

Abstract
Triggerable coatings, such as pH-responsive polymethacrylate copolymers, can be used to protect the active pharmaceutical ingredients contained within oral solid dosage forms from the acidic gastric environment and to facilitate drug delivery directly to the intestine. However, gastrointestinal pH can be highly variable, which can reduce delivery efficiency when using pH-responsive drug delivery technologies. We hypothesized that biomaterials susceptible to proteolysis could be used in combination with other triggerable polymers to develop novel enteric coatings. Bioinformatic analysis suggested that silk fibroin is selectively degradable by enzymes in the small intestine, including chymotrypsin, but resilient to gastric pepsin. Based on the analysis, we developed a silk fibroin-polymethacrylate copolymer coating for oral dosage forms. In vitro and in vivo studies demonstrated that capsules coated with this novel silk fibroin formulation enable pancreatin-dependent drug release. We believe that this novel formulation and extensions thereof have the potential to produce more effective and personalized oral drug delivery systems for vulnerable populations including patients that have impaired and highly variable intestinal physiology.
Keywords: silk fibroin–polymethacrylate copolymers, enteric coating, pancreatin-dependent drug delivery, triggered release, in vivo validation
Introduction
Oral drug delivery remains the preferred route of administration by patients and physicians given its facile self-administration.1 Some active pharmaceutical ingredients (API) need to be delivered to specific parts of the intestinal tract for optimal efficacy; however, premature degradation or absorption can decrease the amount of API that reaches the desired target site. One way to reduce premature degradation and absorption is to include a selectively degradable coating, such as a polymethacrylate copolymer, on the pharmaceutical dosage form. Unfortunately, heterogeneity among patient populations may reduce the effectiveness of these approaches for certain patients.2 For example, polymethacrylate copolymers are typically used to protect drugs until they reach the small intestine. A sharp increase in pH from the stomach to the small intestine causes the polymer to deprotonate and disintegrate. Eudragit S coatings typically degrade in about 1–2 hours following small intestinal transit, which can be used to facilitate delivery to the colon.2 Mixing polymethacrylate copolymers with substrates that are metabolized by the gastrointestinal (GI) microbiome, such as starch, can increase delivery efficiency.3 However, some drugs, such as proton-pump inhibitors, can cause significant changes in the GI environment, such as shifts in intestinal pH,4 a reduction in the activity or presence of digestive enzymes, and microbiota dysbiosis. Such physiological changes affect the degradation behavior of pH-responsive coatings, which can reduce the amount of drug that reaches its target site. For example, in patients suffering from pseudomembranous colitis, an inflammation of the large intestine, the pathogenic bacteria Clostridioides difficile dominates the intestinal microbiota; for these patients, delivery strategies that rely on microbial enzymes (e.g., to break down starch) may be less effective.5
In order to identify alternative coatings that would not be adversely affected by compromised GI environments and could achieve colonic release, we selected 9 polypeptides from 5 different proteins that are currently investigated in pre-clinical studies for composite biomaterials in biomedical applications.13 We computationally screened these polypeptides for their digestibility by different GI enzymes. Silk fibroin (SF) protein emerged as a promising biomaterial candidate because it is predicted to be more rapidly digested by intestinal enzymes compared to gastric enzymes.
Silk has been approved by the United States Food and Drug Administration (FDA) as sutures6, and SF as a coating material is currently pending FDA GRAS evaluation.7 We tested SF coatings with and without dimethyl sulfoxide (DMSO) for their degradation behavior in different simulated gastric conditions to evaluate their potential as a biomaterial for targeted release applications through pancreatin-dependent proteolysis.
Even though various studies have shown the effect of pH on a variety of formulations, including those with SF8–10, gastrointestinal pH is highly variable. Furthermore, some research for SF has revealed that further modifications, such as the addition of PEG and graphene oxide, can improve material properties for pH sensitive and sustained drug delivery.11 Using in vitro dissolution testing, we found that a mixture of Eudragit S100 (ES100) and SF was stable throughout GI transit and disintegrated within 2–3 hours after simulated GI passage. This is consistent with clinically established delivery strategies that apply ES100 for ileocolonic drug delivery. In contrast to using ES100, the combinatory approach does not solely rely on a pH shift but also requires proteolysis, which is an inherent and highly conserved part of digestion that remains functional for a wide range of different pathobiologies.12 Consequently, this novel coating could potentially increase the success rate of intestinal and colonic delivery, especially in clinical scenarios in which pH shifts or microbial dysbiosis is present (e.g., pseudomembranous colitis). We investigated the effectiveness of this delivery approach by conducting in vitro dissolution tests on capsules loaded with mesalamine (5-aminosalicylic acid). Using an in vivo swine model, we demonstrated that coated capsules could successfully release their payload to the colon three hours after the capsule passed through the small intestine.
Materials and Methods
Protein sequence analysis for cleavage sites
A list of candidate biomaterials for coatings, including SFs, collagens, resilins, and elastins, was extracted from the literature.13 The descriptions and amino acid sequences of each protein were extracted from the UniProt Knowledgebase (UniProtKB) database14 with reviewed records (Swiss-Prot15). For each type of protein, we selected the most abundant subtype that was also commercially available. We performed a sequence-based cleavage site prediction on each of the selected proteins using the cleaver R package16 based on the rules from the Expasy PeptideCutter tool17 with 11 different digestive enzymes in the human GI tract (i.e. pepsin (at pH 1.3), trypsin, chymotrypsin (high specificity), caspase 1, 3, 5, 6, 7, 10, glutamyl endopeptidase, and enterokinase). The number of predicted cleavage sites was normalized by the length of the specific candidate protein to account for differences in length.
Silk fibroin solution preparation
Thai silk cocoons of Bombyx mori (Nangnoi Srisaket 1) were obtained from Queen Sirikit Sericulture Center, Nakhon Ratchasima, Thailand. SF solution was prepared according to previously published protocols.18 Briefly, the cocoons were boiled in 0.02 M sodium bicarbonate (Ajax-Finechem, New South Wales, Australia) and thoroughly rinsed with double distilled water (ddH2O) to remove sericin protein. The SF solution was prepared by adding 4 g of the degummed silk fiber to 16 mL of a 9.3 M lithium bromide (Sigma-Aldrich, St. Louis, MO, USA) solution and incubated at 60°C for 4 hours or until it fully dissolved. Dialysis was performed by loading the dissolved silk into Slide-A-Lyzer™ G2 Dialysis Cassettes with a 3.5K molecular weight cut-off (ThermoFisher Scientific, Waltham, MA, USA) and placing the cassettes in ddH2O for 48 hours while stirring at 200 rpm. After dialysis, the cassettes were air-dried overnight to increase the solution concentration to over 7% w/w. The solution was centrifuged twice at 9,000 rpm for 20 minutes at 4°C to remove pelleted silk particulates. The SF solution was diluted to a stock solution of 7% w/w concentration and stored at 4°C.
Film generation
SF films were created using a polystyrene (PS) mold (ThermoFisher Scientific, Waltham, MA, USA). The SF stock solution was poured into the PS mold using a ratio of 1 cm3 mixture to 13.2 cm2 area of the mold. The solution was left on a bench at room temperature for 24 hours to allow the solvent to evaporate. The resulting film was removed from the mold, and the edge was trimmed off using a razor blade. Subsequently, the film was cut into a circular shape (15.5 mm in diameter) for penetration tests. In a separate experiment, the same protocol was used to generate a film from the SF stock solution, which included an additional 0.01% v/v of DMSO (ThermoFisher Scientific, Waltham, MA, USA).
Test solution preparation
Simulated gastric fluid (SGF) was prepared by mixing 4 g of sodium chloride (Sigma-Aldrich, St. Louis, MO, USA) and 14 mL of hydrochloric acid (Sigma-Aldrich, St. Louis, MO, USA) in 2 L of ddH2O, then adjusting the pH to 1.2. Pepsin from porcine gastric mucosa powder (Sigma-Aldrich, St. Louis, MO, USA) was added at a concentration of 3.2 mg/mL prior to the dissolution testing. Simulated intestinal fluid (SIF) was prepared by mixing 1.792 g of sodium hydroxide (Sigma-Aldrich, St. Louis, MO, USA) and 13.61 g of potassium dihydrogen phosphate (Alfa Aesar, Ward Hill, MA, USA) in 2 L of ddH2O, then adjusting the pH to 6.8. Pancreatin from porcine pancreas (Sigma-Aldrich, St. Louis, MO, USA) was added at a concentration of 10 mg/mL prior to the dissolution testing.
Penetration tests
We 3D printed a specific 24-well plate that enabled us to secure the SF films by clamping them between two separated plates using magnets. We tested five conditions consisting of water, SGF, SGF with pepsin, SIF, and SIF with pancreatin. Each test solution was added to the bottom wells. Then, these test solutions with 5 μM Rhodamine B (Sigma-Aldrich, St. Louis, MO, USA) were added to the top of both the SF film and the SF film with the added DMSO (SF/DMSO film). Four replicates for each condition were performed. The films were then incubated at 37°C in a 50 rpm shaker. We used a flatbed scanner to take images of the bottom wells at 0, 1, 2, 3, and 6 hours.
Capsule coating
PENTASA® capsules (Shire Pharmaceuticals, Lexington, MA, USA), microgranules of mesalamine capsules coated with ethyl cellulose, were first dip-coated with one layer of Eudragit L100 (EL100, Evonik, Essen, Germany) to provide a non-water-soluble base coating for additional SF/ES100 coatings. To prepare the initial coat solution, 5 g of EL100 was added to 50 mL of acetone (Sigma-Aldrich, St. Louis, MO, USA) and 50 μL of triethyl citrate (Sigma-Aldrich, St. Louis, MO, USA) and stirred until a whitish-clear liquid was formed. PENTASA® capsules were then dip-coated with 10 layers of a SF/ES100 solution. This outer coat was prepared in a 3:1 ratio of SF/ES100. The ES100 solution was prepared by combining 9.94 g of ES100 (Evonik, Essen, Germany), 4.97 mL of triethyl citrate (Sigma-Aldrich, St. Louis, MO, USA), and 73.4 mL of ddH2O and the pH was adjusted to 7.4.
Release study
The release study was performed using the following release protocol on a Hanson Vision G2 Elite 8 Dissolution Tester in 1L-capacity vessels: 1.5 mL samples of dissolution media were collected every hour from 1–11 hours, with additional collection timepoints at 1 minute, 0.5, 1.5, and 20 hours. For the first hour, the capsules were immersed in 800 mL of SGF and stirred at a speed of 50 rpm, then switched to 800 mL of SIF from the 1-hour timepoint until the 2-hour timepoint with stirring, and then the media was finally switched to 800 mL of SIF with pancreatin until 20 hours (stirring until 11 hours). Solutions were prepared as previously described in “test solution preparation”. To confirm the substrate specificity for pancreatin, a modified release protocol was used in which pancreatin was not added to the SIF. High-performance liquid chromatography was performed as described below to analyze mesalamine concentration at every collection timepoint. The amount of drug released from the capsules was calculated from a standard calibration curve of mesalamine, which used 0.1 M hydrochloric acid and spanned concentrations of 500 μg/mL to 0.24 μg/mL. The experiment was performed on 3 replicates.
High Performance Liquid Chromatography
High Performance Liquid Chromatography (HPLC) was used to determine the drug concentrations from all in vitro release assays. An Agilent 1260 Infinity II HPLC system (Santa Clara, CA, USA) equipped with a quaternary pump, an autosampler, a thermostat, a control module, and a diode array detector was utilized, as described previously. Data processing and analysis were performed using OpenLab CDS ChemStation®.
Mesalamine was separated on an Agilent Zorbax Eclipse XDB C18 analytical column (4.6 mm × 250 mm, 5 μm particles, Santa Clara, CA, USA), maintained at 30°C. The optimized mobile phases A and B consisted of 50 mM of ammonium acetate buffer at a pH of 5.00 and acetonitrile, respectively (Sigma-Aldrich, St. Louis, MO, USA). Isocratic elution was employed over a 6-minute period using a mobile phase composition of 85% A and 15% B. The injection volume was 8 μL, and the selected ultraviolet (UV) detection wavelength was 254 nm at a bandwidth of 4.0 nm, with no reference wavelength and an acquisition rate of 10 Hz.
In vivo experiments
All procedures were conducted in accordance with protocols approved by the Massachusetts Institute of Technology Committee on Animal Care. Following overnight fasting, three separate female Yorkshire pigs weighing approximately 30–50 kg were sedated with TELEZOL® (tiletamine/zolazepam, Zoetis, Parsippany, NJ, USA) at a dose of 5 mg/kg, xylazine at 2 mg/kg, and atropine at 0.04 mg/kg. Endotracheal intubation was performed, and anesthesia was maintained with isoflurane thereafter (1–3% in oxygen). The radiopaque beads were assembled first by emptying out PENTASA® capsules of their mesalamine drug payload and filling them with 3 mm of steel beads (McMaster-Carr, Elmhurst, IL, USA) to mimic the size of the drug granules. The coating protocol for the radiopaque bead capsules was identical to the in vitro SF/ES100 coating protocol. The radiopaque bead capsule delivery systems were placed into the small intestine using an esophageal overtube (US Endoscopy, Mentor, OH, USA) with endoscopic guidance. Radiographic evaluation was performed following a previously described protocol26 at the following timepoints: 1, 2, 3, and 20 hours.
Results
Sequence analysis for cleavage sites
We computationally examined the degradation behavior of nine candidate proteins that could potentially be used to formulate coatings for proteolysis-dependent drug delivery. Potential cleavage sites on these nine proteins were predicted for 11 digestive enzymes. The enzymes caspase 3, 5, 6, 7, 10 and enterokinase did not cleave any of the candidate proteins, while caspase 1 only cleaved resin at a single cleavage site. The four enzymes which were predicted to cleave all the selected proteins were pepsin (at a pH of 1.3), trypsin, chymotrypsin (with high specificity), and glutamyl endopeptidase. In order to evaluate the sensitivity of each protein to a specific digestive enzyme, we normalized the number of predicted cleavage sites by the protein length (Figure 1). The SF heavy chain was the most resistant to pepsin digestion (< 1% of the SF heavy chain sequence had pepsin cleavage sites). To measure proteolytic selectivity, we compared the number of pepsin cleavage sites (shown in orange in the bar plot of Figure 1) to the number of cleavage sites by other digestive enzymes of the small intestine, specifically pancreatic enzymes (trypsin and chymotrypsin) and glutamyl endopeptidase (shown in blue in the bar plot of Figure 1). We quantified selectivity through the ratio of pepsin cleavage sites compared to chymotrypsin cleavage sites. The most selective protein sequence was the SF heavy chain: the ratio between the number of cleavage sites from the intestinal enzymes to gastric pepsin was 7:1. The collagen α−1(i) chain was the second most selective with a 2.8:1 ratio. Furthermore, we ruled out that the cleavage sites were localized to only specific parts of the peptide chain through visual inspection; as shown in Figure 1, the 373 cleavage sites from the intestinal enzymes over the 5,263 amino acids (AA) of SF heavy chain was spread across the complete peptide chain. Together this data suggested that the SF protein might be able to withstand pepsin exposure in the stomach and could potentially provide a scaffold for a pancreatin-dependent delivery coating.
Figure 1.
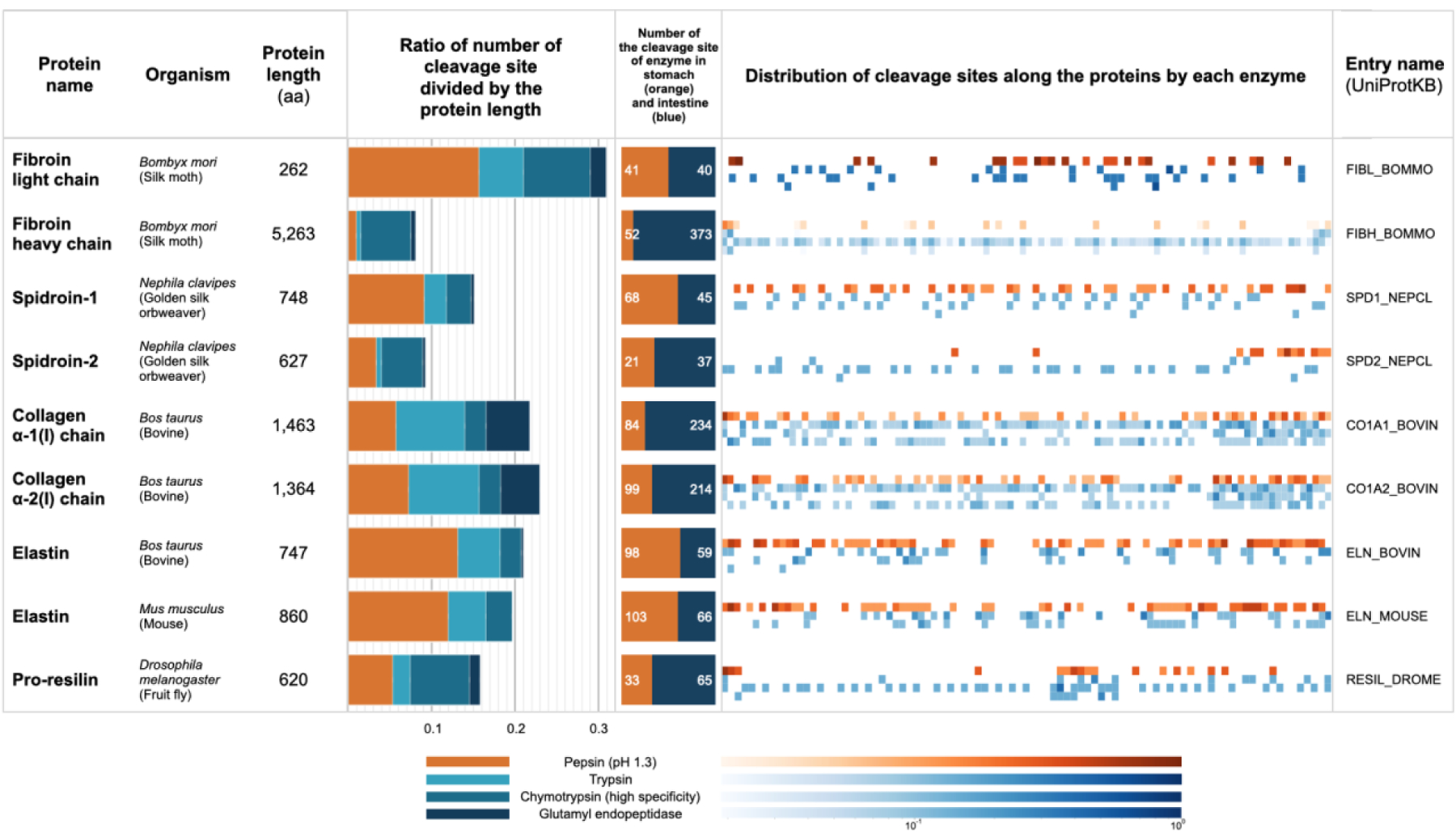
List of the selected protein candidates and the cleavage site analysis for each protein. Note that for the heatmap, the cleavage site data were normalized to the length of 100. The color bar is scaled logarithmically.
Penetration studies of films
We generated SF films through evaporation. The average thickness of the SF film was 0.05 ± 0.01 mm with an average Young’s modulus of 1,233 ± 82 MPa. We next investigated whether the films would be stable in different media by clamping the films between a donor and receiver chamber and testing the susceptibility of the film to degrade in water, SGF, SGF with pepsin, SIF, and SIF with pancreatin. 5 μM Rhodamine B served as a molecular dye to identify leaks in the film. For each penetration test (Supplementary Figure 1), we captured an image of the bottom of each plate. A change of color in the receiver well indicated the presence of rhodamine B in the receiver solution in the bottom plate, which flowed from the top plate by either penetrating the SF film or dissolving the film.
We observed that the SF film dissolved in water, in SGF with pepsin, in SIF, and in SIF with pancreatin. In SGF, the films remained stable but shrunk. Apparently, our SF films were too soluble in aqueous conditions to serve as a suitable coating. We hypothesized that additives could decrease the solubility of the SF film. To test this, we added DMSO to the SF solution and generated another set of films through evaporation. The thickness of the SF/DMSO film was like the SF film: 0.05 ± 0.01 mm. However, the average Young’s modulus of the SF/DMSO film, presented in Supplementary Figure 2, was significantly lower than the SF film: 1,039 ± 88 MPa (unpaired t-test, p<0.0001). We conducted the same penetration tests on these films and observed that the novel films were sufficiently stable and did not solubilize in water. Furthermore, the films also appeared stable to enzymatic digestion by pepsin but not pancreatin, as predicted by our bioinformatic approach. Interestingly, the film did not exhibit solubility in SIF with pancreatin until the upper part of the setting plate was removed at the end of the experiment (6 hours). This was because the film exposed to the SIF with pancreatin was still intact before the setting plate was removed but broke after a small force was applied.
Capsules with silk fibroin-based coating enable pancreatin-dependent drug release
PENTASA® capsules were coated with a single layer of EL100 (to protect the capsule from premature degradation during the coating procedure) and 10 layers of either SF/ES100 or ES100 (Figure 2A). These capsules were used in a standard dissolution protocol to test whether they would release mesalamine when simulating gastric passage by exposing them to SGF with pepsin, then to SIF, and finally to SIF with pancreatin (Figure 2B). Both types of capsules released mesalamine, which was used to calculate the dissolution rate of the capsules. After 20 hours, capsules coated with SF/ES100 released 15% ± 3% of their dose, whereas capsules coated with ES100 released 19% ± 11% of their dose (Figure 2C). This difference is not statistically significant (two-sample t-test, p=0.6506). To test whether pancreatin would indeed affect the dissolution of the capsules, we used the same procedure to coat another set of capsules but then exposed them to a modified dissolution protocol where the capsules were exposed to SGF with pepsin but then to SIF that did not contain pancreatin. Capsules coated with the novel coating (SF/ES100) did not release mesalamine (1.4% ± 0.7%), while capsules coated with the control coating that contained ES100 continued to release mesalamine (35% ± 14%; dissolution after 20 hours, two-sample t-test, p=0.0275; Figure 2D).
Figure 2.
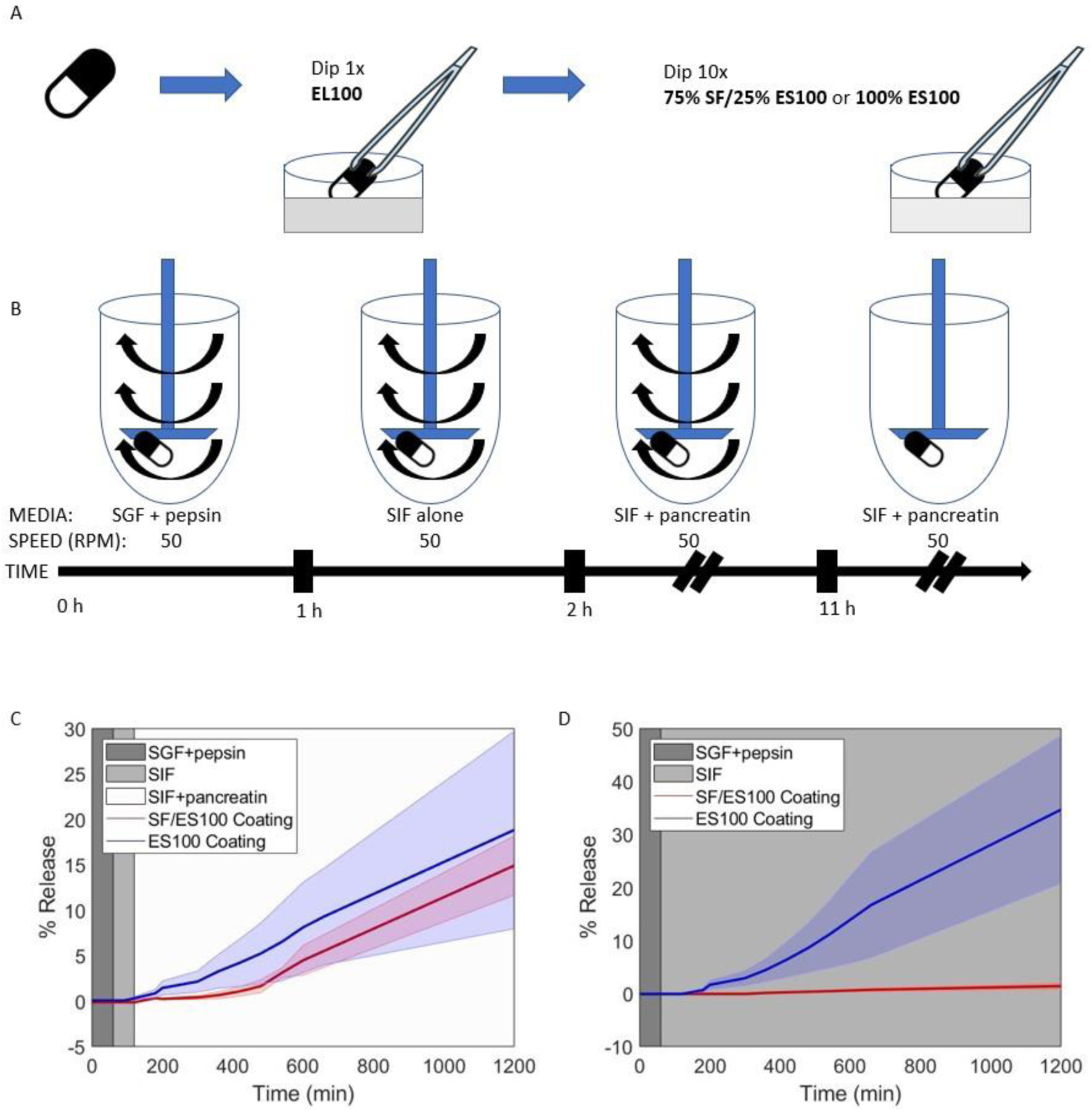
Pentasa pills coated with the silk-based formulation release mesalamine in the presence of pancreatin. (A) Schematic of the experimental set-up for the pill coating protocol. Pills were dipped once in Eudragit L100 and 10 times in pure Eudragit S100 (ES100) or in a 75%/25% mixture of SF/ES100.
(B) Release testing experimental protocol. Coated pills were separately immersed in simulated gastric fluid (SGF) containing pepsin for 1 hour, switched to simulated intestinal fluid (SIF) for 1 hour, then switched to SIF containing pancreatin for 9 hours (with stirring). After 11 hours, stirring ceased and pills sat in SIF with pancreatin until t = 20 hours.
(C) Percent release curves for capsules coated with either silk/ES100 or pure ES100 are shown. Pills went through the standard dissolution protocol outlined in (B).
(D) Percent release curves for capsules coated with either SF/ES100 or pure ES100 are shown. Pills went through a modified release protocol where pancreatin was never added to SIF media after time t = 2 hours.
Disintegration of capsules with silk fibroin-based coating in vivo
Next, we tested whether the capsules with SF-based coating would release an encapsulated payload in vivo. We created capsules that contained radiopaque steel beads to enable tracking of their release through X-ray radiography. Following intestinal placement, the capsules with the SF-based coatings remained stable for three hours in all three animals tested (this timeframe was chosen to reflect intestinal transit time). All capsules disintegrated and released the payload within 24 hours, and the radiopaque beads were distributed throughout the GI tract (Figure 3).
Figure 3.
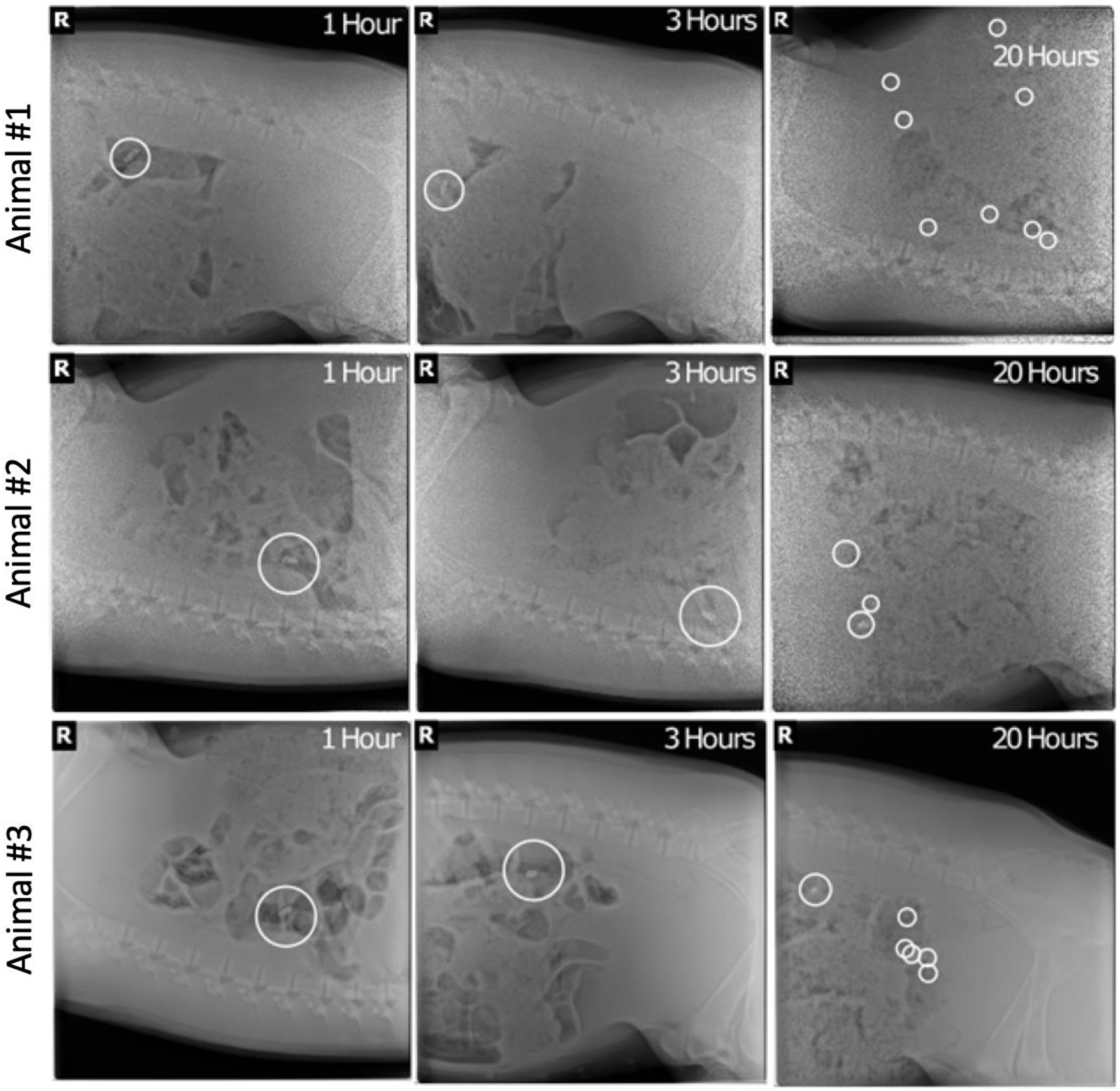
In vivo disintegration of the combined silk-coated capsules following administration into the small intestine (visualized with radiopaque beads).
Discussion
Enteric coatings, which resist dissolution in acidic environments but break down rapidly in more alkaline conditions, are commonly used in formulations to enable the delivery of medications to the small intestine and the colon. However, certain medical conditions and drugs, such as proton pump inhibitors, can significantly alter the pH of the GI tract, reducing the effectiveness of enteric coatings. We hypothesized that proteolytically degradable materials could enable oral drug delivery systems to target the small intestine and colon more effectively and consistently, especially in individuals with compromised GI environments. Computational modeling of cleavage sites on selected proteins by different enzymes indicated that the SF heavy chain would exhibit the most selectivity for degradation by intestinal enzymes. The SF heavy chain was consequently chosen as the leading candidate for a pancreatin-dependent release system. In addition to containing a heavy chain, natural SF (produced by the silkworm Bombyx mori) also contains a light chain and the protein P25 at a molar weight ratio of 6:6:1. Since the SF heavy chain (325 kDa and 5,263 AA) is about 13 times heavier and 20 times longer than the SF light chain (25 kDa and 262 AA), we assume that the degradation kinetics of natural silk are predominantly defined by the cleavage characteristics of the SF heavy chain.
SF film was soluble in water due to the abundant random coil structure in the film. As a result, SF film would not be practical for oral drug delivery. One simple way to decrease the solubility of SF film in water is to increase the β-sheet structure using temperature-controlled water vapor annealing.19 However, water vapor annealing might not be practical during the capsule coating step because the saturated water vapor environment might adversely affect the capsule and the active pharmaceutical ingredient. Instead, we found that by adding DMSO to the SF solution, this film formulation would remain insoluble in water and other testing solutions with the exception of SIF with pancreatin. Because of this, the SF/DMSO film has the potential to be used for a coating for solid oral dosage forms. The penetration technique that we developed enabled us to conduct penetration and solubility tests for multiple film formulations in different solutions in parallel.
However, the SF/DMSO solution dried very slowly, which made it unsuitable for use as a coating. We explored using ES100, which is a commercially available enteric coating, as an adjuvant to facilitate coating. Besides, adding the single subcoating layer of EL100 for a non-water-soluble base did not affect the dissolution due to the more permissive pH range of EL100 (dissolution pH threshold at 6.020) compared to ES100 (dissolution pH threshold at 7.021). Moreover, the subcoatings are a widely used approach to improve characteristics of the desired drug coating.22 Note that the thickness of the coating layers was not sufficient to explain material variability, as the ES100 was significantly thicker than SF/ES100 (Supplementary Figure 3). The variability observed in performance is likely due to the variation introduced in the manufacturing methods. Furthermore, different coating aspects, i.e., a pH-independent subcoating, ratios of SF/ES100, alternative coating protocols (e.g., solvent-free coating), and mechanical dipping platforms or spray coating methods to help mitigate the variability in the coating, could be explored in the future to provide further improvements in performance.
The SF/ES100 coating successfully degraded in the presence of pancreatin but not in other conditions. However, the coating with ES100 of the control pills achieved higher release in SIF compared to SIF with pancreatin. This could be due to the variability observed with manual bench prototyping of the capsules and deeper characterization of alternative processes as noted above could help delineate further temporal difference in dissolution in the future. Furthermore, even though we observed that SF/ES100 coating can support pancreatin-specific release, the release percentage after 20 hours was relatively low. We anticipate that further formulation and capsule coating application across a range of thicknesses could help delineate and tune the release kinetics further in the future.
A porcine in vivo model was selected because the structure, length per body weight ratio, and digestive physiology of its small intestine is similar to humans.23 Intestinal administration of our novel enteric coating capsule informed whether the coatings are susceptible to degradation in the intestinal environment. Given the significantly slower gastric transit in swine compared to humans24, the swine model is suboptimal for the evaluation of gastric release. We tracked the release of radio-opaque steel beads using X-ray radiography which indicated that the coating would remain stable during intestinal transit for 3 hours after intestinal placement. This timeframe was chosen to assure that the coating would remain stable throughout small intestinal transit (2–3 hours) to facilitate colonic delivery.25 We confirmed the degradation in the GI tract by the scattering of the steel beads after 20 hours.
In summary, we have demonstrated that SF-based formulations can be used to coat capsules in order to enable pancreatin-dependent triggered release. This is a novel form of enteric coatings with alternative release mechanisms compared to classical pH-dependent systems. We believe that these coatings have the potential to be leveraged for future fail-safe oral drug delivery systems, even in challenging patient populations with impaired and highly variable intestinal physiology.
Supplementary Material
Acknowledgments
N.N. is financially supported by Chula Engineering Innovation Fund from Faculty of Engineering, Chulalongkorn University. D.R. is a Swiss National Science Foundation Fellow (grants P2EZP3_168827 and P300P2_177833) and supported by the NIH NIGMS grant R35GM151255. G.T. was supported in part by the Division of Gastroenterology at the Brigham and Women’s Hospital, the Karl van Tassel Career Professorship and Department of Mechanical Engineering, Massachusetts Institute of Technology. We thank the Koch Institute Swanson Biotechnology Center and the Animal Imaging and Preclinical Testing core for technical support. This work was supported in part by the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute and by the NIH grant EB000244.
Abbreviations
- API
Active pharmaceutical ingredient
- GI
Gastrointestinal
- SF
Silk fibroin
- EL100
Eudragit L100
- ES100
Eudragit S100
- DMSO
Dimethyl sulfoxide
- ddH2O
Double distilled water
- PS
Polystyrene
- SGF
Simulated gastric fluid
- SIF
Simulated intestinal fluid
- HPLC
High Performance Liquid Chromatography
- AA
Amino acids
References
- 1.Verma M, et al. A gastric resident drug delivery system for prolonged gram-level dosing of tuberculosis treatment. Sci. Transl. Med 2019;11:483. 10.1126/scitranslmed.aau6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibekwe VC, et al. An investigation into the in vivo performance variability of pH responsive polymers for ileo-colonic drug delivery using gamma scintigraphy in humans. J. Pharm. Sci 2006;95:12. 10.1002/jps.20742. [DOI] [PubMed] [Google Scholar]
- 3.Watts P, Smith A. TARGITTM technology: coated starch capsules for site-specific drug delivery into the lower gastrointestinal tract. Expert Opin. Drug Deliv 2005;2:159–167. 10.1517/17425247.2.1.159. [DOI] [PubMed] [Google Scholar]
- 4.Shin JM, Sachs G Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep 2008;10:528–534. 10.1007/s11894-008-0098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sivapragasam N, et al. Novel starch based nano scale enteric coatings from soybean meal for colon-specific delivery. Carbohydr. Polym 2014;111:273–279. 10.1016/j.carbpol.2014.04.091. [DOI] [PubMed] [Google Scholar]
- 6.Thilagavathi G, Viju S. Silk as a suture material. Advances in Silk Science and Technology. 2015;219–232. https://doi: 10.1016/b978-1-78242-311-9.00011-2. [DOI] [Google Scholar]
- 7.FDA. GRN No. 1026: Silk fibroin derived from Bombyx mori cocoons. 2022. Available at: https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory.
- 8.Xi H, Zhao H. Silk fibroin coaxial bead-on-string fiber materials and their drug release behaviors in different pH. J. Mater. Sci 2019; 54:4246–4258. 10.1007/s10853-018-3137-z. [DOI] [Google Scholar]
- 9.Tao X, et al. Synthesis of pH and glucose responsive silk fibroin hydrogels. Int. J. Mol. Sci 2021;22(13):7107. 10.3390/ijms22137107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan J, et al. pH-responsive silk fibroin-based CuO/Ag micro/nano coating endows polyetheretherketone with synergistic antibacterial ability, osteogenesis, and angiogenesis. Acta Biomater. 2020;115:220–234. 10.1016/j.actbio.2020.07.062. [DOI] [PubMed] [Google Scholar]
- 11.Jeshvaghani PA, et al. Synthesis and characterization of a novel, pH-responsive sustained release nanocarrier using polyethylene glycol, graphene oxide, and natural silk fibroin protein by a green nano emulsification method to enhance cancer treatment. Int. J. Biol. Macromol 2023;226:1100–1115. 10.1016/j.ijbiomac.2022.11.226. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Yao W, Rao Y, Lu X, Gao J. pH-Responsive carriers for oral drug delivery: challenges and opportunities of current platforms. Drug Deliv. 2017;24:569–581. 10.1080/10717544.2017.1279238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu X, Cebe P, Weiss AS, Omenetto F, Kaplan DL. Protein-based composite materials. Mater. Today 2012;15:208–215. 10.1016/S1369-7021(12)70091-3. [DOI] [Google Scholar]
- 14.Consortium UniProt. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2018;47:D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boutet E, et al. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Methods Mol. Biol 2016;1374:23–54. 10.1007/978-1-4939-3167-5_2. [DOI] [PubMed] [Google Scholar]
- 16.Gibb S Cleaver: cleavage of polypeptide sequences. R package version 1.26.0. 2020. Available at: https://github.com/sgibb/cleaver/. [Google Scholar]
- 17.Gasteiger E, et al. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook. 2005;571–607. 10.1385/1-59259-890-0:571. [DOI] [PubMed] [Google Scholar]
- 18.Kim U-J, Park J, Kim HJ, Wada M, Kaplan DL. Three-dimensional aqueous-derived biomaterial scaffolds from silk fibroin. Biomaterials. 2005;26:2775–2785. 10.1016/j.biomaterials.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, et al. Regulation of silk material structure by temperature-controlled water vapor annealing. Biomacromolecules. 2011;12(5):1686–1696. 10.1021/bm200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pharma Excipients. EUDRAGIT® L 100. 2023. Available at: https://www.pharmaexcipients.com/product/eudragit-l-100/.
- 21.Pharma Excipients. EUDRAGIT® S 100. 2023. Available at: https://www.pharmaexcipients.com/product/eudragit-s-100/.
- 22.Seo KS, Bajracharya R, Lee SH, Han HK. Pharmaceutical application of tablet film coating. Pharmaceutics. 2020;12(9):853. 10.3390/pharmaceutics12090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler A, Gonzalez L, Blikslager A. Large animal models: the key to translational discovery in digestive disease research. Cell. Mol. Gastroenterol. Hepatol 2016;2:716–724. 10.1016/j.jcmgh.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoyagi N, et al. Gastric emptying of tablets and granules in humans, dogs, pigs, and stomach-emptying-controlled rabbits. J Phar Sci. 1992; 81(12):1170–1174. 10.1002/jps.2600811208. [DOI] [PubMed] [Google Scholar]
- 25.Davis SS, Hardy JG, Fara JW Transit of pharmaceutical dosage forms through the small intestine. Gut. 1986;27:886–892. 10.1136/gut.27.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, et al. Ingestible hydrogel device. Nat. Commun 2019;10:493. 10.1038/s41467-019-08355-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.