

Abstract
The unique palindromic inverted terminal repeats (ITRs) and single-stranded nature of adeno-associated virus (AAV) DNA are major hurdles to current sequencing technologies. Due to these characteristics, sequencing noncanonical AAV genomes present in AAV vector preparations remains challenging. To address this limitation, we developed thorough molecule configuration analysis of noncanonical AAV genomes (TMCA-AAV-seq). TMCA-AAV-seq takes advantage of the documented AAV packaging mechanism in which encapsidation initiates from its 3′ ITR, for AAV-seq library construction. Any AAV genome with a 3′ ITR is converted to a template suitable to adapter addition by a Bst DNA polymerase-mediated extension reaction. This extension reaction helps fix ITR heterogeneity in the AAV population and allows efficient adapter addition to even noncanonical AAV genomes. The resulting library maintains the original AAV genome configurations without introducing undesired changes. Subsequently, long-read sequencing can be performed by the Pacific Biosciences (PacBio) single-molecule, real-time (SMRT) sequencing technology platform. Finally, through comprehensive data analysis, we can recover canonical, noncanonical AAV DNA, and non-AAV vector DNA sequences, along with their molecular configurations. Our method is a robust tool for profiling thorough AAV-population genomes. TMCA-AAVseq can be further extended to all parvoviruses and their derivative vectors.
Keywords: adeno-associated virus, AAV, TMCA-AAVseq, noncanonical AAV genomes, molecular configuration, library construction
Graphical abstract
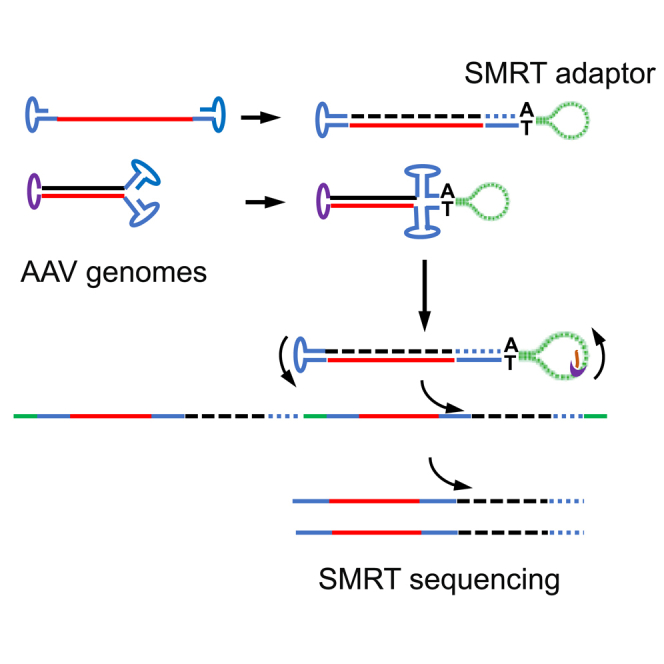
Zhang and colleagues address the sequencing challenges presented by noncanonical AAV genomes, which lack corresponding counterpart molecules for annealing. Using 3′ ITR initiation in AAV encapsidation, they efficiently construct libraries compatible with the SMRT sequencing platform, offering a robust tool for comprehensive AAV-population genome profiling.
Introduction
Recombinant adeno-associated virus (rAAV) vectors offer a promising tool for therapeutic transgene delivery. Products based on AAV vectors are part of the market-approved drugs and not only approved for human gene therapy.1,2,3,4,5,6,7 Recent studies have raised concerns regarding the safety of AAV-mediated gene therapy.8 AAV induces hepatocellular carcinoma (HCC) in diabetic and obese mice dependent on the Pebp1 pathway.9 Also, clonal expansions of transduced liver cells have been reported in a long-term study of AAV gene therapy in dogs with hemophilia A.10 In clinical trials, a patient treated with AAV for 1 year was diagnosed with HCC,8 although later it was stated that the HCC case reported in the Hope B trial was unrelated to the treatment using AAV vectors.11 Noncanonical AAV genomes in rAAV vector preparations are implicated in many undesired biological properties in cell or animal models.12,13,14 It is essential to comprehensively characterize the entire AAV genome population to assess vector quality. Understanding the rAAV population will help improve AAV vector quality control and provide a strong base to design safer and more efficient vectors for gene therapy.
The typical AAV genome is a single-stranded DNA (ssDNA) molecule flanked by two inverted terminal repeats (ITRs). AAV ITRs are highly GC rich (∼70%) and assume a T-hairpin structure.15 The palindromic structure of the ITR within the ssDNA genome presents significant challenges in fully characterizing AAV genome populations.16,17 Popular sequencing platforms such as Illumina NGS and PacBio’s single-molecule, real-time (SMRT) sequencing technology necessitate double-stranded DNA (dsDNA) for sequencing purposes.18,19 Therefore, ssDNA molecules packaged in AAV capsid must be converted into dsDNA before sequencing. Helicos and Nanopore platforms have been developed to study AAV genomes.20,21,22 The Helicos system is limited by its short reads and cannot produce the long reads needed for assessing AAV genome heterogeneity. Nanopore sequencing has been limited by base calling accuracy at the single-molecule level, which contributes to notable incidences of miscalled bases and false indels. Since assessing the heterogeneity of rAAV vectors requires both long read and efficient ITR sequencing, SMRT sequencing provides significant advantages over both Nanopore and Illumina platforms.
In this study, we present a thorough AAV genome sequencing protocol and analysis pipeline to systematically characterize the molecular state of rAAV genomes at the single-vector level. Leveraging the PacBio SMRT platform, our protocol integrates Bst DNA polymerase and A-tailing DNA end modification. This innovative approach effectively surmounts challenges posed by hairpin structures, enabling successful sequencing of wild-type AAV virus and rAAV vectors encompassing normal, partial, and oversized genomes and various other parvoviruses. Profiles obtained through this method reveal the complete population of AAV genomes in the preparation.
Procedure
A scheme of the workflow is presented in Figure 1.
Figure 1.

Flowchart of the library preparation and subsequent sequencing by TMCA-AAVseq
The process commences with the extraction of DNA from both normal and noncanonical vectors. Subsequently, second-strand synthesis is carried out from the 3′ ITR, followed by end repair and modification. Next, adapters designed for SMRT sequencing are ligated with the library DNA molecules. In the accompanying chart, original AAV DNA strands are represented by solid red lines, and newly synthesized strands are depicted with dashed black lines. Normal ITRs are illustrated in blue, and mutant ITRs are shown in pink. The ultimate output of this process comprises high-quality CCSs, ensuring a minimum quality threshold of 0.99 and a minimum of 3 passes. Finally, AAV genome sequence and configuration were retrieved. scAAV genomes were not tested here.
The thorough molecule configuration analysis of noncanonical AAV genomes-sequencing (TMCA-AAVseq) workflow includes (1) DNA extraction from ssAAV vector populations, (2) second-strand synthesis, (3) library preparation, (4) sequencing based on PacBio-SMRT technology, and (5) data analysis using our dedicated bioinformatics pipeline (Figure 2).
Figure 2.

Overview of data analysis using the TMCA-AAVseq protocol
Starting from library preparations of SMRTbell DNA templates (as shown in Figure 1), through SMRT sequencing, to computational analysis with the <TMCA-AAVseq_Blast.py> script, which uses the BlastN algorithm for alignment. Iterative alignment refinement is depicted in the blue box, where the process extracts and aligns the best-matching segments for consecutive rounds of comparison. This iterative approach enables accurate mapping of subgenomic configurations, as indicated in the orange box, showcasing the primary configurations within the AAV vector genome library. For instance, with LOOP = 4, sequences from L1-1 to L3-8 are generated, along with their corresponding alignment results. These alignments allow for the HiFi reads to be precisely matched to reference loci. M denotes segments aligned in the current round, and L∗ represents segments not aligned. Unaligned segments L∗ are carried forward for alignment in subsequent rounds. This can continue recursively to provide a detailed overview of alignment status and subgenome configurations. scAAV genomes were not tested here.
DNA extraction from ssAAV vector preparations primarily exists in dsDNA form, so before second-strand synthesis, DNA was first denatured at 95°C for 5 min and then immediately cooled on ice. Subsequently, Bst 2.0 polymerase was added to the denatured mixture for second-strand synthesis (step 2, Figure 1). The extension product was then purified and end modified by A-tailing using Klenow fragment (3′→5′ exo). Then, dsDNA with blunt/A end are ligated with blunt/A adapters to generate library preparation. The AAV genome library preparation was then subjected to SMRT sequencing. A high-quality library preparation is crucial for the success of next-generation sequencing (NGS), ensuring uniform representation of the original sample. The detailed library preparation procedures are as follows, along with troubleshooting guidance outlined in Table 1.
Table 1.
Dataset and troubleshooting
| Sample dataset | Production method | Platform | # CCS 3 | No. of aligned reads | # Total time, h | # SUM |
|---|---|---|---|---|---|---|
| AAV2-CB-EGFP (4,316 bases) | Triple-plasmid transfections | RSII | 19,494 | 19,369 | ∼0.8 | 83.8 |
| AAV2-CB-Luc (3,152 bases) | Sequel | 155,891 | 155,058 | ∼5.8 | 93.2 | |
| AAV8-hAAT-hLC (4,003 bases) | Sequel | 231,057 | 110,632 | ∼5.5 | 89.6 | |
| AAV8-TTR-hFVIII (5,123 bases) | Sequel | 55,300 | 52,470 | ∼2.6 | 53.6∗ | |
| AAV8-β-actin-hFVIII (4,993 bases) | Sequel | 154,403 | 152,774 | ∼6.0 | 83.0 | |
| AAV2-CMV-Luc (3,153 bases) | Sequel | 154,120 | 29,890 | ∼1.5 | 81.9 | |
| AAV2-hM4D.b (5,133 bases) | Baculovirus system | Sequel | 178,313 | 29,258 | ∼1.5 | 69.1∗ |
| AAV2-Luc.b (3,153 bases) | Sequel | 257,458 | 25,000 | ∼1.0 | 90.1 |
| Troubleshooting step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2–1 | Inefficiency of DNase digestion | Insufficient incubation or low concentration | Increase the length of incubation or DNase concentration. |
| 2–3 | Low DNA extraction and purity | 1. Incomplete lysis of capsid 2. Residual phenol or ethanol |
1. The addition of Proteinase K is recommended in lysis buffer for complete lysis of AAV capsid 2. Further purification of DNA extraction by PCR purification kit |
| 3–1 | Inefficiency of DNA extension Poor quality of extension product |
1. High DNA amount (>1 μg) 2. High incubation temperature (>60°C) 3. Insufficient incubation time (<30 min) |
1. No more than 1 μg DNA/100 μL should be used in the reaction system 2. Incubation can go no higher than 60°C 3. Increase the length of incubation to 1 h |
| 3–3 | Inefficiency of ligation | 1. Blunt-end ligation 2. Insufficient incubation time for ligation |
1. Modify DNA end by tailing A 2. Apply sticky end adapters 3. Increase the length of incubation for ligation to at least 3 h or overnight at room temperature |
| 4 | Low amount or poor-quality DNA library product | 1. Vortex time is too long (>10) during AMPure bead purification, damaging the DNA 2. There are residual beads in the final library product 3. Bead pellet has been overdried before elution |
1. Quick flick or brief vortex to mix the bead solution and library sample 2. To completely remove residual beads in the final product after adding elution by repeating the following step 1–2 times: place the tube back in the magnetic bead rack until the solution clears, and then slowly transfer the supernatant into a new clean tube 3. To avoid overdrying and cracking the bead pellet, air dry for ∼2 min |
| 6–4 | <TMCA_Rearrange.sh>: discarding the invalid data using such VBA code is time-consuming | If processing a large dataset | Scripts could be optimized and parallelized using multiple CPUs |
| 6–5 | <TMCA_Rearrange.sh>: detection of main configurations are calculated with low ratio | The complexity of foreign sequencing integration exists, as well as secondary combinations | We strongly recommend using appropriate online searching analysis tools |
| 6–6 | <TMCA_Visualization.sh>: some defined configuration is detected with low abundance | The number of viral sequences detected in 1 sample is not exactly a real virus abundance, because of sequencing bias | Subsample varying length filtering is needed before format grouping |
# CCS_3: HiFi reads with an accuracy of >99% and with passes ≥3; # total time, h: for processing aligned reads, after Minimap2 alignment process, including BLAST and visualization; # SUM: the overall ratio of AAV genome molecules, including those with full, SBG, ICG, and GDM configurations, as well as their secondary derivative genomes, in the sequencing reads. SUMs of AAV-TTR and AAV-hM4D.b are low (∗), and further details can be found in the limitations section.
AAV vector production and purification
-
(1)
AAV vectors subjected to sequencing are listed in Table 1. AAV2-hM4D.b and AAV2-Luc.b were ordered from Virovek (Houston, TX). AAV2-CB-EGFP, AAV2-CB-Gluc, AAV8-hAAT-hLC, AAV-TTR-hFVIII, AAV8-β-actin-hFVIII, and AAV2-CMV-Cluc were produced by a triple plasmid transfection method in adherent HEK293 cells in our lab. All of the constructs used for AAV vector production by triple plasmid transfection are from our lab stock. The AAV helper plasmid DNA harbors rep and cap coding sequences. The adenovirus helper plasmid DNA harbors the E2A, E4, and VA RNA regions. The AAV vector plasmid DNA harbors a transgene expression cassette flanked by ITRs. AAV helper, adenovirus helper, and transgene plasmids were cotransfected into HEK293 cells via PolyJet transfection reagent. At 72 h after transfection, AAV vectors were harvested and purified by CsCl density gradient ultracentrifugation, as described previously.23,24 The final vector genome titers were determined by quantitative real-time PCR as vector genomes per milliliter (vg/mL).
AAV DNA extraction
-
(1)
Treat 50 μL (1 × 1013 vg/mL) of viral vectors with DNase I (1 U/mL) and incubate for 30 min at 37°C.
-
(2)
Add 1 μL of 0.5 M EDTA (final concentration of 5 mM) and subsequently heat for 20 min at 85°C.
-
(3)
Add 1/2 vol of lysis buffer containing Proteinase K (40 μg/mL) and incubate for 1 h at 56°C. Then, heat for 10 min at 95°C. Add 1 vol of phenol:chloroform:isoamyl alcohol (25:24:1). Vortex thoroughly for ∼20 s. Centrifuge at 16,000 × g for 30 min at 4°C to remove debris.
-
(4)
Carefully transfer the supernatant to a fresh tube. Add 200 μL of 70% ethanol and centrifuge at 16,000 × g for 10 min at 4°C. Collect the pellets and dry in the air at room temperature.
-
(5)
Dissolve DNA in 20 μL Tris-EDTA buffer. Measure DNA concentration using a Nanodrop One spectrophotometer.
Sequencing library preparation
For long-read PacBio SMRT sequencing, rAAV samples were prepared according to SMRTbell procedures. To sequence the AAV vector DNA at the single-virus level, single-stranded vector DNA was first converted to duplex DNA by using AAV ITR as the extension primer with DNA polymerase.
-
(1)
DNA extension: denature 20 μL of 600 ng DNA in 10 μL of 10× ThermoPol Buffer system with 6 μL of 100 mM MgSO4, and 49 μL of double-distilled H2O (ddH2O) at 95°C for 5 min and immediately cool it in an ice-water bath for 10 min. Add 10 μL of 100 nM deoxynucleotide triphosphates (dNTPs), 4 μL Bst 2.0 polymerase, and 1 μL BSA 2.0 (10 mg/mL). Incubate the mixture for 1 h at 50°C. Withdraw 5 μL of the DNA extension product and subject it to electrophoresis on an alkaline gel to evaluate the extension efficiency. Purify the remaining extension product using the GeneJET Gel Extraction Kit for subsequent steps.
-
(2)
End repair: treat the extension product with DNA Polymerase I large (Klenow) fragment to generate blunt ends. DNA should be dissolved in 1× NEBuffer supplemented with 33 μM each dNTP. Add 1 U of Klenow per microgram DNA, and then incubate for 20 min at room temperature. Finally, purify DNA using the GeneJET Gel Extraction Kit.
-
(3)
Add A-tailing: mix the following components in a sterile tube: 30 μL of 1 μg DNA, 5 μL 10× NEB buffer, 1 μL 100 nM dATP, 3 μL Klenow fragment (3′→5′ exo-), and 11 μL ddH2O. Incubate the mixture for 30 min at 37°C. Purify DNA using the GeneJET Gel Extraction Kit.
-
(4)
Ligate T-tailing adapters: mix 30 μL of 600 ng DNA. Add A-tailing with 50 μL 10× Blunt/TA Ligase Master Mix, 2 μL overhang T-tailing adapter (100 μM), and 18 μL of ddH2O, in a total of 100 μL of a reaction system and incubate the mixture for 12 h at room temperature. Purify the ligation product with GeneJET Gel Extraction Kit.
-
(5)
Exonuclease treatment: mix 30 μL of 1 μg ligated DNA with 5 μL 10× Cutsmart buffer, 1 μL Exonuclease III, 1 μL Exonuclease VII, and 13 μL of ddH2O. Incubate the mixture for 60 min at 37°C.
Sequencing library purification
Purify the ligation product by implementing the AMPure PB bead size-selection protocol (https://www.pacb.com/wp-content/uploads/Procedure-Checklist-%E2%80%93-Using-AMPure-PB-Beads-for-Size-Selection.pdf). Warm the AMPure beads to room temperature and mix thoroughly before use. (All of the AMPure PB bead purification steps should be performed at room temperature.) Add 100 μL AMPure PB beads to 100 μL of the ligation product, quick flick to mix or brief vortex to mix, and then let it sit, with an occasional flick to mix.
-
(1)
Quickly spin the tube for 1 s to collect the beads.
-
(2)
Incubate samples on the bench top for 5 min at room temperature.
-
(3)
Spin the tube for 1 s to collect beads.
-
(4)
Place the tube on a bead rack to separate beads from supernatant.
-
(5)
Slowly transfer the clear supernatant to a new tube. Do not disturb the beads.
-
(6)
Add a sufficient volume of freshly prepared 70% ethanol to the tube while in the bead rack (1.5 mL for a 1.5-mL DNA LoBind tube). Slowly dispense the 70% ethanol to wash the beads by turning the tube 180° and allowing the beads to re-collect on the side of the tube. Do not disturb the beads. Incubate at room temperature for 30 s and then pipette and discard the ethanol.
-
(7)
Repeat step 6 once.
-
(8)
Spin the tube and place back in the bead rack. Completely remove the residual ethanol.
-
(9)
Add 50 μL elution buffer volume to the beads.
-
(10)
Mix well via quick flick or brief vortex to elute DNA, and then let it sit for 10 min.
-
(11)
Spin the tube and place the tube back in the magnetic bead rack until the solution clears.
-
(12)
Slowly transfer the supernatant to a clean tube. The supernatant contains the purified DNA library.
-
(13)
Perform a second round of purification by repeating steps 1–8.
-
(14)
Add 10 μL elution buffer volume to the beads and repeat steps 10–12.
-
(15)
Measure the total amount and concentration of DNA by Nanodrop One spectrophotometer for library submission.
-
(16)
Verify the DNA concentration by Qubit for loading the library. An optimal DNA concentration of ∼30 ng/μL is preferable for subsequent sequencing procedures. In general, step 16 can be completed by the sequencing service provider.
-
(17)
Anneal sequencing primer and bind Sequel DNA Polymerase to the SMRTbell library. All SMRT sequencing reactions are performed on the PacBio RSII/Sequel System.
TMCA-AAVseq primary data analysis
TIMING: including circular consensus sequence (CCS) run time (set multithreads = –10, 20,000–25,000 reads/h used for processing HiFi reads in the datasets; Table 1).
-
(1)
Raw data must first be processed using the SMRT Link User Guide and CCS analysis performed for additional guidance for this step. Use recalladapters (with disableAdapterCorrection) and CCS analysis (predicted min-rq = 0.99, minPasses = 3) to obtain high-quality sequencing data for further analysis, <Preparation.sh> STEP 2.
recalladapters -s <subreadset.xmL> -o OUTPUT_subread.bam --disableAdapterCorrection --adapters <adapters.fasta>
ccs --minPasses 3 --min-rq 0.99 --report-file report.txt out_subread.bam outccs3.bam
samtools view outccs3.bam | awk '{OFS="∖t"; print ">"$1"∖n"$10}'> outccs3.fasta
CRITICAL STEP: This will generate your output HiFi FASTA file in the following format: outccs3.fasta
-
(2)
(Optional) If your library is multiplexed, demultiplex barcodes.
-
(3)
(Optional) Obtain the status of the HiFi reads and produce size distribution.
seqkit stats outccs.fasta
seqkit fx2tab -l -n -i -H outccs3.fasta > ccs3len.txt
-
(4)
Store the processed data in the LINUX working directory.
TMCA-AAVseq mapping data analysis
TIMING: ∼1.5–7.5 h, for processing HiFi reads in the datasets (Table 1).
-
(1)
Download the outccs3.fasta in the working directory. Mapping to reference genome by running the following command25: <Preparation.sh> STEP 3. ref.fasta is the FASTA file of your reference sequence. TIMING: within 30 min
minimap2 -d ref.min ref.fasta
minimap2 -ax map-pb ref.min outccs3.fasta > all.sam
samtools sort -@ "$NUM_THREADS" -O bam -o all.sorted.bam all.sam
samtools index all.sorted.bam
samtools faidx ref.fasta
samtools view -bF "$NUM_THREADS" all.sorted.bam > all.F.sorted.bam
samtools fasta all.F.sorted.bam > all.F.fasta
-
(2)
BLAST-based alignments. Run the BLAST by typing the STEP 4 commands in the <Preparation.sh>. ref.fasta and all.F.fasta input files are required.
TIMING: ∼50,000–250,000 reads/h
Note that users first need to choose the loop number.
read -p "number of loop:" num
The output reports in this protocol are processed in the five loop alignments.
? TROUBLESHOOTING
-
(3)
(Optional) Present the size distribution. Load the Flen.txt in the R working directory (“../work_directory”) in Windows.
library(ggplot2)
histogram_plot <- ggplot()
Please note that the adjustment of parameters, such as “bins,” is linked to the status of aligned sequencing data.
-
(4)
Visualization of alignments. Load the BLAST alignment result output files in the working directory “../work_directory/t”). <b∗> input files are required.
TIMING: ∼25,000–30,000 reads/h
setwd("../work_directory/t")
CRITICAL STEP: Run the <Rearrange.sh>.
OUTPUT files are q∗.csv, r∗.csv, urcom.csv, urnew.csv, u.csv.
? TROUBLESHOOTING
-
(5)
Calculate the ratio of specific rAAV configurations based on the BLAST-based alignment process.
TIMING: within 10 min.
Run the the <Visualization.sh> to calculate the main molecule configurations in the sequencing reads (Table 1).
Note that the subgenomic formats need to be adjusted based on one’s project. The filtering subset R commands are generated to calculate the main AAV configurations, including standard single-stranded full-length AAV genomes, self-complementary (sc) full-length AAV genomes, and subgenomes in the AAV population. The subgenomes cover various noncanonical structural AAV DNA molecules such as symmetric or asymmetric snapback genomes (SBGs), genome-deletion mutants (GDMs), incomplete genomes (ICGs), and secondary derivative genomes, as listed in our previous studies.26,27
? TROUBLESHOOTING
-
(6)
(Optional) Subsample the sequencing reads in varying size ranges. Here, we show the SBG ratio in varying size ranges (Figure S1).
target<-subset(u,{filter setting})
Run the Optional commands in the <Format.sh>.
TIMING
The computing time to run the data analysis depends on the number of sequencing reads. The example sets described in this protocol contain ∼20,000–250,000 processed aligned data. To run this protocol, the total computing time consumed to complete the data after CCS processing was ∼0.8–6.0 h (Table 1). Much shorter computing times can be achieved by using more cores in parallel.
Step 1: Primary data analysis, including CCS processed run time (set multithreads = 10): ∼1–8 h.
Step 2: Mapping data analysis: ∼0.8–6.0 h.
-
(1)
BLAST-based alignments: ∼50,000–250,000 reads/h.
-
(2)
Visualization alignments: ∼25,000–30,000 reads/h.
-
(3)
Calculate the ratio of molecule configurations: within 10 min.
-
(4)
Other optional steps: within 10 min.
Anticipated results
The protocol outlined in Figure 2 generates multiple tabular output files during the BLAST-based alignment process in the <Preparation> section. These alignment data enable users to retrieve the specific configurations on sequences by running the scripts in the <Rearrange> and <Visualization> sections, accompanied by the unique sample identifiers. All of the required files needed to run this protocol are deposited at https://github.com/xiangpingyu/TMCA-AAVSeq-primary-data-analysis. We detected and grouped the viral samples used in this protocol into major configurations, with the extent of mapped to an estimated 80% (Table 1). For virus sequencing applications, examples in the form of SMRT-generated post-CCS sequences of rAAV are requested.
Discussion
Various methods for profiling AAV genome populations have been reported.19,20,28,29 Although substantial advancements in accuracy, read length, and throughput have been made with these methods, there remain challenges in achieving a comprehensive understanding of the entire AAV population. Some methods are limited by sequencing platforms. For example, although the Helicos method avoided the need for PCR amplification procedures and was able to sequence AAV genomes, it displays greater efficiency in sequencing relatively shorter templates compared to longer ones.21 Therefore, the Helicos approach lacked the capacity to generate the necessary data for many molecular configurations. The Illumina platform-based method for ssAAV vectors (SSV-seq) successfully identified known contaminations, including randomly packaged host cell sequences, AAV purification-specific DNA impurities, and helper plasmid-derived impurities.19 However, the Illumina DNA library preparation relies on DNA polymerase I and random hexamers for second-strand synthesis. Because this preparation requires DNA fragmentation into short reads and computational sequence assembly, reads are not a direct representation of the native state DNA that entered the system.19,30 The fragmentation and computational assembly results in the inability to identify many noncanonical genomes.
SMRT sequencing uses a phi29-derived DNA polymerase, which possesses strong strand-displacing activity. This sequencing does not require DNA fragmentation and can read along the whole length of the target in one read at accuracies approaching 99.9% when combined with long reads (>20 kb) and circular consensus on multiple passes of a template.18 The previous reported PacBio-SMRT system was tailored for sc vectors or AAV ssDNA that can anneal to one another for adapter ligation.28 As such, the molecules without corresponding counterpart molecules for annealing may not be included in the PacBio libraries. Aberrant AAV genomes with structural alterations, such as mutations, deletions, or inversions in the AAV DNA, are among those molecules that are the most difficult to find an annealing molecule. Owing to this difficulty, many of these species may be missed using the prior method. The rapid transposase-based protocol provided by Oxford Nanopore Technologies is based on amplification-free direct sequencing, thereby simplifying the sample preparation, and potentially eliminating an additional source of bias.31 Although hairpin loops arising from self-annealing on short stretches of a few bases represent potential substrates, it has been demonstrated that ssDNA can be cleaved, resulting in what is called mu-ends.32
Here, we propose TMCA-AAVseq for noncanonical AAV genome analysis. The concept is based on the fact that AAV packaging initiates from a 3′ ITR.33 Therefore, any AAV genome in an AAV capsid will carry a 3′ ITR. Helicos is capable of direct sequencing AAV 3′ end, which confirms that all AAV genomes indeed carry a 3′ ITR.21 In our previous study, we identified the following types of AAV DNA, including (1) the canonical AAV genomes: standard ssAAV DNA and scAAV DNA; (2) noncanonical AAV genomes: various snapback structural DNA, ICG without 5′ ITR, and GDMs; all of these molecules carry a 3′ ITR.21,23,24
The 3′ ITR is capable of self-priming in replication reactions.34 Therefore, any DNA packaged in the AAV capsid can be extended to a dsDNA form using the 3′ ITR. The most critical point for TMCA-AAVseq is that it does not require that the ITR to be perfect. Therefore, molecules that cannot self-anneal or anneal to other species in the library can still be included in the library. The AAV ITR is known to be heterogeneous, and it is not always 145 nt in size.35 This property will exclude some AAV genomes from the PacBio library, which requires a perfect ITR for adapter addition. In TMCA-AAVseq, even if 3′ ITR is mutated or truncated, as long as a stem-loop structure is maintained, second-strand extension for library construction is not affected. Studies have shown that such ITR isoforms can still be extended and included in the sequencing library.28 Although we did not include an scAAV vector in our examples, scAAV is compatible with TMCA-AAVseq. In perfect blunt ends, the adapters can be added directly. In nonblunt duplex ends, the extension reaction will fix the ends so that they can be included in the PacBio library. For additional aberrant molecules that have no matching annealing partners, the 3′ ITR extension reaction will generate a compatible molecule for adapter addition (Figure 1).
TMCA-AAVseq has two additional advantages in sequencing library construction. The first advantage is in the use of Bst DNA polymerase instead of Phi29 polymerase. Bst DNA polymerase navigates through all of the secondary structures present within the template strand, facilitating the generation of complete dsDNA with blunt ends. This reaction can take place at 50°–65°, which can help relieve some problems associated with nonspecific annealing at a low temperature. Second, A-tailing of the blunt end plus Blunt/TA ligation significantly improves ligation efficiency and library quality, and it also eradicates the self-ligation of DNA molecules or adapters. Regarding the efficiency of second-strand synthesis, we could not confirm 100% DNA molecules are converted to be dimer structure. However, we carried out alkaline gel analysis of the extended AAV genomes (Figure S2). The majority of the molecules were converted from monomers into dimers. Again, this suggested that most of the molecules were from the extension of 3′ ITR.
False annealing in the extension reaction may be a concern. For example, when the 3′ ITR is annealed to the 5′ ITR in the classical panhandle configuration, the extension will make blunt ends for the ITR, and the adapter can be added and be sequenced as well. We addressed this possibility in Figure 1. Using Bst polymerase for the extension step at the optimized temperature of 50°C greatly reduces the chance of panhandle configuration. If the ITR is intermolecularly annealed in trans, then the extension reaction may not lead to blunt ends in both 3′ and 5′ ends, and these molecules will be not in the final sequencing library. The higher extension temperature disrupts and minimizes such scenarios.
The TMCA-AAVseq method is an enhanced tool specifically for noncanonical AAV genome configuration analysis. Our technique can overcome some limitations of existing techniques to sequence a diverse range of AAV genomes, including those complex structures. The application of this approach will benefit a nuanced understanding of the AAV population and other parvoviruses and their derivative vectors.
Materials and methods
Reagents
HEK293 cells (American Type Culture Collection, catalog no. CRL-1573)
DMEM-high glucose (Sigma-Aldrich, catalog no. D5671)
Antibiotic-antimycotic 100× (Thermo Fisher, catalog no. 15240062)
Fetal bovine serum (Thermo Fisher, catalog no. 10437036)
Sodium hydroxide (Thermo Fisher, catalog no. 1310-73-2)
NaCl (Thermo Fisher, catalog no. S271)
MgCl2 (Sigma-Aldrich, catalog no. M8266)
KCl (Sigma-Aldrich, catalog no. P8333)
Tris base (Thermo Fisher, catalog no. BP152)
Hydrochloric acid 5 M (VWR, catalog no. BDH7419)
PEG-8000 (Thermo Fisher, catalog no. BP233)
Cesium chloride (Thermo Fisher, catalog no. MP215058980)
Sodium deoxycholate (Sigma-Aldrich, catalog no. D6750)
Pierce universal nuclease for cell lysis (Thermo Fisher, catalog no. 88702)
RNase (Thermo Fisher, catalog no. AAJ61996MC)
PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories, catalog no. SL100688)
Thermo Scientific DNase I Solution (1 U/μL), RNase-free (Thermo Scientific, catalog no. PI89836)
0.5 M EDTA, pH 8.0 (Thermo Scientific, catalog no. R1021)
DirectPCR Lysis Reagent (Mouse Tail) (Viagen, catalog no. 102-T)
Proteinase K Solution (20 mg/mL), RNA grade (Thermo Scientific Cat no. 25530049)
UltraPure Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) (Invitrogen, catalog no. 15593031)
Absolute ethanol, 200 proof, molecular biology grade (Thermo Scientific, catalog no. T038181000)
10× ThermoPol Reaction Buffer (New England Biolabs, catalog no. B9004S)
100 mM MgSO4 solution (New England Biolabs, catalog no. B1003S)
dNTP Set 100 mM Solutions (Thermo Scientific, catalog no. R0181)
Bst 2.0 polymerase (New England Biolabs, catalog no. M0537S)
Klenow fragment (3′→5′ exo-) (New England Biolabs, catalog no. M0212S)
Blunt/TA Ligase Master Mix (New England Biolabs, catalog no. M0367S)
Overhang T-tailing adapter (synthesized by Integrated DNA Technology)
Exonuclease III (New England Biolabs, catalog no. M0206S)
Exonuclease VII (New England Biolabs, catalog no. M0379S)
AMPure XP (Beckman Coulter, catalog no. A63880)
UltraPure Agarose (Invitrogen, catalog no. 16500500)
GeneJET Gel Extraction Kit (Thermo Scientific, catalog no. K0691)
Equipment
Optima L-90K ultracentrifuge (Beckman Coulter, catalog no. 365670)
SW28 rotor (Beckman Coulter, catalog no. 342207)
Ti70 ultracentrifuge rotor (Beckman Coulter, catalog no. 337922)
Open-Top Thinwall Ultra-Clear Tube (Beckman Coulter, 38.5 mL, catalog no. NC9146666)
Quick-Seal Round-Top Ultra-Clear Tube (Beckman Coulter, 16 × 76 mm, 13.5 mL, catalog no. NC9325049)
Conical tubes, 50 mL (Thermo Fisher, catalog no. 14-432-22)
Conical tubes, 250 mL (Corning, catalog no. 430776)
Conical tubes, 500 mL (Corning, catalog no. 89091-000)
Microcentrifuge 5425 (Eppendorf, catalog no. 5405000646)
Needles, 18G 1½ in (Thermo Fisher, catalog no. 14-840-97)
Syringe, 5 mL (Thermo Fisher, catalog no. 14-817-29)
Sterile syringe filter, 0.22 mm (CellTreat, catalog no. 229746)
Metal stand and clamp (Thermo Fisher, catalog no. S24250 and S477653Q)
Sterile 150-mm cell culture plates (Sigma-Aldrich, catalog no. SIAL0599)
Avanti J-E centrifuge (Beckman Coulter, catalog no. 369001)
JS-5.3 centrifuge rotor (Beckman Coulter, catalog no. 368690)
Sartorius Vivaspin 20 Centrifugal Concentrators, PES Membrane 100,000 Da (Sartorius, catalog no. EW-36224-78)
Fisher vortex Genier (Fisher Scientific, catalog no. 12812)
Magnetic Separation Rack (New England Biolabs, catalog no. s1509s)
NanoDrop One/One C Microvolume UV-Vis Spectrophotometer (Thermo Fisher, catalog no. ND-ONE-W)
Computational hardware
This protocol requires a computational server running a Linux-based or Windows-based system with multithreaded processors. Smaller complete PacBio sequencing sets (after machine sequencing) can be run on stand-alone desktops. For large datasets, extensive computations (e.g., mapping very large sequencing data using BLAST) can be optimized and parallelized using multiple central processing units (CPUs). Most scripts are written in Bash, R, and Python.
Software
Install Anaconda (https://anaconda.org/anaconda/git) to perform the corresponding SMRT or R packages. These SMRT packages can be freely git cloned from https://github.com/PacificBiosciences. Download the SMRT_Link documents supports from (https://www.pacb.com/support/software-downloads/).
recalladapters: https://github.com/PacificBiosciences/recalladapters
Minimap2: https://github.com/lh3/minimap2
Seqkit-tools: https://anaconda.org/bioconda/seqkit
Example data
Using TMCA-AAVseq, we tested eight batches of rAAV virus libraries (Table 1). The data are available upon request.26,27
Applications
TMCA-AAVseq can be applied to three main types of experiments. TMCA-AAVseq can serve as a tool for profiling the genome of all wild-type AAV and other parvovirus family members. It can also work as a tool for the quality control of rAAV vectors in the gene therapy field by profiling rAAV-population genomes. Finally, this system can be used to optimize AAV vectors—for instance, it can enhance vector quality or be used in the design of novel vectors.36 By pinpointing vectors that contain excessive aberrant genomes, studies can be developed to improve rAAV efficiency and safety profiles for use in patients.
Expertise Needed to Implement the Protocol
The steps involved in this protocol can be easily performed by a graduate student or postdoctoral researcher with a basic understanding of molecular biology. Nonetheless, TMCA-AAVseq does require long-read sequencing on a PacBio SMRT-Seq instrument. Such instruments are available at sequencing centers and companies globally and need to be operated by trained personnel. In addition, TMCA-AAVseq involves running the provided data analysis software, which is written in Python 3. Running this software and handling the sequencing files generated by PacBio sequencing require knowledge of the UNIX command-line interface and software management in UNIX systems.
Limitations
One limitation of our study is the necessity to convert ssDNA into dsDNA form. The formation of dsDNA is a common challenge for all current NGS platforms. Since native state ssDNA cannot be sequenced, data analysis is complicated by distinguishing extended dsDNA from native dsDNA within a pool. The SMRT sequencing platform is based on phi29-derived DNA polymerase, and its processivity is highly efficient, so that long reads are made possible. However, the AAV-population genome has heterogeneity with different size and molecular structures, presenting a challenge for subsequent data analysis. Due to size selection, this approach is incapable of quantifying every molecular variant within the complete population.
This protocol is aimed to detect the whole virus genome from library samples. To increase the sensitivity of mapping process, the users need to first detect subsample sequencing to make sure of the extent of the loop process to run BLAST-based alignment. An example in this protocol is applied to process five loops to categorize the main AAV virus genome. As detailed in the visualization of alignments, the overall rate of calculated configuration is estimated to be over 80%. In addition, TMCA-AAVseq requires mapping reads to a defined reference genome, but it is not compatible with metagenomic sequencing or analysis of populations with unknown constituents. Users may need to verify foreign addition sequences unaligned to reference in the process of BLAST-based mapping, using the appropriate sequence searching tools.
Acknowledgments
This work was supported by grants from the NIH grants HL114152 and P01HL160472. This project has been funded, in whole or in part, with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N92019D00018.
Author contributions
W.X. and R.W.H. supervised the project; J.Z., P.G., and D.P. developed the protocol for the AAV genome library; X.Y. and J.T. performed the bioinformatic analysis; W.X., J.Z., X.Y., P.G., M.C., and D.P. completed the data analysis; and all of the authors contributed to the writing of this paper.
Declaration of interests
W.X. holds equity in Ivygen Corporation and Nikegen.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101215.
Supplemental information
References
- 1.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fong S., Yates B., Sihn C.-R., Mattis A.N., Mitchell N., Liu S., Russell C.B., Kim B., Lawal A., Rangarajan S., et al. Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A. Nat. Med. 2022;28:789–797. doi: 10.1038/s41591-022-01751-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pipe S.W., Leebeek F.W.G., Recht M., Key N.S., Castaman G., Miesbach W., Lattimore S., Peerlinck K., Van der Valk P., Coppens M., et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023;388:706–718. doi: 10.1056/NEJMoa2211644. [DOI] [PubMed] [Google Scholar]
- 4.Ogbonmide T., Rathore R., Rangrej S.B., Hutchinson S., Lewis M., Ojilere S., Carvalho V., Kelly I. Gene therapy for spinal muscular atrophy (SMA): A review of current challenges and safety considerations for onasemnogene abeparvovec (Zolgensma) Cureus. 2023;15 doi: 10.7759/cureus.36197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornburg C.D. Etranacogene dezaparvovec for hemophilia B gene therapy. Ther. Adv. Rare Dis. 2021;2 doi: 10.1177/26330040211058896. 26330040211058896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lek A., Wong B., Keeler A., Blackwood M., Ma K., Huang S., Sylvia K., Batista A.R., Artinian R., Kokoski D., et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023;389:1203–1210. doi: 10.1056/NEJMoa2307798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwu P.W.L., Kiening K., Anselm I., Compton D.R., Nakajima T., Opladen T., Pearl P.L., Roubertie A., Roujeau T., Muramatsu S.I. Gene therapy in the putamen for curing AADC deficiency and Parkinson's disease. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202114712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davé U.P., Cornetta K. AAV Joins the Rank of Genotoxic Vectors. Mol. Ther. 2021;29:418–419. doi: 10.1016/j.ymthe.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Y., Zhang Z., Gao P., Lai H., Zhong W., Feng N., Yang Y., Yu H., Zhang Y., Han Y., et al. AAV induces hepatic necroptosis and carcinoma in diabetic and obese mice dependent on Pebp1 pathway. EMBO Mol. Med. 2023;15 doi: 10.15252/emmm.202217230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt M., Foster G.R., Coppens M., Thomsen H., Cooper D., Dolmetsch R., Sawyer E.K., Heijink L., Pipe S.W. Liver safety case report from the phase 3 HOPE-B gene therapy trial in adults with hemophilia B. International Society on Thrombosis and Haemostasis; 2021. [Google Scholar]
- 12.Rosas L.E., Grieves J.L., Zaraspe K., La Perle K.M., Fu H., McCarty D.M. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. 2012;20:2098–2110. doi: 10.1038/mt.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulcrone P.L., Zhang J., Pride P.M., Lam A.K., Frabutt D.A., Ball-Kell S.M., Xiao W. Genomic designs of raavs contribute to pathological changes in the livers and spleens of mice. Adv. Cell Gene Ther. 2022;2022 doi: 10.1155/2022/6807904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Troxell B., Jaslow S.L., Tsai I.W., Sullivan C., Draper B.E., Jarrold M.F., Lindsey K., Blue L. Partial genome content within rAAVs impacts performance in a cell assay-dependent manner. Mol. Ther. Methods Clin. Dev. 2023;30:288–302. doi: 10.1016/j.omtm.2023.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earley L.F., Conatser L.M., Lue V.M., Dobbins A.L., Li C., Hirsch M.L., Samulski R.J. Adeno-Associated Virus Serotype-Specific Inverted Terminal Repeat Sequence Role in Vector Transgene Expression. Hum. Gene Ther. 2020;31:151–162. doi: 10.1089/hum.2019.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilmott P., Lisowski L., Alexander I.E., Logan G.J. A user’s guide to the inverted terminal repeats of adeno-associated virus. Hum. Gene Ther. Methods. 2019;30:206–213. doi: 10.1089/hgtb.2019.276. [DOI] [PubMed] [Google Scholar]
- 17.Kieleczawa J. Fundamentals of sequencing of difficult templates--an overview. J. Biomol. Tech. 2006;17:207–217. [PMC free article] [PubMed] [Google Scholar]
- 18.Rhoads A., Au K.F. PacBio Sequencing and Its Applications. Dev. Reprod. Biol. 2015;13:278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecomte E., Tournaire B., Cogné B., Dupont J.B., Lindenbaum P., Martin-Fontaine M., Broucque F., Robin C., Hebben M., Merten O.W., et al. Advanced characterization of DNA molecules in rAAV vector preparations by single-stranded virus next-generation sequencing. Mol. Ther. Nucleic Acids. 2015;4 doi: 10.1038/mtna.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Namkung S., Tran N.T., Manokaran S., He R., Su Q., Xie J., Gao G., Tai P.W.L. Direct ITR-to-ITR Nanopore Sequencing of AAV Vector Genomes. Hum. Gene Ther. 2022;33:1187–1196. doi: 10.1089/hum.2022.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapranov P., Chen L., Dederich D., Dong B., He J., Steinmann K.E., Moore A.R., Thompson J.F., Milos P.M., Xiao W. Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum. Gene Ther. 2012;23:46–55. doi: 10.1089/hum.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart C., Lipson D., Ozsolak F., Raz T., Steinmann K., Thompson J., Milos P.M. Single-molecule sequencing: Sequence methods to enable accurate quantitation. Methods Enzymol. 2010;472:407–430. doi: 10.1016/S0076-6879(10)72002-4. [DOI] [PubMed] [Google Scholar]
- 23.Dong B., Nakai H., Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol. Ther. 2010;18:87–92. doi: 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong B., Moore A.R., Dai J., Roberts S., Chu K., Kapranov P., Moss B., Xiao W. A concept of eliminating nonhomologous recombination for scalable and safe AAV vector generation for human gene therapy. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gkt404. p6609-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J., Guo P., Yu X., Frabutt D.A., Lam A.K., Mulcrone P.L., Chrzanowski M., Firrman J., Pouchnik D., Sang N., et al. Subgenomic particles in rAAV vectors result from DNA lesion/break and non-homologous end joining of vector genomes. Mol. Ther. Nucleic Acids. 2022;29:852–861. doi: 10.1016/j.omtn.2022.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J., Yu X., Guo P., Firrman J., Pouchnik D., Diao Y., Samulski R.J., Xiao W. Satellite subgenomic particles are key regulators of adeno-associated virus life cycle. Viruses. 2021;13:1185. doi: 10.3390/v13061185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated Virus Genome population sequencing achieves full vector genome resolution and reveals human-vector chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran N.T., Heiner C., Weber K., Weiand M., Wilmot D., Xie J., Wang D., Brown A., Manokaran S., Su Q., et al. AAV-Genome Population Sequencing of Vectors Packaging CRISPR Components Reveals Design-Influenced Heterogeneity. Mol. Ther. Methods Clin. Dev. 2020;18:639–651. doi: 10.1016/j.omtm.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan G., Opitz L., Schlapbach R., Rehrauer H. Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci. Rep. 2019;9:2856. doi: 10.1038/s41598-019-39076-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radukic M.T., Brandt D., Haak M., Müller K.M., Kalinowski J. Nanopore sequencing of native adeno-associated virus (AAV) single-stranded DNA using a transposase-based rapid protocol. NAR Genom. Bioinform. 2020;2:lqaa074. doi: 10.1093/nargab/lqaa074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saariaho A.H., Savilahti H. Characteristics of MuA transposase-catalyzed processing of model transposon end DNA hairpin substrates. Nucleic Acids Res. 2006;34:3139–3149. doi: 10.1093/nar/gkl405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King J.A., Dubielzig R., Grimm D., Kleinschmidt J.A. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 2001;20:3282–3291. doi: 10.1093/emboj/20.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava A. Replication of the adeno-associated virus DNA termini in vitro. Intervirology. 1987;27:138–147. doi: 10.1159/000149732. [DOI] [PubMed] [Google Scholar]
- 35.Samulski R.J., Srivastava A., Berns K.I., Muzyczka N. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell. 1983;33:135–143. doi: 10.1016/0092-8674(83)90342-2. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J., Yu X., Herzog R.W., Samulski R.J., Xiao W. Flies in the ointment: AAV vector preparations and tumor risk. Mol. Ther. 2021;29:2637–2639. doi: 10.1016/j.ymthe.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.