

Abstract
Gastric neuroendocrine tumors (G-NET) are rare tumors arising from enterochromaffin-like cells of the gastric mucosa. They belong to a larger group called gastroenteropancreatic neuroendocrine tumors and are classified as low, intermediate, or high-grade tumors based on their proliferative indices. They are further categorized into three subtypes based on their morphologic characteristics, pathogenesis, and behavior. Type 1 and 2 tumors are characterized by elevated serum gastrin and are usually multifocal. They typically occur in the setting of atrophic gastritis or MEN1/Zollinger Ellison syndrome, respectively. Type 2 tumors are associated with the most symptoms, such as abdominal pain and diarrhea. Type 3 tumors are associated with normal serum gastrin, are usually solitary, and occur sporadically. This type has the most aggressive phenotype and metastatic potential. Treatment and prognosis for G-NET is dependent on their type, size, and stage, with Type 1 having the best prognosis and Type 3 having the worst. This review discusses the presentation, work-up, and surgical management of these tumors.
Keywords: gastric neuroendocrine, neuroendocrine carcinoma, high-grade, gastrinoma, NET, G-NET
1. Introduction
Gastric neuroendocrine tumors (G-NET), previously referred to as gastric carcinoids, are rare tumors arising from enterochromaffin-like (ECL) cells of the gastric oxyntic mucosa.1,2 They are part of a larger group of tumors called neuroendocrine neoplasms (NEN) and include well-differentiated neuroendocrine tumors (NET), poorly differentiated NET, poorly differentiated neuroendocrine carcinomas (NEC), and mixed neuroendocrine-non-neuroendocrine neoplasms (MiNEN).3,4
G-NET comprise 1.9-2.2% of all NET and 5-15% of all gastroenteropancreatic NET.5,6 According to recent analyses of the Surveillance, Epidemiology, and End Results (SEER) database in 2020, G-NET were found to have an annual incidence of 4.97 cases per 1,000,000 patients in the USA, with a median age of diagnosis of 59 years and 5-year overall survival of 81.1% for all stages combined.7
The clinical presentation of G-NET is not specific. They are usually diagnosed during an upper gastrointestinal endoscopy, and carcinoid syndrome is seldom seen in this subset of NETs.8,9 After appropriate diagnostic, imaging, and staging procedures, treatment depends on the size, number, depth of invasion, metastasis, and differentiation of the tumor.10 Treatment options include surveillance with or without excision, endoscopic therapy / resection, surgical resection, and systemic therapies. In this paper, we have reviewed the different types of G-NETs and provided insights into their management and surveillance based on the presenting type.
2. Types
As per the World Health Organization (WHO) in 2019, G-NET are classified based on their morphologic characteristics, behavior, and pathogenesis into histamine-producing ECL-cell NET, somatostatin-producing D-cell NET, gastrin-producing G-cell NET and serotonin-producing enterochromaffin (EC) cell NET.4
The majority of G-NET are histamine-producing ECL-cell NET which are categorized into three main types. Type 1 is associated with autoimmune atrophic gastritis, type 2 with Zollinger-Ellison syndrome (ZES) and Multiple endocrine neoplasia type 1 (MEN1) syndrome, and type 3 arises sporadically, usually with a unifocal lesion (Table 1). Rarely, ECL-cell NETs have been associated with an intrinsic defect in the acid secretion of the parietal cells.1,2,4,11–13 Type 1 and type 2 G-NET are characterized by hypergastrinemia, whereas patients with type 3 tumors usually have normal serum gastrin levels.1,2,4,14 Type 3 G-NET are frequently larger in size compared to the other two types and behave more aggressively.2,14 Clinically, patients with G-NET are usually asymptomatic, but they can present with abdominal pain, nausea, vomiting, gastrointestinal bleeding or gastric outlet obstruction.4,15,16
Table 1.
Distinctive characteristics of different types of G-NET.
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| % among G-NET | 80%-90% | 5%-7% | 10%-15% |
| Association with | Atrophic gastritis | MEN1-gastrinomas | - |
| Gastrin levels | Elevated | Elevated | Normal |
| Gastric pH | High | Low | Normal |
| Tumor size | <1cm | <2cm | >2cm |
| Number of tumors | Multifocal | Multifocal | Solitary |
| Sex predominance | Female | Equal | Male |
| Risk of metastasis | 1-3% | 10-30% | 50% |
| Prognosis | Excellent | Very Good | Poor |
3. Evolution of Nomenclature and Staging
In 2000, the WHO defined gastric carcinoids as “well differentiated neoplasms of the diffuse endocrine system”. This change was undertaken to underline the malignant potential of these tumors and also separate them from other entities producing bioactive molecules, such as insulinomas, VIPomas and gastrinomas.1,17 In 2010, the WHO classified well-differentiated gastroenteropancreatic NET into low grade (G1) or intermediate grade (G2), whereas the term high-grade (G3) was used to define poorly differentiated NEC. This grading system utilizes the tumor’s proliferative characteristics to assign the appropriate grade, by taking into consideration the mitotic rate and Ki-67 index.18 Since then, studies have revealed a subgroup of patients with well-differentiated high-grade NET.19 For this reason, in 2019, WHO re-classified G-NET into three grades (low or G1, intermediate or G2 and high or G3) (Table 2). NEC are no longer graded as they are uniformly defined as high-grade, but continue to be separated into small-cell and large-cell subtypes. Well-differentiated NET may be high-grade, despite remaining well-differentiated morphologically, and distinct from poorly differentiated NEC.3,4,20 Mutations in the p53 and RB1 genes are usually present in NEC, differentiating them from G3 NET.4
Table 2.
Classification of gastroenteropancreatic neuronendocrine neoplasms (WHO, 2019) 4.
| Terminology | Differentiation | Grade | Ki-67 index | Mitotic rate (mitoses/10HPF) |
|---|---|---|---|---|
| G1 NET | Well differentiated | Low | <3% | <2 |
| G2 NET | Intermediate | 3-20% | 2-20 | |
| G3 NET | High | >20% | >20 | |
| NEC | Poorly differentiated | High | >20% | >20 |
In cases of disparity between Ki-67 index and mitotic rate, the result that indicates a higher grade tumor should be selected.
G-NET are staged according to the Tumor (T), Node (N) and metastasis (M) staging system of the Union for International Cancer Control (UICC) and the American Joint Committee on Cancer (AJCC) (Table 3,4).4,11,21
Table 3.
TNM staging for gastrointestinal NET per AJCC 8th edition 21.
| T | Primary Tumor | N | Regional lymph nodes |
|---|---|---|---|
| TX | Primary tumor cannot be assessed | Nx | Regional lymph nodes cannot be assessed |
| T0 | No evidence of primary tumor | N0 | No regional lymph node metastasis |
| T1 | Invades the lamina propria or submucosa and less than or equal to 1 cm in size | N1 | Regional lymph node metastasis |
| T2 | Invades the muscularis propria or greater than 1 cm in size | M | Distant metastasis |
| T3 | Invades through the muscularis propria into subserosal tissue without penetration of overlying serosa | M0 | No distant metastasis |
| T4 | Invades visceral peritoneum (serosa) or other organs or adjacent structures | M1 | M1a: Metastasis confined to liver M1b: Metastases in at least one extrahepatic site M1c: Both hepatic and extrahepatic metastases |
Table 4.
Prognostic stage groups for G-NET per AJCC 8th edition.21
| T | N | M | |
|---|---|---|---|
| Stage 1 | T1 | N0 | M0 |
| Stage 2 | T2,T3 | N0 | M0 |
| Stage 3 | T1,T2,T3 T4 |
N0 N1 |
M0 M0 |
| Stage 4 | Any T | Any N | M1 |
4. Type 1 Gastric Neuroendocrine Tumors
Type 1 G-NET comprise 80-90% of all G-NET.2,4,6,12,13 These tumors have a female predominance with a male to female ratio (M:F) of 0.4:1.4,13 This type is associated with chronic autoimmune atrophic gastritis, in which the formation of autoantibodies target the H+/K+ ATPase of the parietal cells in the gastric body and fundus, leading to achlorhydria and the decreased production of intrinsic factor (IF). Critically, only a subset of patients with autoimmune atrophic gastritis develop G-NET with an annual risk of 0.4-0.7%, indicating that other pathogenic processes may be at play, including mutations of the Regalpha gene.22–24 The absence of hydrochloric acid causes a physiologic loss of the normal feedback inhibition via somatostatin producing D cells, which subsequently leads to increased gastrin production by G-cells in the antrum, leading to hypergastrinemia. The continuous production of gastrin causes hyperplasia of the ECL cells, acting as the nidus for the formation of type 1 G-NET via subsequent dysplasia and NET development, which appear as multiple small lesions (Figure 1). Furthermore, the decreased production of IF results in the decreased absorption of vitamin B12, leading to megaloblastic anemia (pernicious or macrocytic anemia).1,2,4,13 Infection with H. Pylori is described as another etiologic factor for development of type 1 tumors.4 Patients with type 1 G-NET have an excellent prognosis, where 5-year overall survival reaches 100%.4,14
Figure 1.
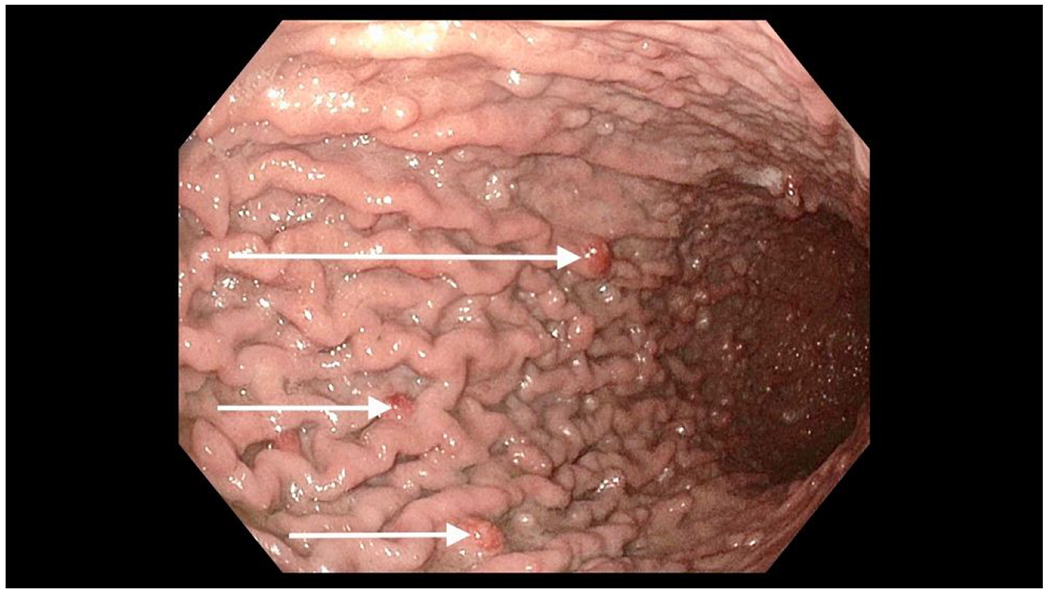
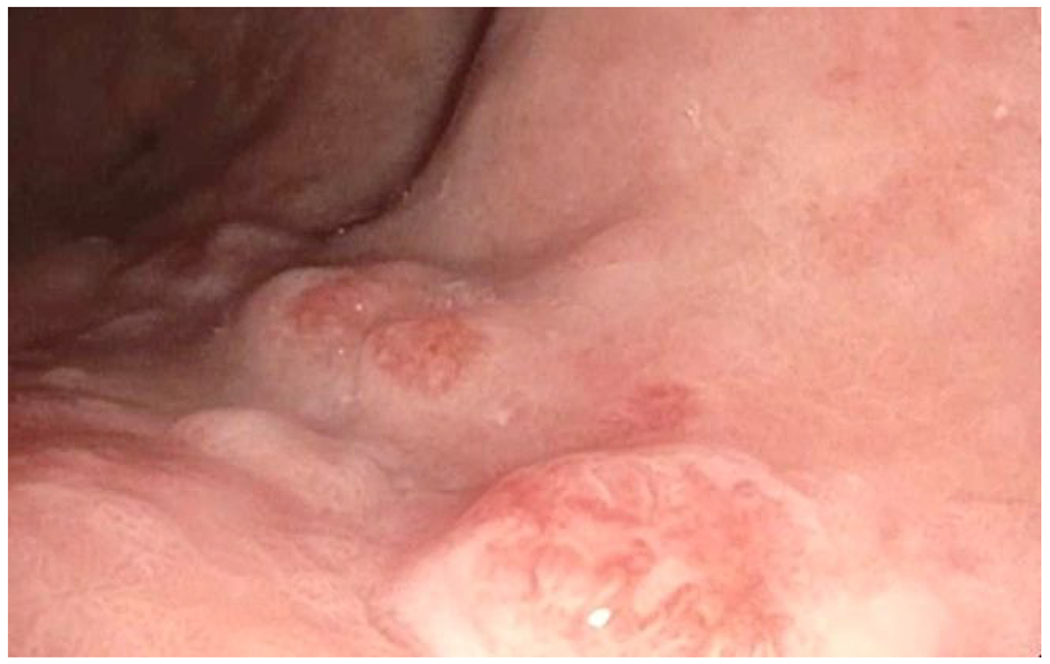
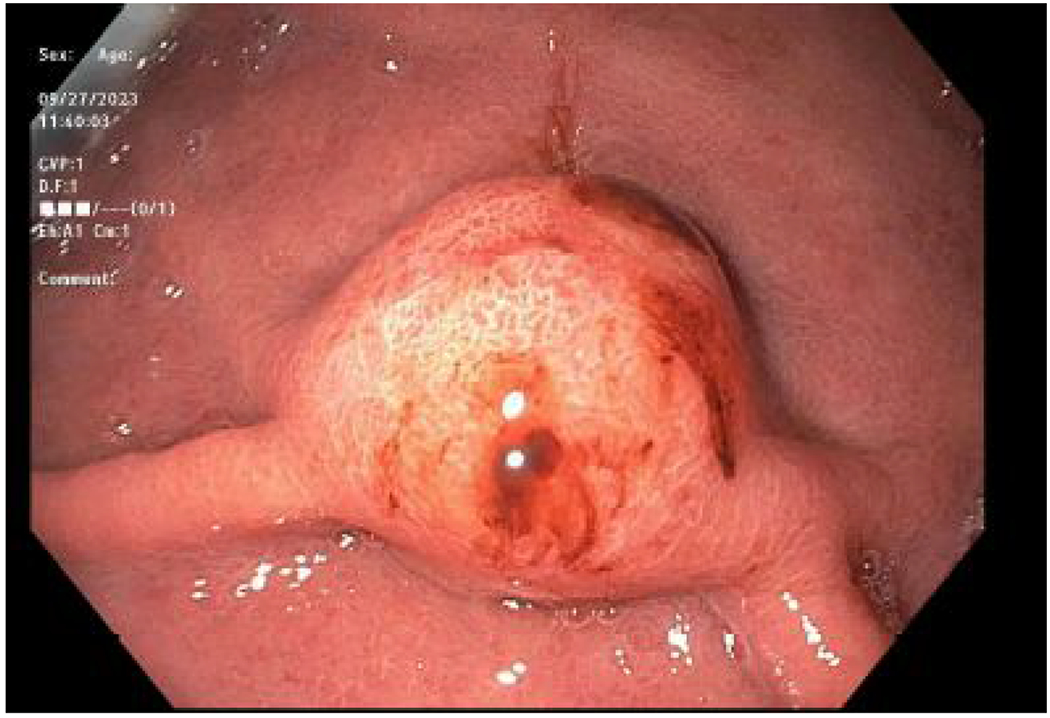
Endoscopy View of Gastric Neuroendocrine Tumors
A. White arrows reveal few of the multiple tumors seen in Type 1 gastric neuroendocrine tumors.
B. Focused and magnified view of Type 1 gastric neuroendocrine tumors.
C. Focused and magnified view of a Type 3 gastric neuroendocrine tumor.
Presentation and histologic evaluation
Type 1 G-NET usually present as multiple sub centimeter polyps or nodules in the gastric body or fundus that rarely metastasize (1-3%).2,4,6,14 Histologically, they are low or intermediate grade monomorphic cells with round nuclei.4,25 The gastric mucosa shows atrophy with intestinal and pseudopyloric metaplasia, ECL-cell hyperplasia and dysplasia, and absence of parietal cells.4
Diagnosis and work-up
Work-up of type 1 G-NET should include endoscopy with gastric polypectomy/biopsy and measurement of serum gastrin levels. High gastric pH, presence of anti-parietal cell and anti-IF antibodies, and low serum B12 supports the diagnosis.26 Proton pump inhibitors (PPIs) may lead to falsely elevated serum gastrin levels, and thus levels should be checked after PPI cessation for at least 1 week. Once the diagnosis has been confirmed, endoscopic ultrasound (EUS) should be considered to evaluate lesions greater than 1cm to identify the depth of invasion and involvement of regional lymph nodes.12 Additional computed tomography (CT) of the chest and multiphasic CT or magnetic resonance imaging (MRI) of the abdomen and pelvis is only recommended in the presence of tumors greater than 2cm, as these lesions have an increased risk of metastasis. Additional imaging with 68Ga-Dotatate positron emission tomography (PET)-CT should only be considered in patients with high-risk or advanced disease. 27
Treatment and follow-up
In the absence of metastatic disease, treatment usually involves endoscopic resection of the most prominent tumor and endoscopic observation of tumors less than 1cm.6,28,29 Endoscopic surveillance should be tailored to the size and number of prominent lesions, annually or every two years with biopsy of the polyps, and mucosal resection for lesions larger than 1cm.11,30,31 In a recent endoscopic surveillance study of 57 patients, 30% of patients who were monitored with surveillance progressed, requiring resection. Patients with disease progression had significantly higher serum gastrin levels than patients without progression. Fifty percent of patients with disease progression required reinterventions with a median follow up of 22 months after the first intervention.32 Another study followed 84 patients with type 1 G-NET after initial endoscopic or surgical intervention. Fifty-two percent of patients developed local recurrence requiring reintervention during a mean follow-up period of 45 months.33 No metastasis or death was reported during this period with a median recurrence-free survival of 31 months. Similar to the previously mentioned study, high serum gastrin at follow up was associated with recurrence.
Endoscopic mucosal resection, endoscopic submucosal dissection, and band and slough technique are acceptable endoscopic techniques with a good safety profile.34–36 Surgical intervention is usually not advised given the benign nature of this disease.1,14 Indications for surgery include tumor extending beyond the submucosa, lymph node involvement, tumors larger than 2cm, recurrent tumors, and in patients with more than six polyps.6,27,31 In these cases where surgery is indicated, the North American Neuroendocrine Tumor Society (NANETS) recommends tumor excision via wedge resection.27,37,38 Antrectomy, particularly minimally invasive antrectomy, to remove the source of gastrin may be used very selectively in symptomatic patients with multifocal disease and can often result in regression of type 1 G-NETs.39–41 Total gastrectomy is largely no longer used in the treatment of type 1 G-NETs, and may be performed rarely in cases with extensive fundal disease or widespread recurrence not amenable to endoscopic resection.42,43
Somatostatin analogs can be used in the setting of recurrence following endoscopic resection or multiple lesions not amenable to endoscopic resection to reduce serum gastrin.44 Netazepide is a gastrin/cholecystokinin-2 receptor antagonist which has shown to reduce the size and number of type 1 G-NET, along with a reduction in plasma chromogranin A levels.45,46 It is a potential alternative systemic treatment strategy either alone or with endoscopic surveillance, although large scale randomized trials are required to validate its effectiveness.
5. Type 2 Gastric Neuroendocrine Tumors
Type 2 tumors represent 5-7% of G-NET.4,6,12,13 A gastrinoma, an ectopic gastrin producing G-cell neoplasm commonly found in the duodenum or pancreas, is responsible for the hypergastrinemia that drives the pathogenesis of type 2 G-NETs. This type is associated with elevated serum gastrin (>1000 pg/ml is considered diagnostic), low gastric pH (hyperchlorhydria, pH <2), and hypertrophic hypersecretory gastropathy that can develop in the setting of MEN-1 syndrome.4 The long-term risk of developing a type 2 G-NET is at least 100 times less in patients with sporadic gastrinomas compared to MEN-1 patients with ZES.47 The risk of metastasis is approximately 10-30%, higher than type 1, but less than type 3.2,4 The 5-year overall survival ranges from 60% to 90%.4
Presentation and histologic evaluation
Type 2 G-NET usually present as multiple tumors in the gastric fundus / body that are less than 2cm in size.2,4,6 Patients may present with symptoms of ZES, including abdominal pain, pepticulcers, and secretory diarrhea due to excessive gastric acid production.48 The diagnosis is often delayed due to the nonspecific nature of these symptoms, with an average time between onset of symptoms to diagnosis being longer than 5 years.49 Histologically, there is evidence of parietal cell hyperplasia and hypertrophy with the presence of monomorphic cells with extracellular mucoid-like material and formation of enlarged trabeculae and ribbons.4
Diagnosis and work-up
Diagnosis should include endoscopy with gastric polypectomy / biopsy, and measurement of serum gastrin and chromogranin A levels. Once the diagnosis is confirmed, EUS may be considered to investigate the depth of the lesion. Patients should undergo multiphasic abdominal CT or MRI and cross-sectional imaging of the chest and pelvis to assess the location of the gastrinoma and evaluate for metastatic disease.28,29 Somatostatin receptor imaging with gallium-68 or 68Ga-Dotatate is a useful study with a higher sensitivity and specificity (91% and 94%, respectively) than somatostatin receptor scintigraphy, as patients with stage IV disease may benefit from systemic therapy if a receptor-positive tumor is identified.50 Type 2 G-NET are associated with MEN1 syndrome, which demonstrates an autosomal dominant pattern of inheritance. Genetic work-up should be performed in patients meeting clinical criteria for MEN1, due to implications related to other MEN1-associated tumors, as well as genetic counseling.11,51,52
Treatment and follow-up
Treatment of type 2 G-NET usually consists of surgical resection of the primary gastrinoma. Appropriate patients with sporadic gastrinoma without unresectable metastatic disease should undergo surgery to remove the gastrinoma.53 Primary gastrinoma resection often results in regression of the type 2 G-NET.54 If resection is not feasible due to metastatic disease or multifocal gastrinomas, endoscopic resection of the most prominent G-NET followed by surveillance is a viable option. Somatostatin analogs should be considered for the management of symptoms or when disease control is required.55 High dose PPIs may help in decreasing gastric acid hypersecretion. In patients with type 2 G-NET as a result of MEN1, resection of the primary gastrinoma is only recommended in the setting of gastrinomas larger than 2cm.53 Medical management of hypergastrinemia is the mainstay of therapy in patients with MEN1 due to the multiplicity of tumors, extra-pancreatic location, co-existent metastatic disease, and low chance of surgical cure. Following resection, imaging surveillance should continue for at least 10 years in the majority of patients.6,11,55 Netazepide as a treatment option has not been validated in type 2 G-NETs.
6. Type 3 Gastric Neuroendocrine Tumors
Type 3 tumors represent 10%-15% of G-NET.4,6,12,13 They develop sporadically and are non-gastrin dependent.2,4,6,14,16 Type 3 tumors have strong male predominance with M:F ratio 2.8:1.4 The 5-year overall survival is <50%.4 Factors associated with worse overall survival are tumor size > 2cm, angio-lymphatic invasion, invasion of the gastric wall, and distant metastasis.14
Presentation and histologic evaluation
Type 3 tumors present as a single, large (usually > 2cm) tumor. Approximately half of patients (51%) have distant metastases at the time of diagnosis.6 Patients usually present with nonspecific symptoms including pain, weight loss, and melena related to tumor progression and metastatic spread. Histologic examination of the normal gastric mucosa is usually unrevealing.2,4,14
Diagnosis and work-up
Endoscopy with histologic examination of the biopsy confirms the diagnosis. Given the aggressive nature of this tumor with a tendency to invade the lymphovascular and submucosal tissue, all patients should be considered for 68Ga-Dotatate PET-CT along with multiphasic CT or MRI abdominal imaging to assess the extent of disease.56 EUS may be considered to determine T-stage, especially for smaller tumors.11,28,29
Treatment and follow-up
Due to its advanced stage at the time of diagnosis and metastatic potential, radical resection, including partial or total gastrectomy, with regional lymphadenectomy is the treatment of choice for non-metastatic lesions >2cm recommended by the European Neuroendocrine Tumor Society (ENETS) and NANETS.27,30 The role of endoscopic resection and surveillance for small and low-grade type 3 G-NET has been explored. A study by Min et al. included 32 patients with type 3 G-NETs, and 22 patients with grade 1 NETs without lymphovascular invasion were treated with wedge or endoscopic resection.57 After a median follow up of 59 months, only patients with tumor size >1.5 cm developed recurrence, and no patients with a grade 1 NET of 1.5 cm or smaller developed recurrence. Another study by Kwon et al. investigated 50 patients with well-differentiated type 3 G-NETs with a mean tumor size of 1cm that were treated with endoscopic resection, and no recurrence was noted in either the pathologically complete resection group (80%) or the incomplete resection group (20%) with a median follow up of 46 (13-60) months.58 A more recent analysis by Hanna et al. compared outcomes between patients presenting with type 1 and type 3 G-NET.59 Patients with type 1 G-NET had a median tumor size of 0.6cm, and patients with type 3 G-NET had a median tumor size of 1.0cm. The majority (88%) of patients with type 1 G-NET had grade 1 lesions, while 53% of patients with type 3 G-NET had grade 2/3 lesions. They found no difference in overall survival between patients presenting with type 1 and type 3 G-NET.
Higher tumor grade and presence of nodal or distant metastases was associated with worse survival. Among type 3 G-NET patients, those with small (< 0.5 cm), grade 1 lesions were less likely to develop metastases (0% versus 33%, p < 0.01) and more likely to survive (100% versus 67%, p < 0.01) at 5 years compared to patients with larger or higher-grade tumors. Patients with grade 1 tumors <0.5cm were largely treated with endoscopic resection (67%).59 Consistent with these studies, the recent National Comprehensive Cancer Network (NCCN) guidelines indicate that small and low grade type 3 G-NETs can be treated with endoscopic resections (< 1 cm) or surgical wedge (< 2 cm).38 Post-operative surveillance with imaging studies should continue for at least 10 years.11
7. Grade 3 G-NET and NEC
Together, grade 3 G-NETs and NEC are classified as high-grade neuroendocrine neoplasms. They are extremely rare, comprising approximately 0.5% of all malignant gastric neoplasms.60 Given their low incidence, there is a paucity of literature regarding the workup and management of high grade gastric NENs. Recently, the North American Neuroendocrine Tumor Society published consensus practice recommendations to provide guidance on best treatment practices for these rare tumors.61
Diagnosis and workup
The workup of G3 G-NET and G-NEC follows a comprehensive approach to establish an accurate diagnosis and assess the extent of the disease. Gastrointestinal endoscopy remains a fundamental step in the evaluation, allowing direct visualization of the tumor and obtaining biopsies for histopathological analysis. Immunohistochemistry is crucial to determine neuroendocrine differentiation and confirm the high-grade nature of the tumor. As previously discussed, G3 G-NETs are well-differentiated or moderately differentiated, whereas G-NEC are poorly differentiated.20 Histologically, G-NEC are nearly identical to gastric adenocarcinoma except that endocrine cells are present in the tumor matrix.62 The diagnostic criteria for G3 NENs includes a Ki-67 proliferation index greater than 20% and/or a mitotic count surpassing 20 per 2mm2, although the majority of NECs significantly surpass these thresholds.63,64 G3 G-NETs are almost always positive for synaptophysin and chromogranin A, while up to 25% of G-NEC are negative for the traditional neuroendocrine markers.61
Due to their aggressive behavior, advanced imaging techniques play a critical role in staging these tumors. EUS provides valuable information on tumor depth and regional lymph node involvement. CT / MRI are essential for assessing local and distant spread. Key imaging characteristics, including larger size, arterial and portal enhancement, ill-defined features, and textural analysis, can be used to differentiate high grade G-NET and NEC from lower-grade lesions.65 PET-CT with 68Ga-Dotatate can also be employed to detect somatostatin receptor expression and assess the potential for targeted therapy, especially in high-grade G-NET. PET-CT with FDG can be used to differentiate G1/G2 lesions from G3. Several studies have shown that the degree of FDG uptake differs in lower versus higher grade lesions, and the degree of uptake is correlated with prognosis.66–68 FDG-PET can be used as the functional imaging of choice in the case of high grade NETs or NEC with low somatostatin receptor expression.61 The utility of brain imaging in the setting of high grade NET or NEC is low. The incidence of brain metastases in extra-pulmonary G3 G-NET and NEC is less than 2%, and patients with a higher disease burden are at higher risk of brain metastasis.69,70 Brain MRI for patients with G3 G-NET or NEC should be reserved for symptomatic patients or patients with a high burden of systemic disease per expert consensus recommendations.61
Treatment and follow-up
Managing G3 G-NET and G-NEC demands an aggressive and individualized treatment approach. The management decisions are influenced by factors such as tumor stage, differentiation, patient’s performance status, and the presence of distant metastases. There is little data to direct the management of G3 G-NET, so treatment recommendations are based on studies that have analyzed the management of G3 NET at other sites along that gastrointestinal tract.71–73 Surgery with thorough lymphadenectomy remains the mainstay of curative treatment for localized G3 G-NET, with a reported median survival of 43-55 months.74 There is currently no consensus regarding the use of definitive, neoadjuvant, or adjuvant radiotherapy or chemotherapy in the management of G3 G-NET. For G-NEC, surgery is recommended for resectable disease, with the understanding of poor prognosis.75–80 Multiple studies compare outcomes between patients undergoing surgical resection of G-NEC versus gastric adenocarcinoma revealing neuroendocrine differentiation as an independent predictor of poor overall survival.81,82 These studies are mainly from Asia, and more studies are needed to examine survival of patients with G-NEC across a more diverse population. A retrospective study of 69 patients with G-NEC by Ma et al. demonstrated that receipt of neoadjuvant chemotherapy was associated with significantly higher overall survival than patients undergoing upfront surgical resection (57.4% vs. 28.5% 5-year OS, p=0.032), indicating a possible benefit.83 Consensus guidelines recommend the consideration of adjuvant therapy after the surgical resection of localized NEC.84 Postoperative surveillance with imaging studies should be completed every 3 months for 3 years, and then every 6-12 months for at least 10 years for patients treated for localized high grade G-NET or G-NEC.61
8. Metastatic G-NET
Patients with metastatic G-NET should have a complete work-up that includes multiphasic CT or MRI abdominal imaging, chest CT, and 68Ga-Dotatate PET-CT. Depending on the disease burden and symptomatology, the following guidelines have been established per the NCCN:85
If complete resection is possible, then patients should undergo resection of the primary tumor and metastasectomy.
If patients are asymptomatic and the disease burden is low, then they can be observed with serial imaging. Alternatively, for somatostatin receptor positive tumors, administration of somatostatin analogs may be considered.
If patients are symptomatic from the primary tumor, then debulking or resection of the primary tumor is recommended.
For patients with unresectable or significant disease burden, somatostatin analogs should be considered as first-line. For this group of patients, who progress on first-line therapy and have significant uptake on Dotatate PET-CT, Peptide Receptor Radionuclide Therapy (PRRT) is considered standard of care. PRRT, a molecular therapy that combines a somatostatin analog with a radionuclide and targets somatostatin receptor positive tumors, is efficacious with some patients achieving complete remission.86,87 Administration of cytotoxic chemotherapy remains controversial and may be considered in patients with progressive disease. Liver-directed therapy (surgical resection, hepatic arterial embolization and ablation) may be considered for control of liver metastases. For symptomatic bone metastases, palliative radiation therapy may be helpful.11,55
9. Other types
Gastrin producing G-cell NET:
These tumors usually present as small non-functioning mucosal or submucosal lesions that develop in the antrum, frequently close to the pylorus. Histologically, they appear as uniform cells with scant cytoplasm and usually demonstrate a trabecular and gyriform pattern.4
Serotonin-producing EC-cell NET:
Usually presents as a non-functioning mass. Rarely, it can be associated with symptoms of carcinoid syndrome. Histologically, they appear as rounded nests of uniform cells with peripheral palisading and eosinophilic cytoplasm. They demonstrate serotonin, somatostatin receptor type 2A (SSTR2A) and caudal type homeobox 2 (CDX2) protein reactivity.4
Somatostatin-producing D-cell NET:
These tumor cells appear as monomorphic cells that are positive for somatostatin, SSTR2A, chromogranin A and synaptophysin.4
10. Future trends
As the endoscopic evaluation of GI symptoms increase and advanced endoscopy becomes more accessible to an increasing proportion of the population, it is likely that the incidence of G-NET will keep increasing. Given the characteristics and unique behavior of G-NET compared to other gastroenteropancreatic NET, it is possible that a G-NET-specific TNM staging and histologic grading system will be developed in the future. In somatostatin receptor positive NET, PRRT has shown exciting results warranting further studies to identify the optimal timing and sequence of therapy.
11. Conclusion
Gastric neuroendocrine tumors pose significant challenges to clinicians due to their diverse origins, outcomes, and treatment demands. The pathogenesis of each is unique: type 1 is associated with hypergastrinemia and atrophic gastritis, type 2 is linked to gastrinoma with Zollinger-Ellison syndrome, while type 3 is sporadic, with no correlation with gastrin levels. The prognosis of gastric neuroendocrine tumors vary significantly: type 1 and 2 tend to have a more favorable outcome with lower metastatic potential, whereas type 3 is associated with a higher risk of metastasis. The role of resection via endoscopic or surgical intervention varies across type. Surveillance often suffices for type 1 lesions, while the surgical management of type 2 is largely focused on treatment of the underlying gastrinoma. Type 3 often requires a more comprehensive surgical approach. In light of these intricacies, it becomes paramount to tailor the management strategy to each patient′s specific gastric neuroendocrine tumor type and symptom presentation. Integrating well-established and innovative treatment methods holds the potential to mitigate disease progression and improve patients′ quality of life.
SYNOPSIS.
Gastric neuroendocrine tumors are rare tumors of the stomach. As endoscopic evaluation of gastrointestinal symptoms increases, it is likely that the incidence of these neoplasms will increase. This review discusses the presentation, work-up, and surgical management of these tumors.
Disclosure:
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number K12 CA237806 from the Emory K12 Clinical Oncology Training Program and Georgia CTSA UL1 TR002378, awarded to MMS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Gilligan CJ, Lawton GP, Tang LH, West AB, Modlin IM. Gastric carcinoid tumors: the biology and therapy of an enigmatic and controversial lesion. Am J Gastroenterol. Mar 1995;90(3):338–52. [PubMed] [Google Scholar]
- 2.Rindi G, Luinetti O, Cornaggia M, Capella C, Solcia E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: a clinicopathologic study. Gastroenterology. Apr 1993;104(4):994–1006. doi: 10.1016/0016-5085(93)90266-f [DOI] [PubMed] [Google Scholar]
- 3.Washington MK, Goldberg RM, Chang GJ, et al. Diagnosis of digestive system tumours. Int J Cancer. Jul 16 2020;doi: 10.1002/ijc.33210 [DOI] [PubMed] [Google Scholar]
- 4.Klimstra DS KG, La Rosa S, Rindi G. Classification of neuroendocrine neoplasms of the digestive system. 5th ed. WHO Classification of Tumours: Digestive System Tumours. International Agency for Research on Cancer; 2019. [Google Scholar]
- 5.Godwin JD 2nd. Carcinoid tumors. An analysis of 2,837 cases. Cancer. Aug 1975;36(2):560–9. doi: 10.1002/1097-0142(197508)36:2<560::aid-cncr2820360235>3.0.co;2-4 [DOI] [PubMed] [Google Scholar]
- 6.Manfredi S, Walter T, Baudin E, et al. Management of gastric neuro-endocrine tumours in a large French national cohort (GTE). Endocrine. Sep 2017;57(3):504–511. doi: 10.1007/s12020-017-1355-9 [DOI] [PubMed] [Google Scholar]
- 7.Hu P, Bai J, Liu M, et al. Trends of incidence and prognosis of gastric neuroendocrine neoplasms: a study based on SEER and our multicenter research. Gastric Cancer. Jul 2020;23(4):591–599. doi: 10.1007/s10120-020-01046-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas D, Tsolakis AV, Grozinsky-Glasberg S, et al. Long-term follow-up of a large series of patients with type 1 gastric carcinoid tumors: data from a multicenter study. Eur J Endocrinol. Feb 2013;168(2):185–93. doi: 10.1530/eje-12-0836 [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Ozao J, Warner R, Divino C. Review of the pathogenesis, diagnosis, and management of type I gastric carcinoid tumor. World J Surg. Aug 2011;35(8):1879–86. doi: 10.1007/s00268-011-1137-0 [DOI] [PubMed] [Google Scholar]
- 10.Liu X, Zhang Z, Huang J, Tan H, Yang Z. Efficacy and Safety of Interferon-Alpha 2b for Patients with Hepatic Epithelioid Hemangioendothelioma: Outcomes of a Case-Series Analysis. Cancer Manag Res. 2021;13:8273–8279. doi: 10.2147/cmar.S334171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.NCCN. National Comprehensive Cancer Network-Neuroendocrine and Adrenal Tumors. Accessed December 10, 2020, https://www.nccn.org/professionals/physician_gls/pdf/neuroendocrine.pdf
- 12.Roberto GA, Rodrigues CMB, Peixoto RD, Younes RN. Gastric neuroendocrine tumor: A practical literature review. World J Gastrointest Oncol. Aug 15 2020;12(8):850–856. doi: 10.4251/wjgo.v12.i8.850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rindi G, Bordi C, Rappel S, La Rosa S, Stolte M, Solcia E. Gastric carcinoids and neuroendocrine carcinomas: pathogenesis, pathology, and behavior. World J Surg. Feb 1996;20(2):168–72. doi: 10.1007/s002689900026 [DOI] [PubMed] [Google Scholar]
- 14.Schindl M, Kaserer K, Niederle B. Treatment of gastric neuroendocrine tumors: the necessity of a type-adapted treatment. Arch Surg. Jan 2001;136(1):49–54. doi: 10.1001/archsurg.136.1.49 [DOI] [PubMed] [Google Scholar]
- 15.Sjoblom SM. Clinical presentation and prognosis of gastrointestinal carcinoid tumours. Scand J Gastroenterol. Sep 1988;23(7):779–87. doi: 10.3109/00365528809090760 [DOI] [PubMed] [Google Scholar]
- 16.Crown A, Kennecke H, Kozarek R, et al. Gastric carcinoids: Does type of surgery or tumor affect survival? Am J Surg. May 2019;217(5):937–942. doi: 10.1016/j.amjsurg.2018.12.057 [DOI] [PubMed] [Google Scholar]
- 17.Capella ES C, Sobin LH, Arnold R. Endocrine tumours of the stomach. World Health Organization Classification of Tumours - Pathology and Genetics of Tumours of the Digestive System. IARCPress; 2000. [Google Scholar]
- 18.van Velthuysen ML, Groen EJ, van der Noort V, van de Pol A, Tesselaar ME, Korse CM. Grading of neuroendocrine neoplasms: mitoses and Ki-67 are both essential. Neuroendocrinology. 2014;100(2-3):221–7. doi: 10.1159/000369275 [DOI] [PubMed] [Google Scholar]
- 19.Coriat R, Walter T, Terris B, Couvelard A, Ruszniewski P. Gastroenteropancreatic Well-Differentiated Grade 3 Neuroendocrine Tumors: Review and Position Statement. Oncologist. Oct 2016;21(10):1191–1199. doi: 10.1634/theoncologist.2015-0476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagtegaal ID, Odze RD, Klimstra D, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. Jan 2020;76(2):182–188. doi: 10.1111/his.13975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amin MB ES, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, et al. (Eds.). AJCC Cancer Staging Manual. 8th ed. Springer International Publishing: American Joint Commission on Cancer; 2017. [Google Scholar]
- 22.Vannella L, Sbrozzi-Vanni A, Lahner E, et al. Development of type I gastric carcinoid in patients with chronic atrophic gastritis. Aliment Pharmacol Ther. Jun 2011;33(12):1361–9. doi: 10.1111/j.1365-2036.2011.04659.x [DOI] [PubMed] [Google Scholar]
- 23.Annibale B, Azzoni C, Corleto VD, et al. Atrophic body gastritis patients with enterochromaffin-like cell dysplasia are at increased risk for the development of type I gastric carcinoid. Eur J Gastroenterol Hepatol. Dec 2001;13(12):1449–56. doi: 10.1097/00042737-200112000-00008 [DOI] [PubMed] [Google Scholar]
- 24.Higham AD, Bishop LA, Dimaline R, et al. Mutations of RegIalpha are associated with enterochromaffin-like cell tumor development in patients with hypergastrinemia. Gastroenterology. Jun 1999;116(6):1310–8. doi: 10.1016/s0016-5085(99)70495-6 [DOI] [PubMed] [Google Scholar]
- 25.Li TT, Qiu F, Qian ZR, Wan J, Qi XK, Wu BY. Classification, clinicopathologic features and treatment of gastric neuroendocrine tumors. World J Gastroenterol. Jan 7 2014;20(1):118–25. doi: 10.3748/wjg.v20.i1.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. Jan 2008;9(1):61–72. doi: 10.1016/s1470-2045(07)70410-2 [DOI] [PubMed] [Google Scholar]
- 27.Kulke MH, Anthony LB, Bushnell DL, et al. NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas. Aug 2010;39(6):735–52. doi: 10.1097/MPA.0b013e3181ebb168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cwikla JB, Buscombe JR, Caplin ME, et al. Diagnostic imaging of carcinoid metastases to the abdomen and pelvis. Med Sci Monit. Jun 2004;10 Suppl 3:9–16. [PubMed] [Google Scholar]
- 29.Kaltsas G, Rockall A, Papadogias D, Reznek R, Grossman AB. Recent advances in radiological and radionuclide imaging and therapy of neuroendocrine tumours. Eur J Endocrinol. Jul 2004;151(1):15–27. doi: 10.1530/eje.0.1510015 [DOI] [PubMed] [Google Scholar]
- 30.Delle Fave G, O’Toole D, Sundin A, et al. ENETS Consensus Guidelines Update for Gastroduodenal Neuroendocrine Neoplasms. Neuroendocrinology. 2016;103(2):119–24. doi: 10.1159/000443168 [DOI] [PubMed] [Google Scholar]
- 31.Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al. ENETS Consensus Guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology. 2012;95(2):74–87. doi: 10.1159/000335595 [DOI] [PubMed] [Google Scholar]
- 32.Chin JL, O’Connell J, Muldoon C, et al. Selective Resection of Type 1 Gastric Neuroendocrine Neoplasms and the Risk of Progression in an Endoscopic Surveillance Programme. Dig Surg. 2021;38(1):38–45. doi: 10.1159/000510962 [DOI] [PubMed] [Google Scholar]
- 33.Daskalakis K, Tsoli M, Karapanagioti A, et al. Recurrence and metastatic potential in Type 1 gastric neuroendocrine neoplasms. 10.1111/cen.14055. Clinical Endocrinology. 2019/October/01 2019;91(4):534–543. doi: 10.1111/cen.14055 [DOI] [PubMed] [Google Scholar]
- 34.Sivandzadeh GR, Ejtehadi F, Shoaee S, et al. Endoscopic mucosal resection: still a reliable therapeutic option for gastrointestinal neuroendocrine tumors. BMC Gastroenterology. 2021/May/24 2021;21(1):238. doi: 10.1186/s12876-021-01821-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawa F, Sako Z, Nguyen T, et al. The band and slough technique is effective for management of diminutive type 1 gastric and duodenal neuroendocrine tumors. Endosc Int Open. Jun 2020;8(6):E717–e721. doi: 10.1055/a-1119-6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato Y, Takeuchi M, Hashimoto S, et al. Usefulness of endoscopic submucosal dissection for type I gastric carcinoid tumors compared with endoscopic mucosal resection. Hepatogastroenterology. Sep 2013;60(126):1524–9. doi: 10.5754/hge121185 [DOI] [PubMed] [Google Scholar]
- 37.Hou W, Schubert ML. Treatment of gastric carcinoids. Curr Treat Options Gastroenterol. Apr 2007;10(2):123–33. doi: 10.1007/s11938-007-0064-5 [DOI] [PubMed] [Google Scholar]
- 38.Kulke MH SM, Benson AB, Bergsland E, Berlin JD, Besh SA et al. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Neuroendocrine Tumors Version 2; 2018. https://www.nccn.org/professionals/physician_gls/PDF/neuroendocrine.pdf. 2018;
- 39.Jenny HE, Ogando PA, Fujitani K, Warner RR, Divino CM. Laparoscopic antrectomy: a safe and definitive treatment in managing type 1 gastric carcinoids. Am J Surg. Apr 2016;211(4):778–82. doi: 10.1016/j.amjsurg.2015.08.040 [DOI] [PubMed] [Google Scholar]
- 40.Kitadani J, Ojima T, Hayata K, et al. Single-incision laparoscopic antrectomy for type I gastric neuroendocrine tumor: a case report. Surg Case Rep. Jan 12 2021;7(1):15. doi: 10.1186/s40792-021-01109-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozao-Choy J, Buch K, Strauchen JA, Warner RR, Divino CM. Laparoscopic antrectomy for the treatment of type I gastric carcinoid tumors. J Surg Res. Jul 2010;162(1):22–5. doi: 10.1016/j.jss.2010.01.005 [DOI] [PubMed] [Google Scholar]
- 42.Borch K, Ahren B, Ahlman H, Falkmer S, Granerus G, Grimelius L. Gastric carcinoids: biologic behavior and prognosis after differentiated treatment in relation to type. Ann Surg. Jul 2005;242(1):64–73. doi: 10.1097/01.sla.0000167862.52309.7d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gladdy RA, Strong VE, Coit D, et al. Defining Surgical Indications for Type I Gastric Carcinoid Tumor. Annals of Surgical Oncology. 2009/September/01 2009;16(11):3154. doi: 10.1245/s10434-009-0687-y [DOI] [PubMed] [Google Scholar]
- 44.Campana D, Nori F, Pezzilli R, et al. Gastric endocrine tumors type I: treatment with long-acting somatostatin analogs. Endocr Relat Cancer. Mar 2008;15(1):337–42. doi: 10.1677/erc-07-0251 [DOI] [PubMed] [Google Scholar]
- 45.Moore AR, Boyce M, Steele IA, Campbell F, Varro A, Pritchard DM. Netazepide, a gastrin receptor antagonist, normalises tumour biomarkers and causes regression of type 1 gastric neuroendocrine tumours in a nonrandomised trial of patients with chronic atrophic gastritis. PLoS One. 2013;8(10):e76462. doi: 10.1371/journal.pone.0076462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fossmark R, Sørdal Ø, Jianu CS, et al. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol Ther. Dec 2012;36(11-12):1067–75. doi: 10.1111/apt.12090 [DOI] [PubMed] [Google Scholar]
- 47.Peghini PL, Annibale B, Azzoni C, et al. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: insights from patients with sporadic gastrinomas. Gastroenterology. Jul 2002;123(1):68–85. doi: 10.1053/gast.2002.34231 [DOI] [PubMed] [Google Scholar]
- 48.Burkitt MD, Pritchard DM. Review article: Pathogenesis and management of gastric carcinoid tumours. Aliment Pharmacol Ther. Nov 1 2006;24(9):1305–20. doi: 10.1111/j.1365-2036.2006.03130.x [DOI] [PubMed] [Google Scholar]
- 49.Rossi RE, Elvevi A, Citterio D, et al. Gastrinoma and Zollinger Ellison syndrome: A roadmap for the management between new and old therapies. World J Gastroenterol. Sep 21 2021;27(35):5890–5907. doi: 10.3748/wjg.v27.i35.5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh S, Poon R, Wong R, Metser U. 68Ga PET Imaging in Patients With Neuroendocrine Tumors: A Systematic Review and Meta-analysis. Clin Nucl Med. Nov 2018;43(11):802–810. doi: 10.1097/RLU.0000000000002276 [DOI] [PubMed] [Google Scholar]
- 51.Giusti F, Marini F, Brandi ML. Multiple Endocrine Neoplasia Type 1. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews((R)). 1993. [Google Scholar]
- 52.Brandi ML, Agarwal SK, Perrier ND, Lines KE, Valk GD, Thakker RV. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr Rev. Nov 28 2020;doi: 10.1210/endrev/bnaa031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cingam SR, Botejue M, Hoilat GJ, Karanchi H. Gastrinoma. StatPearls. 2023. [PubMed] [Google Scholar]
- 54.Dacha S, Razvi M, Massaad J, Cai Q, Wehbi M. Hypergastrinemia. Gastroenterol Rep (Oxf). Aug 2015;3(3):201–8. doi: 10.1093/gastro/gov004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kunz PL. Carcinoid and neuroendocrine tumors: building on success. J Clin Oncol. Jun 1 2015;33(16):1855–63. doi: 10.1200/JCO.2014.60.2532 [DOI] [PubMed] [Google Scholar]
- 56.Basuroy R, Srirajaskanthan R, Prachalias A, Quaglia A, Ramage JK. Review article: the investigation and management of gastric neuroendocrine tumours. Aliment Pharmacol Ther. May 2014;39(10):1071–84. doi: 10.1111/apt.12698 [DOI] [PubMed] [Google Scholar]
- 57.Min BH, Hong M, Lee JH, et al. Clinicopathological features and outcome of type 3 gastric neuroendocrine tumours. Br J Surg. Oct 2018;105(11):1480–1486. doi: 10.1002/bjs.10901 [DOI] [PubMed] [Google Scholar]
- 58.Kwon YH, Jeon SW, Kim GH, et al. Long-term follow up of endoscopic resection for type 3 gastric NET. World J Gastroenterol. Dec 14 2013;19(46):8703–8. doi: 10.3748/wjg.v19.i46.8703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanna A, Kim-Kiselak C, Tang R, et al. Gastric Neuroendocrine Tumors: Reappraisal of Type in Predicting Outcome. Ann Surg Oncol. Dec 2021;28(13):8838–8846. doi: 10.1245/s10434-021-10293-7 [DOI] [PubMed] [Google Scholar]
- 60.Iwasaki K, Barroga E, Enomoto M, et al. Long-term surgical outcomes of gastric neuroendocrine carcinoma and mixed neuroendocrine-non-neuroendocrine neoplasms. World J Surg Oncol. May 24 2022;20(1):165. doi: 10.1186/s12957-022-02625-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eads JR, Halfdanarson TR, Asmis T, et al. Expert Consensus Practice Recommendations of the North American Neuroendocrine Tumor Society for the management of high grade gastroenteropancreatic and gynecologic neuroendocrine neoplasms. Endocr Relat Cancer. Aug 1 2023;30(8)doi: 10.1530/ERC-22-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahmed M. Gastrointestinal neuroendocrine tumors in 2020. World J Gastrointest Oncol. Aug 15 2020;12(8):791–807. doi: 10.4251/wjgo.v12.i8.791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Travis W, Brambilla E, Burke A, Marx A, Nicholson A. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. 2015; [DOI] [PubMed] [Google Scholar]
- 64.Rinde G, Mete O, Uccella S, et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocrine Pathology. 2022;33:115–154. [DOI] [PubMed] [Google Scholar]
- 65.Feng S, Luo Y, Chan T, et al. CT evaluation of gastroenteric neuroendocrine tumors: relationship between CT features and the pathologic classification. AJR American Journal of Roentgenology. 2014;203:W260–W266. [DOI] [PubMed] [Google Scholar]
- 66.Panagiotidis E, Bomanji J. Role of 18F-fluorodeoxyglucose PET in the study of neuroendocrine tumors. PET Clin. Jan 2014;9(1):43–55. doi: 10.1016/j.cpet.2013.08.008 [DOI] [PubMed] [Google Scholar]
- 67.Tomimaru Y, Eguchi H, Tatsumi M, et al. Clinical utility of 2-[(18)F] fluoro-2-deoxy-D-glucose positron emission tomography in predicting World Health Organization grade in pancreatic neuroendocrine tumors. Surgery. Feb 2015;157(2):269–76. doi: 10.1016/j.surg.2014.09.011 [DOI] [PubMed] [Google Scholar]
- 68.Majala S, Seppanen H, Kemppainen J, et al. Prediction of the aggressiveness of non-functional pancreatic neuroendocrine tumors based on the dual-tracer PET/CT. EJNMMI Res. Dec 23 2019;9(1):116. doi: 10.1186/s13550-019-0585-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alese OB, Jiang R, Shaib W, et al. High-Grade Gastrointestinal Neuroendocrine Carcinoma Management and Outcomes: A National Cancer Database Study. Oncologist. Jul 2019;24(7):911–920. doi: 10.1634/theoncologist.2018-0382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akimoto J, Fukuhara H, Suda T, et al. Clinicopathological analysis in patients with neuroendocrine tumors that metastasized to the brain. BMC Cancer. Jan 22 2016;16:36. doi: 10.1186/s12885-015-1999-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dasari A, Shen C, Devabhaktuni A, Nighot R, Sorbye H. Survival according to primary tumor location, stage, and treatment patterns in locoregional gastroenteropancreatic high-grade neuroendocrine carcinomas. Oncologist. 2022;27:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu A, Ueberroth B, McGarrah P, et al. Treatment outcomes of well-differentiated high-grade neuroendocrine tumors. Oncologist. 2021;26:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pommergaard H, Nielsen K, Sorbye H, et al. Surgery of the primary tumor in 201 patients with high-grade gastroenteropancreatic neuroendocrine and mixed neuroendocrine-non-neuroendocrine neoplasms. Journal of Neuroendocrinology. 2021;33. [DOI] [PubMed] [Google Scholar]
- 74.Tang L, Untch B, Reidy D, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clinical Cancer Research. 2016;22:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thornblade LW, Warner SG, Melstrom L, et al. Does surgery provide a survival advantage in non-disseminated poorly differentiated gastroenteropancreatic neuroendocrine neoplasms? Surgery. Jun 2021;169(6):1417–1423. doi: 10.1016/j.surg.2021.01.026 [DOI] [PubMed] [Google Scholar]
- 76.Sorbye H, Grande E, Pavel M, et al. European Neuroendocrine Tumor Society (ENETS) 2023 guidance paper for digestive neuroendocrine carcinoma. J Neuroendocrinol. Mar 2023;35(3):e13249. doi: 10.1111/jne.13249 [DOI] [PubMed] [Google Scholar]
- 77.Smith J, Reidy-Lagunes D. The management of extrapulmonary poorly differentiated (high-grade) neuroendocrine carcinomas. Semin Oncol. Feb 2013;40(1):100–8. doi: 10.1053/j.seminoncol.2012.11.011 [DOI] [PubMed] [Google Scholar]
- 78.Pavel M, Oberg K, Falconi M, et al. Gastroenteropancreatic neuroendocrine neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. Jul 2020;31(7):844–860. doi: 10.1016/j.annonc.2020.03.304 [DOI] [PubMed] [Google Scholar]
- 79.Garcia-Carbonero R, Sorbye H, Baudin E, et al. ENETS Consensus Guidelines for High-Grade Gastroenteropancreatic Neuroendocrine Tumors and Neuroendocrine Carcinomas. Neuroendocrinology. 2016;103(2):186–94. doi: 10.1159/000443172 [DOI] [PubMed] [Google Scholar]
- 80.Dasari A, Shen C, Devabhaktuni A, Nighot R, Sorbye H. Survival According to Primary Tumor Location, Stage, and Treatment Patterns in Locoregional Gastroenteropancreatic High-grade Neuroendocrine Carcinomas. Oncologist. Apr 5 2022;27(4):299–306. doi: 10.1093/oncolo/oyab039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen J, Wang A, Ji K, Bu Z, Ji J. Comparison of overall survival of gastric neoplasms containing neuroendocrine carcinoma components with gastric adenocarcinoma: a propensity score matching study. BMC Cancer. Aug 18 2020;20(1):777. doi: 10.1186/s12885-020-07281-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin J, Zhao Y, Zhou Y, et al. Comparison of Survival and Patterns of Recurrence in Gastric Neuroendocrine Carcinoma, Mixed Adenoneuroendocrine Carcinoma, and Adenocarcinoma. JAMA Netw Open. Jul 1 2021;4(7):e2114180. doi: 10.1001/jamanetworkopen.2021.14180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma F, Wang B, Xue L, et al. Neoadjuvant chemotherapy improves the survival of patients with neuroendocrine carcinoma and mixed adenoneuroendocrine carcinoma of the stomach. J Cancer Res Clin Oncol. Aug 2020;146(8):2135–2142. doi: 10.1007/s00432-020-03214-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sorbye H, Grande E, Pavel M, et al. European Neuroendocrine Tumor Society (ENETS) 2023 guidance paper for digestive neuroendocrine carcinoma. Journal of Neuroendocrinology. 2023;35 [DOI] [PubMed] [Google Scholar]
- 85.Shah MH, Goldner WS, Benson AB, et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. Jul 28 2021;19(7):839–868. doi: 10.6004/jnccn.2021.0032 [DOI] [PubMed] [Google Scholar]
- 86.Ozdirik B, Amthauer H, Schatka I, et al. A rare case of a patient with a high grade neuroendocrine tumor developing neutropenic sepsis after receiving PRRT combined with Capecitabine or Temozolomide: A case report. Mol Clin Oncol. Jan 2021;14(1):20. doi: 10.3892/mco.2020.2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Essen M, Krenning EP, Bakker WH, de Herder WW, van Aken MO, Kwekkeboom DJ. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with foregut carcinoid tumours of bronchial, gastric and thymic origin. Eur J Nucl Med Mol Imaging. Aug 2007;34(8):1219–27. doi: 10.1007/s00259-006-0355-4 [DOI] [PubMed] [Google Scholar]