

Abstract
Background:
The sympathoadrenergic system (SAS) and its major effector PKA (protein kinase A) are activated to maintain cardiac output coping with physiological or pathological stressors. If and how PKA plays a role in physiological (PhCH) and pathological (PaCH) cardiac hypertrophy are not clear.
Methods:
Transgenic mouse models expressing a PKA inhibition peptide (PKAi)-GFP fusion protein in a cardiac-specific and inducible manner (cPKAi) were used to determine the roles of PKA in PhCH during postnatal growth or induced by swimming, and in PaCH induced by transaortic constriction (TAC) or augmented Ca2+ influx. Kinase profiling was used to determine cPKAi specificity. Echocardiography was used to determine cardiac morphology and function. Western blotting and immunostaining were used to measure protein abundance and phosphorylation. Protein synthesis was assessed by puromycin incorporation and protein degradation by measuring protein ubiquitination and proteasome activity. Neonatal rat cardiomyocytes (NRCMs) infected with AdGFP or AdPKAi-GFP were used to determine the effects and mechanisms of cPKAi on myocyte hypertrophy. rAAV9.PKAi-GFP was used to treat TAC mice.
Results:
(1) cPKAi delayed postnatal cardiac growth and blunted exercise-induced PhCH; (2) PKA was activated in hearts after TAC due to activated SAS, the loss of endogenous PKIα, and the stimulation by non-canonical PKA activators; (3) cPKAi ameliorated PaCH induced by TAC and increased Ca2+ influxes and blunted NRCM hypertrophy by isoproterenol and phenylephrine; (5) cPKAi prevented TAC-induced protein synthesis by inhibiting mTOR signaling through reducing Akt activity, but enhancing inhibitory GSK-3α and GSK-3β signals; (6) cPKAi reduced protein degradation by the ubiquitin-proteasome system via decreasing RPN6 phosphorylation; (7) cPKAi increased the expression of antihypertrophic ANP; (8) cPKAi ameliorated established PaCH and improved animal survival.
Conclusions:
Cardiomyocyte PKA is a master regulator of PhCH and PaCH through regulating protein synthesis and degradation. cPKAi can be a novel approach to treat PaCH.
Keywords: Protein kinase A (PKA), physiological cardiac hypertrophy, pathological cardiac hypertrophy, mTOR signaling, protein quality control
Subject Terms: Basic Science Research, Autonomic Nervous System, Animal Models of Human Disease, Cell Signaling/Signal Transduction, Gene Therapy
Introduction
Cardiac hypertrophy (CH) is an adaption developed in response to the increased workload during physiological or pathological stress conditions to meet cardiac output demands. Physiological cardiac hypertrophy (PhCH) occurs with increasing heart size during postnatal growth, pregnancy and athletic training, whereas it does not negatively impact on contractile function and has no adverse effect over time.1 In face of physiological stress, the sympathetic-adrenergic system (SAS) activation is an essential mechanism providing short-term adaptation. Yet whether SAS and its major effector, protein kinase A (PKA), play a role in physiological cardiac hypertrophy remains unclear.2
Persistent activation of the SAS and PKA due to hypertension, myocardial infarction or valvular dysfunction exerts adverse effects on the heart. The original compensatory hypertrophic process can evolve into pathological cardiac hypertrophy (PaCH) and lead to contractile dysfunction, myocardial stiffening, interstitial fibrosis and ultimately heart failure.1 Preventing and treating PaCH have been considered as an important measure to reduce heart failure and arrhythmias in patients with cardiac stress but this need is unmet. Novel approaches to prevent PaCH are warranted.3
PKA is a major cardiac effector of the SAS system responsible for modulating a number of cellular processes in cardiovascular homeostasis and pathogenesis of heart disease by directly phosphorylating distinct protein effectors or regulating the transcription of specific genes.2 Persistent stimulation of β1-adrenergic signaling leads to overactivation of PKA and PaCH.4 PKA is also activated by some factors other than cAMP, including reactive oxygen species (ROS), angiotensin II and endothelin I, lipopolysaccharide (LPS) and inflammatory factors. These factors are referred as non-canonical PKA activators and often upregulated when the heart is stressed.2 It has been reported that PKA catalytic subunit α overexpression results in dilated cardiomyopathy.5 PKA regulatory subunit knockout often elicits PKA overactivation6, 7 or abnormal localization of PKA8 causing detrimental cardiac effects, in which reducing PKA activity could have beneficial effect9. PKA could enhance cardiomyocyte hypertrophy through regulating nuclear PKA and thus cAMP response element-binding protein (CREB)-mediated transcription of hypertrophic genes.10 In contrast, it has been reported that PKA phosphorylates HDAC5 to inhibit its translocation out of the nucleus and thus blunt cardiac hypertrophy.11 Nuclear PKA activation induces HDAC4 proteolysis to release an antihypertrophic fragment inhibiting MEF2 activity.12 More interestingly, the inhibition of PDE3 and PDE4 increases a pool of cAMP to cause cardiac hypertrophy.13 In consistent, activation of PDE2 blunted catecholamine-induced cAMP/PKA overactivation to protect the infarcted hearts.14 However, the inhibition of PDE2 increases another pool of cAMP to inhibit cardiac hypertrophy via PKA-dependent NFAT phosphorylation.13 Therefore, the roles of PKA in PaCH have been controversial and not clearly elucidated because previously cardiomyocyte specific complete PKA knockout animal models were not available.
It has been believed that different signaling cascades are distinctively responsible for the two types of cardiac hypertrophy, PhCH and PaCH. The best characterized signaling cascade mediating PhCH is the IGF1- PI3K-Akt pathway (growth factor signaling) and the classical signaling for PaCH is the overactivation of GPCR signaling.1 Both forms of cardiac hypertrophy require the activation of the mTORC/protein synthesis signaling. Yet the upstream regulators of protein synthesis during cardiac hypertrophy have not been fully characterized. As the activation of the SAS and its effector PKA are involved in both physiological and pathological hypertrophy, we hypothesize that the activation of SAS and PKA could be the common regulators of these two forms of cardiac hypertrophy. This has never been tested because of the lack of cardiac specific PKA inhibitors and the difficulty to genetically ablate all cardiac PKA catalytic subunits.15 To achieve cardiomyocyte-specific PKA inhibition, we used the PKA inhibitory domain of PKA inhibitory peptide α (PKIα) which is consisting of the N-terminus (amino acids 1–25) of PKAi and a highly specific and potent PKA inhibitor (IC50 in nM range).15, 16 It has been widely used for specifically inhibiting PKA. To stabilize this inhibitory peptide in the cell, it was fused with GFP (PKAi-GFP). Our previous study clearly showed that PKAi-GFP expressed in cardiomyocytes could effectively inhibit PKA in cellular compartments such as the membrane, SR/ER, cytosol, myofilaments and the nucleus.17, 18
To gain insights into the roles of PKA in PhCH and PaCH, we inhibited PKA activity and its activation in cardiomyocyte-specific (controlled by αMHC promoter) and inducible (by tetracycline-controlled transactivator) manner with a double transgenic (DTG) mouse model overexpressing PKAi-GFP.17 Our study showed that PKA is a master regulator of both PhCH and PaCH by regulating protein synthesis, degradation and homeostasis for the first time. Furthermore, our study showed that selective inhibition of PKA would be a novel strategy to ameliorate PaCH and to prevent ensuing heart failure.
Methods
Data Availability.
A transgenic mouse model overexpressing PKAi-GFP with a cardiac-specific and inducible system was established and the middle expression line was selected as described previously.17 Four to five-month-old sex matched (equal amount of male and female) mice were used in this study. Animal studies were approved by the Institutional Animal Care and Use Committee of Temple University and Tianjin Medical University. Single point kinase profiling for PKAi-GFP was performed by ThermoFisher. Adult mouse ventricular myocytes and neonatal rat cardiomyocytes (NRCM) were isolated as reported.17 Adult mouse left ventricular myocyte (AMLVMs) contractions and calcium transients were measured with the Ionoptix Calcium and Contractility System as described previously.18 Myocyte protein/DNA ratio and cell surface area were measured. Basal and maximal PKA activity stimulated with 1μM cAMP, and 20S proteasome activity in control and cPKAi DTG whole heart extract were assessed with commercial kits according to the manufactures’ instructions, respectively. Transverse aortic constriction (TAC) surgery was performed as described previously.19 Control and PKAi DTG mice were subjected with swimming for 3 weeks. rAAV9.cPKAi-GFP was produced and used for gene therapy for PaCH. Animal survival was determined, and heart morphology and function were followed with echocardiography. Cardiac tissue histology was performed as described in detail as previously.19 Standard Western blotting was used to determine protein abundance and phosphorylation levels. The qRT-PCR was used to detect the mRNA expression levels. Protein synthesis and degradation were analyzed by puromycin injection and ubiquitinated protein measurements, respectively. Detailed information of materials and reagents used in this study was provided in the Major Resources Table in the Supplemental Materials.
Data were examined for normality with Shapiro-Wilk test and Normal QQ plot in GraphPad prism 8.4.3 and were reported as mean±SEM. The Student’s t test was performed for comparing 2 groups. One-way or two-way ANOVA followed by post-hoc tests with Bonferroni correction was performed for multiple group comparisons. Nested and Factorial Design ANOVA was used for determining the significance between myocyte parameters with SAS 9.0 (SAS Institute Inc, Cary, NC). Kaplan-Meier analysis was used to determine survival difference with GraphPad Prism 8.4.3. Nonparametric tests such as Mann-Whitney test, Wilcoxon signed-rank test, Kruskal-Wallis test and Aligned Ranks Transformation were used for datasets with sample number below six or for non-normal datasets. A P value of <0.05 was considered significant. Detailed methodology was provided in the Online Data Supplement.
Results
1. cPKAi-GFP blocks PKA specifically
We purified a total of 3.57μg PKAi-GFP (estimated from ELISA) from 624.0mg LV tissue from 10 cPKAi-GFP medium expression DTG hearts with magnetic beads. Assuming cardiac tissue density is 1.06g/mL, 81% of cardiac tissue is myocyte volume and intracellular fluid is 60% of the wet cardiac tissue20, the estimate of PKAi-GFP (MW = 30.22KD) concentration in cardiomyocytes is 0.41μM. We purified PKAi-GFP from AdPKAi-GFP infected HEK293 cells for kinase profiling. At 0.5μM, a concentration close to estimated intracellular PKAi-GFP concentration of our medium expression mouse hearts, a single point kinase profiling for 272 kinases was done with ThermoFisher’s Z’-LYTE™ Kinase Assay Kits. As shown in Supplemental Figure 1 and Supplemental Table 1, at this concentration, only PKA (99% inhibition) and PRKX (36% inhibition; PRKX is an analog of PKA that is not abundantly expressed in the adult mouse heart21) were significantly inhibited while other kinases’ activities were not significantly changed (range: −5%~9%; mean: 0.57%; standard deviation 4.64%) (Figure S1A and Table S1). PKG activation by 8-Br-PET-cGMP in isolated cPKAi-DTG cardiomyocytes or hearts was not affected by the expression of PKAi-GFP in myocytes and was independent from the level of PKGI expression (Figure S1B-E).
2. Physiological hypertrophy is reduced by cPKAi
Four transgenic mouse lines with high (HE), medium (ME), low (LE) and very low (VLE) expression of PKAi-GFP were generated with the inhibition of 95%, 57%, 20% and 10% of maximum PKA activity induced by 1μM cAMP.18 Immunostaining showed that the expression of PKAi-GFP in VLE DTG mice was mosaic in the ventricular tissue (Figure 1A). Among cardiomyocytes isolated from these VLE hearts at the age of 4 months when the transgene expression was fully induced in the heart, some were green with PKAi-GFP expressed (PKAi-GFP+) but some were not green (PKAi-GFP−). Compared with the PKAi-GFP– VMs, PKAi-GFP+ VMs had significantly smaller surface area (Figure 1B and C). In the PKAi-GFP high expression mouse line, at the age of 4 months DTG mice had smaller heart weight to body weight ratio (HW/BW, Figure 1D and E) and myocyte surface area (Figure 1 F and G). To test if there was a dose-effect relationship between cPKAi expression and myocyte size, we isolated myocytes from a low expression mouse line and measured surface area of the myocytes. The myocytes could be categorized into three groups, fully green and medium/low green and no expression levels (Figure S2). Clearly, the myocyte size inversely correlated with the expression levels of PKAi-GFP (Figure S2B). These results showed that cPKAi could inhibit physiological myocyte growth during development.
Figure 1.

Cardiac PKAi-GFP (protein kinase A inhibitor α amino acids 1–25 fused with the green fluorescent protein) reduces ventricular myocyte developmental growth and blunts physiological cardiac hypertrophy induced by exercise. A. An immunostaining image of GFP expression in a VLE mouse heart tissue showing mosaic expression of GFP. B. Images of green (PKAi-GFP+) and nongreen (PKAi-GFP−) cardiomyocytes isolated from the same heart taken under bright light and GFP-excitation light. Myocytes were isolated from 4-month-old control and DTG mice with mosaic PKAi-GFP expression. C. Surface area of green and non-green myocytes isolated from the same hearts. D. An immunostaining image of GFP expression in a HE mouse heart tissue section showing high expression of GFP homogenously in all ventricular myocytes. E. The HW/BW ratio of 4-month-old control and PKAi-GFP high expression DTG mice. F-G. Surface areas of isolated ventricular myocytes of control and PKAi-GFP HE DTG mice. H. Control and PKAi-GFP ME DTG mice were subjected to swimming, 2 hours in the morning and 2 hours in the afternoon (totally 4 hours/day) for 3 weeks. I-L. Body weights and thickness of interventricular septum (IVS; d), left ventricular posterior (free) wall (LVPW; d) during diastole, corrected LV mass measured with echocardiography, showing increased thickness of these walls in control mice but not in TG mice subjected to exercise. M. HW/BW ratios of hearts of control and PKAi-GFP medium expression mice, showing cPKAi blunted exercise induced cardiac hypertrophy. P values were reported above the comparison lines. Animal numbers were added in the bars. Student’s t test was used in C; Mann-Whitney test was used for E and G; two-way ANOVA with post-hoc test (Bonferroni correction) was used in I through M. The scale bars in all images were 100 μm.
Furthermore, we set to determine whether cardiomyocyte PKA is required for exercise-induced PhCH. Exercise causes β-AR system activation and we suspected that PKA linked physiologically increased contractility demand with PhCH. We forced both control and PKAi-GFP ME DTG mice to swim for 3 weeks to test this hypothesis (Figure 1H). At the end of the experiment, animals showed no statistically significant differences in their body weights whether they were sedentary or swimming (Figure 1I). Echocardiography analysis showed that exercise increased the thickness of interventricular septum (IVS; d) and left ventricular posterior (free) wall (LVPW; d) during diastole, and corrected LV mass in control mice but not in PKAi-GFP DTG mice (Figure 1J through 1L). The HW/BW ratio was smaller in DTG mice than in control mice after finishing the swimming training (Figure 1M). Long-term exercise increased myocyte cross-sectional area (Figure S3A and B) and cardiac function in WT mice but not in cPKAi DTG mice (Figrue S3). Together, these data demonstrated that cPKAi blunted exercise induced PhCH.
3. PKA is activated in hearts stressed with TAC
It is well known that pathological cardiac stress would activate the SAS system. Pressure overload due to hypertension is a common cause of cardiac hypertrophy. We found that TAC-induced cardiac hypertrophy were significant at 2 weeks post pressure overload in control mice (Figure 2A). PKA activity measured by ELISA was significantly increased in the cardiac homogenates of mice subjected to TAC at two days post TAC then was less but significantly increased at 5 days after TAC. PKA activity tended to be greater in TAC hearts than that in sham-operated mice thereafter (Figure 2B). Maximum PKA activity in cardiac homogenates stimulated with 1 μmol/L cAMP was greater in mice subjected to TAC during the first 2 days but only showed a tendency of greater PKA activity thereafter (Figure 2C), which could be due to the decrease of PKIα and slightly increased PKA C subunit expression (Figure S4). Once the specificity of PKIα antibody was confirmed (Figure S5), immunostaining showed that TAC reduced PKIα in the nucleus and in the cytosol (Figure 2E).
Figure 2.

Increased PKA activity in the heart after pressure overload (TAC) due to SAS activation, noncanonical activator (ROS and AngII) stimulation, and the loss of endogenous PKIα. A. HW/BW ratio at different time points after TAC. B. Basal PKA activity in cardiac tissue homogenates of sham or TAC mice at different times after surgeries. C. Maximal PKA activity activated by 1 μM exogenous cAMP in cardiac tissue homogenates as in B. D. PKA activation by H2O2 and angiotensin II (AngII) was blocked by cPKAi. E. Immunostaining of PKIα in sham and TAC cardiac tissue sections. P values were reported above the comparison lines. Animal numbers were added in or next to the bars. Kruskal-Wallis test was used for A; aligned rank transform followed by two-way ANOVA with post-hoc (Bonferroni correction) was used for B-D. Scale bars in E were 25 μm.
The increased PKA activity in WT TAC hearts within 2 weeks of TAC could be due to strongly activated SAS system with reduced PKIα (but not PKIβ and PKIγ) and significantly increased expression of PKA catalytic subunits (Figure 2E, Figure S4). The lower level of PKA activation by TAC in mouse hearts after 2 weeks of TAC could be due to desensitization of the β-adrenergic system, less decreases of PKIα and less increases of PKA catalytic subunits (Figure S4), non-cAMP dependent ROS- and AngII-mediated low-level PKA activation (Figure 2D).2 ROS (H2O2) and angiotensin II (AngII) induced PKA activation could be abolished by PKAi-GFP in isolated mouse VMs (Figure 2D).
To probe PKA activities in different cellular compartments, PKA-dependent phosphorylation of sarcolemmal membrane protein Cavα1c, ER/SR protein phospholamban (PLB), myofilament troponin I (TnI) and the nuclear protein CREB (cAMP response element binding protein) was determined by western blotting. TAC increased phosphorylation of Cav1.2 α1c at all times examined though the increase reached significance only at 4h, 5d and 2 weeks after TAC. PLB and TnI phosphorylation in TAC hearts had similar trends in that they were greatly increased at 4 hours after TAC but significantly reduced at 2 days after TAC, then increased at 5 days again. Afterwards, there was no significant difference between sham and TAC hearts, though the phosphorylation level showed a trend of decrease in TAC hearts at 9 weeks. CREB had higher phosphorylation in TAC hearts than in sham hearts within the first 5 days post TAC but then no change afterward (Figure S6). These results suggest PKA activation in different compartments during PaCH development was regulated differently.
The activity and compartmental distribution of activated PKA C subunit can be regulated by endogenous PKA inhibitor peptides (PKIs).15 Our data showed all three PKI isoforms (PKIα, PKIβ and PKIγ) were expressed but PKIβ (both mRNA and proteins) was less abundant in adult mouse hearts. Significantly decreased expression of PKIα but not PKIβ and PKIγ was found in mouse hearts post TAC surgery (Figure S4). Immunochemical staining of PKIα in cardiac tissues showed its decrease in the cytosol but more dramatically in the nucleus in TAC mice compared to in sham operated mice (Figure 2E). These results were in consistent with a decrease of PKIα mRNA in TAC hearts at all time points (Figure S4C). This could account for differential regulation of PKA activity in different cellular compartments of cardiomyocytes in hearts stressed with TAC.
4. PKAi-GFP DTG mice have better survival, blunted cardiac hypertrophy and ameliorated cardiac function after TAC
Although it is well established that chronic stimulation of β1-AR induces PaCH,4 whether PKA plays an important role in PaCH remains elusive. We tested the role of PKA in PaCH by studying the effect of cPKAi in PaCH induced by pressure overload using the TAC model (Figure 3). Pressure gradients across the transverse aortic constriction determined with Pulse Wave Doppler were similar in control TAC mice and in PKAi-GFP medium expression DTG TAC mice (Figure 3A). Echocardiography was performed serially in control mice and DTG mice subjected to TAC weekly. Heart rates were controlled to be similar between groups with anesthesia when ECHO was done (Figure 3C). TAC significantly decreased stroke volume, ejection fraction and cardiac output; but significantly increased LV diastolic diameter, end diastolic volume, end systolic volume and LV mass in control mice but not in PKAi-GFP DTG mice (Figure 3D–J). Hemodynamically, cPKAi improved both cardiac diastolic (less end diastolic pressure and greater -dp/dt) and systolic function (greater maximum dp/dt, contractility index) after TAC (Figure S7). The benefits of cPKAi in TAC hearts could be explained by less depression of cardiomyocyte calcium transient and contraction in cPKAi TAC LV myocytes (Figure 3 M & N). At 8 weeks post TAC, cPKAi significantly reduced HW/BW ratio (Figure 3K) and lung weight to body weight ratio (Figure 3L), an index of lung edema suggesting cardiac decompensation. Analysis of cardiac tissue showed that TAC significantly increased cross-sectional area of myocytes (Figure 3O and P) and fibrosis (Figure 3Q and R) in control mice but not in PKAi-GFP DTG mice subjected to TAC. These results could explain a better survival of PKAi-GFP DTG mice after TAC (Figure 3B).
Figure 3. Medium level of PKAi-GFP expression blunts PaCH and improves cardiac function after TAC.

A. Pressure gradients across the transverse aortic constriction were similar in control TAC mice (60.5±2.5mmHg) and in PKAi-GFP/ttA DTG TAC mice (62.3±3.1mmHg). B. PKAi-GFP DTG mice survived better after TAC. When echocardiography (ECHO) was performed, the heart rates were controlled to be similar (C). TAC significantly decreased ejection fraction (D) and increased LV diastolic diameter (E) in control mice but not in DTG mice. F-I. TAC significantly increased end diastolic volume (F) and end systolic volume (G) in control mice but not in DTG mice, thus leading to a decreased stroke volume (H) and a worse cardiac output (I). J-L. At 8 weeks post TAC, DTG mice had reduced cardiac hypertrophy (corrected LV mass (J), HW/BW ratio (K) and lung weight to body weight ratio (L), an index of cardiac decompensation. M. Contractions of myocytes isolated from control and DTG hearts subjected to sham and TAC surgeries for 8 weeks. Ma, Representative cardiomyocyte contraction traces. Mb, Fractional shortening amplitudes. Mc, Time to peak. Md, Time to half relaxation. Me, maximal contraction/relaxation rate. N. Ca2+ transients of myocytes isolated from control and DTG hearts subjected to sham and TAC surgeries for 8 weeks. Na, Representative cardiomyocyte Ca2+ transients traces. Nb, Diastolic Ca2+. Nc, Systolic Ca2+. Nd, Ca2+ transient amplitudes. Ne, Time to peak. Nf, Time to half relaxation. Ng, Ca2+ transient decay time constant (Tau). O & P. Myocyte cross sectional area of papillary muscles of control and TG hearts after TAC. Q & R. Masson’s Trichrome staining of cardiac tissue of sham or TAC operated control and PKAi-GFP DTG animals. TAC significantly induced cardiac fibrosis in control mice but not in PKAi-GFP DTG mice. P values were reported above the comparison lines. Animal numbers were added in or next to the bars for A-L, P and R. The numbers next to the bars in M and N panels were myocyte numbers from 3 or 4 animals depicted in M.a. Student’s t test was used for A; the difference in survival rates was determined by Kaplan-Meier survival analysis (B); aligned rank transform followed by two-way ANOVA with post-hoc test (Bonferroni correction) was used for C-L, P and R; Nested ANOVA analyses were done for M and N pannels .
5. cPKAi alleviates increased Ca2+ influx-mediated pathological cardiac hypertrophy
It has been postulated that β-adrenergic overstimulation induces PaCH via enhancing L-type Ca2+ channel mediated Ca2+ influx, which directly regulates cardiac hypertrophic signaling.12, 22, 23 To test if cPKAi could still reduce cardiac hypertrophy induced by direct increases in Ca2+ influxes that was not regulated by PKA24, PKAi-GFP TG mice were crossbred with cardiomyocyte specific Cavβ2a/ttA DTG mice, as established previously.12 We first confirmed that PKA was not activated by increased Ca2+ influx as it did not increase the phosphorylation levels of PKA targets (Cavα1c, PLB, MyBPC3, and TnI) in Cavβ2a/ttA DTG hearts, agreeing with our previous findings19. However, cPKAi did decrease the basal level phosphorylation of some of these PKA targets (Figure S8). We found that triple TG (TTG) mice survived better than Cavβ2a DTG mice (Figure 4A). Echocardiography was performed when the heart rates were controlled to be similar (Figure 4B). cPKAi significantly reduced the dilation of the LV chamber (Figure 4C) without affecting fractional shortening (Figure 4D), ejection fraction (Figure 4E) and LV posterior wall (LVPW) thickening (an index of contractility), resulting less stroke volume (Figure 4F) and cardiac output (Figure 4H). cPKAi reduced diastolic LV posterior wall (LVPW) thickness (Figure 4J) but did not affect diastolic septum thickness (Figure 4I), causing a reduced corrected LV mass (Figure 4K) in agreement with a diminished HW/BW ratio in PKAi-GFP/Cavβ2a/ttA TTG mice (Figure 4L). As no significant deterioration of cardiac function in these mouse lines, the reduction of cardiac hypertrophy, and possible SR Ca2+ overload and associated arrhythmias25 could account for a reduction of animal mortality (Figure 4A).
Figure 4. cPKAi reduces PaCH induced by increased Ca2+ influx.
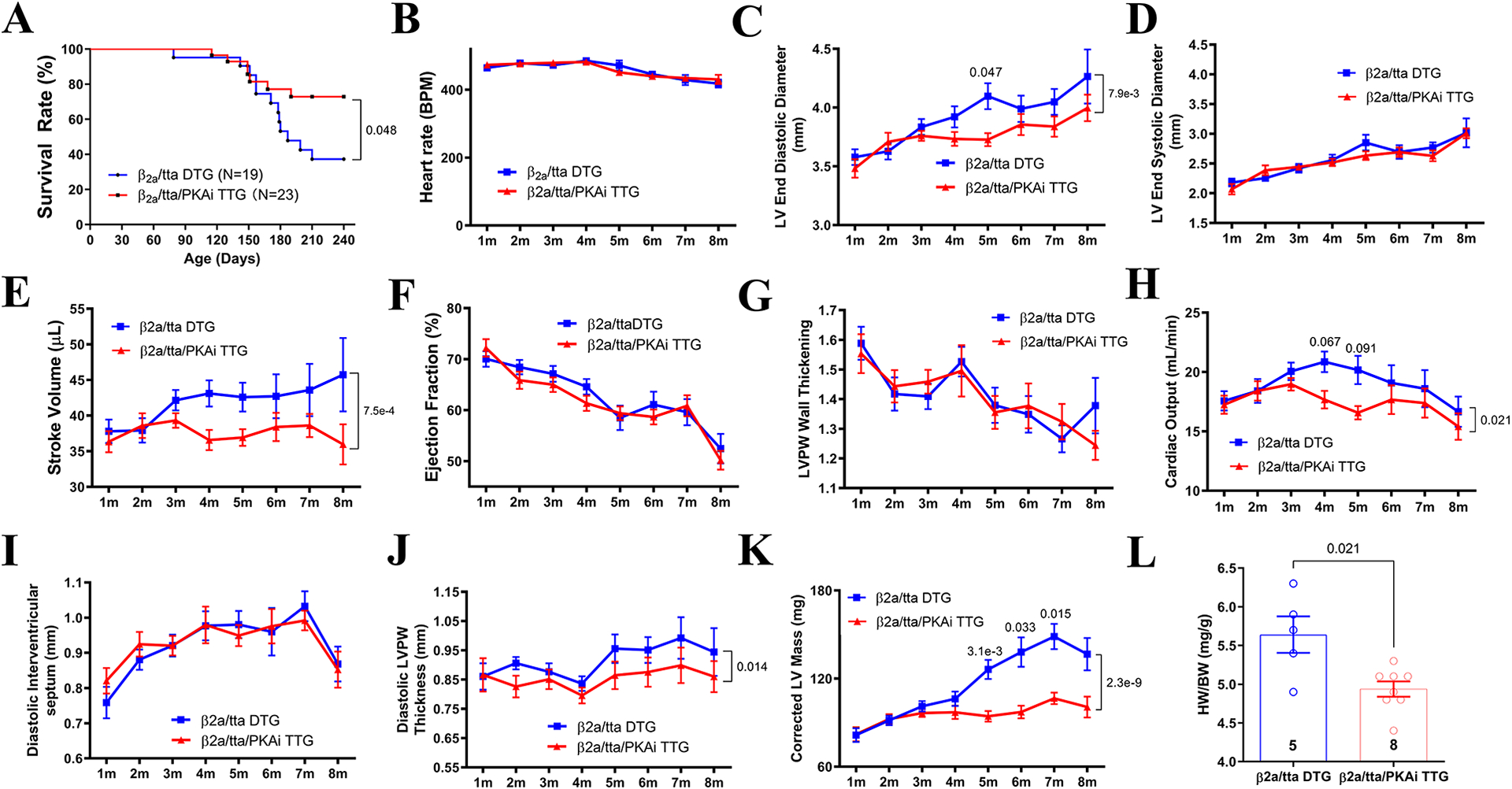
PKAi-GFP ME TG mice were crossbred with Cavβ2a/ttA DTG mice. A. Triple TG (TTG) mice survived better than Cavβ2a/ttA DTG mice. Heart rates were controlled similar between groups for ECHO (B). cPKAi significantly reduced the dilation of the LV chamber (C), without affecting LV end systolic diameter (D). cPKAi significantly blunted the dilation, leading to a decreased stroke volume (E) and cardiac output (H) without affecting LVPW wall thickening (G) and ejection fraction (F). cPKAi did not affect diastolic septum thickness (I) but blunted LVPW thickening induced by increased ICa-L (J), resulting into a reduced corrected LV mass (K). Gravimetric analysis showed a reduced HW/BW (L) by cPKAi in TTG animals euthanized at 8m of age. P values were reported above the comparison lines. Animal numbers were added in or next to the bars; animal numbers used in A-L varied as animals died during experiments. Data from these animals were not excluded. The difference in survival rates was determined by Kaplan-Meier survival analysis; two-way ANOVA with post-hoc test (Bonferroni correction) was used in C through K; Mann-Whitney test was used in L.
6. cPKAi inhibition on protein synthesis, a common mechanism for exercise-, TAC- and increased Ca2+ influx- induced cardiac hypertrophy.
As cPKAi inhibited PaCH, of which protein synthesis is critical,26 we determined if cPKAi reduced PaCH by inhibiting protein synthesis.26 De novo protein synthesis rate was quantitated by the incorporation of puromycin into nascent proteins. TAC increased protein synthesis in control mice but not in cPKAi DTG mice (Figure 5A). Numerous studies have illustrated a vital role of mTOR signaling pathway in protein synthesis1, which could be regulated by Akt, GSK-3α, GSK-3β, and PKA.27 GSK-3s are normally active to inhibit mTOR while PKA phosphorylates and inactivates them and thus activates mTOR.27, 28 Our results showed that TAC induced an increase of mTOR phosphorylation at Ser2448 and Ser2481 and thus its activation, probably by activated Akt (increased phosphorylation at Thr308 site) and inactivating inhibitory GSK-3α (increased phosphorylation of Ser21) and GSK-3β (increased phosphorylation of Ser9) (Figure 5C and D). Accordingly, downstream to mTORC1 stimulatory phosphorylation of 4E-BP1, ribosome protein S6 and p70S6K were increased while inhibitory phosphorylation of eEF2 tended to be decreased in control TAC mice (Figure S9). cPKAi reversed all these effects of TAC on protein synthesis signaling (Figure 5C & D and Figure S9). These findings demonstrated a master regulatory role of PKA in cardiac protein synthesis during TAC-induced PaCH.
Figure 5. cPKAi blunts protein synthesis induced by TAC.

A & B. De novo protein synthesis was quantitated by the incorporation of puromycin into nascent proteins (darker lanes); S: sham; T: TAC. C & D. Total and phosphorylation of key regulatory molecules involved in protein synthesis in control hearts and in PKAi-GFP DTG hearts subjected to pressure overload (TAC) or sham surgery. P values were reported above the comparison lines. Animal numbers were added in or next to the bars. Aligned rank transform followed by two-way ANOVA with post-hoc test (Bonferroni correction) was used in B; two-way ANOVA with post-hoc test (Bonferroni correction) was used in other comparisons in this figure.
As cPKAi also inhibited PaCH induced by increased Ca2+ influx that could not be regulated by PKA. Protein synthesis signaling was also determined in control, Cavβ2a/ttA DTG (increased Ca2+ influx), cPKAi/ttA DTG and cPKAi/Cavβ2a/ttA TTG hearts. mTOR phosphorylation on both S2481 and S2448 was enhanced by increased Ca2+ influx but this increase was reduced by cPKAi in cPKAi/Cavβ2a/ttA TTG. Upstream to mTOR, Cavβ2a overexpression (OE) significantly increased pS473-Akt and tended to increase pS21-GSK3ɑ and pS9-GSK3β. cPKAi on top of Cavβ2a OE did not affect GSK3s’ phosphorylation but decreased Akt phosphorylation on both T308 and T473 sites (Figure S10). These results suggested that increased Ca2+ influx could activate protein synthesis signaling that was decreased by cPKAi even though subtle signaling differences existed between increased Ca2+ influx and TAC-induced protein synthesis (e.g., pT308-Akt not increased in Cavβ2a OE model). Since in Cavβ2a overexpression did not activate PKA, our results suggested that basal PKA activity18 could play an important role in protein synthesis regulation.
As cPKAi also inhibited physiological hypertrophy, in which protein synthesis also plays an important role, we studied if exercise activated protein synthesis signaling and if cPKAi blunted that. Increased Akt/mTOR activity/phosphorylation and its downstream protein synthesis signaling were observed in control hearts after long-term swimming but not in exercised cPKAi-DTG hearts, suggesting the PKA regulated mTOR/protein synthesis in physiological hypertrophy as well (Figure S11). However, we did not see the increases in GSK-3α and GSK-3β phosphorylation. We suspect that the effects of exercise on GSK-3s were transient because the activation of SAS/β-adrenergic/PKA signaling in the exercising hearts was transient and we collected the samples after the exercise when SAS activation was already gone. It seemed that Akt/mTORC1 and its downstream signaling had sustained response to exercise as reported in cardiac and skeletal muscles after exercise29, 30. As such, we used ISO to simulate the effects of acute exercise on protein synthesis signaling and found predicted activation of protein synthesis signaling by ISO both upstream and downstream to mTOR (Figure S12). The phosphorylation/activity of a key regulator of proteasome activity, Rpn6, was also augmented by ISO in control hearts but not in cPKAi DTG hearts (Figure S12 C&D).
7. Protein degradation is reduced in DTG hearts after TAC.
Normally, when there is increased protein synthesis, there is increased protein degradation to achieve proteostasis. However, in PaCH, there is derangement of protein quality control (PQC), leading to accumulation of misfolded proteins.31 We found that ubiquitination and polyubiquitination of proteins were augmented in the hearts subjected to TAC compared to the hearts subjected to sham surgeries at 7 days and 8 weeks after surgeries. cPKAi blunted the increased ubiquitination and polyubiquitination of proteins at both times (Figure 6A and B). Furthermore, 20S proteasome activities were increased by TAC in control mice but not in PKAi-GFP DTG mice (Figure 6C). In accordance, the phosphorylation of RPN6 by PKA, a key mediator for PKA-regulation of proteasome activity,32 was decreased by cPKAi (Figure 6D).
Figure 6. Protein degradation is reduced in cPKAi hearts after TAC.

A & B. Total ubiquitination (A) and poly ubiquitination (B) of proteins was increased by TAC in control mice, which was blunted by cPKAi. C. 20S proteasome activities were increased by TAC in control mice but not in cPKAi DTG mice. D. The phosphorylation of Rpn6 by PKA, a key mediator of PKA-regulated proteasome activity, was increased by TAC in control mice but not in PKAi-GFP DTG mice. P values were reported above the comparison lines. Animal numbers were added in or next to the bars. Aligned rank transform followed by two-way ANOVA with post-hoc test (Bonferroni correction) was used in C.
8. PKAi-GFP inhibits hypertrophic stimuli-induced NRCM hypertrophy partially by increasing antihypertrophic ANP expression.
ISO and PE were applied to neonatal rat cardiomyocytes (NRCMs) with or without PKAi-GFP expression to test the role of PKA in cardiomyocyte hypertrophy. AdPKAi-GFP infected NRCMs displayed almost no increases in surface area and protein/DNA ratio after ISO or PE stimulation (Figure 7A and B, Figure S13). In PKAi-GFP overexpressing NRCMs, cPKAi DTG hearts and serum, the expression of ANP mRNA, proANP or ANP, an antihypertrophic factor, was increased even without any hypertrophic stimulation (Figure 7C–E), respectively. When ANP in the culture medium was neutralized with an antibody, the antihypertrophic effect of PKAi-GFP against PE- and angiotensin II induced hypertrophy was diminished (Figure 7F). Our results also showed that the ANP antibody could diminish the cGMP-responses of NRCM to both endogenous and exogenous ANP (Figure S14 and S15). These results indicated that cPKAi inhibits NRCM hypertrophy probably partly through enhancing antihypertrophic ANP expression. In addition, PKAi-GFP was able to blunt CaMKII activation induced by ISO, PE and AngII, even though PKAi-GFP did not block CaMKII directly shown by our kinase profiling study and previous cellular study.17
Figure 7. cPKAi inhibits cardiomyocyte hypertrophy partially through increasing antihypertrophic ANP expression.

Neonatal rat cardiomyocytes (NRCMs) were infected with AdGFP or AdPKAi-GFP and then treated with isoproterenol (ISO), phenylephrine (PE) or angiotensin II (AngII). NCRM hypertrophy induced by ISO, PE and AngII as indicated by the increases in myocyte surface area (A) and protein synthesis (protein/DNA ratio, B) was prevented by PKAi-GFP. Without hypertrophic stimulation, cPKAi increased ANP mRNA expression in NRCMs (C), pro-ANP expression in hearts of PKAi-GFP DTG mice (D) and ANP concentration in serum of PKAi-GFP DTG mice (E). F&G. The PKAi-GFP mediated antihypertrophic effect was blunted by ANP neutralizing antibody. F. The representative images of GFP positive NRCMs treated with or without ANP neutralizing antibody when challenge with hypertrophic stimuli (F). Cell images were taken with adjusted exposure time to make the image fluorescence brightness to the same to facilitate surface area measurements. The quantitation of cell surface area of NRCMs (G). P value was shown above each bracket. Aligned rank transform followed by two-way ANOVA with post-hoc test (Bonferroni correction) was used in A-C and G;Mann-Whitney test was used in D and E. The scale bars in F are 25μm. The numbers next to datasets in D & E are animal numbers used and in A, B, C, G are independent experiment times performed.
9. cPKAi reverses established PaCH.
Our study showed that cPKAi before the imposition of pressure overload could prevent PaCH. Here we determined whether cPKAi induction after the establishment of PaCH could reverse PaCH using the PKAi-GFP HE TG mice and rAAV9.PKAi-GFP mediated gene therapy. In cPKAi DTG mice, PKAi-GFP expression was induced at 5 weeks after TAC surgery via removing doxycycline at 2 weeks after TAC. cPKAi induction after TAC improved the animal survival after TAC (Figure 8A). TAC surgery had the same effect on both control and DTG mice when the PKAi-GFP did not express. After the induction of the PKAi-GFP expression, the DTG mice showed normalization of LV mass (Figure 8B) and HW/BW ratio (Figure 8E), which indicated partial reversal of PaCH by cPKAi. Furthermore, the decreased LV ejection fraction (Figure 8C), increased diastolic LV internal diameter (Figure 8D) and lung weight to body weight ratio (Figure 8F) induced by TAC in DTG mice seemed to be halted or even partially reversed after the PKAi-GFP expression, preventing cardiac decompensation.
Figure 8. cPKAi reverses PaCH induced by TAC.
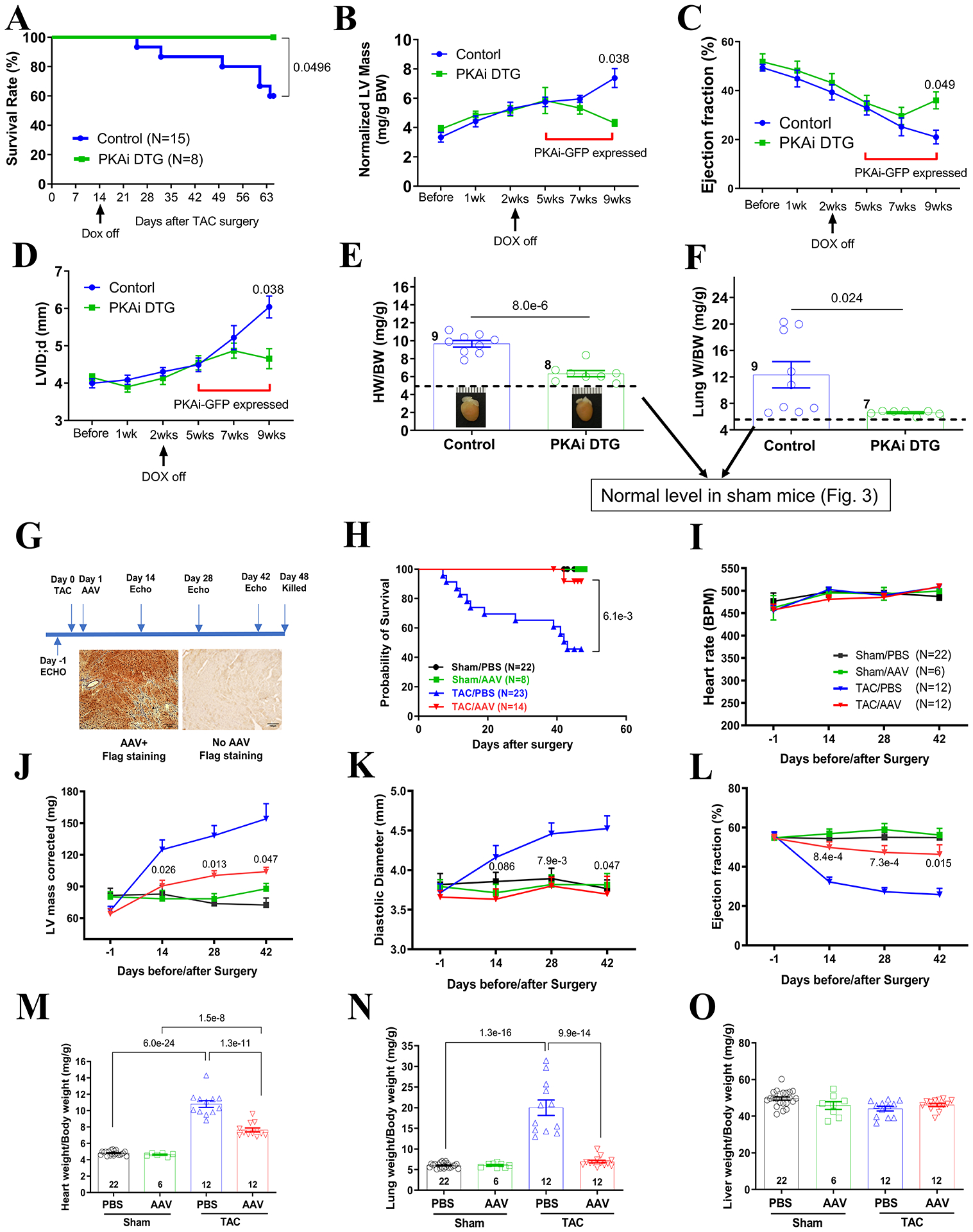
A-F. In the PKAi-GFP HE DTG mice, the expression of PKAi-GFP was kept off by feeding the mice on Dox-containing water and PaCH was induced by TAC for 2 weeks. Then PKAi-GFP expression was induced by removing the Dox-containing water. cPKAi at 5 weeks after TAC prevented animal death (A), partially reversed cardiac hypertrophy (B, LV mass determined by ECHO; E, HW/BW), and halted cardiac functional decompensation (C, D and F). G-O. AAV mediated cPKAi gene therapy to treat PaCH. G. The scheme of the gene therapy approach and PKAi-GFP expression in the heart showing almost all myocytes were expressing PKAi-GFP-6x FLAG. Animal survival was significantly improved with a single dose of AAV (H). Heart rates were similar for ECHO (I). rAAV9.PKAi-GFP treatment reduced corrected LV mass (J) and the dilation of the LV chamber (K) and improved ejection fraction after TAC surgeries (L). Gravimetric analysis showed there was a reduced HW/BW (M) and a reduced LungW/BW (N) in the rAAV9.PKAi-GFP-TAC group. There was nothing different of LiverW/BW (O) among all the groups. P value was shown above each bracket. Survival rate difference was determined by Kaplan-Meier survival analysis in A and H; two-way ANOVA with post-hoc test (Bonferroni correction) was used in B through D and I through O;Student’s t test was used in E&F. The scale bars in F are 100μm. The numbers next to datasets are animal numbers used.
We also used rAAV9 mediated gene therapy method to test the potential of using cPKAi to treat PaCH (Figure 8G). The expression of PKAi-GFP was restricted in cardiomyocytes as it was under the control of cTNT promoter. We injected a single dose of AAV9.cTNT-PKAi-GFP at 2 × 1011 vg/mouse one day after TAC. It takes about 14 days for full expression of PKAi-GFP mediated by AAV9. Animal survival was significantly improved (Figure 8H) and PaCH was significantly reduced (Figure 8 J and M). There was less cardiac dilation (Figure 8K) and functional deterioration (Figure 8L) and pulmonary edema (Figure 8N) in the rAAV9.PKAi-GFP group. These results strongly support that cPKAi via gene therapy approach was feasible.
Discussion
Cardiac hypertrophy represents an adaptive response of the heart to various physiological or pathological stress.1 In face of cardiac stress, the heart maintains cardiac output first by activating the SAS and then by hypertrophy with enlargement of myocytes, increased total protein synthesis, sarcomere addition and reorganization and thickening of the wall.1 In the present study, we provided compelling evidence for a critical role of protein kinase A (PKA) in regulating both physiological and pathological cardiac hypertrophy through activating protein synthesis and degradation signaling. In accordance, cPKAi exerted protective effects on pressure-overloaded hearts by blunting pathological hypertrophy and ameliorating cardiac function. This could be due to enhanced expression of antihypertrophic ANP signaling and better protein quality control, in addition to the prevention of myocyte Ca2+ overload, the activation of cardioprotective EPAC/ERK signaling and anti-apoptosis effects. These beneficial effects of cPKAi also worked for established PaCH, i.e., cPKAi reverses PaCH. Thus, our study provides a novel strategy for PaCH therapy.
The role of PKA in physiological cardiac hypertrophy
PKA activity is pivotal for physiological adaptation of the heart, however, it remains unknown whether PKA is necessary for physiological cardiac growth. The cardiomyocytes undergo rapid hypertrophic growth during early postnatal life. We observed that cPKAi decreased the postnatal cardiomyocyte size during the first 4 months. However, given enough time, cPKAi cardiomyocytes would reach the same size as normal cardiomyocytes.18 Our results also clearly documented that cPKAi blunted exercise-induced PhCH, highlighting a critical role of cardiomyocyte PKA activation in PhCH for the first time. It is possible that intermittent cardiomyocyte PKA activation during normal activities or exercise confers the cue for physiological cardiac growth. It is well known that IGF1-PI3K(p110α)-Akt/mTORC pathway plays an important role in physiological growth.1 Our study suggests that PKA also regulates Akt/mTORC1 signaling to regulate protein synthesis and thus these two signaling pathways could be synergistic or converging.
PKA activation in the heart after pressure overload
Prolonged β-adrenergic signaling stimulation induces PaCH.4 Here we showed that PKA was dramatically activated in the heart during the first two weeks and tended to be increased till 9 weeks after TAC. Furthermore, our results showed that PKA in different cellular compartments remained activated till different times after TAC. These findings suggest that PKA is activated to adapt to the stress by phosphorylating key molecules involved in contractility regulation to maintain cardiac output and systemic hemodynamics at least at the early stage after pressure overload.
Our results suggest that the increased PKA activity during PaCH could be due to multiple mechanisms: Firstly, the activation of the SAS system especially during early phase of stress imposition strongly activates PKA. Even after desensitization of β-adrenergic signaling, residual β-adrenergic stimulation could still cause a low level of PKA activation. Secondly, the loss of endogenous PKA inhibitory peptide (PKIα rather than PKIβ and PKIγ) could augment PKA activation, even when PKA was maximally activated by cAMP. Lastly, the increases of nonclassical PKA activators such as ROS33 and AngII34 might induce PKA activation even during late stage of PaCH, which can be inhibited by cPKAi to bring about some beneficial effects. Other nonclassical PKA activators such as TGF-β35, and endothelin-1 (ET-1)34 might play a role as well.33, 34
PKA as a master regulator of PaCH
Whether PKA contributes to PaCH remains elusive.36 Here, we illustrated that cPKAi blunted PaCH induced by pressure overload without notable functional deficit, instead with improved heart function and survival rate. cPKAi also prevented cardiomyocyte hypertrophy induced by α-adrenergic (PE) agonists using cultured NRCMs. Generally, it is accepted that α-adrenergic induction of cardiomyocyte hypertrophy is through phospholipase mediated activation PKC and Ca2+ signaling.1 Furthermore, our study demonstrated cPKAi even diminished PaCH induced by direct increase of Ca2+ influx. PKAi-GFP was very specific for PKA and it inhibited PKA activation and associated mTOR protein synthesis signaling, and induced antihypertrophic ANP in these animal models and cell culture models showing both activated PKA and basal PKA were important for cardiac growth. It also has other protective effects such as anti-Ca2+ overload and anti-arrhythmias, EPAC/ERK1/2 activation and anti-apoptosis, anti-fibrosis17. The protective effect of cPKAi on PaCH was most likely due to PKA inhibition. Our findings demonstrated an unexpected essential role of PKA in all these forms of PaCH or myocyte hypertrophy. This agrees with that global PKA-Cβ null mice are resistant to Ang II-induced cardiac hypertrophy.37
There could be multiple mechanisms accounting for the amelioration of PaCH by cPKAi: First, cPKAi could blunt the increases of Ca2+ influx, Ca2+ uptake into the SR and SR Ca2+ overload, myocyte death and arrhythmias caused by sympathetic activation during cardiac stress18. In NRCMs, PKAi-GFP blunted CaMKII activation induced by ISO, PE and AngII (Figure S13), probably through a PKA-dependent indirect Ca2+-regulated mechanism. Previously, we have clearly showed that calcium influx through Cav1.2 is a proximal signal for PaCH,12 myocyte death and fibrosis38, arrhythmias25 and heart failure38. However, it seems that preventing Ca2+ influx increases by cPKAi is only part of the reason for its blunting PaCH as cPKAi was able to reduce PaCH induced by increased Ca2+ influxes through Cav1.2 that is not regulated by PKA. cPKAi induced antihypertrophic ANP and protein synthesis inhibition could be other two beneficial mechanisms. In addition, our previous study showed that cPKAi could prevent myocyte Ca2+ overload and preserve an EPAC/ERK mediated cardiac protection to reduce cardiomyocyte death. At last, myocyte Ca2+ transients and contractions after TAC were better maintained. These benefits could account for the ameliorated cardiac remodeling and function in cPKAi DTG mice subjected to TAC.
PKA-regulated protein synthesis is essential for both physiological and pathological cardiac hypertrophy
Of note, regardless of the initial cardiac hypertrophic inducers, the increase in protein synthesis is critical for hypertrophic hearts to increase myocyte volume and cardiac mass to cope with increased cardiac stress. Cardiac hypertrophy leads to increased expression of not only specific proteins involved in hypertrophy such as ANP and BNP but also many structural and functional proteins (e.g., sarcomeric proteins), metabolic enzymes and other proteins.39 We showed that PKA regulated mTORC1 signaling is critical for regulating both physiological and pathological protein synthesis during cardiac hypertrophy. Akt acts as a positive regulator while GSK-3s are negative regulators of mTORC1 signaling.39 By specifically inhibiting cardiomyocyte PKA, it was shown that PKA was a major regulator of GSK-3α/β phosphorylation during PaCH even many kinases have been reported to phosphorylate GSK-3α/β. Our study revealed a previously unappreciated role of PKA in promoting protein synthesis in physiological and pathological hypertrophy through regulating mTORC1 in GSK3- and Akt-dependent manners, which is a unified mechanism for both PhCH and PaCH. There could be a difference in the nature of physiological and pathological stresses, of which the former is intermittent while the latter is persistent, leading to slightly different protein synthesis signaling. This difference was reflected in GSK-3s’ inactivation was not shown after finishing swimming but could be observed if simulated with β-adrenergic stimulation. Interestingly, even though cPKAi blunts protein synthesis induced by TAC, some signaling proteins were expressed at high abundance (but low phosphorylation level) in cPKAi-DTG sham and TAC hearts, suggesting a dissociated ensemble protein synthesis and specific protein synthesis or decreased degradation of these signaling molecules.
Protein quality control in PaCH
During protein synthesis, there are always misfolded or unfolded proteins. When the protein synthesis is increased in hypertrophied hearts, the UPS activity is increased to maintain protein homeostasis.31 Previous studies have shown that the development of PaCH was accompanied by upregulated proteasome activity, while targeting the ubiquitin-proteasome system (UPS) could delay PaCH progression.40 The relative decrease of protein degradation and the loss of proteostasis have been proposed to contribute to cardiac decompensation.41 Here, our data illustrated that while cPKAi blunted PaCH and prevented decompensation, it simultaneously reduced protein synthesis and degradation after TAC, probably leading to better protein quality control. PKA promotes 20S proteasome activity via the key molecule Rpn6. In this case, we are not clear if there is a feedback mechanism to coordinate protein synthesis and degradation. Here we highlight the critical role of PKA in protein quality control to maintain the protein homeostasis.
Cardiac hypertrophic growth is not necessary for hypertension adaption
It has been long believed that the acute adaptive cardiac hypertrophy is a critical process to maintain cardiovascular homeostasis in hypertension and protects the heart from stress.3 However, our data showed an unexpected finding that TAC mice survived better and had better cardiac function without cardiac hypertrophy even they were under persistent pressure overload. This could be related to better cardiomyocyte function characterized. Our study suggests that PaCH may display adverse effects in the end partially by imposing metabolic stress for increased protein synthesis. cPKAi reduces both protein synthesis and degradation to maintain proteostasis, thereby reducing excess energy consumption for futile protein synthesis. Thus, hearts have more energy to maintain cardiac contraction to maintain cardiac output, having improved cardiac function.
cPKAi as a novel strategy for PaCH treatment
PaCH greatly increases the incidence of lethal arrhythmias and heart failure, and thus becomes an independent risk factor for them1, and it calls for earlier interventions with PaCH to prevent heart failure and arrhythmias in patients. However, to date, beyond clinical agents that alleviate hypertension and other causes of PaCH, few effective drugs are available to treat PaCH directly. Thus, new prevention and treatment modalities for PaCH based on new mechanisms at the cellular and molecular levels are still needed. Our inspiring results, especially the gene therapy results, demonstrated cPKAi could reverse established PaCH, improved cardiac function and animal survival. In addition, the expression of an antihypertrophic molecule, ANP, was already induced by cPKAi without hypertrophic stimulation in both cultured NRCMs and hearts of PKAi-GFP DTG mice. This could be due to the accumulation of NFAT in the nucleus as PKA functions as a primming kinase to make NFAT phosphorylated and exported from the nucleus42.
Recently, combining neprilysin inhibitor with angiotensin receptor blocker (ARNi) was shown as an effective heart failure treatment taking advantage of the increased ANP in the blood by neprilysin inhibitor43 and it also reduced cardiac hypertrophy in hypertensive patients44. However, as neprilysin has many targets other than ANP, it has been concerned that neprilysin inhibitor may cause long-term severe side effects such as aggravating Alzheimer’s disease as neprilysin also cleaves Aβ protein45. As PKA is universally expressed in many types of cells and plays a plethora of functions, PKAi must be restricted in a cardiomyocyte specific and controllable manner, for example, using gene therapy.
It has been shown that β-blockers may reduce PaCH. However, β-blockers may confer side effects in some patients and some patients could be resistant to these drugs. cPKAi could be a surrogate or an alternative for patients irresponsive to β-blockers (e.g., due to βAR polymorphisms) or in patients where β-blockers are contra-indicated (e.g., diabetic patients, obstructive pulmonary disease patients). Moreover, selective inhibition of PKA could preserve the cardioprotective features of the SAS/β-adrenergic signaling, via protective cAMP/EPAC/ERK1/2 signaling as we previously reported.17 We have also shown that long-term cardiomyocyte PKA activity ablation doesn’t cause adverse effects in the heart.18 Our kinase profiling results showed that cPKAi was very specific for PKA inhibition. Our results also showed that the cardiac protective roles of cPKAi in PaCH, suggesting that cPKAi could be an effective PaCH therapy. cPKAi, similar to PKA catalytic subunit knockout9, could be an effective treatment for cardiac diseases/manifestations of inherited syndrome caused by the deficiency of PKA regulatory subunit. Thus, cPKAi treatment has the potential to be a novel approach as an effective alternative or a supplementary method for PaCH therapy. Nonetheless, using cPKAi to ameliorate PaCH in human beings still has a long way to go.
Conclusion
The present study shows that PKA regulates postnatal physiological cardiomyocyte growth, exercise induced PhCH, pressure-overload-induced or increased Ca2+ influx induced PaCH via unified PKA regulated protein synthesis and degradation mechanisms, though there could be subtle differences. During cardiac stress, PKA activity could be enhanced by SAS activation and/or noncanonical PKA activators, and concomitant decrease of endogenous PKIα hearts. cPKAi could prevent PKA overactivation, inhibit basal PKA activity, and induce antihypertrophic molecule expression to prevent or treat PaCH, which warrants further test in more translational settings.
Novelty and Significance.
What is known?
The sympathetic-adrenergic system (SAS) is activated to maintain cardiac output in the face of physiological and pathological cardiac stress and protein kinase A (PKA) is a major cardiac effector of the SAS.
Persistent stimulation of β1-adrenergic signaling may lead to pathological cardiac hypertrophy (PaCH).
It has been believed that different signaling cascades are distinctively responsible for physiological cardiac hypertrophy (PhCH) and PaCH, respectively.
What new information does this article contribute?
Cardiac PKA is activated by both canonical SAS/cAMP signaling and noncanonical activators including ROS, angiotensin II when the heart is stressed.
Cardiomyocyte PKA plays an essential role in both physiological and pathological cardiac growth/hypertrophy by regulating both protein synthesis and degradation.
Cardiomyocyte-specific PKA inhibition (cPKAi) via transgene or gene therapy was able to prevent or reverse adverse cardiac remodeling and PaCH, showing that cardiac hypertrophy and β-adrenergic mediated contractility enhancement are not necessary for dealing with high afterload.
SAS is initially activated to maintain cardiac output in the setting of physiological and pathological cardiac stress. However, whether its major cardiac effector PKA plays a role in PhCH and PaCH are not known due to the lack of cardiac specific PKA inhibitors and the difficulty of genetically ablating all cardiac PKA catalytic subunits in a myocyte-specific manner. We made a transgenic mouse model expressing a PKA inhibition peptide (PKAi)-GFP fusion protein in a cardiac-specific and inducible manner to achieve a functional knockout of PKA activity (cPKAi). This study reveals an essential role for cardiomyocyte PKA in physiological and pathological cardiac growth/hypertrophy. cPKAi was able to prevent or reverse PaCH to improve animal survival, cardiac function and prevent adverse cardiac remodeling in mice subjected to pressure overload. The core mechanisms include PKA regulated calcium signaling, protein synthesis, and antihypertrophic molecule expression. We found PKA activation increases both protein synthesis and the UPS system activity to maintain protein homeostasis. Furthermore, after the establishment of pathological cardiac hypertrophy, cPKAi was able to halt or reverse adverse hypertrophy, indicating cPKAi via gene therapy could be a novel approach to treat cardiac hypertrophy.
Acknowledgements
The pRPN6 antibody was gifted to Dr. X Wang’s group by Dr. Goldberg’s group.
Sources of Funding
This study was supported by the Ministry of Science and Technology of China (Grant No. 2021YFF0702103), National Natural Science Foundation of China (Grant No. 31471089 and 31671178), and NIH (Grant No. HL140071 01A1).
Nonstandard Abbreviations and Acronyms
- AMLVMs
adult mouse left ventricular myocytes
- AngII
angiotensin II
- cPKAi
cardiomyocyte-specific PKA inhibition
- CREB
cAMP response element binding protein
- DTG
double transgenic
- ER/SR
endoplasmic/sarcoplasmic reticulum
- IGF-1
insulin-like growth factor
- ISO
isoproterenol
- LV
left ventricle
- NRCM
neonatal rat cardiomyocyte
- PaCH
pathological cardiac hypertrophy
- PDE
phosphodiesterase
- PE
phenylephrine
- PhCH
physiological cardiac hypertrophy
- PKA
protein kinase A
- PKAi
PKA inhibitory domain of PKI
- PKI
PKA inhibition peptide
- PQC
protein quality control
- RPN
regulatory particle non-ATPase 6 (RPN6)
- SAS
the sympathetic/adrenergic system
- STG
single transgenic
- TAC
transaortic constriction
- TTG
triple transgenic
- UPS
ubiquitin-proteasome system
- VASP
vasodilator-stimulated phosphoprotein
Footnotes
References
- 1.Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407 [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Chen J, Fontes SK, Bautista EN, Cheng Z. Physiological and pathological roles of protein kinase a in the heart. Cardiovasc Res. 2022;118:386–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: A new therapeutic target? Circulation. 2004;109:1580–1589 [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Gareri C, Rockman HA. G-protein-coupled receptors in heart disease. Circ Res. 2018;123:716–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997–1004 [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Chen J, Xia P, Stratakis CA, Cheng Z. Loss of pka regulatory subunit 1alpha aggravates cardiomyocyte necrosis and myocardial ischemia/reperfusion injury. J Biol Chem. 2021;297:100850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin Z, Jones GN, Towns WH 2nd, Zhang X, Abel ED, Binkley PF, Jarjoura D, Kirschner LS. Heart-specific ablation of prkar1a causes failure of heart development and myxomagenesis. Circulation. 2008;117:1414–1422 [DOI] [PubMed] [Google Scholar]
- 8.Zakhary DR, Moravec CS, Stewart RW, Bond M. Protein kinase a (pka)-dependent troponin-i phosphorylation and pka regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–510 [DOI] [PubMed] [Google Scholar]
- 9.Yin Z, Pringle DR, Jones GN, Kelly KM, Kirschner LS. Differential role of pka catalytic subunits in mediating phenotypes caused by knockout of the carney complex gene prkar1a. Molecular endocrinology (Baltimore, Md.). 2011;25:1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Burmeister BT, Johnson KR, Baillie GS, Karginov AV, Skidgel RA, O’Bryan JP, Carnegie GK. Ucr1c is a novel activator of phosphodiesterase 4 (pde4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal. 2015;27:908–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG. Pka phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2010;107:15467–15472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, Olson EN. Selective repression of mef2 activity by pka-dependent proteolysis of hdac4. J Cell Biol. 2011;195:403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M. Cardiac hypertrophy is inhibited by a local pool of camp regulated by phosphodiesterase 2. Circ Res. 2015;117:707–719 [DOI] [PubMed] [Google Scholar]
- 14.Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lammle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechene P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R, El-Armouche A. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res. 2017;120:120–132 [DOI] [PubMed] [Google Scholar]
- 15.Liu C, Ke P, Zhang J, Zhang X, Chen X. Protein kinase inhibitor peptide as a tool to specifically inhibit protein kinase a. Front Physiol. 2020;11:574030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalton GD, Dewey WL. Protein kinase inhibitor peptide (pki): A family of endogenous neuropeptides that modulate neuronal camp-dependent protein kinase function. Neuropeptides. 2006;40:23–34 [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Wang WE, Zhang X, Li Y, Chen B, Liu C, Ai X, Zhang X, Tian Y, Zhang C, Tang M, Szeto C, Hua X, Xie M, Zeng C, Wu Y, Zhou L, Zhu W, Yu D, Houser SR, Chen X. Cardiomyocyte pka ablation enhances basal contractility while eliminates cardiac beta-adrenergic response without adverse effects on the heart. Circ Res. 2019;124:1760–1777 [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Nakayama H, Zhang X, Ai X, Harris DM, Tang M, Zhang H, Szeto C, Stockbower K, Berretta RM, Eckhart AD, Koch WJ, Molkentin JD, Houser SR. Calcium influx through cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011;50:460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Wilson RM, Kubo H, Berretta RM, Harris DM, Zhang X, Jaleel N, MacDonnell SM, Bearzi C, Tillmanns J, Trofimova I, Hosoda T, Mosna F, Cribbs L, Leri A, Kajstura J, Anversa P, Houser SR. Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ Res. 2007;100:536–544 [DOI] [PubMed] [Google Scholar]
- 21.Li W, Yu ZX, Kotin RM. Profiles of prkx expression in developmental mouse embryo and human tissues. J Histochem Cytochem. 2005;53:1003–1009 [DOI] [PubMed] [Google Scholar]
- 22.Houser SR, Molkentin JD. Does contractile ca2+ control calcineurin-nfat signaling and pathological hypertrophy in cardiac myocytes? Sci Signal. 2008;1:pe31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: Role of calcineurin/nfat as ca2+ signal integrators. Proc Natl Acad Sci U S A. 2008;105:2859–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang M, Zhang X, Li Y, Guan Y, Ai X, Szeto C, Nakayama H, Zhang H, Ge S, Molkentin JD, Houser SR, Chen X. Enhanced basal contractility but reduced excitation-contraction coupling efficiency and beta-adrenergic reserve of hearts with increased cav1.2 activity. Am J Physiol Heart Circ Physiol. 2010;299:H519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Ai X, Nakayama H, Chen B, Harris DM, Tang M, Xie Y, Szeto C, Li Y, Li Y, Zhang H, Eckhart AD, Koch WJ, Molkentin JD, Chen X. Persistent increases in ca(2+) influx through cav1.2 shortens action potential and causes ca(2+) overload-induced afterdepolarizations and arrhythmias. Basic Res Cardiol. 2016;111:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89:1401–1438 [DOI] [PubMed] [Google Scholar]
- 27.Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mtorc1 and mtorc2. Physiol Rev. 2021;101:1371–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang X, Yu SX, Lu Y, Bast RC Jr., Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase a. Proc Natl Acad Sci U S A. 2000;97:11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Figueiredo VC. Revisiting the roles of protein synthesis during skeletal muscle hypertrophy induced by exercise. Am J Physiol Regul Integr Comp Physiol. 2019;317:R709–R718 [DOI] [PubMed] [Google Scholar]
- 30.Ma Z, Qi J, Meng S, Wen B, Zhang J. Swimming exercise training-induced left ventricular hypertrophy involves micrornas and synergistic regulation of the pi3k/akt/mtor signaling pathway. Eur J Appl Physiol. 2013;113:2473–2486 [DOI] [PubMed] [Google Scholar]
- 31.Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14:637–653 [DOI] [PubMed] [Google Scholar]
- 32.VerPlank JJS, Lokireddy S, Zhao J, Goldberg AL. 26s proteasomes are rapidly activated by diverse hormones and physiological states that raise camp and cause rpn6 phosphorylation. Proc Natl Acad Sci U S A. 2019;116:4228–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haushalter KJ, Schilling JM, Song Y, Sastri M, Perkins GA, Strack S, Taylor SS, Patel HH. Cardiac ischemia-reperfusion injury induces ros-dependent loss of pka regulatory subunit rialpha. Am J Physiol Heart Circ Physiol. 2019;317:H1231–H1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dulin NO, Niu J, Browning DD, Ye RD, Voyno-Yasenetskaya T. Cyclic amp-independent activation of protein kinase a by vasoactive peptides. J Biol Chem. 2001;276:20827–20830 [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Duan CJ, Binkley C, Li G, Uhler MD, Logsdon CD, Simeone DM. A transforming growth factor beta-induced smad3/smad4 complex directly activates protein kinase a. Mol Cell Biol. 2004;24:2169–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray AJ. Pharmacological pka inhibition: All may not be what it seems. Sci Signal. 2008;1:re4. [DOI] [PubMed] [Google Scholar]
- 37.Enns LC, Bible KL, Emond MJ, Ladiges WC. Mice lacking the cbeta subunit of pka are resistant to angiotensin ii-induced cardiac hypertrophy and dysfunction. BMC Res Notes. 2010;3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mtor signaling in the cardiovascular system. Circ Res. 2018;122:489–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drews O, Taegtmeyer H. Targeting the ubiquitin-proteasome system in heart disease: The basis for new therapeutic strategies. Antioxid Redox Signal. 2014;21:2322–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ Jr., Rothermel BA, Hill JA. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shou J, Jing J, Xie J, You L, Jing Z, Yao J, Han W, Pan H. Nuclear factor of activated t cells in cancer development and treatment. Cancer Lett. 2015;361:174–184 [DOI] [PubMed] [Google Scholar]
- 43.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR, Investigators P-H, Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004 [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto K, Rakugi H. Angiotensin receptor-neprilysin inhibitors: Comprehensive review and implications in hypertension treatment. Hypertens Res. 2021;44:1239–1250 [DOI] [PubMed] [Google Scholar]
- 45.Bozkurt B, Nair AP, Misra A, Scott CZ, Mahar JH, Fedson S. Neprilysin inhibitors in heart failure: The science, mechanism of action, clinical studies, and unanswered questions. JACC Basic Transl Sci. 2023;8:88–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F [DOI] [PubMed] [Google Scholar]
- 47.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207 [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Zhao M, Shi Q, Xu B, Zhu C, Li M, Mir V, Bers DM, Xiang YK. Monoamine Oxidases Desensitize Intracellular beta(1)AR Signaling in Heart Failure. Circ Res. 2021;129:965–967. doi: 10.1161/CIRCRESAHA.121.319546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lokireddy S, Kukushkin NV, Goldberg AL. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015;112:E7176–7185. doi: 10.1073/pnas.1522332112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. Faseb j. 2011;25:1028–1039. doi: 10.1096/fj.10-168799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A transgenic mouse model overexpressing PKAi-GFP with a cardiac-specific and inducible system was established and the middle expression line was selected as described previously.17 Four to five-month-old sex matched (equal amount of male and female) mice were used in this study. Animal studies were approved by the Institutional Animal Care and Use Committee of Temple University and Tianjin Medical University. Single point kinase profiling for PKAi-GFP was performed by ThermoFisher. Adult mouse ventricular myocytes and neonatal rat cardiomyocytes (NRCM) were isolated as reported.17 Adult mouse left ventricular myocyte (AMLVMs) contractions and calcium transients were measured with the Ionoptix Calcium and Contractility System as described previously.18 Myocyte protein/DNA ratio and cell surface area were measured. Basal and maximal PKA activity stimulated with 1μM cAMP, and 20S proteasome activity in control and cPKAi DTG whole heart extract were assessed with commercial kits according to the manufactures’ instructions, respectively. Transverse aortic constriction (TAC) surgery was performed as described previously.19 Control and PKAi DTG mice were subjected with swimming for 3 weeks. rAAV9.cPKAi-GFP was produced and used for gene therapy for PaCH. Animal survival was determined, and heart morphology and function were followed with echocardiography. Cardiac tissue histology was performed as described in detail as previously.19 Standard Western blotting was used to determine protein abundance and phosphorylation levels. The qRT-PCR was used to detect the mRNA expression levels. Protein synthesis and degradation were analyzed by puromycin injection and ubiquitinated protein measurements, respectively. Detailed information of materials and reagents used in this study was provided in the Major Resources Table in the Supplemental Materials.
Data were examined for normality with Shapiro-Wilk test and Normal QQ plot in GraphPad prism 8.4.3 and were reported as mean±SEM. The Student’s t test was performed for comparing 2 groups. One-way or two-way ANOVA followed by post-hoc tests with Bonferroni correction was performed for multiple group comparisons. Nested and Factorial Design ANOVA was used for determining the significance between myocyte parameters with SAS 9.0 (SAS Institute Inc, Cary, NC). Kaplan-Meier analysis was used to determine survival difference with GraphPad Prism 8.4.3. Nonparametric tests such as Mann-Whitney test, Wilcoxon signed-rank test, Kruskal-Wallis test and Aligned Ranks Transformation were used for datasets with sample number below six or for non-normal datasets. A P value of <0.05 was considered significant. Detailed methodology was provided in the Online Data Supplement.