

Abstract
Background:
Several evolutionary explanations have been proposed for why chronic pain is a major clinical problem. One is that some mechanisms important for driving chronic pain, while maladaptive for modern humans, were adaptive because they enhanced survival. Evidence is reviewed for persistent nociceptor hyperactivity (PNH), known to promote chronic pain in rodents and humans, being an evolutionarily adaptive response to significant bodily injury, and primitive molecular mechanisms related to cellular injury and stress being exapted (co-opted or repurposed) to drive PNH and consequent pain.
Summary:
PNH in a snail (Aplysia californica), squid (Doryteuthis pealeii), fruit fly (Drosophila melanogaster), mice, rats, and humans has been documented as long-lasting enhancement of action potential discharge evoked by peripheral stimuli, and in some of these species as persistent extrinsically driven ongoing activity and/or intrinsic spontaneous activity (OA and SA, respectively). In mammals, OA and SA are often initiated within the protected nociceptor soma long after an inducing injury. Generation of OA or SA in nociceptor somata may be very rare in invertebrates, but prolonged afterdischarge in nociceptor somata readily occurs in sensitized Aplysia. Evidence for the adaptiveness of injury-induced PNH has come from observations of decreased survival of injured squid exposed to predators when PNH is blocked, from plausible survival benefits of chronic sensitization after severe injuries such as amputation, and from the functional coherence and intricacy of mammalian PNH mechanisms. Major contributions of cAMP-PKA signaling (with associated calcium signaling) to the maintenance of PNH both in mammals and molluscs suggests that this ancient stress signaling system was exapted early during the evolution of nociceptors to drive hyperactivity following bodily injury. Vertebrates have retained core cAMP-PKA signaling modules for PNH, while adding new extracellular modulators (e.g., opioids) and cAMP-regulated ion channels (e.g., TRPV1 and Nav1.8 channels).
Key Messages:
Evidence from multiple phyla indicates that PNH is a physiological adaptation that decreases the risk of attacks on injured animals. Core cAMP-PKA signaling modules make major contributions to the maintenance of PNH in molluscs and mammals. This conserved signaling has been linked to ancient cellular responses to stress, which may have been exapted in early nociceptors to drive protective hyperactivity that can persist while bodily functions recover after significant injury.
Keywords: Aplysia, Cyclic AMP signaling, Excitability, Mammals, Spontaneous activity
Introduction
Why do one in five adults (one in three above the age of 65) [1] suffer from chronic pain? This is a question of clear clinical importance and substantial social significance, given the societal burden and economic costs of uncontrolled pain plus the links between chronic pain and opioid abuse [2, 3]. However, it is also an interesting question from the perspective of evolutionary biology. Evolutionary considerations have led to several suggested answers to the question of why chronic pain is so common in people. These include assertions that 1) chronic pain today is promoted by a mismatch between humans’ more protected modern environments and sedentary lifestyles compared to those of their ancestors [4], 2) excessively sensitive and prolonged responses to stimuli that threaten bodily integrity are less costly than insufficient responses to noxious stimuli, resulting evolutionarily in a bias towards persistent and frequent pain (smoke detector principle) [5], 3) pain and other chronic ailments increase with age as injuries and illnesses accumulate, and these are not subject to countervailing evolutionary selection pressures past the child-bearing phase of life [6], and 4) some mechanisms that drive chronic pain were selected during evolution because they enhanced survival and thus reproductive success.
These reasons for why chronic pain is so common in modern humans are not mutually exclusive, all four are likely true. Here I review evidence for the fourth reason (chronic pain is promoted by evolutionarily adaptive traits), focusing on one physiological alteration, persistent nociceptor hyperactivity (PNH). I extend previous reviews supporting the evolutionary adaptiveness of PNH after significant bodily injury [7, 8] and consider evidence that cAMP signaling, which has primordial links to stress responses, may have been exapted in nociceptors to drive biologically adaptive hyperactivity that promotes clinically maladaptive chronic pain.
Persistent Nociceptor Hyperactivity as an Adaptive Response to Consequential Injury
Chronic pain depends upon numerous interacting mechanisms in many parts of the human body and nervous system, including the spinal cord and brain [9–12]. While central alterations are clearly important for chronic pain [13–15], my focus is on a peripheral mechanism, PNH – a sustained enhancement of action potential [AP] generation in nociceptors (Fig. 1), which can activate central sensitization mechanisms and may often be essential for maintaining central alterations [16, 17]. Nociceptor alterations in addition to hyperactivity are important for driving central pain mechanisms, including potentiation of a nociceptor’s synapses and sprouting of its peripheral and central terminals [18, 19]. However, these additional mechanisms in nociceptors are beyond the scope of this review. PNH is quite interesting from an evolutionary perspective because the availability of detailed information about specific molecular contributions to the same well-defined cellular alteration (hyperactivity) in an identified cell type (nociceptor) with the same type-defining functions (nociception and nociceptive sensitization) across distantly related lineages enables plausible conjectures about the exaptation of ancient molecular mechanisms for functions that are of great importance to humans.
Fig. 1.
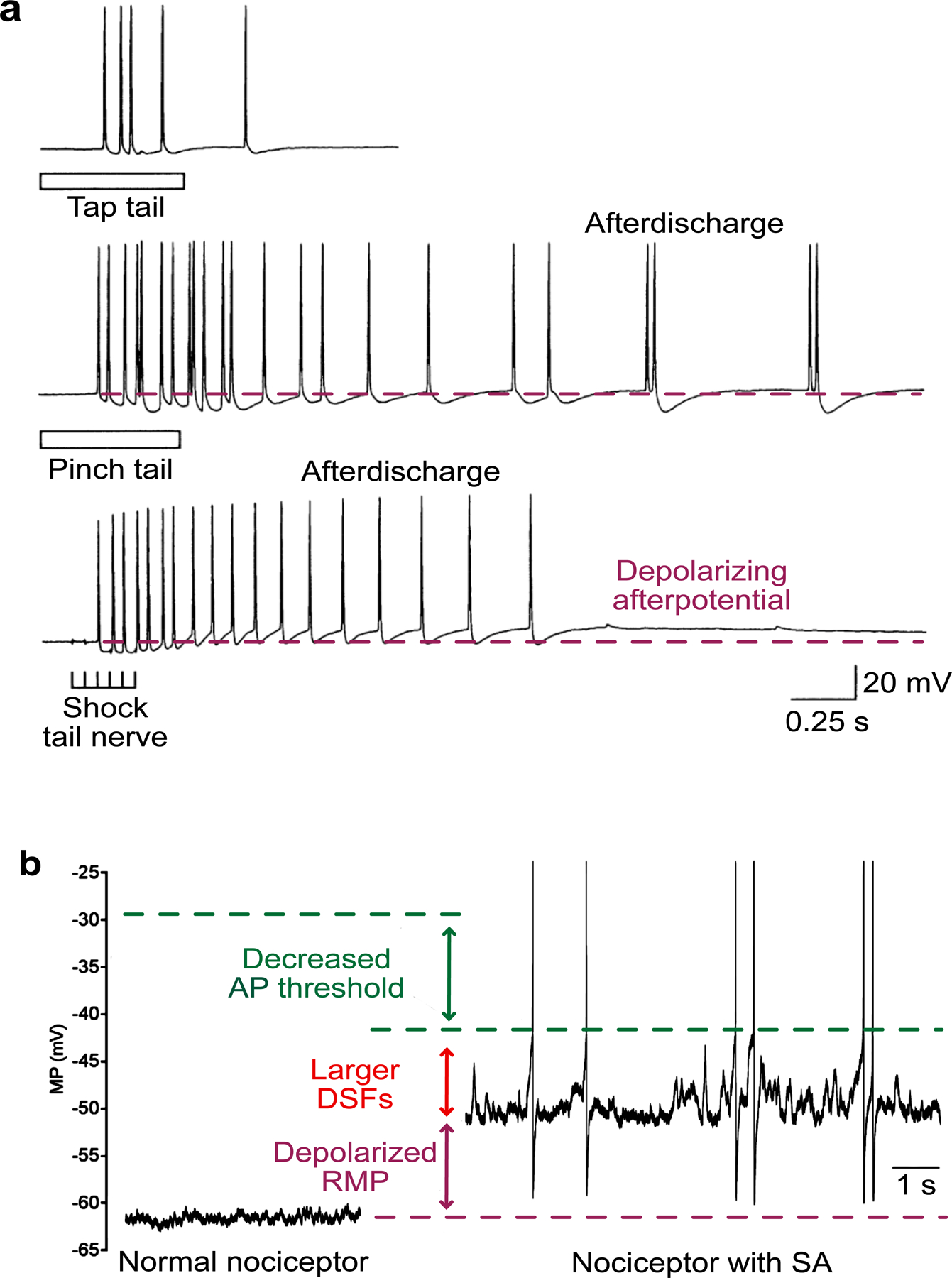
Two types of somal hyperactivity in nociceptors. a Afterdischarge generated by three kinds of peripheral stimulation delivered sequentially to the same Aplysia nociceptor in a semi-intact preparation with the tail attached to central ganglia containing the nociceptor somata: top panel, 0.5-s tap (75 g/mm2) to the neuron’s receptive field on the animal’s tail; middle panel, ~0.5-s pinch to the same receptive field; bottom panel, 0.25-s train of 2-ms shocks (20 Hz) to the nerve containing the nociceptor’s axon. The dashed purple line indicates the resting membrane potential (RMP) for comparison to the depolarizing afterpotential that can bring the soma membrane to action potential (AP) threshold. Afterdischarge when this potential is small, as shown here immediately after tail pinch, is generated peripheral to the soma, probably within the distant receptive field. The larger depolarizing afterpotential evoked in the soma by subsequent tail nerve shock triggers afterdischarge as a long-lasting consequence of the prior pinch. Modified from [51]. b Examples of RMP and one form of ongoing activity (OA) in nociceptors dissociated from dorsal root ganglia excised from a previously uninjured rat (left) and from a rat 2 months after spinal cord injury. Compared to the isolated control nociceptor shown on the left, the nociceptor from the previously injured rat exhibited OA at RMP (likely intrinsically generated spontaneous activity, SA), depolarization of RMP, enhanced depolarizing spontaneous fluctuations (DSFs) of membrane potential (MP), and decreased voltage threshold for AP initiation. The dashed purple line indicates RMP in the control nociceptor, and the dashed green line indicates AP threshold for each nociceptor. Modified from [52].
Nociceptors are defined as primary sensory neurons that are specialized to detect signals of injury and inflammation. They have been identified not only in multiple classes of chordates (mammals, birds, reptiles, fish) but also in annelids, molluscs, nematodes, and arthropods [20–26]. As reviewed below, nociceptor hyperactivity persisting for days, weeks, or months after noxious events has been found in invertebrates, mammals, and humans. PNH has been observed as increases in evoked discharge and in the development of spontaneous discharge in peripheral terminals, injured axons, and cell bodies (somata) of nociceptors.
PNH Manifested as Sensitization and Ongoing Activity in Nociceptor Peripheral Terminals
Sensitization of the peripheral branches of nociceptors associated with hyperalgesia (enhanced pain evoked by normally painful stimuli) was the first type of injury-induced PNH discovered, with the threshold for eliciting APs by heat stimuli decreasing and the number of APs generated by subthreshold thermal stimuli increasing in recordings from anesthetized cats [27, 28], monkeys [29] and rodents [reviewed by 30], and from humans [31]. Increased AP discharge during heating and possibly cooling following damaging ultraviolet irradiation that sensitizes defensive behavior has been shown in polymodal nociceptors (class IV) of Drosophila larvae [32, 33]. It is not known if this hyperactivity originates in peripheral branches of the nociceptors or in the soma, which is located close beneath the skin of a larva in the center of the peripheral arbor. Peripheral sensitization of electrical activity by noxious stimulation or injury, expressed as increased AP discharge to suprathreshold mechanical stimuli has been described in molluscs (Fig. 1a) [34–36], humans [31], and rodents [e.g., 37, 38–40]. Interestingly, acute sensitization of AP discharge after noxious stimulation appears to be rare or absent in fish nociceptors [41]. While sensitization of molluscan and mammalian nociceptors can be long lasting, persisting for 1–2 months after nerve injury in Aplysia (a substantial fraction of its 1-year lifespan) [34, 42, 43] and for months, years, or longer in rodents and humans [e.g., 44, 45], PNH often resolves after tissue healing and nerve regeneration [40, 43]. The eventual recovery of normal excitability that can occur after repair of injuries severe enough to damage innervating nerves is consistent with the inference that long-lasting PNH and behavioral sensitization function to protect organisms during prolonged healing following serious injury [20, 46].
The primary nociceptors that have been examined electrophysiologically in various species, including leeches, snails, fish, mice, rats, guinea pigs, cats, monkeys, and humans, are usually electrically silent; APs are generated when evoked by noxious stimuli delivered to their receptive fields but otherwise APs are absent or infrequent (Fig. 1a) [25]. Polymodal nociceptors in vertebrates [e.g., 27], the leech [47, 48], and Drosophila [32, 49] sometimes show low-frequency spontaneous discharge under basal recording conditions, which in some polymodal nociceptors may be caused by stimulation of thermosensitive ion channels. During nociceptive sensitization, another type of PNH – afterdischarge – can occur, in which stimuli that normally evoke a few APs evoke higher-frequency, longer-lasting discharge. For example, sensitized Aplysia nociceptors that usually respond to a moderate intensity tap stimulus with a few APs will respond with many APs generated in the periphery and sometimes initiated as well in the soma; responding as if to an intense pinch (Fig. 1a) [43, 50, 51].
In some species, significant injury and/or inflammation can result in yet another type of PNH: persistent generation of continuing discharge in the peripheral terminals and axons (especially at neuromas) of some nociceptors in the absence of additional mechanical or thermal stimulation. I use the term ongoing activity (OA) to describe any continuing discharge that does not depend upon brief, transient depolarization from extrinsic inputs; e.g., from conventional sensory generator potentials or excitatory postsynaptic potentials. Many investigators have used the term spontaneous activity (SA) for any form of OA, but it is useful mechanistically to define SA as a subtype of OA that is generated by intrinsic alterations [52] resulting from a change in cellular state that outlasts immediate effects of triggering events such as axonal membrane rupture or transient exposure to sensitizing neuromodulators. Persistent, low-frequency, irregular OA (possibly involving intrinsic SA) in presumed nociceptors occurs in rodents after peripheral nerve injury [53–56] or spinal cord injury (SCI) [39], and it also occurs in squid after bodily injury caused by attacks from other squid [36]. Enhanced OA in class IV nociceptors in Drosophila larvae was observed across a wide range of temperatures 24 hours after epidermal damage resulting from ultraviolet irradiation [32]. In rodent nociceptors, persistent OA can be driven by long-lasting peripheral inflammation [55, 57, 58]. Furthermore, peripheral generation of persistent OA in C-fibers (composed primarily of nociceptors) is prominent in various human neuropathic conditions [45, 59, 60]. Blocking OA in peripheral nerves of human patients effectively interrupts spontaneous neuropathic pain [16] and, more surprisingly, can also block poststroke pain [61].
PNH Manifested as Ongoing Activity and Afterdischarge in Nociceptor Somata
After nerve injury, PNH may involve APs generated ‘ectopically’ in the nociceptor cell body (soma) as well as more peripherally. In Aplysia nociceptors, somally generated afterdischarge can be evoked immediately and potentiated persistently by noxious peripheral stimulation (Fig. 1a) or by nerve injury, with the afterdischarge adding substantially to the number of APs reaching the central nervous system (CNS) and excite motor neurons mediating defensive behavior [43, 62, 63]. Afterdischarge is produced by a depolarizing afterpotential generated in or near the nociceptor soma, which requires Ca2+ influx [43, 51]. Unlike unmyelinated C-fiber nociceptors, which have very small receptive fields (e.g., ~1 mm diameter in cats) [27], Aplysia nociceptors’ receptive fields are quite large relative to the size of the body (diameters often >2 cm in individuals weighing ~250 g) [34, 64], similar to receptive field sizes of Aδ nociceptors in cats [65]. Consequently, Aplysia nociceptors and Aδ nociceptors are much less likely to be disconnected completely from their receptive fields during injury. It may be that in small C-fiber nociceptors of mammals, the most prominent form of somal hyperactivity is OA rather than afterdischarge because a C-fiber nociceptor innervating severely injured tissue is likely to lose its entire receptive field and with it the usual source of afferent AP discharge that triggers afterdischarge in the central soma.
While PNH manifested as OA and afterdischarge generated in the receptive fields and neuromas of mammalian nociceptors has long been evident [reviewed by 66], only recently has OA initiated in mammalian nociceptor somata been recognized as a major form of PNH and driver of persistent pain. Most in situ and in vivo studies of somally generated OA have been on sensory neurons other than nociceptors; specifically, large, low-threshold, A-fiber mechanoreceptors (LTMRs) [e.g., 67, 68–72, 55, 73]. In these studies, little or no somally initiated OA was found in C-fiber nociceptors under neuropathic conditions. However, technical limitations of the three methods used to record somally generated OA in situ or in vivo have precluded accurate detection of single APs in the thin axons and small somata of C-fiber nociceptors. First, teased fiber recording from nerves is an indirect monitor of APs initiated in distant somata, and even when conditions are optimal, only APs in the largest C-fibers may be detectable. Second, sharp electrode recording is more informative but suffers from a widely overlooked limitation – even very sharp microelectrodes decrease input resistance dramatically (often >10-fold). Although this inadvertent shunting may have relatively little effect on the generation of APs (because enormous voltage-gated Na+ currents provide a large safety factor for producing overshooting APs – see Figure 1a) or on resting membrane potential (RMP, because of powerful homeostatic mechanisms), the decreased input resistance during sharp electrode recording will make normally small spontaneous depolarizations proportionally smaller and much less likely to reach AP threshold. Third, Ca2+ imaging is necessarily a coarse and indirect monitor of APs and, while it continually improves, its limited sensitivity and temporal resolution has hampered detection of single APs. For example, ongoing clustered OA initiated in DRG neuron somata in vivo was recently reported to be correlated with spontaneous, paroxysmal pain in Pirt-Cre/GCaMP6s mice [74]. However, each imaging frame used to measure Ca2+ transients produced by APs was 238 ms, far longer than the 1–5 ms duration of single APs. All these technical limitations mean the incidence and importance of ectopic somal OA in C-fiber nociceptors may often be underestimated.
On the other hand, strong evidence for OA (including intrinsic SA, Fig. 1b) generated within nociceptor somata has come from dissociated DRG neurons recorded with the whole-cell, gigaseal patch technique. Although this method also has limitations, including the dialysis of soluble intracellular constituents that may be important for the phenomena under investigation, it provides unmatched fidelity for measurement of detailed electrophysiological properties, such as small spontaneous fluctuations of RMP. This method revealed SA continuously generated in mammalian nociceptor somata isolated after various neuropathic conditions, including chronic constriction injury (CCI) of the sciatic nerve in rats [75], spinal cord injury (SCI) in rats [44] (Fig. 1b) and mice [76], paclitaxel treatment in rats [77], cisplatin treatment in mice [78], and compression of spinal nerves by tumors in humans [79]. These findings show that diverse neuropathic conditions induce hyperexcitability that (quite remarkably) survives the cellular trauma and stresses caused by subsequent DRG excision, dissociation, and neuronal culturing [80]. Countering the logical possibility that somally generated SA might only be an artifact of dissociation are the findings that 1) neuropathy-induced somal SA was preserved when recording from a DRG neuron in vivo and then after it was isolated from the DRG [69], 2) the irregular pattern and low discharge frequency of ongoing discharge (perhaps extrinsically driven OA plus intrinsic SA) after SCI when recorded in vivo from dorsal root filaments (in or near C-fiber and Aδ somata within a DRG) show remarkable parallels to intrinsic SA generated in dissociated nociceptor somata after SCI [44], and 3) SA in dissociated nociceptor somata after SCI continues to be regulated by potent cell signaling pathways (including cAMP and ERK pathways; see below) under whole-cell recording conditions [81, 82]. Thus, somally initiated SA appears to be a robust nociceptor specialization that can operate similarly in vivo and in dissociated rodent and human DRG neurons.
Multiple ion channels contribute to the generation of somal SA in dissociated rodent nociceptors, including voltage-gated Na+ and Ca2+ channels and TRP channels [83]. One of these, Nav1.8 (which is expressed almost entirely in primary somatosensory neurons, mainly nociceptors), was shown to be required for SA in sensory axons after peripheral nerve transection in mice [84]. After SCI in rats, both SA in dissociated nociceptors and correlated measures of spontaneous pain, heat hyperalgesia, and mechanical allodynia were eliminated by antisense oligodeoxynucleotide knockdown of Nav1.8 [85].
While it seems likely that spontaneous discharge recorded in dissociated neurons represents, at least in part, intrinsic SA driven by a persisting hyperexcitable state in DRG neurons rather than continuing stimulation by extrinsic factors, these cultures contain heterogeneous cell types that may continually release excitatory factors. Indeed, in the presence but not the absence of conditioned medium from DRG neuron cultures prepared from SCI rats, neurons cultured from naïve control rats exhibited OA when artificially depolarized to −45 mV for 30 seconds [86]. This SCI-dependent extrinsic excitatory factor present in the culture medium drove OA at −45 mV but was not sufficient to drive OA at RMP, suggesting that the extrinsic excitants work together with persistent intrinsic hyperexcitability after SCI to drive OA at RMP. Interestingly, the OA produced by conditioned medium was prevented by an inhibitor of a widely expressed proinflammatory cytokine, macrophage migration inhibitory factor (MIF) [86], known to chronically increase in humans after SCI [87]. An important open question concerns the degree to which PNI driving ongoing pain in any condition represents persistent intrinsic alterations of nociceptors versus persistently altered exposure of the nociceptors to extrinsic modulators such as cytokines, and how the relative contributions of each may change over time.
Clinically Maladaptive but Evolutionarily Adaptive PNH Is Linked to Pain and Anxiety
Pain and associated PNH that persist after healing are assumed almost universally to be purely pathological, maladaptive consequences of injury or disease [11, 88, 89]. In the modern world, chronic pain seems to bring no benefit to those afflicted, and it not only diminishes quality of life, it also is linked to many health risks, including substance abuse and suicide [1]. Nevertheless, several arguments suggest that, while maladaptive from a modern clinical perspective, the PNH that often drives persistent pain has been subject to positive evolutionary selection. These arguments have been detailed elsewhere [7, 8, 90] and are briefly summarized here. The first argument was an extension of arguments for the adaptive value of acute nociceptive sensitization, namely that nociceptor hyperactivity protects a wounded animal, and especially the injured site, until healing has occurred, and it helps to compensate for lost sensory function in tissue denervated by injury [20, 46, 91]. This argument was based on the obvious need to protect injured sites from additional stress during movements of an animal, on the long periods needed for recovery from severe sublethal injury, and on numerous field observations showing that injury increases the risk of attack from predators that often selectively target wounded prey [e.g., 92]. Direct evidence for the adaptiveness of nociceptor hyperactivity came from squid in which bodily injury had been shown to induce widespread, long-lasting nociceptor sensitization and OA [36]. Staged encounters with predatory fish revealed that the fish selectively target previously injured squid, which showed heightened vigilance to the predators (initiating earlier escape responses) but were still killed more frequently than uninjured squid [93]. However, squid in which the injury site was transiently anesthetized (previously shown to prevent nociceptor sensitization and OA [36]) during the experimental injury later showed significantly higher mortality than squid that had not received local anesthesia at the time of injury. This indicated that injury-induced PNH can increase survival under conditions of high predation risk by driving continuing hypervigilance [93]. Analogous results [94] were found in mice using a spared nerve injury pain model that can induce somal OA in DRG neurons [74]. When injured mice had to choose between long and short routes to a food reward, they avoided the short route when it exposed them to the smell of fox urine, suggesting that ongoing awareness of chronic injury driven by nociceptor OA altered their behavior to reduce predatory risk [94]. Such injury-induced hypervigilance appears functionally similar to generalized anxiety in humans [20]. Anxiety is a common comorbidity of chronic pain in humans and rodents [95] and it seems likely that PNH may promote ongoing generalized anxiety as well as ongoing pain.
The second argument extends the first by noting that predators often look for signs of physical impairment, including indicators that may not resolve after healing (e.g., limping after amputation) and that result in life-long behavioral alterations detectable by predators and aggressive conspecifics [7, 90]. Amputees in various species have been reported to survive and reproduce long after injury. Importantly, in human patients, ongoing phantom limb pain and non-painful phantom sensations (both of which can be permanent) are continuously driven by sensory neuron OA initiated and generated within DRGs [96]. This finding supports the idea that PNH (and possibly OA in some low-threshold mechanosensory neurons) promotes hypervigilance that might be prompted by ongoing pain and which, during our evolutionary history, enhanced the survival of severely and sometimes permanently injured individuals having detectable physical impairments that increase their risk of attack by predators and members of their own species [7, 8]. Most species are not social (most invertebrates don’t provide care for their young), and they are unlikely to benefit from communication of pain states to others (humans and domesticated animals being notable exceptions). In asocial and minimally social species, including many mammals, ongoing pain does not appear to be manifested in obvious behavioral cues (e.g., ongoing vocalization, clear grimacing) that could invite attack [97, 98].
The third argument is for the adaptiveness of OA in the mammalian nociceptor soma [7, 8, 52, 90, 99], and is based on arguments for a complex ‘functional design’ [100, 101] of primary nociceptors in mammals that enables potentially beneficial somal generation of OA after bodily injury. First, mammalian nociceptor somata can be continuing sources of OA after severe injury because they are likely to survive, residing in sensory ganglia that are centrally located and protected by bone and dura. Second, nociceptor somata are in a unique position to integrate numerous local and systemic signals of bodily injury when ‘deciding’ whether to generate OA because they are exposed to humoral signals both in the blood (sensory ganglia lack an effective vascular permeability barrier) and in cerebrospinal fluid, in addition to injury- and inflammation-related electrical and molecular signals conveyed from their peripheral and central terminals by action potentials and axonal transport. Third, OA itself shows functional specialization by virtue of multiple, redundant mechanisms [52, 83, 99]. In particular, changes in RMP, input resistance, AP threshold, and the frequency of large depolarizing spontaneous fluctuations (DSFs) of membrane potential drive OA in diverse persistent pain conditions in rodents and humans (Fig. 1b) [44, 52, 76, 78, 86, 99, 102]. These changes involve complementary contributions from numerous ion channels [83, 85, 103, 104] and multiple cell signaling pathways [81, 82, 86]. Although PNH might be induced in modern humans by various pathological conditions that had nothing to do with its selection during evolution, such as chemotherapy or SCI (SCI is almost always fatal without modern medical support), these arguments suggest that PNH is a physiological specialization that was selected in part for its ability to increase survival after injury or disease that persistently increased the risk of being attacked.
Exaptation and Mechanisms of Persistent Nociceptor Hyperactivity
The existence of PNH in multiple species across several phyla, coupled with increasing knowledge about underlying mechanisms, enables a consideration of primitive cellular and molecular plasticity mechanisms that may have been exapted in the evolution of PNH. The term ‘exaptation’ was coined by Stephen Jay Gould and Elizabeth Vrba, who defined it as a trait (previously evolutionarily neutral or having a different function) that was co-opted for a new function [105]. They termed the preexisting trait an ‘aptation’, which they considered less loaded teleologically than the older but equivalent word ‘preadaptation,’ which is used by some evolutionary biologists [e.g., 106, 107, 108]. The concept of exaptation – repurposing preexisting traits for new functions - was recognized by Charles Darwin [109] and many other evolutionary biologists as a fundamental evolutionary process [107], and the idea of exaptation followed by progressive adaptation (e.g., scales being exapted into insulating feathers and then further adapted for flight) has become a core principle of evolution that has been applied at anatomical, physiological, and molecular levels of organization [e.g., 106, 110].
Possible exaptation of early adaptive cellular responses to injury for plasticity in nociceptors and other neurons
Among neurons, primary somatosensory neurons, including nociceptors, are unusual in interfacing directly with one of the strongest evolutionary selection pressures – bodily injury. Because damage to cells and tissues has always had a direct impact on survival, it seems safe to assume that adaptations to reduce damage and promote recovery from injury were among the earliest to appear during evolution. Furthermore, the survival benefits of rapid detection of injury probably drove the early evolution of molecular sensors of noxious events [111] and, after the appearance of animals, the evolution of nociceptive neurons specialized to detect incipient injury and trigger adaptive responses. Activation of primeval nociceptors may have directly excited muscle or ciliary cells for withdrawal and escape (and possibly stimulated innate immune cells to combat microbes that enter open wounds [112]), and, as more complex behaviors and circuits evolved, nociceptors excited interneurons controlling defensive behaviors and their integration with other activities [20]. Two general arguments have been made for primitive cellular reactions to injury being exapted for neuronal mechanisms that later became important for pain.
One argument was spurred indirectly by evidence for independent origins of neurons and nervous systems in different lineages [113]. Moroz argued that neurons could have evolved independently in different animals because injury-induced repair, regeneration of asymmetric morphological processes, and secretion of cellular metabolites and peptides were readily exapted for new functions – such as directed chemical signaling -- that now define neurons [114]. Rupturing a cell releases intracellular molecules, including glutamate, ATP, and peptides, which are used as secreted signals in diverse contemporary eukaryotes. These organisms include some unicellular species, animals that lack neurons (placozoa and sponges), and by most if not all animals with nervous systems [108, 115–117]. Even plants use glutamate as a wound signal [118]. While biological demands in addition to responding adaptively to injury were certainly important for the evolution of neuronal signaling [119, 120], various observations support the hypothesis that some injury-related molecules and receptors were exapted for chemical signaling specializations during the early evolution of neurons and nervous systems in different animal lineages [108].
A second argument was suggested by a discovery in nociceptive sensory neurons of Aplysia. Persistent hyperexcitability and synaptic potentiation induced rapidly by activity-dependent learning mechanisms were found to be indistinguishable from delayed alterations induced by crushing a nerve containing the axons of the tested sensory neurons – under conditions that prevented AP generation and acute release of neuromodulators [63]. The fact that the same hyperfunctional neuronal state could be induced in primary nociceptors either by molecular injury signals conveyed by axonal transport after axotomy [121–123] or by activity-dependent signals important for learning in Aplysia and mammals [124, 125] suggested a link between mechanisms underlying adaptive neuronal responses to injury and mechanisms underlying learning and memory. Given the probable universality of injury as a mediator of natural selection and the ubiquity of predation as a major source of injury during most of animal evolution [20, 126], a further suggestion was that adaptive responses to cellular injury present in the earliest animals [111] and perhaps in their single-celled ancestors [127] were exapted in some of the earliest sensory neurons to detect bodily injury and to produce hyperfunctional alterations after tissue injury, with the adaptive alterations likely to include regeneration and sprouting into denervated tissue, hyperexcitability, and increased excitatory synaptic transmission to cells with defensive functions [91]. Mechanisms of nociceptor hyperexcitability at peripheral injury sites might have then been exapted in larger animals to more central nociceptor somata that were more likely to survive peripheral injury. Even in very simple nervous systems, hyperfunctional alterations in sensory neurons might serve both to compensate for loss of function in a damaged region and to sensitize protective behavior in a dangerous environment. Subsequently, injury-dependent plasticity mechanisms might have been exapted for additional functions in other neuronal types as nervous systems and behavior became more complex. Notably, ancient injury-related mechanisms might have been exapted for learning and memory [18, 20, 91, 128].
Possible exaptation of ancient injury-linked cell signaling pathways for PNH
Despite the enormous range of form and function across the eukaryotic kingdoms, core cellular constituents and processes are highly conserved, and the evolution of new cellular functions in animals largely depended on the exaptation and re-exaptation of proteins and other molecules that already existed when this kingdom first emerged [110, 129]. However, stimulus transduction and cell signaling pathways have diversified greatly in animals (metazoans), raising the possibility that signaling pathways critical for adaptive responses to injury diverged significantly across different metazoan lineages. On the other hand, strong continuing selection pressures, such as injury, can maintain ancient molecular adaptations [129]. Two of the signaling systems that appeared early in evolution, long before the emergence of animals, are Ca2+ signaling and cAMP signaling. These interacting signaling pathways are associated with many functions in a wide range of organisms, including responses to cellular injury, such as wound repair and regeneration, as well as responses to other kinds of organismic stress [130–136]. Both signaling pathways are also closely linked to synaptic plasticity [135, 137–139]. Ca2+-dependent processes for cellular repair probably appeared at the dawn of eucaryotic evolution 1.5–2 billion years ago, when early eukaryotic cells lost the protective, rigid cell walls of prokaryotes, and these early repair processes have been highly conserved in metazoans [111, 140]. One ancient repair process was Ca2+-induced vesicle fusion that seals ruptured plasma membrane in response to Ca2+ entry through membrane breaches and sometimes also through voltage-gated Ca2+ channels (activated by the depolarization resulting from membrane damage), a process that may have been exapted in early animals to trigger chemical synaptic transmission [111, 127].
Given the plausibility of primitive cellular injury and stress responses being exapted for use as mechanisms of learning and memory, it is interesting that early discoveries in Aplysia sensory neurons implicated cooperative activation of Ca2+ signaling and cAMP signaling as important for synaptic plasticity, leading to the first proposed cell signaling mechanism of associative learning [141, 142]. In Aplysia, the coincidence of activity-dependent Ca2+ signaling and stimulation of a G protein-coupled receptor (GPCR) by serotonin (5-HT) produced cooperative activation of an adenylyl cyclase (AC) homologous with AC1 in mammals to amplify cAMP synthesis and produce short- and long-term memory-like effects [143]. The same coincident activation of AC1 by Ca2+ signaling and neuromodulators to promote learning and memory occurs in Drosophila brain [144, 145] and the mammalian brain [124]. Integrated activation of these and associated cell signaling pathways (notably ERK signaling, well known for its roles in cellular differentiation, growth, and survival in response to growth factors) is now recognized as fundamental to the induction and consolidation of many forms of learning and memory in mammals [137–139].
cAMP signaling in nociceptors may represent a fundamental exaptation for PNH
It is often overlooked that the Aplysia sensory neurons used for studies of associative learning [141, 142, 146–148] function as primary nociceptors, with direct exposure to peripheral trauma [35, 128, 149]. Studies of plasticity in these nociceptive neurons have usually been modeled on nonassociative or associative aversive learning paradigms. In all cases an aversive stimulus that activates nociceptors (often electric shock to skin or nerve) or the presumed major extracellular modulator released by noxious stimulation, serotonin (5-HT), has been used. This makes these Aplysia studies at least partly equivalent to nociceptive sensitization and hyperalgesia paradigms in mammalian pain studies. In addition to revealing critical roles for PKA, ERK, and other protein kinases in potentiatiing nociceptor synapses [125, 150, 151], Aplysia studies showed that PKA, ERK, tyrosine kinase, MNK, and PKG signaling contribute to nociceptor hyperexcitability [151–158]. All these protein kinases have been associated with pain in mammals, but I will focus on PKA and cAMP signaling (Fig. 2) because of how much is known about cAMP-PKA contributions to nociceptive plasticity in mammals and invertebrates.
Fig. 2.
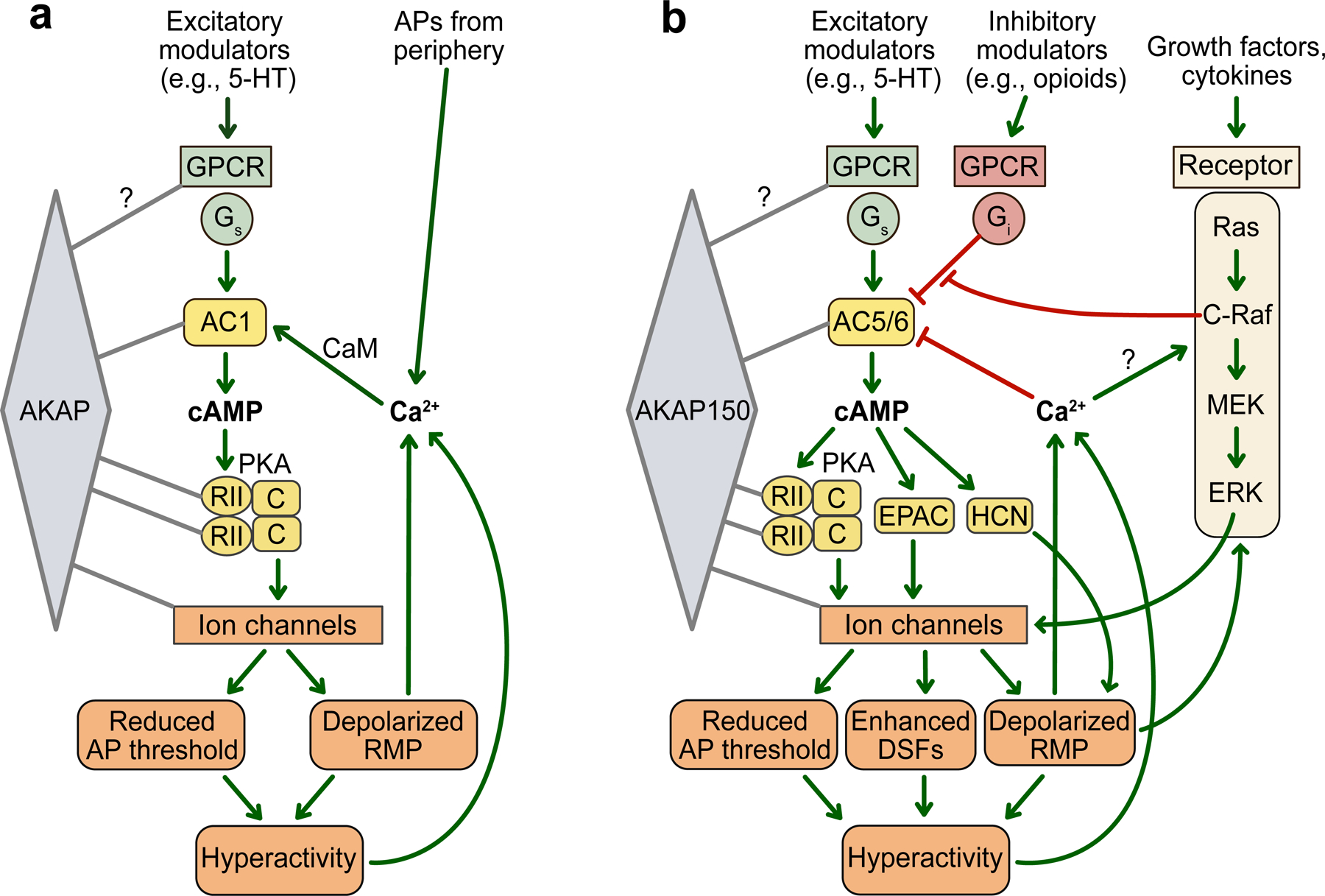
Core cAMP signaling modules described in Aplysia and rodent nociceptors that might have been exapted during early animal evolution to promote hyperactivity and persistent pain after bodily injury. a Scaffolded AC1-centered signaling implicated in the induction and possible maintenance of PNH in Aplysia. Some evidence suggests that AC1 signaling might also contribute to PNH in mouse Aδ nociceptors and possibly in Drosophila nociceptors (see text). The yellow elements are close orthologs in Aplysia and rodent nociceptors. It is not yet known whether other cAMP signaling elements (e.g., EPAC or HCN channels) function in Aplysia nociceptors. Most cAMP research on these neurons has focused on 5-HT stimulation, and the ion channels phosphorylated by PKA have not been identified, except for a TREK-1-like leak K+ channel inhibited by PKA. b AC5/6-associated signaling that contributes to the maintenance of PNH in rodent nociceptors. AC5/6 is also prominently expressed in Aplysia nociceptors, but their potential contribution to PNH in these neurons has not been explored. In rodents, numerous excitatory and inhibitory modulators and GPCRs can stimulate or inhibit AC5/6 in primary sensory neurons. AC5/6 is inhibited by Ca2+ and by opioids and other modulators coupled to Gi, and the Gi-mediated inhibition can be reduced by depolarization of RMP, at least in part through C-raf in the ERK pathway. Among the ion channels regulated by scaffolded PKA in these neurons are TRPV1, Nav1.8, Cav1.2, and KCNQ channels (not shown). Neither other scaffolded proteins such as phosphodiesterases and phosphatases nor pathways regulating DNA transcription and mRNA translation downstream from AC5/6 or ERK are shown. Activating effects are indicated by green arrows, inhibitory effects by red bars. 5-HT, serotonin; AC5, adenylyl cyclase 5; AC5/6, adenylyl cyclase 5 and/or AC6; AKAP150, A-kinase anchoring protein 150; AP, action potential; DSF, depolarizing spontaneous fluctuation; C, catalytic subunit of PKA; CaM, calmodulin; C-raf, Raf-1 proto-oncogene; cAMP, cyclic adenosine monophosphate; EPAC, exchange protein directly activated by cAMP; ERK, extracellular signal-related kinase; GPCR, G-protein-coupled receptor; Gi, inhibitory G-protein Gαi; Gs, stimulatory G-protein Gαs ; HCN, hyperpolarization-activated, cyclic nucleotide-gated channel; MEK, mitogen-activated protein kinase kinase; PKA, protein kinase A; RII; regulatory subunit II of PKA; Ras, “rat sarcoma virus” GTPase.
Drosophila melanogaster has been used for many seminal investigations of nociception and nociceptive sensitization. Although multiple alterations in Drosophila class IV polymodal nociceptors and other neurons have been reported during nociceptive sensitization [26, 32, 159–162], very little attention has been paid to cAMP signaling in these neurons. However, photoactivation of a membrane-anchored AC selectively expressed in class IV nociceptors in Drosophila larvae can elicit defensive behavior [163]. This finding plus observations of enhanced AP discharge in polymodal nociceptors during heating after damaging ultraviolet irradiation [32] and of cAMP-PKA stimulation driving hyperactivity in other Drosophila neurons [164] suggest that cAMP might promote PNH in insect nociceptors after tissue injury, even though central sensitization mechanisms may be more important than peripheral sensitization mechanisms for nociceptive behavioral alterations in insects [8, 165–168].
In Aplysia nociceptors (Fig. 2a), ongoing PKA activity contributes to the maintenance of somal hyperexcitability after nerve injury [154]. However, more attention has been paid in Aplysia to cAMP signaling for the induction of nociceptor alterations [125, 153]. Ca2+-calmodulin (CaM) directly activates AC1. Ca2+ enters through voltage-gated Ca2+ channels during bursts of APs, and a coincidence of bursts with excitatory neuromodulatory input (notably, from 5-HT) enhances cAMP synthesis [169, 170] by AC1 [143] and produces long-lasting depolarization of RMP [171, 172], which increases excitability by bringing membrane potential closer to AP threshold (see Fig. 1a). Aplysia nociceptors also express AC5 and/or highly similar AC6 as well as AC1 [143], but possible roles of AC5/6 in Aplysia PNH have yet to be explored. These ACs activate PKA in Aplysia, which is tethered to a scaffolding A-kinase anchoring protein (AKAP) by a regulatory subunit (RII) of PKA (Fig. 2A) that is a close ortholog of mammalian RII [173] (Fig. 2).
In mammalian nociceptors, more is known about AC5/6 (see below) than AC1, and mRNA for AC5 and AC6 is more highly expressed than mRNA for AC1 both in rat DRGs [81] and in most rodent and primate nociceptors [174, 175]. However, indirect evidence suggests a contribution of nociceptor AC1 to sensitization in mice [176]. This could be explained by contributions from the relatively small population of myelinated Aδ nociceptors, which express more AC1 than do nociceptors in unmyelinated C-fiber subpopulations in mammals [174, 175], and which display striking similarities to Aplysia nociceptors in rapid axonal conduction relative to other nociceptor types in the same species, large receptive fields, sensitivity to a wide range of intensities of mechanical stimuli, and sensitization of mechanosensory responses by noxious mechanical stimulation [35, 65, 177].
In rodents (Fig. 2b), at least three downstream signaling effectors of cAMP are reported to enhance nociceptor function in pain models: 1) PKA in mechanical and heat hypersensitivity in inflammatory and neuropathic models [72, 81, 178–181], 2) EPAC in inflammatory and SCI models [76, 182–184], and 3) HCN channels in inflammatory models [185, 186]. While many excitatory modulators are likely to stimulate cAMP-dependent hyperactivity in various mammalian nociceptors, it is interesting that at least one of these, 5-HT, is the same as in Aplysia [187–189]. The most direct links between cAMP signaling and PNH have been shown in rodent dissociated nociceptor somata months after SCI (Fig. 2b). In rats and mice, ongoing EPAC activity was required for SCI-induced SA, depolarization of RMP, reduction of AP threshold, and enhanced DSFs [76]. In rat nociceptors, SCI-induced SA and depolarization of RMP required continuing activation of PKA by adenylyl cyclases (AC5 and/or highly similar AC6) scaffolded to A-kinase anchoring protein 150 (AKAP150) in rodents or its ortholog AKAP79 in humans [81]. As in Aplysia nociceptors, the RII regulatory subunit binds PKA to an AKAP in rodent primary somatosensory neurons [190, 191], in this case to AKAP150. Ion channels in rodent sensory neurons known to be scaffolded with and/or phosphorylated by PKA include TRPV1, Nav1.8, Cav1.2, and KCNQ channels [192–194].
Unexpectedly, SCI decreased the sensitivity of AC to inhibitory Gi in isolated DRG membranes [81], suggesting that an additional hyperactive effect in vivo might result from induced resistance of AC5 and/or AC6 in nociceptors to endogenous and clinically applied opioids (Fig. 2b). Indeed, SCI reduced the in vitro inhibitory effects of mu-opioid agonists on PKA activated by forskolin treatment [82]. One of the hyperexcitable alterations after SCI is sustained depolarization of RMP [44, 52]. Experimental depolarization was found to activate C-Raf (upstream to ERK signaling), and C-Raf activation decreased the inhibition of AC by the G protein Gi [82] (Fig. 2b). Blocking MEK (downstream from C-Raf and upstream of ERK) blocked SA, DSF enhancement, and sustained depolarization of RMP. Thus, both cAMP signaling and ERK signaling are likely to depolarize nociceptor RMP after SCI, and cAMP signaling via PKA and EPAC probably contributes to the C-Raf-MEK-ERK effects by helping to depolarize RMP, while C-Raf activation by depolarization in the presence of endogenous or exogenous opioids should enhance cAMP signaling by reducing ongoing opioid inhibition of AC. These positive feedback loops and others involving translational and transcriptional regulation by these pathways (not shown in Fig. 2) are likely to help maintain PNH. Associated negative feedback loops, including inhibition of AC5/6 by Ca2+ (Fig. 2b) and AKAP-scaffolded phosphodiesterases and phosphatases (not shown) are probably important in nociceptors for protecting against inappropriate or excessive hyperactivity and possible excitotoxicity [86, 195, 196].
Because the last common ancestor of molluscs and chordates lived ~600 million years ago, it is striking that the same isoforms of AC (AC1 and AC5/6) and PKA regulatory subunit RII are expressed in nociceptors that exhibit prominent cAMP-dependent PNH both in Aplysia and rodents. AKAP-scaffolded cAMP signaling arose near the onset of multicellular eukaryotic evolution [197] and it would have been available to modulate early nociceptors as they evolved to detect, integrate, and respond to information about injury to an organism. It is also notable that PNH depends upon ACs that are directly modulated by Ca2+, perhaps reflecting primordial properties of Ca2+ both as a signal of cellular injury and as a potential cellular toxin, which might have shaped fundamental nociceptive functions of AC1 and AC5/6, respectively. Thus, AKAP-scaffolded cAMP signaling may have been exapted from even more ancient injury- and stress-related functions of this signaling to drive PNH in ancient animals, prior to the divergence of molluscs and chordates (or exapted independently in each lineage later in evolution for common hyperexcitability functions). In contrast, the identities of the ion channels involved in PNH differ markedly across phyla. For example, PKA-regulated Nav1.8 and TRPV1 channels are important for SA in rodent nociceptors [83, 85, 102], but ion channels have diverged substantially over the course of evolution [198, 199]. Indeed, Nav1.8 and TRPV1 are unique to chordates and thus cannot contribute to PNH in Aplysia or Drosophila. Other channels may have more ancient, conserved roles. For example, PKA can also contribute to PNH by reducing background activity in K+ leak channels in rodents and Aplysia (TREK-1 and TREK-1-like channels, respectively) under sensitizing conditions [200–204]. Another late evolutionary development is inhibitory regulation of cAMP signaling by opioids and opioid receptors, which only occur in the vertebrate lineage, although other Gi-coupled GPCRs found on mammalian nociceptors, such as somatostatin receptors, are found in multiple phyla [205, 206]. Thus, while core cAMP signaling mechanisms linked to PNH have been highly conserved, downstream ion channels and upstream GPCRs in nociceptors may exhibit greater evolutionary diversification.
Conclusions
An evolutionary perspective can provide insights into why chronic pain is so prevalent and difficult to treat. One of several explanations for the pervasiveness of chronic pain in modern life is that some mechanisms in primary nociceptors that predispose humans to chronic pain were selected during evolution because they promoted the survival of seriously injured individuals, and these mechanisms might also be activated pathologically in other neuropathic conditions. One physiological mechanism, PNH, has been associated with enhanced anti-predator responses after injury in squid and mice, indicating its adaptive value. The few species in which PNH has been investigated mechanistically (primarily Aplysia, rats, mice, and humans) have revealed many similarities and some differences between Aplysia and mammals. Evidence for essential contributions of scaffolded cAMP-PKA signaling (with associated Ca2+ signaling) to the maintenance of PNH in rodents and Aplysia suggest that ancient cell signaling modules linked to primordial organismic injury responses were exapted during the early evolution of nociceptors to drive persistent hyperactivity and adaptive behavior following significant bodily injury. Much remains to be learned about the pain-maintaining roles of these pathways and other mechanisms underlying PNH, but current knowledge about PNH mechanisms already has therapeutic implications. One is that the cellular signaling pathways found to be important for PNH thus far (cAMP, Ca2+, and ERK signaling) are important for many organismic functions and thus may be poor candidates for targeting chronic pain without unwanted side effects. In addition, redundancy has been found in the complex physiological and molecular mechanisms of PNH in mammals, which may impede effective therapeutic targeting. The complexity and functional coherence of these PNH mechanisms in mammals are consistent with shaping and maintenance by strong, continuing evolutionary pressures. On the other hand, recent evolution may offer new molecular targets to treat pain in vertebrates. For example, PNH in mammals depends upon the activity of three ion channels that are mainly expressed in nociceptors (Nav1.8, Nav1.9, and, with somewhat less specificity, Nav1.7) [207] and these channels first appeared in tetrapod vertebrates [208]. Specific inhibition of one or more of these recently evolved channels might suppress PNH with few side effects. For example, experimental targeting of Nav1.8 (whose activity is enhanced by cAMP-PKA signaling) alleviates persistent pain behavior in several rodent neuropathic pain models at least in part by blocking nociceptor SA [84, 85, 209]. Recent results from phase 2 clinical trials for a highly selective Nav1.8 blocker [210] encourage a strategy of treating chronic pain by inhibiting ion channels that are important for PNH and are preferentially expressed in nociceptors.
Acknowledgements
The author is grateful to Dr. Carmen Dessauer for her leadership of the mammalian cell signaling studies discussed in this review and for her valuable comments on the manuscript, and to Drs. Alexis Bavencoffe, Anibal Garza Carbajal, Jinbin Tian, and Michael Zhu for their many contributions to the research and ideas presented here.
Funding Sources
Scholarship by the author during the preparation of this article and some of the research reviewed was supported by NIH grants NS091759 to Carmen W. Dessauer and Edgar T. Walters, and NS111521 to Edgar T. Walters and Michael X. Zhu, and by the Fondren Chair in Cellular Signaling.
Footnotes
Conflict of Interest Statement
The author has no conflicts of interest to declare.
References
- 1.Rikard SM, Strahan AE, Schmit KM, Guy GP. Chronic Pain Among Adults - United States, 2019–2021. MMWR Morb Mortal Wkly Rep 2023; 72:379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulliford M Opioid use, chronic pain and deprivation. EClinicalMedicine 2020; 21:100341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo YF, Baillargeon J, Raji MA. Overdose deaths from nonprescribed prescription opioids, heroin, and other synthetic opioids in Medicare beneficiaries. J Subst Abuse Treat 2021; 124:108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams ACC. What can evolutionary theory tell us about chronic pain. Pain 2016; 157:788–790. [DOI] [PubMed] [Google Scholar]
- 5.Nesse RM, Schulkin J. An evolutionary medicine perspective on pain and its disorders. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stearns SC, Medzhitov R (2016) Evolutionary Medicine Sinauer Associates, Inc.: Sunderland, MA. [Google Scholar]
- 7.Walters ET. Adaptive mechanisms driving maladaptive pain: how chronic ongoing activity in primary nociceptors can enhance evolutionary fitness after severe injury. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters ET, Crook RJ, Neely GG, Price TJ, Smith ESJ. Persistent nociceptor hyperactivity as a painful evolutionary adaptation. Trends Neurosci 2023; 46:211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuner R, Kuner T. Cellular Circuits in the Brain and Their Modulation in Acute and Chronic Pain. Physiol Rev 2021; 101:213–258. [DOI] [PubMed] [Google Scholar]
- 10.Seifert O, Baerwald C. Interaction of pain and chronic inflammation. Z Rheumatol 2021; 80:205–213. [DOI] [PubMed] [Google Scholar]
- 11.Finnerup NB, Kuner R, Jensen TS. Neuropathic Pain: From Mechanisms to Treatment. Physiol Rev 2021; 101:259–301. [DOI] [PubMed] [Google Scholar]
- 12.Chen T, Wang J, Wang YQ, Chu YX. Current Understanding of the Neural Circuitry in the Comorbidity of Chronic Pain and Anxiety. Neural Plast 2022; 2022:4217593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011; 152:S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eller-Smith OC, Nicol AL, Christianson JA. Potential Mechanisms Underlying Centralized Pain and Emerging Therapeutic Interventions. Front Cell Neurosci 2018; 12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuo M Cortical plasticity as synaptic mechanism for chronic pain. J Neural Transm (Vienna) 2020; 127:567–573. [DOI] [PubMed] [Google Scholar]
- 16.Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrom JB, Jensen TS. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014; 155:1272–1279. [DOI] [PubMed] [Google Scholar]
- 17.Brazenor GA, Malham GM, Teddy PJ. Can Central Sensitization After Injury Persist as an Autonomous Pain Generator? A Comprehensive Search for Evidence. Pain Med 2022; 23:1283–1298. [DOI] [PubMed] [Google Scholar]
- 18.Price TJ, Inyang KE. Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain. Prog Mol Biol Transl Sci 2015; 131:409–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walters ET (2021) Nociceptors and chronic pain. In: Oxford Research Encyclopedia of Neuroscience (Murray S, ed), Oxford University Press. [Google Scholar]
- 20.Walters ET. Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia, and analgesia. Int Rev Neurobiol 1994; 36:325–427. [DOI] [PubMed] [Google Scholar]
- 21.Tobin DM, Bargmann CI. Invertebrate nociception: behaviors, neurons and molecules. J Neurobiol 2004; 61:161–174. [DOI] [PubMed] [Google Scholar]
- 22.Smith ES, Lewin GR. Nociceptors: a phylogenetic view. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 2009; 195:1089–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burrell BD. Comparative biology of pain: What invertebrates can tell us about how nociception works. J Neurophysiol 2017; 117:1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sneddon LU. Evolution of nociception and pain: evidence from fish models. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walters ET (2020) Evolutionary aspects of nociception and pain. In: The Senses: A Comprehensive Reference, Vol 5, Pain (Fritzsch B, Pogatzki-Zahn E, Schaible H-G, eds), pp 463–480. Elsevier, Academic Press. [Google Scholar]
- 26.He J, Li B, Han S, Zhang Y, Liu K, Yi S, Liu Y, Xiu M. Drosophila as a Model to Study the Mechanism of Nociception. Front Physiol 2022; 13:854124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bessou P, Perl ER. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. J Neurophysiol 1969; 32:1025–1043. [DOI] [PubMed] [Google Scholar]
- 28.Perl ER. Cutaneous polymodal receptors: characteristics and plasticity. Prog Brain Res 1996; 113:21–37. [DOI] [PubMed] [Google Scholar]
- 29.Beitel RE, Dubner R. Response of unmyelinated (C) polymodal nociceptors to thermal stimuli applied to monkey’s face. J Neurophysiol 1976; 39:1160–1175. [DOI] [PubMed] [Google Scholar]
- 30.Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med 2010; 16:1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torebjörk E Human microneurography and intraneural microstimulation in the study of neuropathic pain. Muscle Nerve 1993; 16:1063–1065. [DOI] [PubMed] [Google Scholar]
- 32.Im SH, Takle K, Jo J, Babcock DT, Ma Z, Xiang Y, Galko MJ. Tachykinin acts upstream of autocrine Hedgehog signaling during nociceptive sensitization in Drosophila. Elife 2015; 4:e10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner HN, Patel AA, Cox DN, Galko MJ. Injury-induced cold sensitization in Drosophila larvae involves behavioral shifts that require the TRP channel Brv1. PLoS One 2018; 13:e0209577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Billy AJ, Walters ET. Long-term expansion and sensitization of mechanosensory receptive fields in Aplysia support an activity-dependent model of whole-cell sensory plasticity. J Neurosci 1989; 9:1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Illich PA, Walters ET. Mechanosensory neurons innervating Aplysia siphon encode noxious stimuli and display nociceptive sensitization. J Neurosci 1997; 17:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crook RJ, Hanlon RT, Walters ET. Squid have nociceptors that display widespread long-term sensitization and spontaneous activity after bodily injury. J Neurosci 2013; 33:10021–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrew D, Greenspan JD. Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. J Neurophysiol 1999; 82:2649–2656. [DOI] [PubMed] [Google Scholar]
- 38.Shim B, Kim DW, Kim BH, Nam TS, Leem JW, Chung JM. Mechanical and heat sensitization of cutaneous nociceptors in rats with experimental peripheral neuropathy. Neuroscience 2005; 132:193–201. [DOI] [PubMed] [Google Scholar]
- 39.Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 2009; 147:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boada MD, Martin TJ, Parker R, Houle TT, Eisenach JC, Ririe DG. Recovery from nerve injury induced behavioral hypersensitivity in rats parallels resolution of abnormal primary sensory afferent signaling. Pain 2020; 161:949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashley PJ, Sneddon LU, McCrohan CR. Nociception in fish: stimulus-response properties of receptors on the head of trout Oncorhynchus mykiss. Brain Res 2007; 1166:47–54. [DOI] [PubMed] [Google Scholar]
- 42.Dulin MF, Steffensen I, Morris CE, Walters ET. Recovery of function, peripheral sensitization and sensory neurone activation by novel pathways following axonal injury in Aplysia californica. J Exp Biol 1995; 198:2055–2066. [DOI] [PubMed] [Google Scholar]
- 43.Gasull X, Liao X, Dulin MF, Phelps C, Walters ET. Evidence that long-term hyperexcitability of the sensory neuron soma induced by nerve injury in Aplysia is adaptive. J Neurophysiol 2005; 94:2218–2230. [DOI] [PubMed] [Google Scholar]
- 44.Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, Walters ET. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci 2010; 30:14870–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serra J, Bostock H, Sola R, Aleu J, Garcia E, Cokic B, Navarro X, Quiles C. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 2012; 153:42–55. [DOI] [PubMed] [Google Scholar]
- 46.Wall PD. On the relation of injury to pain. The John J. Bonica lecture. Pain 1979; 6:253–264. [DOI] [PubMed] [Google Scholar]
- 47.Pastor J, Soria B, Belmonte C. Properties of the nociceptive neurons of the leech segmental ganglion. J Neurophysiol 1996; 75:2268–2279. [DOI] [PubMed] [Google Scholar]
- 48.Summers T, Holec S, Burrell BD. Physiological and behavioral evidence of a capsaicin-sensitive TRPV-like channel in the medicinal leech. J Exp Biol 2014; 217:4167–4173. [DOI] [PubMed] [Google Scholar]
- 49.Himmel NJ, Sakurai A, Patel AA, Bhattacharjee S, Letcher JM, Benson MN, Gray TR, Cymbalyuk GS, Cox DN. Chloride-dependent mechanisms of multimodal sensory discrimination and nociceptive sensitization in Drosophila. Elife 2023; 12:e76863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walters ET, Byrne JH, Carew TJ, Kandel ER. Mechanoafferent neurons innervating tail of Aplysia. II. Modulation by sensitizing stimulation. J Neurophysiol 1983; 50:1543–1559. [DOI] [PubMed] [Google Scholar]
- 51.Clatworthy AL, Walters ET. Rapid amplification and facilitation of mechanosensory discharge in Aplysia by noxious stimulation. J Neurophysiol 1993; 70:1181–1194. [DOI] [PubMed] [Google Scholar]
- 52.Odem MA, Bavencoffe AG, Cassidy RM, Lopez ER, Tian J, Dessauer CW, Walters ET. Isolated nociceptors reveal multiple specializations for generating irregular ongoing activity associated with ongoing pain. Pain 2018; 159:2347–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, Meyer RA. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci 2001; 21:RC140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorodetskaya N, Constantin C, Janig W. Ectopic activity in cutaneous regenerating afferent nerve fibers following nerve lesion in the rat. Eur J Neurosci 2003; 18:2487–2497. [DOI] [PubMed] [Google Scholar]
- 55.Djouhri L, Koutsikou S, Fang X, McMullan S, Lawson SN. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J Neurosci 2006; 26:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roza C, Bernal L. Electrophysiological characterization of ectopic spontaneous discharge in axotomized and intact fibers upon nerve transection: a role in spontaneous pain. Pflugers Arch 2022; 474:387–396. [DOI] [PubMed] [Google Scholar]
- 57.Moalem G, Grafe P, Tracey DJ. Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience 2005; 134:1399–1411. [DOI] [PubMed] [Google Scholar]
- 58.Xiao WH, Bennett GJ. Persistent low-frequency spontaneous discharge in A-fiber and C-fiber primary afferent neurons during an inflammatory pain condition. Anesthesiology 2007; 107:813–821. [DOI] [PubMed] [Google Scholar]
- 59.Kleggetveit IP, Namer B, Schmidt R, Helås T, Rückel M, Ørstavik K, Schmelz M, Jørum E. High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 2012; 153:2040–2047. [DOI] [PubMed] [Google Scholar]
- 60.Becker AK, Babes A, Düll MM, Khalil M, Kender Z, Gröner J, Namer B, Reeh PW, Sauer SK. Spontaneous activity of specific C-nociceptor subtypes from diabetic patients and mice: Involvement of reactive dicarbonyl compounds and (sensitized) transient receptor potential channel A1. J Peripher Nerv Syst 2023; 28:202–225. [DOI] [PubMed] [Google Scholar]
- 61.Haroutounian S, Ford AL, Frey K, Nikolajsen L, Finnerup NB, Neiner A, Kharasch ED, Karlsson P, Bottros MM. How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: a prospective, open-label pilot study. Pain 2018; 159:1317–1324. [DOI] [PubMed] [Google Scholar]
- 62.Walters ET. Multiple sensory neuronal correlates of site-specific sensitization in Aplysia. J Neurosci 1987; 7:408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walters ET, Alizadeh H, Castro GA. Similar neuronal alterations induced by axonal injury and learning in Aplysia. Science 1991; 253:797–799. [DOI] [PubMed] [Google Scholar]
- 64.Walters ET, Bodnarova M, Billy AJ, Dulin MF, Díaz-Ríos M, Miller MW, Moroz LL. Somatotopic organization and functional properties of mechanosensory neurons expressing sensorin-A mRNA in Aplysia californica. J Comp Neurol 2004; 471:219–240. [DOI] [PubMed] [Google Scholar]
- 65.Burgess PR, Perl ER. Myelinated afferent fibres responding specifically to noxious stimulation of the skin. J Physiol 1967; 190:541–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roza C, Bernal L. Electrophysiological characterization of ectopic spontaneous discharge in axotomized and intact fibers upon nerve transection: a role in spontaneous pain. Pflugers Arch 2022; 474:387–396. [DOI] [PubMed] [Google Scholar]
- 67.Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 1983; 17:321–339. [DOI] [PubMed] [Google Scholar]
- 68.Amir R, Liu CN, Kocsis JD, Devor M. Oscillatory mechanism in primary sensory neurones. Brain 2002; 125:421–435. [DOI] [PubMed] [Google Scholar]
- 69.Ma C, LaMotte RH. Multiple sites for generation of ectopic spontaneous activity in neurons of the chronically compressed dorsal root ganglion. J Neurosci 2007; 27:14059–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devor M Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res 2009; 196:115–128. [DOI] [PubMed] [Google Scholar]
- 71.Xie W, Strong JA, Zhang JM. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 2015; 291:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song XJ, Wang ZB, Gan Q, Walters ET. cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J Neurophysiol 2006; 95:479–492. [DOI] [PubMed] [Google Scholar]
- 73.Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 2014; 120:1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng Q, Xie W, Lückemeyer DD, Lay M, Wang XW, Dong X, Limjunyawong N, Ye Y, Zhou FQ, Strong JA, Zhang JM, Dong X. Synchronized cluster firing, a distinct form of sensory neuron activation, drives spontaneous pain. Neuron 2022; 110:209–220.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain 1996; 65:235–242. [DOI] [PubMed] [Google Scholar]
- 76.Berkey SC, Herrera JJ, Odem MA, Rahman S, Cheruvu SS, Cheng X, Walters ET, Dessauer CW, Bavencoffe AG. EPAC1 and EPAC2 promote nociceptor hyperactivity associated with chronic pain after spinal cord injury. Neurobiol Pain 2020; 7:100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, Johansson CA, Kosturakis AK, Edwards DD, Zhang H, Dougherty PM. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017; 158:417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Laumet G, Bavencoffe A, Edralin JD, Huo XJ, Walters ET, Dantzer R, Heijnen CJ, Kavelaars A. Interleukin-10 resolves pain hypersensitivity induced by cisplatin by reversing sensory neuron hyperexcitability. Pain 2020; 161:2344–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, Johansson CA, Zhang H, Kim YH, Zhang B, Dussor G, Kim TH, Price TJ, Dougherty PM. Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 2019; 142:1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng JH, Walters ET, Song XJ. Dissociation of dorsal root ganglion neurons induces hyperexcitability that is maintained by increased responsiveness to cAMP and cGMP. J Neurophysiol 2007; 97:15–25. [DOI] [PubMed] [Google Scholar]
- 81.Bavencoffe A, Li Y, Wu Z, Yang Q, Herrera J, Kennedy EJ, Walters ET, Dessauer CW. Persistent Electrical Activity in Primary Nociceptors after Spinal Cord Injury Is Maintained by Scaffolded Adenylyl Cyclase and Protein Kinase A and Is Associated with Altered Adenylyl Cyclase Regulation. J Neurosci 2016; 36:1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garza Carbajal A, Bavencoffe A, Walters ET, Dessauer CW. Depolarization-Dependent C-Raf Signaling Promotes Hyperexcitability and Reduces Opioid Sensitivity of Isolated Nociceptors after Spinal Cord Injury. J Neurosci 2020; 40:6522–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tian J, Bavencoffe AG, Zhu MX, Walters ET. Readiness of nociceptor cell bodies to generate spontaneous activity results from background activity of diverse ion channels and high input resistance. bioRxiv 2023; [DOI] [PMC free article] [PubMed]
- 84.Roza C, Laird JM, Souslova V, Wood JN, Cervero F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J Physiol 2003; 550:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci 2014; 34:10765–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bavencoffe AG, Spence EA, Zhu MY, Garza-Carbajal A, Chu KE, Bloom OE, Dessauer CW, Walters ET. Macrophage Migration Inhibitory Factor (MIF) Makes Complex Contributions to Pain-Related Hyperactivity of Nociceptors after Spinal Cord Injury. J Neurosci 2022; JN–RM. [DOI] [PMC free article] [PubMed]
- 87.Stein A, Panjwani A, Sison C, Rosen L, Chugh R, Metz C, Bank M, Bloom O. Pilot study: elevated circulating levels of the proinflammatory cytokine macrophage migration inhibitory factor in patients with chronic spinal cord injury. Arch Phys Med Rehabil 2013; 94:1498–1507. [DOI] [PubMed] [Google Scholar]
- 88.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009; 32:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Monteiro BP, Lascelles BDX, Murrell J, Robertson S, Steagall PVM, Wright B. 2022 WSAVA guidelines for the recognition, assessment and treatment of pain. Journal of Small Animal Practice 2023; 64:177–254. [Google Scholar]
- 90.Walters ET. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front Physiol 2012; 3:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walters ET. A functional, cellular, and evolutionary model of nociceptive plasticity in Aplysia. Biol Bull 1991; 180:241–251. [DOI] [PubMed] [Google Scholar]
- 92.Curio E (2012) The Ethology of Predation Springer Science & Business Media: [Google Scholar]
- 93.Crook RJ, Dickson K, Hanlon RT, Walters ET. Nociceptive sensitization reduces predation risk. Curr Biol 2014; 24:1121–1125. [DOI] [PubMed] [Google Scholar]
- 94.Lister KC, Bouchard SM, Markova T, Aternali A, Denecli P, Pimentel SD, Majeed M, Austin JS, de C Williams AC, Mogil JS. Chronic pain produces hypervigilance to predator odor in mice. Curr Biol 2020; 30:R866–R867. [DOI] [PubMed] [Google Scholar]
- 95.Kremer M, Becker LJ, Barrot M, Yalcin I. How to study anxiety and depression in rodent models of chronic pain. Eur J Neurosci 2021; 53:236–270. [DOI] [PubMed] [Google Scholar]
- 96.Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M. Peripheral nervous system origin of phantom limb pain. Pain 2014; 155:1384–1391. [DOI] [PubMed] [Google Scholar]
- 97.Finlay BL, Syal S. The pain of altruism. Trends Cogn Sci 2014; 18:615–617. [DOI] [PubMed] [Google Scholar]
- 98.Williams ACC. Persistence of pain in humans and other mammals. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.North RY, Odem MA, Li Y, Tatsui CE, Cassidy RM, Dougherty PM, Walters ET. Electrophysiological Alterations Driving Pain-Associated Spontaneous Activity in Human Sensory Neuron Somata Parallel Alterations Described in Spontaneously Active Rodent Nociceptors. J Pain 2022; 23:1343–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Williams GC (1966) Adaptation and natural selection Princeton University Press: Princeton, NJ. [Google Scholar]
- 101.Andrews PW, Gangestad SW, Matthews D. Adaptationism--how to carry out an exaptationist program. Behav Brain Sci 2002; 25:489–504; discussion 504. [DOI] [PubMed] [Google Scholar]
- 102.Wu Z, Yang Q, Crook RJ, O’Neil RG, Walters ET. TRPV1 channels make major contributions to behavioral hypersensitivity and spontaneous activity in nociceptors after spinal cord injury. Pain 2013; 154:2130–2141. [DOI] [PubMed] [Google Scholar]
- 103.Ritter DM, Zemel BM, Hala TJ, O’Leary ME, Lepore AC, Covarrubias M. Dysregulation of Kv3.4 channels in dorsal root ganglia following spinal cord injury. J Neurosci 2015; 35:1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu H, Lauzadis J, Gunaratna K, Sipple E, Kaczocha M, Puopolo M. Inhibition of T-Type Calcium Channels With TTA-P2 Reduces Chronic Neuropathic Pain Following Spinal Cord Injury in Rats. J Pain 2023; 24:1681–1695. [DOI] [PubMed] [Google Scholar]
- 105.Gould SJ, Vrba ES. Exaptation—a missing term in the science of form. Paleobiology 1982; 8:4–15. [Google Scholar]
- 106.Futuyma D (2013) Evolution Sinauer: Sunderland, MA. [Google Scholar]
- 107.Casinos A From Cuénot’s préadaptation to Gould and Vrba’s exaptation: a review. Biological Journal of the Linnean Society 2017; 121:239–247. [Google Scholar]
- 108.Moroz LL, Romanova DY, Kohn AB. Neural versus alternative integrative systems: molecular insights into origins of neurotransmitters. Philos Trans R Soc Lond B Biol Sci 2021; 376:20190762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Darwin C (1859) On the Origin of Species by Means of Natural Selection
- 110.Frenkel-Pinter M, Petrov AS, Matange K, Travisano M, Glass JB, Williams LD. Adaptation and Exaptation: From Small Molecules to Feathers. J Mol Evol 2022; 90:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brunet T, Arendt D. From damage response to action potentials: early evolution of neural and contractile modules in stem eukaryotes. Philos Trans R Soc Lond B Biol Sci 2016; 371:20150043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kraus A, Buckley KM, Salinas I. Sensing the world and its dangers: An evolutionary perspective in neuroimmunology. Elife 2021; 10:e66706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moroz LL, Kohn AB. Independent origins of neurons and synapses: insights from ctenophores. Philos Trans R Soc Lond B Biol Sci 2016; 371:20150041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moroz LL. On the independent origins of complex brains and neurons. Brain Behav Evol 2009; 74:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moroz LL, Romanova DY, Nikitin MA, Sohn D, Kohn AB, Neveu E, Varoqueaux F, Fasshauer D. The diversification and lineage-specific expansion of nitric oxide signaling in Placozoa: insights in the evolution of gaseous transmission. Sci Rep 2020; 10:13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moroz LL, Nikitin MA, Poličar PG, Kohn AB, Romanova DY. Evolution of glutamatergic signaling and synapses. Neuropharmacology 2021; 199:108740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nikitin MA, Romanova DY, Borman SI, Moroz LL. Amino acids integrate behaviors in nerveless placozoans. Front Neurosci 2023; 17:1125624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Toyota M, Spencer D, Sawai-Toyota S, Jiaqi W, Zhang T, Koo AJ, Howe GA, Gilroy S. Glutamate triggers long-distance, calcium-based plant defense signaling. Science 2018; 361:1112–1115. [DOI] [PubMed] [Google Scholar]
- 119.Jékely G The chemical brain hypothesis for the origin of nervous systems. Philos Trans R Soc Lond B Biol Sci 2021; 376:20190761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Najle SR, Grau-Bové X, Elek A, Navarrete C, Cianferoni D, Chiva C, Cañas-Armenteros D, Mallabiabarrena A, Kamm K, Sabidó E, Gruber-Vodicka H, Schierwater B, Serrano L, Sebé-Pedrós A. Stepwise emergence of the neuronal gene expression program in early animal evolution. Cell 2023; S0092-8674(23)00917. [DOI] [PMC free article] [PubMed]
- 121.Gunstream JD, Castro GA, Walters ET. Retrograde transport of plasticity signals in Aplysia sensory neurons following axonal injury. J Neurosci 1995; 15:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ambron RT, Zhang XP, Gunstream JD, Povelones M, Walters ET. Intrinsic injury signals enhance growth, survival, and excitability of Aplysia neurons. J Neurosci 1996; 16:7469–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ambron RT, Walters ET. Priming events and retrograde injury signals. A new perspective on the cellular and molecular biology of nerve regeneration. Mol Neurobiol 1996; 13:61–79. [DOI] [PubMed] [Google Scholar]
- 124.Poser S, Storm DR. Role of Ca2+-stimulated adenylyl cyclases in LTP and memory formation. Int J Dev Neurosci 2001; 19:387–394. [DOI] [PubMed] [Google Scholar]
- 125.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science 2001; 294:1030–1038. [DOI] [PubMed] [Google Scholar]
- 126.Vermeij GJ (1987) Evolution and escalation : an ecological history of life Princeton University Press: Princeton, N.J. [Google Scholar]
- 127.Cooper ST, McNeil PL. Membrane Repair: Mechanisms and Pathophysiology. Physiol Rev 2015; 95:1205–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walters ET, Moroz LL. Molluscan memory of injury: evolutionary insights into chronic pain and neurological disorders. Brain Behav Evol 2009; 74:206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gerhart J, Kirschner M (1997) Cells, Embryos and Evolution Wiley-Blackwell: [Google Scholar]
- 130.Cooper PD, Dennis SR, Woodman JD, Cowlings A, Donnelly C. Effect of opioid compounds on feeding and activity of the cockroach, Periplaneta americana. Comp Biochem Physiol C Toxicol Pharmacol 2010; 151:298–302. [DOI] [PubMed] [Google Scholar]
- 131.Verkhratsky A, Parpura V. Calcium signalling and calcium channels: evolution and general principles. Eur J Pharmacol 2014; 739:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Knapp GS, McDonough KA. Cyclic AMP Signaling in Mycobacteria. Microbiol Spectr 2014; 2 [DOI] [PubMed] [Google Scholar]
- 133.Plattner H, Verkhratsky A. The ancient roots of calcium signalling evolutionary tree. Cell Calcium 2015; 57:123–132. [DOI] [PubMed] [Google Scholar]
- 134.Batty NJ, Fenrich KK, Fouad K. The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci Lett 2017; 652:56–63. [DOI] [PubMed] [Google Scholar]
- 135.Wild AR, Dell’Acqua ML. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol Ther 2018; 185:99–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aslam M, Ladilov Y. Emerging Role of cAMP/AMPK Signaling. Cells 2022; 11:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang H, Zhang M. The role of Ca²⁺-stimulated adenylyl cyclases in bidirectional synaptic plasticity and brain function. Rev Neurosci 2012; 23:67–78. [DOI] [PubMed] [Google Scholar]
- 138.Giese KP, Mizuno K. The roles of protein kinases in learning and memory. Learn Mem 2013; 20:540–552. [DOI] [PubMed] [Google Scholar]
- 139.Kaushik M, Kaushik P, Parvez S. Memory related molecular signatures: The pivots for memory consolidation and Alzheimer’s related memory decline. Ageing Res Rev 2022; 76:101577. [DOI] [PubMed] [Google Scholar]
- 140.Kretsinger RH. Structure and evolution of calcium-modulated proteins. CRC Crit Rev Biochem 1980; 8:119–174. [DOI] [PubMed] [Google Scholar]
- 141.Hawkins RD, Abrams TW, Carew TJ, Kandel ER. A cellular mechanism of classical conditioning in Aplysia: activity-dependent amplification of presynaptic facilitation. Science 1983; 219:400–405. [DOI] [PubMed] [Google Scholar]
- 142.Walters ET, Byrne JH. Associative conditioning of single sensory neurons suggests a cellular mechanism for learning. Science 1983; 219:405–408. [DOI] [PubMed] [Google Scholar]
- 143.Lin AH, Cohen JE, Wan Q, Niu K, Shrestha P, Bernstein SL, Abrams TW. Serotonin stimulation of cAMP-dependent plasticity in Aplysia sensory neurons is mediated by calmodulin-sensitive adenylyl cyclase. Proc Natl Acad Sci U S A 2010; 107:15607–15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Livingstone MS, Sziber PP, Quinn WG. Loss of calcium/calmodulin responsiveness in adenylate cyclase of rutabaga, a Drosophila learning mutant. Cell 1984; 37:205–215. [DOI] [PubMed] [Google Scholar]
- 145.Davis RL. Physiology and biochemistry of Drosophila learning mutants. Physiol Rev 1996; 76:299–317. [DOI] [PubMed] [Google Scholar]
- 146.Murphy GG, Glanzman DL. Mediation of classical conditioning in Aplysia californica by long-term potentiation of sensorimotor synapses. Science 1997; 278:467–471. [DOI] [PubMed] [Google Scholar]
- 147.Antonov I, Antonova I, Kandel ER, Hawkins RD. The contribution of activity-dependent synaptic plasticity to classical conditioning in Aplysia. J Neurosci 2001; 21:6413–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hawkins RD, Byrne JH. Associative learning in invertebrates. Cold Spring Harb Perspect Biol 2015; 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Walters ET, Byrne JH, Carew TJ, Kandel ER. Mechanoafferent neurons innervating tail of Aplysia. I. Response properties and synaptic connections. J Neurophysiol 1983; 50:1522–1542. [DOI] [PubMed] [Google Scholar]
- 150.Ormond J, Hislop J, Zhao Y, Webb N, Vaillaincourt F, Dyer JR, Ferraro G, Barker P, Martin KC, Sossin WS. ApTrkl, a Trk-like receptor, mediates serotonin- dependent ERK activation and long-term facilitation in Aplysia sensory neurons. Neuron 2004; 44:715–728. [DOI] [PubMed] [Google Scholar]
- 151.Zhang Y, Smolen PD, Cleary LJ, Byrne JH. Quantitative description of the interactions among kinase cascades underlying long-term plasticity of Aplysia sensory neurons. Sci Rep 2021; 11:14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Klein M, Hochner B, Kandel ER. Facilitatory transmitters and cAMP can modulate accommodation as well as transmitter release in Aplysia sensory neurons: Evidence for parallel processing in a single cell. Proc Natl Acad Sci U S A 1986; 83:7994–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Scholz KP, Byrne JH. Intracellular injection of cAMP induces a long-term reduction of neuronal K+ currents. Science 1988; 240:1664–1666. [DOI] [PubMed] [Google Scholar]
- 154.Liao X, Gunstream JD, Lewin MR, Ambron RT, Walters ET. Activation of protein kinase A contributes to the expression but not the induction of long-term hyperexcitability caused by axotomy of Aplysia sensory neurons. J Neurosci 1999; 19:1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lewin MR, Walters ET. Cyclic GMP pathway is critical for inducing long-term sensitization of nociceptive sensory neurons. Nat Neurosci 1999; 2:18–23. [DOI] [PubMed] [Google Scholar]
- 156.Purcell AL, Carew TJ. Modulation of excitability in Aplysia tail sensory neurons by tyrosine kinases. J Neurophysiol 2001; 85:2398–2411. [DOI] [PubMed] [Google Scholar]
- 157.Chin J, Liu RY, Cleary LJ, Eskin A, Byrne JH. TGF-beta1-induced long-term changes in neuronal excitability in aplysia sensory neurons depend on MAPK. J Neurophysiol 2006; 95:3286–3290. [DOI] [PubMed] [Google Scholar]
- 158.Mihail SM, Wangzhou A, Kunjilwar KK, Moy JK, Dussor G, Walters ET, Price TJ. MNK-eIF4E signalling is a highly conserved mechanism for sensory neuron axonal plasticity: evidence from Aplysia californica. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Babcock DT, Landry C, Galko MJ. Cytokine signaling mediates UV-induced nociceptive sensitization in Drosophila larvae. Curr Biol 2009; 19:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Babcock DT, Shi S, Jo J, Shaw M, Gutstein HB, Galko MJ. Hedgehog signaling regulates nociceptive sensitization. Curr Biol 2011; 21:1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Follansbee TL, Gjelsvik KJ, Brann CL, McParland AL, Longhurst CA, Galko MJ, Ganter GK. Drosophila nociceptive sensitization requires BMP signaling via the canonical SMAD pathway. J Neurosci 2017; 37:8524–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Khuong TM, Hamoudi Z, Manion J, Loo L, Muralidharan A, Neely GG. Peripheral straightjacket (α2δ Ca2+ channel subunit) expression is required for neuropathic sensitization in Drosophila. Philos Trans R Soc Lond B Biol Sci 2019; 374:20190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang S, Constantin OM, Sachidanandan D, Hofmann H, Kunz TC, Kozjak-Pavlovic V, Oertner TG, Nagel G, Kittel RJ, Gee CE, Gao S. PACmn for improved optogenetic control of intracellular cAMP. BMC Biol 2021; 19:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Global Lee D. and local missions of cAMP signaling in neural plasticity, learning, and memory. Front Pharmacol 2015; 6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tabuena DR, Solis A, Geraldi K, Moffatt CA, Fuse M. Central neural alterations predominate in an insect model of nociceptive sensitization. J Comp Neurol 2017; 525:1176–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khuong TM, Wang QP, Manion J, Oyston LJ, Lau MT, Towler H, Lin YQ, Neely GG. Nerve injury drives a heightened state of vigilance and neuropathic sensitization in Drosophila. Sci Adv 2019; 5:eaaw4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mukherjee R, Trimmer BA. Local and generalized sensitization of thermally evoked defensive behavior in caterpillars. J Comp Neurol 2020; 528:805–815. [DOI] [PubMed] [Google Scholar]
- 168.Gu P, Wang F, Shang Y, Liu J, Gong J, Xie W, Han J, Xiang Y. Nociception and hypersensitivity involve distinct neurons and molecular transducers in Drosophila. Proc Natl Acad Sci U S A 2022; 119:e2113645119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abrams TW. Activity-dependent presynaptic facilitation: an associative mechanism in Aplysia. Cell Mol Neurobiol 1985; 5:123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ocorr KA, Walters ET, Byrne JH. Associative conditioning analog selectively increases cAMP levels of tail sensory neurons in Aplysia. Proc Natl Acad Sci U S A 1985; 82:2548–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Walters ET, Byrne JH. Slow depolarization produced by associative conditioning of Aplysia sensory neurons may enhance Ca2+ entry. Brain Res 1983; 280:165–168. [DOI] [PubMed] [Google Scholar]
- 172.Walters ET, Byrne JH. Long-term enhancement produced by activity-dependent modulation of Aplysia sensory neurons. J Neurosci 1985; 5:662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu J, Hu JY, Schacher S, Schwartz JH. The two regulatory subunits of aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci 2004; 24:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015; 18:145–153. [DOI] [PubMed] [Google Scholar]
- 175.Jung M, Dourado M, Maksymetz J, Jacobson A, Laufer BI, Baca M, Foreman O, Hackos DH, Riol-Blanco L, Kaminker JS. Cross-species transcriptomic atlas of dorsal root ganglia reveals species-specific programs for sensory function. Nat Commun 2023; 14:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giacoletti G, Price T, Hoelz LVB, Shremo Msdi A, Cossin S, Vazquez-Falto K, Amorim Fernandes TV, Santos de Pontes V, Wang H, Boechat N, Nornoo A, Brust TF. A Selective Adenylyl Cyclase 1 Inhibitor Relieves Pain Without Causing Tolerance. Front Pharmacol 2022; 13:935588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Reeh PW, Bayer J, Kocher L, Handwerker HO. Sensitization of nociceptive cutaneous nerve fibers from the rat’s tail by noxious mechanical stimulation. Exp Brain Res 1987; 65:505–512. [DOI] [PubMed] [Google Scholar]
- 178.Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 1999; 19:2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M. PKA/AKAP/VR-1 module: A common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci 2002; 22:4740–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brackley AD, Gomez R, Guerrero KA, Akopian AN, Glucksman MJ, Du J, Carlton SM, Jeske NA. A-Kinase Anchoring Protein 79/150 Scaffolds Transient Receptor Potential A 1 Phosphorylation and Sensitization by Metabotropic Glutamate Receptor Activation. Sci Rep 2017; 7:1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schaefer I, Verkest C, Vespermann L, Mair T, Voß H, Zeitzschel N, Lechner SG. PKA mediates modality-specific modulation of the mechanically gated ion channel PIEZO2. J Biol Chem 2023; 299:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Eijkelkamp N, Wang H, Garza-Carbajal A, Willemen HL, Zwartkruis FJ, Wood JN, Dantzer R, Kelley KW, Heijnen CJ, Kavelaars A. Low nociceptor GRK2 prolongs prostaglandin E2 hyperalgesia via biased cAMP signaling to Epac/Rap1, protein kinase Cepsilon, and MEK/ERK. J Neurosci 2010; 30:12806–12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang H, Heijnen CJ, van Velthoven CT, Willemen HL, Ishikawa Y, Zhang X, Sood AK, Vroon A, Eijkelkamp N, Kavelaars A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J Clin Invest 2013; 123:5023–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Huang LY, Gu Y. Epac and Nociceptor Sensitization. Mol Pain 2017; 13:1744806917716234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 2011; 333:1462–1466. [DOI] [PubMed] [Google Scholar]
- 186.Weng X, Smith T, Sathish J, Djouhri L. Chronic inflammatory pain is associated with increased excitability and hyperpolarization-activated current (Ih) in C- but not Aδ-nociceptors. Pain 2012; 153:900–914. [DOI] [PubMed] [Google Scholar]
- 187.Taiwo YO, Heller PH, Levine JD. Mediation of serotonin hyperalgesia by the cAMP second messenger system. Neuroscience 1992; 48:479–483. [DOI] [PubMed] [Google Scholar]
- 188.Cardenas CG, Del Mar LP, Cooper BY, Scroggs RS. 5HT4 receptors couple positively to tetrodotoxin-insensitive sodium channels in a subpopulation of capsaicin-sensitive rat sensory neurons. J Neurosci 1997; 17:7181–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lopez ER, Carbajal AG, Tian JB, Bavencoffe A, Zhu MX, Dessauer CW, Walters ET. Serotonin enhances depolarizing spontaneous fluctuations, excitability, and ongoing activity in isolated rat DRG neurons via 5-HT4 receptors and cAMP-dependent mechanisms. Neuropharmacology 2021; 184:108408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jeske NA, Diogenes A, Ruparel NB, Fehrenbacher JC, Henry M, Akopian AN, Hargreaves KM. A-kinase anchoring protein mediates TRPV1 thermal hyperalgesia through PKA phosphorylation of TRPV1. Pain 2008; 138:604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Isensee J, Diskar M, Waldherr S, Buschow R, Hasenauer J, Prinz A, Allgöwer F, Herberg FW, Hucho T. Pain modulators regulate the dynamics of PKA-RII phosphorylation in subgroups of sensory neurons. J Cell Sci 2014; 127:216–229. [DOI] [PubMed] [Google Scholar]
- 192.Fischer MJ, McNaughton PA. How anchoring proteins shape pain. Pharmacol Ther 2014; 143:316–322. [DOI] [PubMed] [Google Scholar]
- 193.Zhang J, Carver CM, Choveau FS, Shapiro MS. Clustering and Functional Coupling of Diverse Ion Channels and Signaling Proteins Revealed by Super-resolution STORM Microscopy in Neurons. Neuron 2016; 92:461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Salzer I, Ray S, Schicker K, Boehm S. Nociceptor Signalling through ion Channel Regulation via GPCRs. Int J Mol Sci 2019; 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hucho T, Suckow V, Joseph EK, Kuhn J, Schmoranzer J, Dina OA, Chen X, Karst M, Bernateck M, Levine JD, Ropers HH. Ca++/CaMKII switches nociceptor-sensitizing stimuli into desensitizing stimuli. J Neurochem 2012; 123:589–601. [DOI] [PubMed] [Google Scholar]
- 196.McIlvried LA, Del Rosario JS, Pullen MY, Wangzhou A, Sheahan TD, Shepherd AJ, Slivicki RA, Lemen JA, Price TJ, Copits BA, Gereau RW. Intrinsic Homeostatic Plasticity in Mouse and Human Sensory Neurons. bioRxiv 2023; 2023.06.13.544829.
- 197.Peng M, Aye TT, Snel B, van Breukelen B, Scholten A, Heck AJ. Spatial Organization in Protein Kinase A Signaling Emerged at the Base of Animal Evolution. J Proteome Res 2015; 14:2976–2987. [DOI] [PubMed] [Google Scholar]
- 198.Peng G, Shi X, Kadowaki T. Evolution of TRP channels inferred by their classification in diverse animal species. Mol Phylogenet Evol 2015; 84:145–157. [DOI] [PubMed] [Google Scholar]
- 199.Kasimova MA, Granata D, Carnevale V. Voltage-Gated Sodium Channels: Evolutionary History and Distinctive Sequence Features. Curr Top Membr 2016; 78:261–286. [DOI] [PubMed] [Google Scholar]
- 200.Siegelbaum SA, Camardo JS, Kandel ER. Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature 1982; 299:413–417. [DOI] [PubMed] [Google Scholar]
- 201.Shuster MJ, Camardo JS, Siegelbaum SA, Kandel ER. Cyclic AMP-dependent protein kinase closes the serotonin-sensitive K+ channels of Aplysia sensory neurones in cell-free membrane patches. Nature 1985; 313:392–395. [DOI] [PubMed] [Google Scholar]
- 202.Ungless MA, Gasull X, Walters ET. Long-term alteration of S-type potassium current and passive membrane properties in aplysia sensory neurons following axotomy. J Neurophysiol 2002; 87:2408–2420. [DOI] [PubMed] [Google Scholar]
- 203.Alloui A, Zimmermann K, Mamet J, Duprat F, Noël J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J 2006; 25:2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.García G, Méndez-Reséndiz KA, Oviedo N, Murbartián J. PKC- and PKA-dependent phosphorylation modulates TREK-1 function in naïve and neuropathic rats. J Neurochem 2021; 157:2039–2054. [DOI] [PubMed] [Google Scholar]
- 205.Elphick MR, Mirabeau O, Larhammar D. Evolution of neuropeptide signalling systems. J Exp Biol 2018; 221:jeb151092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Huang AY, Taylor AMW, Ghogha A, Pribadi M, Wang Q, Kim TSJ, Cahill CM, Coppola G, Evans CJ. Genetic and functional analysis of a Pacific hagfish opioid system. J Neurosci Res 2022; 100:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol Rev 2019; 99:1079–1151. [DOI] [PubMed] [Google Scholar]
- 208.Zakon HH. Adaptive evolution of voltage-gated sodium channels: the first 800 million years. Proc Natl Acad Sci U S A 2012; 109 Suppl 1:10619–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 2002; 95:143–152. [DOI] [PubMed] [Google Scholar]
- 210.Jones J, Correll DJ, Lechner SM, Jazic I, Miao X, Shaw D, Simard C, Osteen JD, Hare B, Beaton A, Bertoch T, Buvanendran A, Habib AS, Pizzi LJ, Pollak RA, Weiner SG, Bozic C, Negulescu P, White PF, VX21-548-101 AVX-5-1TG. Selective Inhibition of NaV1.8 with VX-548 for Acute Pain. N Engl J Med 2023; 389:393–405. [DOI] [PubMed] [Google Scholar]