literature. Reprod Biol Endocrinol 16, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borja-Aburto VH et al. Blood lead levels measured prospectively and risk of spontaneous abortion. Am J Epidemiol 150, 590–597 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Cole DC et al. Environmental contaminant levels and fecundability among non-smoking couples. Reprod Toxicol 22, 13–19 (2006). [DOI] [PubMed] [Google Scholar]
- 33.da Cunha Martins A, Carneiro MFH, Grotto D, Adeyemi JA & Barbosa F Arsenic, cadmium, and mercury-induced hypertension: mechanisms and epidemiological findings. 10.1080/10937404.2018.1432025 21, 61–82 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Tsuji JS, Perez V, Garry MR & Alexander DD Association of low-level arsenic exposure in drinking water with cardiovascular disease: A systematic review and risk assessment. Toxicology 323, 78–94 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Nigra AE, Ruiz-Hernandez A, Redon J, Navas-Acien A & Tellez-Plaza M Environmental Metals and Cardiovascular Disease in Adults: A Systematic Review Beyond Lead and Cadmium. Curr Environ Health Rep 3, 416–433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu K et al. Associations of exposure to lead and cadmium with risk of all-cause and cardiovascular disease mortality among patients with type 2 diabetes. Environ Sci Pollut Res Int 29, 76805–76815 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Mehta LS et al. On behalf of the American Heart Association Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Stroke Council Cardiovascular Considerations in Caring for Pregnant Patients A Scientific Statement From the American Heart Association. Circulation 141, 884–903 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Hoyert DL Health E-stat, February 2022. (2020) doi: 10.15620/cdc:103855. [DOI] [Google Scholar]
- 39.Parikh NI et al. Adverse Pregnancy Outcomes and Cardiovascular Disease Risk: Unique Opportunities for Cardiovascular Disease Prevention in Women: A Scientific Statement From the American Heart Association. Circulation 143, E902–E916 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Quintana MT et al. Cardiomyocyte-Specific Human Bcl2-Associated Anthanogene 3 P209L Expression Induces Mitochondrial Fragmentation, Bcl2-Associated Anthanogene 3 Haploinsufficiency, and Activates p38 Signaling. Am J Pathol 186, 1989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia N et al. Cardioprotective effects of granulocyte colony-stimulating factor in angiotensin II-induced cardiac remodelling. Clin Exp Pharmacol Physiol 36, 262–266 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Lin BL et al. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 7, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaguchi K et al. Indoxyl Sulfate Activates NLRP3 Inflammasome to Induce Cardiac Contractile Dysfunction Accompanied by Myocardial Fibrosis and Hypertrophy. Cardiovasc Toxicol 22, 365–377 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Long V & Fiset C Contribution of estrogen to the pregnancy-induced increase in cardiac automaticity. J Mol Cell Cardiol 147, 27–34 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Eghbali M et al. Molecular and functional signature of heart hypertrophy during pregnancy. Circ Res 96, 1208–1216 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Chung E, Yeung F & Leinwand LA Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J Appl Physiol (1985) 112, 1564–1575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fulghum KL et al. Metabolic Signatures of Pregnancy-Induced Cardiac Growth. Am J Physiol Heart Circ Physiol 323, 1–56 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorn GW & Mann DL Signaling pathways involved in left ventricular remodeling: Summation. J Card Fail 8, (2002). [DOI] [PubMed] [Google Scholar]
- 49.Oldfield CJ, Duhamel TA & Dhalla NS Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol 98, 74–84 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Davey JC, Bodwell JE, Gosse JA & Hamilton JW Arsenic as an Endocrine Disruptor: Effects of Arsenic on Estrogen Receptor–Mediated Gene Expression In Vivo and in Cell Culture. Toxicological Sciences 98, 75–86 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Chatterjee A & Chatterji U Arsenic abrogates the estrogen-signaling pathway in the rat uterus. Reproductive Biology and Endocrinology 8, 1–11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kitchin KT & Wallace K Arsenite binding to synthetic peptides based on the Zn finger region and the estrogen binding region of the human estrogen receptor-α. Toxicol Appl Pharmacol 206, 66–72 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Kaltreider RC, Davis AM, Lariviere JP & Hamilton JW Arsenic alters the function of the glucocorticoid receptor as a transcription factor. Environ Health Perspect 109, 245–251 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akram Z et al. Adverse effects of arsenic exposure on uterine function and structure in female rat. Exp Toxicol Pathol 62, 451–459 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Chen P et al. Arsenic exposure during juvenile and puberty significantly affected reproductive system development of female SD rats. Ecotoxicol Environ Saf 242, (2022). [DOI] [PubMed] [Google Scholar]
- 56.Bodwell JE, Gosse JA, Nomikos AP & Hamilton JW Arsenic disruption of steroid receptor gene activation: Complex dose - Response effects are shared by several steroid receptors. Chem Res Toxicol 19, 1619–1629 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haines CD, Harvey PA & Leinwand LA Estrogens Mediate Cardiac Hypertrophy in a Stimulus-Dependent Manner. Endocrinology 153, 4480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longo LD Maternal blood volume and cardiac output during pregnancy: a hypothesis of endocrinologic control. Am J Physiol 245, (1983). [DOI] [PubMed] [Google Scholar]
- 59.Sala C et al. Atrial Natriuretic Peptide and Hemodynamic Changes During Normal Human Pregnancy. Hypertension 25, 631–636 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Castro LC, Hobel CJ & Gornbein J Plasma levels of atrial natriuretic peptide in normal and hyprtensive pregnancies: A meta-analysis. Am J Obstet Gynecol 171, 1642–1651 (1994). [DOI] [PubMed] [Google Scholar]
- 61.Oyagbemi AA et al. Sodium arsenite-induced cardiovascular and renal dysfunction in rat via oxidative stress and protein kinase B (Akt/PKB) signaling pathway. Redox Rep 22, 467–477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fournier A et al. Atrial natriuretic factor in pregnancy and pregnancy-induced hypertension. 10.1139/y91-237 69, 1601–1608 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Thomsen Jø. K., Fogh-Andersen N, Jaszczak P & Glese J Atrial natriuretic peptide (ANP) decrease during normal pregnancy as related to hemodynamic changes and volume regulation. Acta Obstet Gynecol Scand 72, 103–110 (1993). [DOI] [PubMed] [Google Scholar]
- 64.Epstein FH, Levin ER, Gardner DG & Samson WK Natriuretic Peptides. 10.1056/NEJM199807303390507 339, 321–328 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Prasad P, Sarkar N & Sinha D Effect of low- and high-level groundwater arsenic on peripheral blood and lung function of exposed rural women. Regul Toxicol Pharmacol 115, (2020). [DOI] [PubMed] [Google Scholar]
- 66.Liu H et al. The Relationship Between Preeclampsia and Arsenic Concentration in the Peripheral Blood. Biol Trace Elem Res 200, 3965–3974 (2022). [DOI] [PubMed] [Google Scholar]
- 67.Wulfsohn D, Nyengaard JR & Tang Y Postnatal growth of cardiomyocytes in the left ventricle of the rat. Anat Rec A Discov Mol Cell Evol Biol 277A, 236–247 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Han SS et al. Investigating the Mechanism of Hyperglycemia-Induced Fetal Cardiac Hypertrophy. PLoS One 10, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vaughan OR et al. Maternal obesity causes fetal cardiac hypertrophy and alters adult offspring myocardial metabolism in mice. J Physiol 600, 3169–3191 (2022). [DOI] [PubMed] [Google Scholar]
- 70.Depla AL et al. Effect of maternal diabetes on fetal heart function on echocardiography: systematic review and meta-analysis. Ultrasound Obstet Gynecol 57, 539–550 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narayanan K et al. Left Ventricular Diameter and Risk Stratification for Sudden Cardiac Death. Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rudnai T et al. Arsenic in drinking water and congenital heart anomalies in Hungary. Int J Hyg Environ Health 217, 813–818 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Jin X et al. Maternal exposure to arsenic and cadmium and the risk of congenital heart defects in offspring. Reproductive Toxicology 59, 109–116 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Richter F et al. Maternal exposure to arsenic in drinking water and risk of congenital heart disease in the offspring. Environ Int 160, (2022). [DOI] [PubMed] [Google Scholar]
- 75.Osorio-Yáñez C et al. Carotid intima-media thickness and plasma asymmetric dimethylarginine in Mexican children exposed to inorganic arsenic. Environ Health Perspect 121, 1090–1096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rahman M et al. Increased childhood mortality and arsenic in drinking water in Matlab, Bangladesh: a population-based cohort study. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuan Y et al. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. Am J Epidemiol 166, 1381–1391 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Rosenberg HG Systemic arterial disease with myocardial infarction. Report on two infants. Circulation 47, 270–275 (1973). [DOI] [PubMed] [Google Scholar]
- 79.Ahmad SA et al. Arsenic in drinking water and pregnancy outcomes. Environ Health Perspect 109, 629–631 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quansah R et al. Association of arsenic with adverse pregnancy outcomes/infant mortality: a systematic review and meta-analysis. Environ Health Perspect 123, 412–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Milton AH et al. Chronic arsenic exposure and adverse pregnancy outcomes in bangladesh. Epidemiology 16, 82–86 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Kozul-Horvath CD, Zandbergen F, Jackson BP, Enelow RI & Hamilton JW Effects of low-dose drinking water arsenic on mouse fetal and postnatal growth and development. PLoS One 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chakraborty A et al. Epigenetic modifications from arsenic exposure: A comprehensive review. Science of The Total Environment 810, 151218 (2022). [DOI] [PubMed] [Google Scholar]
- 84.Khan F, Momtaz S, Niaz K, Hassan FI & Abdollahi M Epigenetic mechanisms underlying the toxic effects associated with arsenic exposure and the development of diabetes. Food and Chemical Toxicology 107, 406–417 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Rahman ML et al. Regulation of birthweight by placenta-derived miRNAs: evidence from an arsenic-exposed birth cohort in Bangladesh. Epigenetics 13, 573–590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bozack AK et al. Cord blood DNA methylation of DNMT3A mediates the association between in utero arsenic exposure and birth outcomes: Results from a prospective birth cohort in Bangladesh. Environ Res 183, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vahter M Species Differences in the Metabolism of Arsenic Compounds. Appl Organomet Chem 8, 175–182 (1994). [Google Scholar]
- 88.Drobna Z, Styblo M & Thomas DJ An Overview of Arsenic Metabolism and Toxicity. doi: 10.1002/0471140856.tx0431s42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kobayashi Y, Agusa T, Kobayashi Y & Agusa T Arsenic Metabolism and Toxicity in Humans and Animals: Racial and Species Differences. 13–28 (2019) doi: 10.1007/978-981-13-2565-6_2. [DOI] [Google Scholar]
- 90.Jeong SH, Ahn C, Kwon JS, Kim KM & Jeung EB Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies. Toxics 11, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bustamante J, Nutt L, Orrenius S & Gogvadze V Arsenic stimulates release of cytochrome c from isolated mitochondria via induction of mitochondrial permeability transition. Toxicol Appl Pharmacol 207, 110–116 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Wadgaonkar P & Chen F Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin Cancer Biol 76, 258–266 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guidarelli A, Fiorani M, Cerioni L & Cantoni O Calcium signals between the ryanodine receptor- and mitochondria critically regulate the effects of arsenite on mitochondrial superoxide formation and on the ensuing survival vs apoptotic signaling. Redox Biol 20, 285–295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prakash C, Soni M & Kumar V Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. Journal of Applied Toxicology 36, 179–188 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Guidarelli A, Fiorani M, Cerioni L, Scotti M & Cantoni O Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition. BioFactors 43, 673–684 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Sugden MC, Changani KK, Bentley J & Holness MJ Cardiac glucose metabolism during pregnancy. Biochem Soc Trans 20, (1992). [DOI] [PubMed] [Google Scholar]
- 97.Kolwicz SC & Tian R Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90, 194–201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu LX et al. PDK4 Inhibits Cardiac Pyruvate Oxidation in Late Pregnancy. Circ Res 121, 1370–1378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu LX & Arany Z Maternal cardiac metabolism in pregnancy. Cardiovasc Res 101, 545–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Y et al. Cardiac Remodeling During Pregnancy With Metabolic Syndrome: Prologue of Pathological Remodeling. Circulation 143, 699–712 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roth DM, Swaney JS, Dalton ND, Gilpin EA & Ross J Impact of anesthesia on cardiac function during echocardiography in mice. Am J Physiol Heart Circ Physiol 282, (2002). [DOI] [PubMed] [Google Scholar]
- 102.Pachon RE, Scharf BA, Vatner DE & Vatner SF Best anesthetics for assessing left ventricular systolic function by echocardiography in mice. Am J Physiol Heart Circ Physiol 308, H1525–H1529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.