

Abstract
Background
Although hepatitis B virus (HBV) infection is a major risk factor for hepatic cancer, the majority of HBV carriers do not develop this lethal disease. Additional molecular alterations are thus implicated in the process of liver tumorigenesis. Since phosphatase and tensin homolog (PTEN) is decreased in approximately half of liver cancers, we investigated the significance of PTEN deficiency in HBV-related hepatocarcinogenesis.
Methods
HBV-positive human liver cancer tissues were checked for PTEN expression. Transgenic HBV, Alb-Cre and Ptenfl/fl mice were inter-crossed to generate WT, HBV, Pten−/− and HBV; Pten−/− mice. Immunoblotting, histological analysis and qRT-PCR were used to study these livers. Gp73−/− mice were then mated with HBV; Pten−/− mice to illustrate the role of hepatic tumor biomarker golgi membrane protein 73 (GP73)/ golgi membrane protein 1 (GOLM1) in hepatic oncogenesis.
Results
Pten deletion and HBV transgene synergistically aggravated liver injury, inflammation, fibrosis and development of mixed hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC). GP73 was augmented in HBV; Pten−/− livers. Knockout of GP73 blunted the synergistic effect of deficient Pten and transgenic HBV on liver injury, inflammation, fibrosis and cancer development.
Conclusions
This mixed HCC-ICC mouse model mimics liver cancer patients harboring HBV infection and PTEN/AKT signaling pathway alteration. Targeting GP73 is a promising therapeutic strategy for cancer patients with HBV infection and PTEN alteration.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-024-04976-4.
Keywords: Hepatocellular carcinoma, Intrahepatic cholangiocarcinoma, Hepatitis B virus, Pten, GP73
Background
Liver cancer is the third leading cause of cancer death worldwide [1, 2]. It comprises hepatocellular carcinoma (HCC), intrahepatic cholangiocarcinoma (ICC), mixed HCC-ICC and other neoplasms [3, 4]. HCC is the dominant primary liver cancer type, accounting for 90% of all cases. ICC is the second common malignancy in liver, with an incidence of approximately 6% of all primary hepatic tumors. It has poorer prognosis than HCC. Hepatobiliary cancer is an infrequent subtype of primary liver cancers, which exhibits much worse prognosis than either HCC or ICC [5, 6]. Once diagnosed, the treatment is limited and the outcome is poor [7]. Since its pathological mechanism is largely unknown, clinically relevant animal models are in need to mimic the initiation and progression of mixed HCC-ICC.
Viral infection is a major risk factor for primary liver cancer. Following chronic infection of hepatitis B virus (HBV) or hepatitis C virus (HCV), hepatocytes experience injury, inflammation, fibrosis, cirrhosis, and eventually carcinogenesis [6–8]. In the United States, Europe, and Japan, most of liver cancers are associated with HCV infection. Due to excellent virological response with antiviral drugs, the risk of developing HCV infection-associated liver cancer has declined substantially. In contrast, HBV infection is still the most prominent risk factor for HCC development [7]. In China, some regions of Asia and Africa, HBV-related liver cancer is prevalent [9]. Half of global liver cancer occurs in China because up to 10% of its population are HBV carriers [10–12]. Although liver cancer development is highly correlated with HBV infection, only a small percentage of HBV carriers finally develop liver cancer after a long latency [13, 14]. This suggests that additional molecular alterations are involved in HBV-related liver cancer progression. Therefore, illumination of the essential molecular events that triggered this malignant transformation may help us gain insight into the mechanisms and potential treatment for HBV-associated liver cancer.
More than half of hepatic cancers exhibit reduced expression of phosphatase and tensin homolog (PTEN) [15, 16]. Low expression of PTEN confers poor prognosis of HCC patients [17]. PTEN exercises its tumor suppressor function mainly by antagonizing phosphatidylinositol 3-kinase (PI3K) pathway through dephosphorylation of PIP3 [18]. Loss of PTEN activates mechanistic target of rapamycin (mTOR) through active AKT serine/threonine kinase (AKT)-mediated suppression of TSC1 and 2 complex, a suppressor of mTOR [19, 20]. Liver-specific Pten deletion mice develop liver cell adenomas at 11 months of age and HCC by 18.5–19.5 months of age [21]. The long latency of cancer initiation and development implies that additional genetic and/or epigenetic alterations likely take place in the liver in the absence of Pten. It remains elusive for the role of Pten loss in HBV-associated liver cancer initiation and progression.
Golgi membrane protein 73 (GP73)/Golgi membrane protein 1 (GOLM1) is a type II Golgi transmembrane protein [22]. It is a serum marker for liver cancer. Serum GP73 level increases gradually during the process of hepatic HBV infection, cirrhosis and cancer [23–25]. With subcutaneous tumor formation and tail-vein injection assays, we demonstrate that overexpression of GP73 is critical for xenografting HCC tumorigenesis and metastasis triggered by mTOR activation in immune-deficient nude mice [26]. Using human HCC cell lines in culture and in nude mice, Ye et al. showed that GP73 promoted HCC metastasis by modulating epidermal growth factor receptor (EGFR)/RTK cell-surface recycling. Furthermore, the abundance of GP73 is well correlated with human HCC metastasis and poor survival of HCC patients [27]. Although augmented GP73 is highly correlated with HBV infection and HCC, the involvement of GP73 in HBV-mediated primary liver cancer development in immune-competent mice remains to be documented.
The genome of HBV encodes four core proteins, including HBV surface antigen (HBsAg), core antigen (HBcAg), DNA polymerase, and X protein (HBx) [28]. Since mouse does not have receptor for HBV, HBV infection does not occur in mice. HBV transgenic mice which contain pre-S, S, and X domains of HBV genome develop primary liver cancer around the age of 12 months [29, 30]. Even though HBV infection is not involved in the disease progression in transgenic mice, severe liver injury and inflammation still simulate the pathogenesis in liver cancer patients with HBV infection. Thus, this mouse model is widely used to illustrate the pathologic and molecular events of HBV-mediated tumorigenesis [31–33]. To simulate liver cancer patients with both HBV infection and activation of PI3K/AKT/mTOR signaling pathway in mice, we disrupted Pten in the livers of HBV transgenic mice and investigated the role of GP73 in liver tumor development of HBV; Pten−/− mice.
Methods
Generation of HBV; Pten−/− and HBV; Pten−/−; GP73−/− mice
HBV mice (stock no. 002226) were described previously [29]. Alb-Cre (stock no. 003574), Ptenfl/fl (stock no. 006068) and EIIA-Cre (stock no. 003724) mice were from Jackson Laboratory (Bar Harbor, ME, USA). HBV and Alb-Cre mice were respectively crossed with Ptenfl/fl mice to generate HBV; Ptenfl/fl and Alb-Cre; Ptenfl/fl mice. By intercrossing HBV; Ptenfl/fl mice with Alb-Cre; Ptenfl/fl mice, HBV; Alb-Cre; Ptenfl/fl (HBV; Pten−/−), Alb-Cre; Ptenfl/fl (Pten−/−), HBV; Ptenfl/fl (HBV) and Ptenfl/fl (WT) mice were generated. GP73fl/+ mice were developed by Beijing Biocytogen (China) and were mated with EIIA-Cre mice to generate global GP73 knockout (GP73−/−) mice. To obtain HBV; Pten−/−; GP73−/− mice, HBV; Pten−/− mice were crossed with GP73−/− mice. All mice were in C57BL/6 background and maintained in a pathogen-free facility under a 12-h light/dark cycle with appropriate temperature and humidity. The genotypes of mice were determined by PCR analysis of tail genomic DNA. The primers were shown in Table 1. The animal experiments were approved by Animal Care and Use Committee of Peking Union Medical College and performed in accordance with international guidelines. All of the mice used for this study are male unless as specified.
Table 1.
Genotyping primer sequences
| Primer name | Sequences |
|---|---|
| Cre-F | TGGGCGGCATGGTGCAAGTT |
| Cre-R | CGGTGCTAACCAGCGTTTTC |
| HBV-1 | AACATGGAGAACATCACATC |
| HBV-2 | AGCGATAACCAGGACAAGTT |
| HBV-3 | ATGTACTGGTCCCGCATGGC |
| HBV-4 | TTTGCAGGACTCCTACCGG |
Human liver cancer specimens
Human liver cancer tissues and adjacent normal liver tissues were obtained from the patients undergoing resection at Peking Union Medical College Hospital. Written informed contents were obtained from all patients. All of these patients were HBV positive.
Histological analysis
Mouse liver tissues were fixed in 4% formalin. 5 μm of paraffin-embedded sections of the same liver lobes from different mice were subjected to hematoxylin–eosin (H&E) staining through the following procedures: 1. Dewaxing and hydration with environmental friendly Dewaxing Transparent Liquid, Anhydrous ethanol, 75% Ethyl alcohol and tap water; 2. Fixing with tissue fixation solution; 3. Hematoxylin staining; 4. Eosin staining; 5. Dehydration and sealing. Liver fibrosis was determined by Masson’s trichrome staining kit (Sigma-Aldrich, HT15, St Louis, USA), according to the manufacturers’ protocols. For immune-histochemical analysis, the paraffin sections were subjected to dewaxing to water, antigen repair, endogenous peroxidase blocking, serum closure, primary antibody and secondary antibody incubation, DAB color development and nuclus restaining. Antibody information was listed as follow: CK19 (1:500 dilution, Servicebio, 11197, Wuhan, China), Heppar-1 (1:500 dilution, ZSGB-BIO, 0131, Shanghai, China), CD34 (1:500 dilution, Abcam, 81289, Cambridge, UK), and Cleaved-caspase 3 (1:200 dilution, Cell Signaling Technology, #9664, Danvers, USA). Quantification of fibrotic areas and microvessel areas was determined by Image J software.
Immunoblotting
Immunoblotting of liver tissues was processed according to the protocols as described previously [26]. Liver tissues were prepared in lysis buffer (2% SDS, 10% glycerol, 10 mM Tris (pH 6.8), and 100 mM DTT) containing Protease and Phosphatase Inhibitor Cocktail (#P002, New cell & molecular, Suzhou, China). Equal amounts of total proteins were separated on 8–12% SDS-PAGE gels, followed by immunoblotting onto the PVDF membranes. Membranes were first incubated with primary antibodies overnight at 4 °C, then incubated with secondary antibodies for 2 h at room temperature. Primary antibodies against PTEN (#9559), pAKT (#4060), AKT (#4691), cleaved-caspase 3 (#9664), JNK (#9252), pSTAT3 (#9131), STAT3 (#9137) were purchased from Cell Signaling Technology, pJNK (131499) was from Abcam, mouse GP73 (customized), GAPDH (AC001) were from Abclonal (Wuhan, China), and human GP73 (66331-1-Ig) was from Proteintech (Rosemont, USA). The detection of protein bands was carried out using LI-COR Odyssey Infrared Scanner.
RNA preparation and quantitative Real-Time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from mouse liver tissues using Trizol (Invitrogen, Waltham, USA) and Ultrapure RNA kit (CWBIO, Taizhou, China), according to the manufacturers’ protocols. 1 μg of total RNA was reverse-transcribed using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (TOYOBO, FSQ-301, Osaka, Japan). qRT-PCR was performed on a Bio-Rad IQ5 machine with SYBR Green PCR Master Mix (TransGen Biotech, AQ141, Beijing, China). The procedures of qPCR assay were as follow: 1. 95 ℃ for 3 min; 2. 95 ℃ for 15 s; 3. 60℃ for 1 min + Plate Read; 4. GOTO 2, 40 cycles; 5. Melt Curve 55–95 ℃, increment 0.5 ℃ For 0:10 + Plate Read. GAPDH served as internal control. The primer sequences were listed in Table 2.
Table 2.
qPCR primer sequences
| Gene name | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| Tgfb1 | CTTCCCGAATGTCTGACGTA | GACCGCAACAACGCCATCT |
| Ctgf | TGACCTGGAGGAAAACATTAAGA | AGCCCTGTATGTCTTCACACTG |
| Il6 | TTCCATCCAGTTGCCTTCTTGG | TTCTCATTTCCACGATTTCCCAG |
| Gapdh | CATGGAGAAGGCTGGGGCTC | AACGGATACATTGGGGGTAG |
Biochemical analysis
Blood of mice was collected and centrifuged at 3000 rpm for 5 min. Supernatants were collected and sent to the clinical laboratory at Peking Union Medical College Hospital for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) measurement.
Statistical analysis
Relative mRNA expression, tumor free, mouse survival, tumor burden and histological results were analyzed by GraphPad Prism 9.0 software. Data were shown as mean ± SD. Tumor free or survival was compared using a log-rank (Mantel-Cox) test. Statistical significance between two groups was subjected to Student’s two-tailed t tests. One-way ANOVA was used to determine the difference more than two groups. *p < 0.05 was considered statistically significant.
Results
PTEN expression is reduced in HBV-related liver cancer
To explore the relevance of PTEN in HBV-related liver cancer, the expression of PTEN was checked in 36 pairs of tumors and adjacent tissues of HBV-positive HCC patients. Comparing with adjacent tissues, PTEN expression was decreased in 72.2% (26/36) cancer samples (Fig. 1).
Fig. 1.

Reduction of PTEN expression in human liver cancer tissues. Immunoblotting analysis of human liver cancer and adjacent tissues. T tumor, N adjacent tissues
Knocking out Pten accelerates HBV-induced liver cancer development
We next explored the involvement of PTEN in the progression of HBV-related HCC. Initially, we noticed reduced PTEN and increased pAKT in 4-month-old HBV transgenic mouse livers comparing with wildtype (WT) mouse livers. Suppressed PTEN expression and increased AKT phosphorylation were more prominent in 9-month-old HBV livers (Fig. 2A). Furthermore, PTEN decreased and pAKT increased in 14-month-old HBV transgenic mouse liver tumor compared to adjacent tissues (Fig. 2B). We thus investigated the role of PTEN in HBV-mediated hepatocarcinogenesis. Ptenfl/fl mice were crossed with HBV and Alb-Cre mice to generate HBV; Ptenfl/fl and Alb-Cre; Ptenfl/fl mice. After mating HBV; Ptenfl/fl mice with Alb-Cre; Ptenfl/fl mice, mice with 4 genotypes were generated, including Ptenfl/fl (WT), HBV; Ptenfl/fl (HBV), Alb-Cre; Ptenfl/fl (Pten−/−), and HBV; Alb-Cre; Ptenfl/fl (HBV; Pten−/−) mice (Additional file 1: Fig. S1). At age of 5 months, neither Pten−/− nor HBV mice developed visible liver tumors while small lumps were observed in HBV; Pten−/− mouse livers (Fig. 2C). At 8 months age, all (25/25) of HBV; Pten−/− mice presented multiple macroscopic tumor foci, whereas none of HBV mice and only 8.3% (2/24) Pten−/− mice exhibited visible tumors (Fig. 2C, D). All of (24/24) Pten−/− or (21/21) HBV mice developed tumors by 11 months or 14 months, respectively (Fig. 2D). Comparing with WT, HBV, and Pten−/− mice, the liver weight to body weight ratio was highest in HBV; Pten−/− mice at all indicated time points (p < 0.05, Fig. 2E). A total of 9.85 ± 4.63 tumors were presented in 9-month-old HBV; Pten−/− mice, while only 0.67 ± 1.07 were formed tumors in Pten−/− mice (p < 0.001, Fig. 2F). The maximal tumor sizes were 9.71 ± 4.25 mm in 9-month-old HBV; Pten−/− mice and 1.90 ± 2.25 mm in Pten−/− mice (p < 0.001, Fig. 2G). HBV; Pten−/− mice began to die at the age of 9 months and none of these mice survived beyond 14 months. Only 22% (4/18) of Pten−/− mice and 21% (4/19) of HBV mice died up to 14 months (Fig. 2H). Taken together, Pten deficiency accelerates the initiation and progression of HBV-induced liver cancers and confers these tumor-bearing mice with poor prognosis.
Fig. 2.

Pten deficiency promotes liver tumor formation of HBV transgenic mice. A Immunoblotting analysis of the livers of WT mice and HBV transgenic mice. B Immunoblotting analysis of liver tumors and their adjacent liver tissues in HBV transgenic mice. T: tumor, N: adjacent non-tumor tissues. C Representative images of livers from WT, HBV, Pten−/−, and HBV; Pten−/− mice. D Tumor free of WT, HBV, Pten−/−, and HBV; Pten−/− mice (n = 21–25). E Ratio of liver weight to body weight for WT, HBV, Pten−/−, and HBV; Pten−/− mice with various ages (n = 3–6). F Number (> 3 mm) and G maximal size of liver tumors in 9-month-old Pten−/− (n = 12), and HBV; Pten−/− (n = 13) mice. H Survival of WT, HBV, Pten−/−, and HBV; Pten.−/− mice (n = 18–20). Data were shown as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001
Enhanced fibrosis, micro-vessel formation and combined HCC-ICC in HBV; Pten−/− mouse livers
To document liver cancer progression in mice, we performed histological analysis of WT, HBV, Pten−/−, and HBV; Pten−/− livers at different stages. Four-month-old HBV; Pten−/− livers were presented with characteristics of bile duct hyperplasia and inflammatory cell infiltration. The pathological difference between HBV; Pten−/− livers and other genotypic livers became wider at the age of 9 months (Fig. 3A). Masson and CD34 staining showed increased liver fibrosis and micro-vessel in HBV; Pten−/− livers (p < 0.001, Fig. 3B, C). Comparing with other three genotypic livers, HBV; Pten−/− livers had higher expression of fibrosis-associated genes Tgfb1 and Ctgf (p < 0.05, Fig. 3D). HCCs and ICCs were more abundant in 9-month-old HBV; Pten−/− mice as tumor samples were positive for hepatocyte paraffin 1 (Heppar-1) (HCC biomarker) and CK19 (ICC biomarker) (Fig. 3E) [34]. Collectively, Pten deletion accelerates the initiation and progression of HCC and ICC in HBV mice.
Fig. 3.
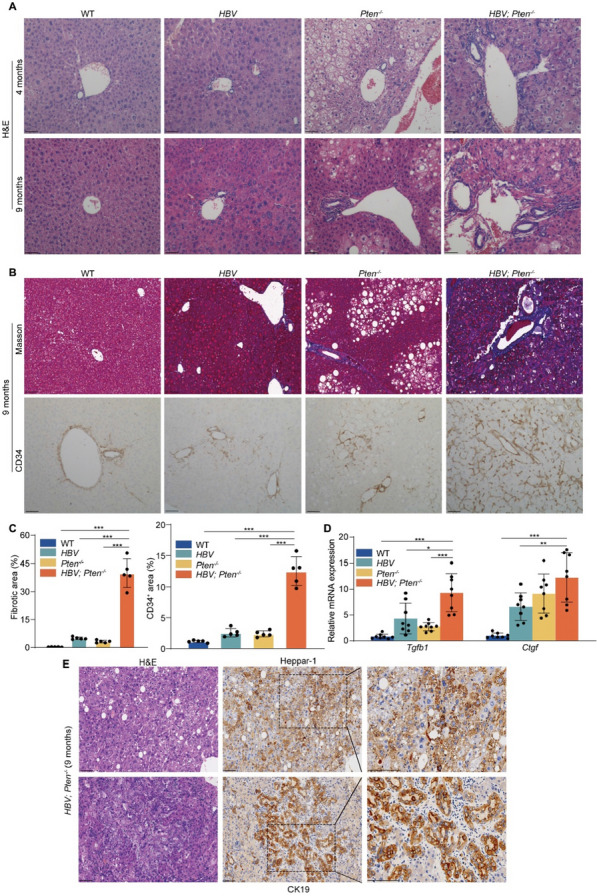
Tumors in HBV; Pten−/− mice are mixed HCC-ICC. A H&E staining of livers from WT, HBV, Pten−/− and HBV; Pten−/− mice. B Masson’s trichrome and CD34 staining of 9-month-old WT, HBV, Pten−/−, and HBV; Pten−/− mouse livers. C Quantitation of Masson’s trichrome and CD34-positive area (n = 5). D Tgfb1 and Ctgf mRNA expression in 9-month-old WT, HBV, Pten−/−, and HBV; Pten−/− livers (n = 8–10). E H&E, Heppar-1 and CK19 staining of 9-month-old HBV; Pten.−/− livers. Scale bar, 100 μm. Data were shown as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001
HBV; Pten−/− mice exhibit enhanced hepatocyte injury, apoptosis and inflammation
To assess liver injury, ALT and AST were analyzed in the serum of mice with 4 genotypes at the ages of 4 and 9 months. ALT and AST were higher in HBV or Pten−/− mice than in WT mice (p < 0.01, Fig. 4A). The highest levels of ALT and AST were observed in HBV; Pten−/− mice (p < 0.01, Fig. 4A). Augmented Cleaved-caspase 3 and pJNK in 4-month-old HBV; Pten−/− mouse livers indicated increased apoptosis (p < 0.01, Fig. 4B, C). Additionally, elevated signal transducer and activator of transcription 3 (STAT3) activity and interleukin 6 (IL6) level indicated severer inflammation in HBV; Pten−/− mouse livers (p < 0.001, Fig. 4D, E). Collectively, Pten deficiency boosts liver injury, hepatocyte apoptosis and inflammation in HBV transgenic mice.
Fig. 4.

Increased hepatocyte injury, apoptosis and inflammation in HBV; Pten−/− livers. A Serum ALT and AST of WT, HBV, Pten−/−, and HBV; Pten−/− mice. B Cleaved-caspase 3 staining of 4-month-old WT, HBV, Pten−/−, and HBV; Pten−/− livers. C and D Immunoblotting analysis of 4-month-old WT, HBV, Pten−/−, and HBV; Pten.−/− livers. E IL6 mRNA level was determined by qRT-PCR (n = 7–10). Scale bar, 100 μm. Data were shown as mean ± SD, **p < 0.01, ***p < 0.001
Removal of GP73 suppresses liver injury, inflammation, and tumor development in HBV; Pten−/− mice
To explore the involvement of GP73 in the HCC progression in HBV; Pten−/− mice, we checked the protein expression of GP73 in the mouse livers with different genotypes. GP73 abundance was slightly elevated in HBV or Pten−/− livers and dramatically increased in HBV; Pten−/− livers (Fig. 5A, upper panel). GP73 level was further increased in liver tumors relative to adjacent tissues of 9-month-old HBV; Pten−/− mice (Fig. 5A, lower panel).
Fig. 5.

GP73 knockout blocks tumor development of HBV; Pten−/− mice. A Immunoblotting analysis of 4-month-old WT, HBV, Pten−/−, and HBV; Pten−/− livers (upper panel) and tumors and adjacent liver tissues from 9-month-old HBV; Pten−/− mice (lower panel). T: tumor, N: adjacent tissues. B–D 9-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73−/− mice. B The ratio of liver weight to body weight of mice (n = 15). C Representative images of livers. D Number (> 3 mm) and maximal size of liver tumors (n = 15). E Immunoblotting analysis of GP73 and cleaved-caspase 3 of 4-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73−/− livers. F Masson’s trichrome, CD34 and Cleaved Caspase 3 staining of 9-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73−/− livers. G Serum ALT and AST of 4-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73−/− mice (n = 10–11). H Immunoblotting analysis of 4-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73−/− livers. I qRT-PCR analysis of IL6 mRNA level in 4-month-old HBV; Pten−/− and HBV; Pten−/−; Gp73.−/− livers (n = 10). Scale bar, 100 μm. Data were shown as mean ± SD, **p < 0.01, ***p < 0.001
To determine whether overexpressed GP73 contributes to accelerated tumor development in HBV; Pten−/− mice, global GP73 knockout (Gp73−/−) mice were crossed with HBV; Pten−/− mice to generate HBV; Pten−/− and HBV; Pten−/−; Gp73−/− mice. The liver to body weight ratio of HBV; Pten−/−; Gp73−/− mice was 20% lower than that of HBV; Pten−/− mice (p < 0.01, Fig. 5B). Furthermore, GP73 knockout profoundly reduced the tumor number (HBV; Pten−/− vs HBV; Pten−/−; Gp73−/−: 11 ± 4.23 vs 4.87 ± 3.18) and size (HBV; Pten−/− vs HBV; Pten−/−; Gp73−/−: 10.54 ± 3.73 vs 4.98 ± 2.50) of HBV; Pten−/− mice (p < 0.01, Fig. 5C, D). Thus, knockout of GP73 prevents hepatocarcinogenesis of HBV; Pten−/− mice.
To further analyze the role of GP73 in liver tumorigenesis, we examined the molecular and pathologic changes imposed by GP73 knockout in the livers of HBV; Pten−/− mice. Comparing with HBV; Pten−/− livers, decreased cleaved-caspase 3 indicated reduced cell apoptosis in HBV; Pten−/−; Gp73−/− livers (Fig. 5E, F). GP73 knockout also alleviated fibrosis and micro-vessel formation in HBV; Pten−/− livers (Fig. 5F). In addition, liver function was improved in HBV; Pten−/−; Gp73−/− mice (p < 0.01, Fig. 5G). The enhanced STAT3 phosphorylation and augmented IL6 in HBV; Pten−/− livers were reversed by GP73 knockout (p < 0.001, Fig. 5H, I). Our data suggest that GP73 promotes liver cancer through participation of hepatocyte injury, apoptosis and inflammation in HBV; Pten−/− mice.
Association between the expression of PTEN and GP73 and the prognosis of HCC patients
To seek the clinical relevance of PTEN and GP73 in human liver cancer, we analyzed patient survival and the expression of PTEN and GP73 in liver cancer database from KM Plotter website. We found that reduced PTEN and elevated GP73 associated with poor prognosis of HCC patients (Fig. 6).
Fig. 6.

Association between the expression of PTEN and GP73 and the prognosis of HCC patients. A–B The overall survival of HCC patients with various expression of PTEN A (p = 0.0048) or GP73 B (p = 0.017) from KM Plotter website
Discussion
To investigate the involvement of aberrant PTEN-PI3K/AKT/mTOR signaling cascade in HBV-associated liver cancer, we studied cancer development of liver-specific HBV transgenic and Pten deletion mice. HBV transgenic mice developed tumors between 10 and 14 months while hepatic Pten disruption caused tumors between 8 to 11 months. In contrast, HBV; Pten−/− mice developed mixed HCC-ICC between 5 to 8 months. All HBV; Pten−/− mice died between 9 to 14 months, whereas 22% (4/18) of Pten−/− mice died between 12 to 14 months and 21% (4/19) of HBV mice died between 12.5 and 14 months. Boosted GP73 was critical for the accelerated liver tumor formation as removal of GP73 alleviated the tumor burden in HBV; Pten−/− mice.
Although prolonged HBV infection is the most frequent etiology in liver cancer, only a small fraction of HBV carriers eventually develop liver cancer after hepatic injury, inflammation, fibrosis and cirrhosis [9]. Additional molecular alterations are thus required for the malignant transformation of HBV infection. Various alterations of proto-oncogenes and tumor suppressors cause malfunction of PTEN-PI3K/AKT/mTOR signaling pathway in hepatic cancer [15, 16]. PTEN is the second most frequently altered tumor suppressor in human cancers [35]. PTEN expression is reduced in liver cancer which may be due to PTEN genetic mutations, transcriptional and post-transcriptional regulation alterations, and protein degradation [36]. Patients with reduced PTEN expression in liver cancer had poor prognosis [17]. Loss of Pten in mouse liver causes HCC around 18.5 months of age [21]. Pten inactivation causes ICC more frequently than HCC [37]. PTEN is a suppressor of AKT activity. AKT pathway alterations were observed in 96.5% of the tumors in HBV transgenic mice [31]. We and others identified that activation of β-catenin, AKT and c-jun are the important contributors of HCC development of HBV transgenic mice [31, 32, 38]. In this study, we found that liver-specific Pten deletion accelerated HBV-mediated liver tumor initiation and progression. In line with the reported findings [29, 30], HBV mice developed infrequent HCC around the age of 14 months. Half of Pten−/− mice developed tumors by the age of 9 months. In contrast, liver tumors emerged at the age of 5 months and became malignant by the age of 8 months in HBV; Pten−/− mice. These tumors exhibited pathologic characteristics of combined HCC-ICC. Compared to WT, HBV or Pten−/− livers, HBV; Pten−/− livers had enhanced apoptosis and severer liver injury. Increased phosphorylation of JNK was observed in HBV; Pten−/− livers. Since JNK activation is a well-known apoptotic inducer in liver [39], it may partly contribute to the enhanced cell death in HBV; Pten−/− livers. HBV; Pten−/− mice not only illuminate the significance of Pten inactivation in HBV-associated liver tumor development but also represent a suitable model to investigate the pathologic and molecular mechanisms for mixed HCC-ICC.
Liver injury is usually followed by sustained inflammation and fibrosis. Fibrosis is a major pathological process of liver cancer [40]. IL-6 plays important role in chronic inflammation and has been demonstrated to promote diethylnitrosamine (DEN)-induced HCC development [41]. IL6 was increased in HBV; Pten−/− livers. STAT3, a downstream target of IL6 [42], was also activated in HBV; Pten−/− livers. Although no significant fibrosis was found in the livers of HBV or Pten−/− mice, HBV; Pten−/− livers had severe fibrosis. Thus, transgenic HBV and Pten deficiency synergistically promote hepatocyte death, liver injury, inflammation and fibrosis.
We present here that reduced PTEN and elevated GP73 associated with poor prognosis of HCC patients. GP73 expression is elevated by virus infection [22]. We and others have demonstrated that serum GP73 is a biomarker for liver cancer as its level increases during the progression of HBV infection, hepatic cirrhosis and tumor formation [23–25]. We reported that GP73 is an mTOR downstream effector and critical for xenografting HCC development and metastasis in immune-deficient nude mice [26]. GP73 also promotes xenografting HCC metastasis by regulating EGFR/RTK cell-surface recycling [27]. However, these studies were conducted on cellular experiments and xenografting tumors in nude mice. The role of GP73 in orthotropic liver cancer should be studied in immune-competent mice. We presented here that GP73 expression was enhanced in the livers and even higher in hepatic tumors of HBV; Pten−/− mice. To study the involvement of GP73 in HBV-mediated orthotropic liver cancer development in immune-competent mice, we deleted GP73 in HBV; Pten−/− mice. Compared with HBV; Pten−/− mice, HBV; Pten−/−; Gp73−/− mice had smaller and fewer tumor foci, indicating that GP73 is critical for the accelerated liver tumor formation in HBV; Pten−/− mice. In accordance with the important role of GP73 in inflammation, knockout of GP73 reduced hepatocyte apoptosis and liver injury of HBV; Pten−/− mice. Fibrosis was suppressed when GP73 was knocked out in HBV; Pten−/− mice. Activated STAT3 and up-regulated IL6 were inhibited in HBV; Pten−/−; Gp73−/− livers. These results explain why GP73 knockout blunted the synergistic effect of HBV transgene and Pten deficiency in promoting liver tumor initiation and progression. Therefore, potential inhibitors for GP73 expression and/or function such as GP73 antibodies may benefit liver cancer patients [26, 43].
Conclusions
Our study provides the evidence of synergistic effect of HBV transgene and Pten deficiency in promoting mixed HCC-ICC initiation and progression in mice. Knockout of GP73 alleviates liver injury, inflammation and fibrosis, finally resulting in suppression of liver tumor development. HBV; Pten−/− mouse is a good animal model for drug screening of liver cancer with PTEN dysregulation and HBV infection. Since GP73 is a promising target for liver cancer treatment, therapeutic regimens against GP73 should be developed and tested preclinically and clinically.
Supplementary Information
Additional file 1: Figure S1. PCR genotyping of mouse tail genomic DNA.
Acknowledgements
Not applicable.
Abbreviations
- HBV
Hepatitis B virus
- HCC
Hepatocellular carcinoma
- ICC
Intrahepatic cholangiocarcinoma
- HCV
Hepatitis C virus
- PTEN
Phosphatase and tensin homolog
- PI3K
Phosphatidylinositol 3-kinase
- GP73
Golgi membrane protein 73
- GOLM1
Golgi membrane protein 1
- H&E
Hematoxylin–eosin
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- DEN
Diethylnitrosamine
- mTOR
Mechanistic target of rapamycin kinase
- AKT
AKT serine/threonine kinase
- TSC1/TSC2
TSC complex subunit 1/2
- EGFR
Epidermal growth factor receptor
- STAT3
Signal transducer and activator of transcription 3
- IL6
Interleukin 6
Author contributions
FH, JG, FL and HZ designed the study and coordinated experiments. FH, JG, FL, NZ, MH, XG, SY, PC, YW, QM, QZ, LL, YJ, DJ, ZH, XZ and HZ performed experiments and analyzed the data. FH, MH, FL and HZ wrote, reviewed and edited the manuscript. YM, HY, and YW provided human tissues. HW provided the described mouse model. All authors reviewed the manuscript.
Funding
This work was supported by Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS) (2021-1-I2M-018), Haihe Laboratory of Cell Ecosystem Innovation Fund (22HHXBSS00012), National Natural Science Foundation of China (81730078, 81872287), Beijing Natural Science Foundation (IS23095) and State Key Laboratory Special Fund (2060204).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Declarations
Ethics approval and consent to participate
All animal studies were performed following the protocols approved by the Animal Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College (ethical code: ACUC-A01-2022-042).
Consent for publication
Consent to publish has been obtained from all authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Fuqiang Huang, Jing Guo, Na Zhao, and Mengjie Hou have contributed equally to this work.
Contributor Information
Fangming Liu, Email: lfmpumc@163.com.
Hongbing Zhang, Email: hbzhang@ibms.pumc.edu.cn.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Singal AG, Kanwal F, Llovet JM. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat Rev Clin Oncol. 2023;20:864–884. doi: 10.1038/s41571-023-00825-3. [DOI] [PubMed] [Google Scholar]
- 3.Beaufrère A, Calderaro J, Paradis V, et al. Combined hepatocellular-cholangiocarcinoma: an update. J Hepatol. 2021;74:1212–1224. doi: 10.1016/j.jhep.2021.01.035. [DOI] [PubMed] [Google Scholar]
- 4.Rumgay H, Ferlay J, de Martel C, Georges D, Ibrahim AS, Zheng R, Wei W, Lemmens V, Soerjomataram I. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108–118. doi: 10.1016/j.ejca.2021.11.023. [DOI] [PubMed] [Google Scholar]
- 5.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152:745–761. doi: 10.1053/j.gastro.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345–1362. doi: 10.1016/S0140-6736(22)01200-4. [DOI] [PubMed] [Google Scholar]
- 7.Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 8.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 9.Hsu YC, Huang DQ, Nguyen MH. Global burden of hepatitis B virus: current status, missed opportunities and a call for action. Nat Rev Gastroenterol Hepatol. 2023;20:524–537. doi: 10.1038/s41575-023-00760-9. [DOI] [PubMed] [Google Scholar]
- 10.Yu C, Song C, Lv J, Zhu M, Yu C, Guo Y, Yang L, Chen Y, Chen Z, Jiang T, et al. Prediction and clinical utility of a liver cancer risk model in Chinese adults: a prospective cohort study of 0.5 million people. Int J Cancer. 2021;148:2924–2934. doi: 10.1002/ijc.33487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L, Yang Y, Li Y, Li Y, Zhang B, Shi R. The past, present, and future of liver cancer research in China. Cancer Lett. 2023;574:216334. doi: 10.1016/j.canlet.2023.216334. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Cao M, Lei L, Yang F, Li H, Yan X, He S, Zhang S, Teng Y, Xia C, Chen W. Burden of liver cancer: from epidemiology to prevention. Chin J Cancer Res. 2022;34:554–566. doi: 10.21147/j.issn.1000-9604.2022.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosaka T, Suzuki F, Kobayashi M, Seko Y, Kawamura Y, Sezaki H, Akuta N, Suzuki Y, Saitoh S, Arase Y, et al. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology. 2013;58:98–107. doi: 10.1002/hep.26180. [DOI] [PubMed] [Google Scholar]
- 14.Jiang Y, Han Q, Zhao H, Zhang J. The mechanisms of HBV-induced hepatocellular carcinoma. J Hepatocell Carcinoma. 2021;8:435–450. doi: 10.2147/JHC.S307962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu TH, Huang CC, Lin PR, Chang HW, Ger LP, Lin YW, Changchien CS, Lee CM, Tai MH. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer. 2003;97:1929–1940. doi: 10.1002/cncr.11266. [DOI] [PubMed] [Google Scholar]
- 16.Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X, Li R, Jing R, Zuo B, Zheng Q. Genome-wide CRISPR knockout screens identify ADAMTSL3 and PTEN genes as suppressors of HCC proliferation and metastasis, respectively. J Cancer Res Clin Oncol. 2020;146:1509–1521. doi: 10.1007/s00432-020-03207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22:138. doi: 10.1186/s12943-023-01827-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 20.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 21.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Investig. 2004;113:1774–1783. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kladney RD, Bulla GA, Guo L, Mason AL, Tollefson AE, Simon DJ, Koutoubi Z, Fimmel CJ. GP73, a novel Golgi-localized protein upregulated by viral infection. Gene. 2000;249:53–65. doi: 10.1016/S0378-1119(00)00136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao Y, Yang H, Xu H, Lu X, Sang X, Du S, Zhao H, Chen W, Xu Y, Chi T, et al. Golgi protein 73 (GOLPH2) is a valuable serum marker for hepatocellular carcinoma. Gut. 2010;59:1687–1693. doi: 10.1136/gut.2010.214916. [DOI] [PubMed] [Google Scholar]
- 24.Block TM, Comunale MA, Lowman M, Steel LF, Romano PR, Fimmel C, Tennant BC, London WT, Evans AA, Blumberg BS, et al. Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc Natl Acad Sci U S A. 2005;102:779–784. doi: 10.1073/pnas.0408928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marrero JA, Romano PR, Nikolaeva O, Steel L, Mehta A, Fimmel CJ, Comunale MA, D'Amelio A, Lok AS, Block TM. GP73, a resident Golgi glycoprotein, is a novel serum marker for hepatocellular carcinoma. J Hepatol. 2005;43:1007–1012. doi: 10.1016/j.jhep.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Wang Y, Tao J, Shi Y, Gai X, Huang F, Ma Q, Zhou Z, Chen H, Zhang H, et al. mTORC1 up-regulates GP73 to promote proliferation and migration of hepatocellular carcinoma cells and growth of xenograft tumors in mice. Gastroenterology. 2015;149:741–752.e714. doi: 10.1053/j.gastro.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Ye QH, Zhu WW, Zhang JB, Qin Y, Lu M, Lin GL, Guo L, Zhang B, Lin ZH, Roessler S, et al. GOLM1 modulates EGFR/RTK cell-surface recycling to drive hepatocellular carcinoma metastasis. Cancer Cell. 2016;30:444–458. doi: 10.1016/j.ccell.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Revill PA, Tu T, Netter HJ, Yuen LKW, Locarnini SA, Littlejohn M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat Rev Gastroenterol Hepatol. 2020;17:618–634. doi: 10.1038/s41575-020-0296-6. [DOI] [PubMed] [Google Scholar]
- 29.Chisari FV, Filippi P, McLachlan A, Milich DR, Riggs M, Lee S, Palmiter RD, Pinkert CA, Brinster RL. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J Virol. 1986;60:880–887. doi: 10.1128/jvi.60.3.880-887.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59:1145–1156. doi: 10.1016/0092-8674(89)90770-8. [DOI] [PubMed] [Google Scholar]
- 31.Bard-Chapeau EA, Nguyen AT, Rust AG, Sayadi A, Lee P, Chua BQ, New LS, de Jong J, Ward JM, Chin CK, et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet. 2014;46:24–32. doi: 10.1038/ng.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trierweiler C, Hockenjos B, Zatloukal K, Thimme R, Blum HE, Wagner EF, Hasselblatt P. The transcription factor c-JUN/AP-1 promotes HBV-related liver tumorigenesis in mice. Cell Death Differ. 2016;23:576–582. doi: 10.1038/cdd.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Zhao W, Cheng L, Guo M, Li D, Li X, Tan Y, Ma S, Li S, Yang Y, et al. CD137-mediated pathogenesis from chronic hepatitis to hepatocellular carcinoma in hepatitis B virus-transgenic mice. J Immunol. 2010;185:7654–7662. doi: 10.4049/jimmunol.1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi WT, Kakar S. Immunohistochemistry in the diagnosis of hepatocellular carcinoma. Gastroenterol Clin North Am. 2017;46:311–325. doi: 10.1016/j.gtc.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Cetintas VB, Batada NN. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J Transl Med. 2020;18:45. doi: 10.1186/s12967-020-02219-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 37.Kenerson HL, Yeh MM, Kazami M, Jiang X, Riehle KJ, McIntyre RL, Park JO, Kwon S, Campbell JS, Yeung RS. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology. 2013;144:1055–1065. doi: 10.1053/j.gastro.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu F, Gai X, Wu Y, Zhang B, Wu X, Cheng R, Tang B, Shang K, Zhao N, Deng W, et al. Oncogenic β-catenin stimulation of AKT2-CAD-mediated pyrimidine synthesis is targetable vulnerability in liver cancer. Proc Natl Acad Sci U S A. 2022;119:e2202157119. doi: 10.1073/pnas.2202157119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan D, Huang S, Berger E, Liu L, Gross N, Heinzmann F, Ringelhan M, Connor TO, Stadler M, Meister M, et al. Kupffer cell-derived Tnf triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell. 2017;31:771–789.e776. doi: 10.1016/j.ccell.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loomba R, Friedman Sl, Shulman Gl. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang S, Tanaka T, Nagasaki M, Kishimoto T. Targeting Interleukin-6 signaling in clinic. Immunity. 2019;50:1007–1023. doi: 10.1016/j.immuni.2019.03.026. [DOI] [PubMed] [Google Scholar]
- 43.Wan L, Gao Q, Deng Y, Ke Y, Ma E, Yang H, Lin H, Li H, Yang Y, Gong J, et al. GP73 is a glucogenic hormone contributing to SARS-CoV-2-induced hyperglycemia. Nat Metab. 2022;4:29–43. doi: 10.1038/s42255-021-00508-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. PCR genotyping of mouse tail genomic DNA.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.