

Abstract
T‐lineage acute lymphoblastic leukemia (T‐ALL) is an aggressive hematological malignancy that accounts for 10%–15% of pediatric and 25% of adult ALL cases. Although the prognosis of T‐ALL has improved over time, the outcome of T‐ALL patients with primary resistant or relapsed leukemia remains poor. Therefore, further progress in the treatment of T‐ALL requires a better understanding of its biology and the development of more effective precision oncologic therapies. The proto‐oncogene MYB is highly expressed in diverse hematologic malignancies, including T‐ALLs with genomic aberrations that further potentiate its expression and activity. Previous studies have associated MYB with a malignant role in the pathogenesis of several cancers. However, its role in the induction and maintenance of T‐ALL remains relatively poorly understood. In this study, we found that an increased copy number of MYB is associated with higher MYB expression levels, and might be associated with inferior event‐free survival of pediatric T‐ALL patients. Using our previously described conditional Myb overexpression mice, we generated two distinct MYB‐driven T‐ALL mouse models. We demonstrated that the overexpression of Myb synergizes with Pten deletion but not with the overexpression of Lmo2 to accelerate the development of T‐cell lymphoblastic leukemias. We also showed that MYB is a dependency factor in T‐ALL since RNA interference of Myb blocked cell cycle progression and induced apoptosis in both human and murine T‐ALL cell lines. Finally, we provide preclinical evidence that targeting the transcriptional activity of MYB can be a useful therapeutic strategy for the treatment of T‐ALL.
INTRODUCTION
T‐cell acute lymphoblastic leukemia (T‐ALL) is an aggressive hematological malignancy characterized by the diffuse distribution of lymphoblasts of the T‐lineage in the bone marrow. 1 T‐ALL accounts for 15% of pediatric and 25% of adult ALL cases. 2 , 3 Despite an improved survival rate due to the introduction of intensified chemotherapy and bone marrow transplantation, still 20% of T‐ALL patients are primarily refractory to the standard treatment or relapse and have a poor clinical outcome with no alternative therapeutic options. 4 In addition, the intensified chemotherapy regimen coincides with severe short‐ and long‐term side effects. 2 , 5 Therefore, further progress in the treatment of T‐ALL may require a better understanding of its biology and the development of more effective precision oncology therapeutics.
T‐ALL patients can be classified into different molecular genetic subgroups based on the expression or genomic alterations of specific oncogenic transcription factors. 6 , 7 The largest subset of T‐ALL patients is characterized by the overexpression of TAL1/2, which often co‐occurs with aberrant levels of LMO1/2. These TAL‐LMO T‐ALLs tend to display a mature T‐cell immunophenotype and expression profile. Other subgroups of T‐ALL are associated with the ectopic expression of either TLX1, TLX3, HOXA9/10, or NKX2‐1. Additionally, the early T‐cell precursor ALL (ETP‐ALL) is a heterogeneous subgroup of immature leukemias that often display high expression of LYL1 in combination with LMO2. 1 Besides mutually exclusive aberrant expression of transcription factor oncogenes, T‐ALL patients harbor a variety of genomic aberrations that ultimately result in the activation of oncogenic signaling pathways and loss of tumor suppressors. Interestingly, PI3K/AKT/mTOR‐activating mutations occur more commonly in mature TAL‐LMO T‐ALLs, whereas RAS/MEK/ERK‐ and JAK/STAT‐activating mutations are associated with immature, HOXA, TLX1, TLX3, and NKX2‐1/2 cases. 6 , 8 , 9 , 10
MYB encodes the transcription factor MYB, considered a master regulator of differentiation, proliferation, and survival of hematopoietic progenitor cells. 11 , 12 MYB is composed of three main domains: an N‐terminal DNA‐binding domain, a central transactivation domain, and a C‐terminal negative auto‐regulatory domain. 13 This transcription factor specifically recognizes a 5′‐YAAC[GT]G‐3′ DNA sequence within the regulatory regions of its targets and has been demonstrated to recruit several histone‐ and gene‐regulatory proteins, including the coactivators CREB‐binding protein (CBP) and p300. 13 , 14 Using mouse models, it was shown that genetic depletion of Myb results in embryonic lethality due to failure of fetal hepatic hematopoiesis. 15 Furthermore, a conditional deletion approach revealed that Myb is required for early T‐cell differentiation, specifically at the beta‐selection checkpoint in the double‐negative 3 CD4−CD8−CD44−CD25+ (DN3) stage, for the survival of CD4+CD8+ double‐positive (DP) thymocytes, and for the differentiation of CD4+CD8− single positive (SP) cells. 16 In conclusion, MYB is highly expressed in various hematopoietic cell types, including in the T‐cell lineage, where it is required for proper differentiation, proliferation, and survival.
Moreover, MYB has been implicated in various hematological malignancies. 17 MYB was identified as the cellular proto‐oncogene ortholog from the retroviral oncogene (v‐Myb), originally found in the avian myeloblastosis virus and the E26 avian leukemia virus. 17 , 18 , 19 Moreover, mice or chickens that develop leukemias or lymphomas upon retroviral infection often contain clusters of proviral insertion sites mapped in close proximity to Myb. 20 , 21 v‐Myb encodes truncated MYB protein variants that lack the negative regulatory domain and exhibit constitutive transcriptional activity. The oncogenic potential of v‐Myb was validated in vivo, as its overexpression in hematopoietic cells results in malignant transformation and spontaneous formation of leukemias and lymphomas in mice. 22 Recently, we have reported that hematopoietic‐specific overexpression of the full‐length cellular Myb is also sufficient to drive spontaneous B‐cell neoplasms and myeloid malignancies in mice. 23 Besides its role in tumor initiation, numerous studies have shown that MYB is also essential for the maintenance of leukemias. 24 , 25 , 26 , 27 In T‐ALL, MYB is aberrantly activated in a subset of patients through T‐cell receptor‐driven translocations (1.6%) or genomic duplications (2.8%–15%) of the MYB locus. 28 , 29 , 30 Furthermore, multiple indel mutations and gene fusions that stabilize and promote the activity of MYB protein have also been reported in T‐ALL. 6 In addition, about 5% of T‐ALL patients contain mutations in noncoding DNA sequences that create de novo MYB binding sites, which recruit MYB and result in the generation of a super‐enhancer complex that drives the expression of oncogenes, such as TAL1, LMO1, and LMO2. 31 , 32 , 33 Therefore, MYB targeting therapies, including the ones that target the MYB‐driven oncogenic enhancers, are being exploited in myeloid 34 , 35 , 36 and recently also in lymphoid 37 , 38 leukemias. Nevertheless, the exact contribution of MYB to the pathogenesis, maintenance, and induction of T‐ALL is still rather unclear.
Here, we showed that MYB‐rearranged T‐ALL patients have increased MYB expression which might be associated with poorer event‐free survival. Moreover, we used a previously published conditional Myb overexpression mouse model 23 to mimic the gain of MYB in two murine T‐ALL models that are based on either deletion of Pten 39 or overexpression of Lmo2. 40 We found that Myb overexpression synergizes with the Pten loss to form aggressive transplantable T‐ALL. Furthermore, we also examined the role of MYB in the maintenance of T‐ALL and assessed the potential of peptidomimetic blocking the transcriptional activity of MYB as a therapeutic approach for T‐cell leukemia.
MATERIAL AND METHODS
Data mining
We analyzed data from the PeCan (pediatric cancers) knowledgebase in the St Jude Cloud 41 , 42 for the expression levels of MYB across different cancers and in 315 T‐ALL patients, 6 , 7 as well as the prevalence of MYB‐duplicated T‐ALL cases, and the event‐free survival of 265 of these patients. For the comparison of MYB levels across T‐ALL subtypes and healthy thymocytes, publicly available polyA+ RNA‐seq raw data of 24 thymocyte samples 43 (PRJNA741323) and 60 T‐ALL patients in a French cohort 44 (PRJNA434176) were downloaded from the Sequence Read Archive (SRA). Furthermore, data from 265 T‐ALL patients from a USA cohort 6 (PRJNA89529) were available in the database of Genotypes and Phenotypes (phs000218/phs000464). The data were normalized in R with the DESeq2 package.
Murine T‐ALL models
Mice with conditional MYB overexpression were previously reported. 23 To obtain the murine T‐ALL models, the R26‐Myb tg/tg mice were crossed with either Pten fl/fl ; Lck‐Cre tg/+ (Pten Lck ) mice 45 , 46 or CD2‐Lmo2 mice 47 to generate R26‐Myb tg/tg ; Pten fl/fl ; Lck‐Cre tg/+ (Myb/Pten Lck ) or R26‐Myb tg/tg ; Lck‐Cre tg/+ ; CD2‐Lmo2 (Myb Lck /Lmo2 CD2 ) mouse models, respectively. We monitored an aging cohort of Pten Lck versus Myb/Pten Lck and Lmo2 CD2 versus Myb Lck /Lmo2 CD2 mice and plotted their survival using a Kaplan–Meier survival curve while testing for statistically significant differences using the log‐rank Mantel–Cox test. All animal experiments were approved by the Animal Experiments Ethics Committee of the University Hospital of Ghent.
Myc break apart fluorescence in situ hybridization (FISH)
Cytogenetic cell suspensions (methanol/acetic acid fixed cells) or unstained slides were made from primary transplants of Pten Lck or Myb/Pten Lck tumor samples or primary Pten Lck or Lmo2 CD2 murine cell lines. For FISH analysis, the following Myc break apart probes (Empire genomics) were used: RP23‐98D8 (green, located telomeric to Myc) and RP23‐55P19 (red, located centromeric to Myc).
FISH analysis was performed as previously described. 48 Fluorescent images were captured using a Zeiss axioplan epifluorescence microscope (Carl Zeiss) (eight‐position filter wheel) equipped with a CCD camera and by using the ISIS software (Metasystems).
Cell lines
Primary murine T‐ALL cell lines, including MyPL1 and MyPL2 cells, were derived from leukemia‐burdened R26‐Myb tg/tg ; Pten fl/fl ; Lck‐Cre tg/+ (Myb/Pten Lck ) mice. In brief, thymomas were dissected, cut into small tumor fragments using two needles, collected in RPMI 1640 medium (Thermo Fisher Scientific) containing 15% heat‐inactivated fetal bovine serum (FBS), 0.1 mM nonessential amino acids (Thermo Fisher Scientific), 2 mM l‐glutamine (Gibco), 10 mM β‐mercaptoethanol (Thermo Fisher Scientific), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco), and 5 ng/mL recombinant murine interleukin‐7 (mIL‐7; PeproTech), and cultured at 37°C in a humidified atmosphere with 5% CO2. Cells in the suspension were split regularly and became independent of mIL‐7 after 4–8 weeks.
Jurkat, RPMI‐8402, Loucy, and P12‐Ichikawa cell lines were purchased from the German collection of microorganisms and cell cultures (DSMZ). For MYB knockdown experiments, RPMI‐8402 and Jurkat were cultured in RPMI 1640 medium supplemented with 10% and 15% tetracycline‐free fetal bovine serum, respectively, supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l‐glutamine at 37°C and 5% CO2. For peptidomimetic blockade experiments, standard FBS was used instead at 10% (Loucy and KOPT‐K1) and 20% (P12‐Ichikawa) concentrations.
Lentiviral vectors
For MYB silencing, we used a pLVTSH lentiviral construct with a dox‐inducible MYB Short hairpin RNA (shRNA) 49 that was generated by Prof. Dr. Tom Gonda and kindly provided by Dr. M. De Dominici. Lentiviral particles were produced by transfecting the pLVTSH plasmid and packing vectors pMD2.G and psPAX into HEK‐293T cells using jetPEI (Polyplus). The culture medium containing viral particles was collected 48 h posttransfection and was used to transduce the mouse and human T‐ALL cells by spinoculation in the presence of 8 µg/mL polybrene (Sigma‐Aldrich). Transduced cells were fluorescence‐activated cell sorting (FACS)‐sorted based on green fluorescent protein (GFP) expression using a BD FACSAria Fusion Flow Cytometer (BD Biosciences). Primary murine T‐ALL cell lines, MyPL1 and MyPL2, and human T‐ALL cell lines, Jurkat and RPMI‐8402, with inducible MYB knockdown were named iMyb MyPL1 and iMyb MyPL2, iMYB Jurkat and iMYB RPMI‐8402, respectively.
CellTiter‐Glo, cell cycle analysis, and apoptosis assay
For induction of MYB knockdown, 1 µg/mL of doxycycline (DOX) was added to the culture medium and refreshed daily. Manual counting and CellTiter‐Glo assay (Promega) were performed daily to estimate the proportion of DOX‐treated cells relative to the control untreated condition. Control and DOX‐treated cells were split using the same dilution factor to maintain comparability. Cell cycle analyses were performed on live cells isolated with Ficoll‐Paque (Cytiva) that were posteriorly permeabilized and fixed in 50% ethanol, followed by RNAse treatment (0,5 mg/mL), propidium iodide staining (40 µg/mL), and flow cytometry determination of DNA content. Apoptosis assays were performed using propidium iodide and annexin‐V (BD Biosciences) following the protocol of the manufacturer.
In vivo MYB dependency and T‐ALL Patient‐derived xenografts (PDXs)
For the in vivo Myb/MYB knockdown, 4 × 105 iMyb MyPL1 cells or 9 × 105 iMYB Jurkat cells were injected in NOD.Cg‐Prkdcscid Il2rgtm1Wjl/SzJ (NSG) or in the equivalent NOD‐Prkdcscid‐IL2rgTm1/Rj (NXG) mice (purchased from Jackson Laboratory or Janvier labs, respectively), and randomized in a “control” and “treatment” group, that received either a standard or a doxycycline‐containing diet (200 mg/kg, Envigo), respectively. The doxycycline‐containing food was given from day 4 after transplantation until the end of the experiment. Longitudinal monitoring of disease progression was performed with iMyb MyPL1 cells by injecting NSG mice with 200 µL of a 15 mg/mL firefly D‐luciferin potassium salt solution (PerkinElmer) intraperitoneally. The mice were anesthetized by inhalation of 5% isoflurane and in vivo bioluminescence was imaged 10 min after luciferin injection using an IVIS lumina II imaging system (PerkinElmer). The average bioluminescence signal (radiance) in each mouse was calculated via the region of interest tool in the Living Image software (PerkinElmer). For the iMYB Jurkat in vivo experiment, five mice from each group were killed on day 42, and their bone marrow cells were analyzed for expression of human CD45 and GFP by flow cytometry. The remaining six mice were used for survival analysis and monitored for weight loss and other signs of illness. Once the humane endpoints were reached, the mice were killed by cervical dislocation, and the affected organs (spleen and bone marrow) were collected. Viable cells were also frozen for posterior analyses, and the survival data of these mice were plotted and analyzed as reported above.
The generation of T‐ALL patient‐derived xenografts (PDX) was achieved by injecting lymphoprep‐purified primary T‐ALL cells from the blood or bone marrow of T‐ALL patients in the tail vein of NSG or NOD.Cg‐Prkdc scid Il2rg tm1Wjl Tg(CMV‐IL3, CSF2, KITLG) 1Eav/MloySzJ (NSGS) mice. To expand the patient cells, primary PDX cells were further passaged in NSG/NSGS mice. All human samples were acquired with written informed consent according to the Declaration of Helsinki, and the studies were approved by the ethical committee review board of the Department of Pediatric Hemato‐Oncology at Ghent University Hospital (Ghent, Belgium).
In vitro/ex vivo therapy and drug response profiling
Murine and human T‐ALL cells were cultured at a density of 10,000 cells/mL and treated with either the peptidomimetic MYBMIM 36 or the inactive control peptide TG3, which were synthetized by LifeTein. The peptides were solubilized in water and added to the culture medium in concentrations ranging from 10 to 40 µM, and the relative cell viability was assessed after 48 h by CellTiter‐Glo.
Immunoblot
Cells were lysed at a density of 20,000/µL in RIPA buffer supplemented with the protease inhibitor cocktail (Roche). The protein content of the lysates was quantified using Pierce BCA Protein Assay (Thermo Fisher Scientific), and 12 µg of protein per sample was run on 10% precast polyacrylamide gels (BioRad) at a constant voltage of 100 V for 90 min. Afterwards, the protein was transferred onto nitrocellulose membranes (BioRad), and the membrane was incubated with primary antibodies targeting MYB (clone D2R4Y, Cell Signaling Technology), Vinculin (clone hVIN‐1, Sigma‐Aldrich), or β‐Actin (clone 2D4H5, Thermo Fisher Scientific), and HRP‐linked secondary antibodies (Cell Signaling Technology). Images were obtained by chemiluminescent reaction with West Dura substrate (Thermo Fisher Scientific), acquired using ChemiDoc (BioRad), and processed and analyzed using the ImageLab software (BioRad).
To assess the histone H3 tail modifications, nuclear extraction was performed using triton extraction buffer with 0.5% Triton X‐100, supplemented with 5 mM sodium butyrate to prevent histone deacetylation, and with the protease/phosphatase inhibitor cocktail (Roche). Histones were extracted out of the nuclei using 0.4 M chloridic acid (HCl), and the pH of the solution was neutralized with 0.5 M Sodium phosphate dibasic (Na2HPO4). The levels of histone H3 lysine 27 acetylation (ab4729) and trimethylation (07‐449) were measured and normalized against the levels of total histone H3 (ab1791) using the immunoblotting protocol described above.
Chromatin immunoprecipitation sequencing
A total of 20 million MyPL1 and MyPL2 cells were cross‐linked with 1% formaldehyde (Sigma‐Aldrich, F1635) for 10 min at room temperature, followed by a 5‐min treatment with 0.125 M glycine (Sigma‐Aldrich, G‐8790). Nuclei were isolated and chromatin was purified by chemical lysis. The purified chromatin was fragmented into 200–300 bp fragments by sonication (Covaris, S220) using the truChIP chromatin shearing kit (Covaris). Chromatin immunoprecipitation was performed by incubating the chromatin fraction overnight with 100 μL of protein‐A coated beads (Thermo‐Scientific, 53,139) and 8 μg of the H3K27Ac antibody (Abcam, ab4729). The next day, beads were washed to remove nonspecific binding events and enriched chromatin fragments were eluted from the beads, followed by reverse cross‐linking by incubation at 65°C overnight. DNA was subsequently purified by phenol/chloroform extraction, assisted by phase lock gel tubes (5Prime). DNA obtained from the ChIP assays was adaptor‐ligated, amplified, and analyzed by Illumina sequencing. Raw sequencing data were mapped to the mouse reference genome (GRCm38) using Bowtie. Peak calling was performed using MACS2. 50
RNA sequencing
iMyb MyPL1 cells were cultured with or without 1 µg/mL of doxycycline hyclate in the culture medium to induce knockdown of Myb, and a million cells per condition were collected for four consecutive days. The cells were washed in phosphate‐buffered saline (PBS), lysed in RLT buffer, and the RNA was purified using the RNeasy kit (Qiagen). RNA quality was assessed on a Fragment Analyzer (Agilent), the RNA was converted to complementary DNA (cDNA), and the barcoded cDNA libraries were amplified (Lexicon) and analyzed by next‐generation sequencing (Illumina). For RNA‐seq data analysis, quality control was performed using fastQC, and the reads were aligned to the murine genome GRCm38 using Tophat2. 51 DESeq2 52 was used for differential expression analysis of RNA‐seq data. Expression data sets are available in the Gene Expression Omnibus (accession no. GSE253616).
RESULTS
MYB duplication confers poor outcomes in human T‐ALL
We started by exploring the expression and mutation status of MYB in the context of human T‐ALL. Within 5510 pediatric cancer samples from the PeCan database, 41 , 42 hematological malignancies, in particular B‐ and T‐ALLs, show the highest levels of MYB expression (Figure 1A). In contrast, MYB mRNA is absent or low in most solid tumors. Within the T‐cell lineage, MYB is highly expressed throughout different stages of normal thymocyte differentiation and in all T‐ALL subgroups (Figure 1B). In line with previous reports, we found that the average MYB RNA levels were consistently higher in the HOXA and TAL‐LMO subgroups, 8 , 53 , 54 as observed in T‐ALL patients from St‐Jude 6 , 7 (USA, n = 265) and Saint‐Louis 44 (France, n = 60) hospitals. This high MYB expression in T‐ALL may partially be explained by the presence of translocations and genomic duplications involving the MYB locus. 28 , 29 , 54 Eight percent (n = 33/398) of the T‐ALL patients in the PeCan database presented with a copy number gain (CNG) of MYB (Figure 1C). We noted that the prevalence of MYB CNG was not evenly distributed over the T‐ALL genetic subgroups (Figure 1C). The incidence was highest at 28.6% in the NKX2‐1 subgroup (n = 4/14), followed by 25.9% in T‐ALL of undetermined subgroup (n = 7/27), 23.5% in HOXA (n = 7/34), and 19.2% in TLX1 T‐ALLs (n = 5/26). Significantly fewer MYB CNGs were seen in the remaining subgroups, comprising 5.6% of TAL‐LMO (n = 6/107), 5.6% of LMO2‐LYL1 (n = 1/18), and 4.3% of TLX3 cases (n = 2/46). These findings were validated in a cohort of 489 T‐ALLs from the Center for Hemato‐Oncology Research, which included 209 pediatric and 280 adult T‐ALL cases (Supporting Information S2: Table S1). Using comprehensive interphase fluorescence in situ hybridization (CI‐FISH), 55 we detected four MYB translocations and 26 MYB tandem duplications. 54 Once again, MYB CNGs were found in the homeobox‐related subgroups (HOXA, n = 10; TLX1/NKX2.1 n = 9; TLX3, n = 2; undetermined, n = 5), while no case was found in TAL‐LMO T‐ALLs (Supporting Information S2: Tables S1 and S2). In line with previous reports, 28 , 29 , 54 we observed a significant increase in the median MYB expression in MYB CNG T‐ALLs from the PeCan database (Figure 1D). Notably, the low prevalence of MYB alterations in TAL‐LMO cases in the PeCan database and their absence in the validation cohort seem to hint that the high MYB levels in TAL‐LMO T‐ALLs are sustained by mechanisms other than genetic alterations. Regarding the prognostic implications of MYB CNG in T‐ALL, it was not possible to draw any conclusions in this Italian cohort (Supporting Information S2: Tables S3 and S4). In the Pecan database, however, MYB CNG was associated with a decreased event‐free survival compared to T‐ALLs without MYB CNG (p = 0.008; Figure 1E). Interestingly, no significant difference was found in the overall survival of these patients (p = 0.17; Supporting Information S1: Figure S1A). When we stratified patients based on the genetic subgroup, we observed that MYB CNG was significantly associated with poor prognosis, both for event‐free and overall survival, in HOXA T‐ALL (Supporting Information S1: Figure S1B,C). For the remaining subgroups, we did not find an association between the presence of MYB CNG and prognosis, possibly due to the low number of these cases per subgroup. Combined, these data indicate that MYB CNGs are not evenly distributed among the genetic subtypes of T‐ALL and that the presence of MYB CNG may be associated with poor outcome in T‐ALL.
Figure 1.
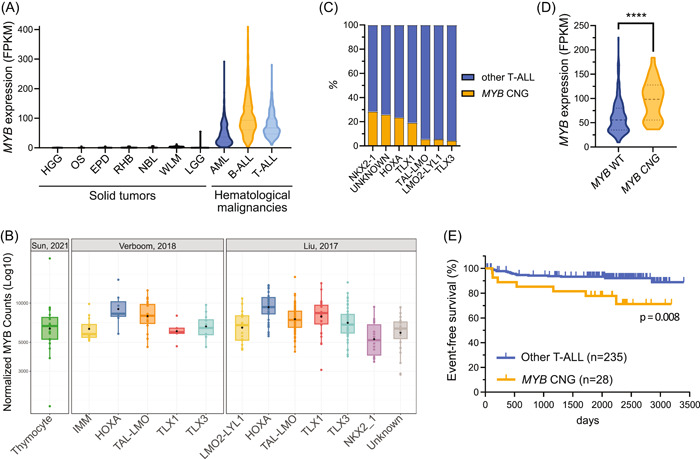
MYB is highly expressed in T‐ALL and MYB copy number gains (CNGs) are associated with unfavorable outcome. (A) Violin plot representing MYB messenger RNA (mRNA) expression across different types of solid (in black) and hematological (colored) cancers from the PeCan database. Tumor types shown were selected on the basis of having a minimum of 50 samples in the PeCan database. 6 , 7 (B) Box and whiskers plot comparing MYB mRNA expression in normal thymocytes representing multiple differentiation stages, 43 and in two T‐ALL cohorts from Saint‐Louis Hospital (n = 60; France), 44 and from St. Jude Children's Research Hospital (n = 265; Memphis, USA; PeCan database). 6 , 7 (C) Stacked bar chart displaying the percentage of MYB copy number gain (MYB CNG) cases in each T‐ALL genetic subgroup from the PeCan cohort. 6 , 7 NKX2‐1, n = 4/14 (28.6%); Unknown, n = 7/27 (25.9%); HOXA, n = 7/34 (23.5%); TLX1, n = 5/26 (19.2%); TAL‐LMO, n = 6/105 (5.6%); LMO2‐LYL1, n = 1/18 (5.6%); TLX3, n = 2/46 (4.3%). (D) Violin plot of MYB mRNA expression in T‐ALLs with or without MYB copy number gain from the PeCan cohort. 6 , 7 A Mann–Whitney test assessed that MYB mRNA levels were significantly higher in T‐ALL with MYB copy number gain. ****p < 0.0001. (E) Kaplan–Meier survival curve of T‐ALL cases with or without MYB copy number gains, showing that patients bearing T‐ALL with MYB copy number gain suffer from reduced event‐free survival (p = 0.008). AML, acute myeloid leukemia; B‐ALL, B‐cell acute lymphoblastic leukemia; EPD, ependymoma; HGG, high‐grade glioma; IMM, immature T‐ALL; LGG, low‐grade glioma; NBL, neuroblastoma; OS, osteosarcoma; RHB, rhabdomyosarcoma; T‐ALL, T‐cell acute lymphoblastic leukemia; WLM, Wilms' tumor.
Myb overexpression cooperates with the loss of Pten in the murine T‐ALL model
To model and functionally evaluate the effects of elevated MYB expression observed in T‐ALL patients with MYB CNG, we used our previously reported R26‐Myb tg mouse model that enables conditional Myb overexpression. 23 When Myb was overexpressed in all hematopoietic cell lineages using Vav‐iCre, these mice only developed spontaneous myeloid malignancies and B‐cell neoplasms, and no tumors of the T‐cell lineage were observed in this model. 23 Additionally, no spontaneous T‐cell malignancies were seen when using the T‐cell‐restricted Lck‐Cre. Of note, we also found no obvious differences in T‐cell proportions and changes in T‐cell differentiation within the thymus of 8‐week‐old mice that overexpress Myb compared to their cre‐negative littermates (Supporting Information S1: Figure S2). Taken together, these findings suggest that overexpression of Myb alone is not sufficient to drive T‐cell malignancies in mice, and additional genetic events are necessary.
In the next step, we evaluated the effects of Myb overexpression in a T‐ALL‐prone background by intercrossing our R26‐Myb tg overexpression with previously published spontaneous mouse T‐ALL models. 39 , 40 Besides high MYB levels, TAL‐LMO T‐ALLs have a higher frequency of inactivating mutations affecting the tumor suppressor PTEN and activating mutations in other genes in the PI3K pathway. 56 , 57 Due to this apparent association between high MYB expression and hyperactivation of the PI3K pathway in TAL‐LMO T‐ALL, we crossed our R26‐Myb tg/tg mice with Pten fl/fl ; Lck‐Cre tg/+ (hereafter named Pten Lck ) mice to generate the R26‐Myb tg/tg ; Pten fl/fl ; Lck‐Cre tg/+ (hereafter named Myb/Pten Lck ) mice, featuring biallelic ROSA26‐driven overexpression of Myb and deletion of Pten specifically in T‐cells (Figure 2A). Additionally, to evaluate whether the functional effects of MYB overexpression is context‐dependent, we also intercrossed our R26‐Myb tg/tg mice with CD2‐LMO2 tg (hereafter named Lmo2 CD2 ) animals to generate a spontaneous Lmo2‐driven immature T‐ALL mouse model with concomitant overexpression of Myb (hereafter named Myb Lck /Lmo2 CD2 ; Figure 2B). We monitored aging cohorts of Pten Lck or Lmo2 CD2 mice with or without Myb overexpression and found that Myb accelerated the onset of murine T‐ALL driven by the loss of Pten (Figure 2C) but not by Lmo2 overexpression (Figure 2D). Similar to what we suggested above, based on the expression and incidence of MYB CNGs in human T‐ALL, these mouse models seem to demonstrate that the functional effects of Myb gain are dependent on the cellular and molecular context.
Figure 2.
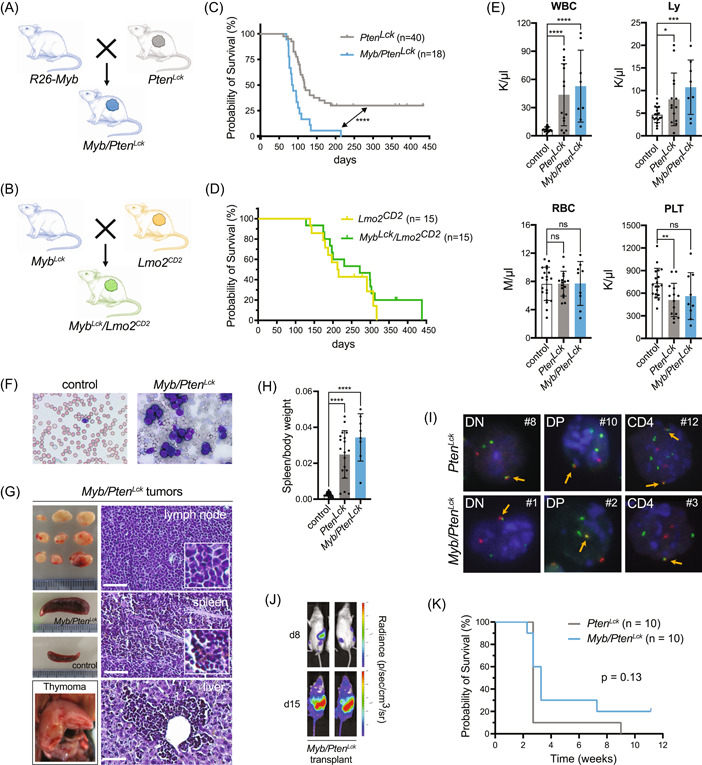
Myb accelerates Pten‐null T‐cell leukemia in mice. (A) Schematic representation of breeding of R26‐Myb mice, that feature R26‐driven conditional Myb overexpression, with Ptenfl/fl ; Lck‐Cretg/+ (PtenLck ) mice, in order to obtain R26‐Mybtg/tg ; Ptenfl/fl ; Lck‐Cretg/+ (Myb/PtenLck ) mice. (B) Schematic representation of breeding of R26‐Myb mice with CD2‐LMO2tg (Lmo2CD2 ) mice, to generate R26‐Mybtg/tg ; Lck‐Cretg/+ ; CD2‐LMO2tg (MybLck/Lmo2CD2 ) mice. (C) Kaplan–Meier survival curve of PtenLck (in gray) and Myb/PtenLck (in blue) mice. A log‐rank Mantel–Cox test showed that survival of Myb/PtenLck mice was significantly lower. ****p < 0.0001. (D) Kaplan–Meier survival curve of Lmo2CD2 (in yellow) and MybLck/Lmo2CD2 mice (in green). A log‐rank Mantel–Cox test could not show a significant difference between the groups. p = 0.39. (E) Bar chart of peripheral blood (PB) values of nonrecombined cre‐negative control mice, or mice. An unpaired t test revealed a significant increase in circulating white blood cells and lymphocytes of tumor‐carrying and mice compared to nonrecombined controls. No statistical difference was found in the number of RBCs among the models, but a significant decrease in the number of platelets was found in mice, compared to control mice. Each dot represents a different mouse. ****p < 0.0001, ***p = 0.0005, **p = 0.0045, and *p = 0.0267. Error bars denote SEM. (F) Giemsa staining of PB of cre‐negative controls and mice demonstrating an increased number of circulating lymphoblasts in mice. (G) Micrographs of lymphoma, splenomegaly, and thymoma samples of a representative mouse with T‐cell malignancies, and normal spleen of control cre‐negative littermate. Hematoxylin and eosin (H&E) staining of paraformaldehyde‐fixed paraffin sections of lymph node, spleen, and liver of mice. Scale bar: 50 µm. (H) Bar plot depicting the spleen‐to‐body weight ratio of and mice that developed neoplasm and age‐matched cre‐negative littermate control mice. An unpaired t test revealed a significant increase in the spleen‐to‐body weight ratio of and mice compared to controls. ****p < 0.0001. (I) break apart fluorescence in situ hybridization (FISH; see details in Supporting Information S1: Figure S3A) in interphase nuclei of double negative (DN, CD4−CD8−), double positive (DP, CD4+CD8+), or single positive CD4+ primary or T‐ALL transplants. Orange arrows indicate colocalization of the red and green probes that flank the locus, indicative of the normal nontranslocated locus. The tumor number is indicated with a # and corresponds to the tumor list in Supporting Information S2: Table S5. (J) Bioluminescence of NSG mice that were transplanted with 106 tumor cells. Measurements on experimental day 8 and day 15 show a steep increase in bioluminescent signal. (K) Kaplan–Meier survival curve of NSG mice injected with 106 primary tumor cells from 10 and 10 T‐ALLs. A log‐rank Mantel–Cox test showed that there is no significant difference in survival of NSG mice that were either transplanted with or T‐ALLs. p = 0.13. Ly, lymphocytes; ns, not significant; RBC, red blood cells; WBC, white blood cells.
Pten Lck mice suffered from T‐cell malignancies in 70% of cases (n = 28/40), with a median survival of 117 days (Figure 2C, in gray). In combination with Myb overexpression, all (n = 18/18) Myb/Pten Lck mice developed T‐cell malignancies significantly faster, with a median survival of 86 days (Figure 2C, in blue). At the time of sacrifice, both Pten Lck and Myb/Pten Lck mice models showed elevated levels of white blood cells and lymphocytes in peripheral blood (PB), while red blood cells were unaffected and platelets were slightly reduced (Figure 2E). An accumulation of lymphoblasts was detected in the PB of Myb/Pten Lck mice, indicative of T‐ALL (Figure 2F). Similar to Pten Lck mice, 39 Myb/Pten Lck mice frequently developed splenomegaly, thymomas, and leukemic infiltrations in lymph nodes and liver (Figure 2G,H). Histological analysis revealed that Myb/Pten Lck tumors were composed of diffuse sheets of small‐ to intermediate‐sized cells that display a high nucleus‐to‐cytoplasm ratio, as well as infiltration of the liver with leukemic cells via the portal veins, which are typical features of lymphoid leukemias (Figure 2G). Detailed flow cytometric analysis of Pten Lck and Myb/Pten Lck tumors revealed a rather similar spectrum of immunophenotypes, ranging from more immature double‐negative CD4−CD8− (DN) to mature single positive CD4+CD8− malignancies (Supporting Information S2: Table S5). The observed DN phenotypes are in contrast with previously reported Pten‐deficient models that develop predominantly DP T‐ALL/T‐cell lymphomas and harbor TCRα‐Myc translocations. 58 , 59 Therefore, we determined whether TCRα‐Myc translocations occur in Myb‐driven T‐ALL by performing Myc break‐apart FISH (Supporting Information S1: Figure S3A) experiments using both Pten Lck and Myb/Pten Lck murine T‐ALLs and Myb/Pten Lck and Lmo2 CD2 cell lines. Nearly all primary Pten Lck and Myb/Pten Lck murine T‐ALLs and Myb/Pten Lck T‐ALL cell lines harbored Myc translocations, irrespective of Myb overexpression or immunophenotype (Figure 2I and Supporting Information S1: Figure S3A,B and Supporting Information S2: Table S5). In contrast, the two Lmo2 CD2 cell lines did not harbor Myc translocations, not even the one with endogenous loss of Pten expression (Supporting Information S1: Figure S3B,C).
Of note, when Pten Lck and Myb/Pten Lck tumors are transplanted into immunodeficient NXG mice, they form secondary T‐ALL within 2–3 weeks (Figure 2J,K and Supporting Information S2: Table S5), confirming the aggressive nature of these leukemias. In conclusion, Myb overexpression synergizes with T‐cell‐specific loss of Pten in mice to accelerate the formation of aggressive and transplantable murine T‐ALL.
Myb is a dependency factor in Pten‐null murine T‐ALL
Next, we wondered whether MYB is essential for T‐ALL maintenance and would, as such, be a promising anti‐T‐ALL therapeutic target. For this, we isolated two primary Myb/Pten Lck murine T‐ALL cell lines, named MyPL1 and MyPL2 (Figure 3A), that represent a double‐positive CD4+CD8+ and a double‐negative CD4−CD8− T‐ALL cell line with high Myb expression, respectively (Figure 3B). To study the effects of loss of Myb in these T‐ALL models, we transduced MyPL1 and MyPL2 with a previously described doxycycline‐inducible lentiviral shRNA vector that targets Myb (iMyb; Figure 3A). 49 When iMyb MYPL1 and MyPL2 cells were treated with doxycycline, we observed a strong decrease in MYB protein levels (Figure 3C), and a substantial reduction in the number of cells compared to noninduced cells (Figure 3D). Additionally, we analyzed the in vivo anti‐leukemic effects of Myb loss in this Myb‐driven Pten‐null T‐ALL model. To this end, iMyb MYPL1 cells were transplanted into immunocompromised NSG mice, and at four days posttransplantation, the mice were randomly assigned to two groups that received either a standard diet or a doxycycline‐containing diet (Figure 3E). Upon in vivo Myb knockdown, we observed a significant delay in tumor progression, illustrated by lower bioluminescence (Figure 3F,G) and prolonged survival of the mice fed with doxycycline‐containing food (median survival of 47 days instead of 27 days; Figure 3H). However, despite a delay in disease onset, the majority of treated animals still developed leukemia. To determine whether the expanding tumor cells expressed Myb, we collected and analyzed tumor samples from control and doxycycline‐treated mice (Figure 3I and Supporting Information S1: Figure S4A), and we found that, irrespective of the diet, all tumors expressed high levels of MYB (Figure S4B). Flow cytometry analysis revealed that all obtained tumors displayed an identical immunophenotype to the parental iMyb MyPL1 cell line (Supporting Information S1: Figure S4C) and that the tumors arising in the doxycycline‐treated group were GFP‐negative (Figure 3I and Supporting Information S1: Figure S4D). This finding indicates that cells with Myb knockdown (GFP‐positive cells) were not expanding in vivo and were overgrown by cells without expression of the shRNA vector (GFP‐negative cells; Supporting Information S1: Figure S4D). These data demonstrate that MYB expression is essential for the in vivo expansion of murine T‐ALL.
Figure 3.
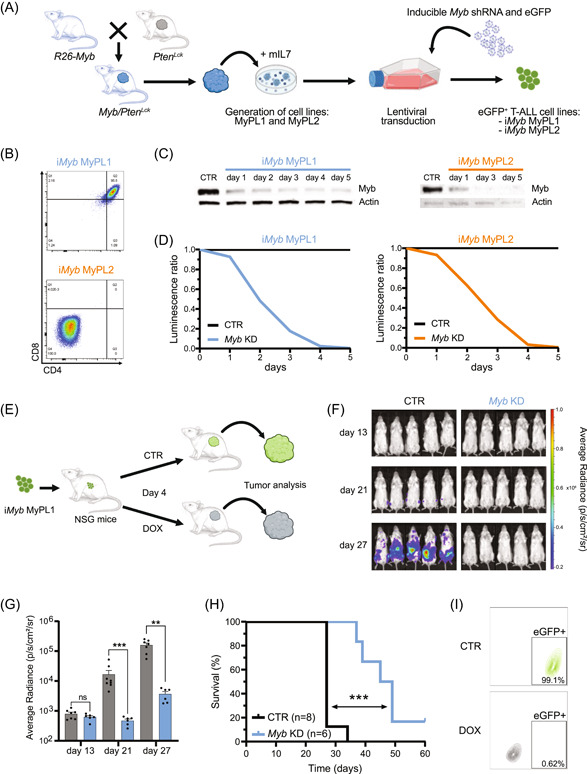
MYB is essential for the expansion of murine T‐ALL. (A) Schematic representation of the generation of two mouse cell lines, MyPL1 and MyPL2, that were derived from thymomas of R26‐Myb tg/tg ; Pten fl/fl ; Lck‐Cre tg/+ mice that developed T‐ALL. Immortalized cell lines were posteriorly lentivirally transduced with a tetracycline‐inducible vector to express a shRNA targeting Myb (iMyb) and a constitutive eGFP reporter, thus generating iMyb MyPL1 and iMyb MyPL2 cells. (B) Pseudocolor scatterplot of flow cytometry analysis of CD8 (horizontal axis) and CD4 (vertical axis) surface markers on iMyb MyPL1 (double positive CD4+CD8+) and iMyb MyPL2 (double negative CD4−CD8−) cells. (C) Immunoblotting analysis of Myb protein levels in iMyb MyPL1 and iMyb MyPL2 cells. Following doxycycline treatment, both cell lines showed a strong and progressive reduction in Myb protein levels. The control condition (CTR) consists of iMyb MyPL1 or iMyb MyPL2 cells that were cultured without doxycycline in the medium; D1–D5 are 5 consecutive days of treatment with 1 µg/mL of doxycycline hyclate in the culture medium; Actin protein levels were used as loading control. (D) Line chart depicting the ratio of luminescence detected with the CellTiter‐Glo luminescent cell viability assay, normalized to the control condition. Loss of MYB caused a drastic reduction in the number of cells over time, compared to the control. A representative result from three independent experiments is shown. (E) Schematic representation of in vivo Myb knockdown in iMyb MyPL1. This murine T‐ALL cell line (4 × 105/mouse) was transplanted intravenously into NSG mice that were randomly assigned, on experimental day 4, to a treatment group that was fed a diet with 200 mg/kg doxycycline (DOX) or to a control group with a standard diet (CTR). (F) Bioluminescence in NSG mice that were transplanted with iMyb MyPL1. Bioluminescence measurements on days 13, 21, and 27 posttransplantation show a steep bioluminescence increase in the control group, but not in the treatment group. (G) Bar chart showing the bioluminescence quantification in control mice (n = 8, in gray) and doxycycline‐treated mice (n = 6, in blue) from panel F. The knockdown of Myb significantly delayed the bioluminescence increase of iMyb MyPL1 cells in NSG mice. A Mann–Whitney test was used for statistical analysis at each timepoint. ns, nonsignificant, p = 0.45; ***p = 0.0007; **p = 0.0012. (H) Kaplan–Meier survival curve of NSG mice that were transplanted with iMyb MYPL1 cells and were treated as described in panel F. The doxycycline‐treated mice survived longer than control mice (median survival of 47 days versus 27 days, respectively), and a log‐rank Mantel–Cox test showed that the difference between groups was statistically significant. ***p = 0.0002. (I) Contour plot of flow cytometry analysis of iMyb MyPL1 tumor cells from control (CTR, in green) or doxycycline‐treated (DOX, in gray) mice showing the eGFP signal on the horizontal axis and the forward scatter area on the vertical axis.
Molecular and transcriptional changes upon Myb knockdown in murine T‐ALL
MYB has been described as a pioneering factor that can open closed chromatin and promote transcription by facilitating H3K27 acetylation (H3K27ac) through its coactivators CBP/p300 and by recruiting key members of the oncogenic T‐ALL core regulatory circuit. 31 , 60 , 61 We performed H3K27ac ChIPseq on MyPL1 and MyPL2 cells, and we identified a total of 607 genes in the vicinity of super‐enhancer regions, 147 of which were found in both cell lines (Figure 4A,B). Analysis with Enrichr 62 , 63 showed that these 607 genes were strongly associated with the T‐cell lineage, and with genes that are downregulated in mouse thymocytes with Myb knockout (Figure 4C–E). Upon Myb knockdown in iMyb MyPL1 cells, we observed a strong reduction in the global levels of the activating H3K27ac histone mark (Figure 4F,G). Overall, these findings suggest that loss of Myb might decrease CBP/p300‐mediated H3K27 acetylation and super‐enhancer‐driven expression of oncogenic networks. These results are consistent with earlier studies on myeloid leukemia, in which the expression of MYB‐dependent genes was decreased by blocking the interaction between MYB and CBP/p300. 36
Figure 4.
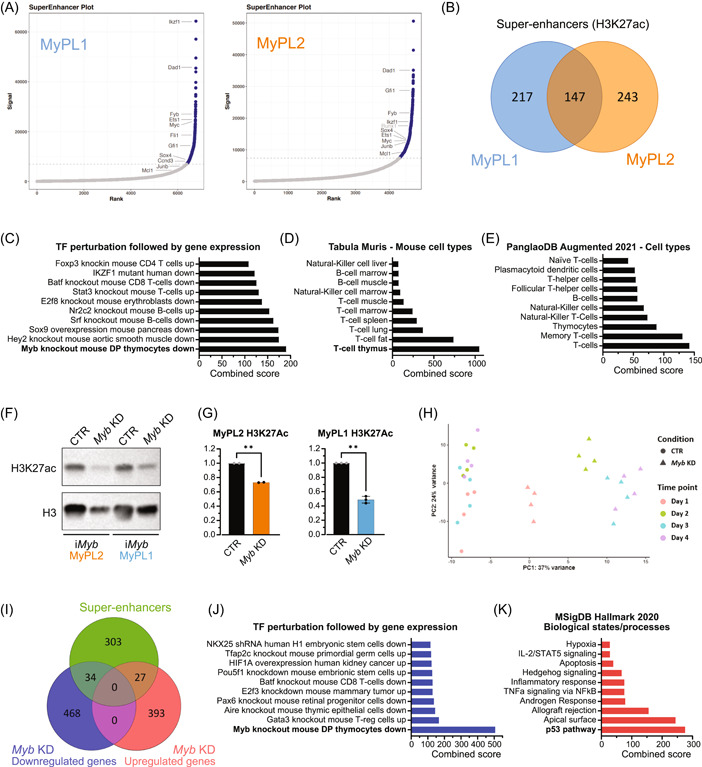
Loss of Myb decreases H3K27 acetylation and downregulates genes in MYB‐driven super‐enhancers. (A) Hockey stick plot showing the levels of H3K27 acetylation in regulatory regions in the vicinity of genes, highlighting some of the super‐enhancer regions (in blue) found in MyPL1 (CD4+CD8+) and MyPL2 (CD4−CD8−) mouse T‐ALL cell lines. (B) Venn diagram showing the overlap between the super‐enhancers found in MyPL1 and MyPL2. Despite representing cell types in distinct differentiation stages, MyPL1 and MyPL2 T‐ALL cell lines share around 24.2% (147/607) of the super‐enhancers, including genes highlighted in panel A, such as Ets1, Myc, Dad1, Ikzf1, and Sox4. (C–E) Bar graphs of enrichment analysis using the 607 super‐enhancer regions found in MyPL1 and MyPL2 cells. The super‐enhancers found match significantly the genes that are downregulated in mouse thymocytes upon knockout of Myb (C), and appear to be strongly related to T‐cell identity (D, E). (F) Western blot analysis showing the global levels of H3K27 acetylation (H3K27ac) in iMyb MyPL1 and iMyb MyPL2 cells that were either treated with 1 µg/mL doxycycline (Myb KD) for 48 h or not (CTR). Total histone H3 (H3) levels were used as a loading control. (G) Bar graphs showing the normalized (against H3) quantification of epigenetic marks from panel F. **p (iMyb MyPL2) = 0.0063 and *p (iMyb MyPL1) = 0.0029. (H) Principal component (PC) analysis of RNAseq data of iMyb MyPL1 cells that were either treated with 1 µg/mL doxycycline (Myb KD, triangle) or not treated (CTR, circle) for 4 consecutive days and four replicates. Replicates cluster per day of treatment, and transcriptomic changes caused by Myb knockdown increase over time. (I) Venn diagram showing the overlap between the super‐enhancers found in MyPL1 and the up‐ or downregulated genes upon Myb knockdown. (J) Bar graph of enrichment analysis using the 34 genes in super‐enhancer regions that are downregulated following Myb knockdown in iMyb MyPL1 cells. These genes match significantly the genes that are downregulated upon loss of Myb in mouse thymocytes, showing that MYB drives specifically super‐enhancers in genes such as Ccnd3, Stat5a, Ets1, and Ezh2 and promotes their expression. (K) Bar graph of enrichment analysis using the 27 genes in super‐enhancer regions that are upregulated following Myb knockdown in iMyb MyPL1 cells. This gene signature is significantly associated with the p53 pathway.
To better understand the transcriptional consequences of Myb knockdown in murine T‐ALL, we treated iMyb MyPL1 cells with doxycycline for four consecutive days and collected samples daily for RNA sequencing. The knockdown of Myb resulted in progressive transcriptomic changes over time (Figure 4H), peaking on days 3 and 4, with around 2400 genes significantly upregulated or downregulated (Supporting Information S2: Table S6). We then performed a strict selection of all the genes that were upregulated or downregulated at all days (p‐adj < 0.005), and compared that list to the 364 super‐enhancers identified in MyPL1 (Figure 4I). We found that 34 genes in super‐enhancer regions were downregulated upon Myb knockdown, including Gata3, Ets1, Stat5a, and Ezh2, and were very significantly associated with loss of Myb in mouse thymocytes (p‐adj = 1.18 × 10−9, combined score = 505.7; Figure 4J). On the other hand, 27 genes in super‐enhancer regions were upregulated after Myb knockdown, and several of these genes seem to be involved in the p53 pathway (p‐adj = 6.42 × 10−6, combined score = 275.6; Figure 4K) or in the regulation of the cell cycle, including Cdkn1a (p21), Mxd4, Trp53inp1, Btg2, and Sh3bgrl2. Taken together, these results highlight the antileukemic effects resulting from the loss of MYB in T‐ALL.
MYB is essential for cell cycle and survival in murine T‐ALL
Focusing on the RNAseq data alone, we observed that the differentially expressed genes significantly match the knockout of Myb in murine thymocytes, both for downregulated (p‐adj = 7.05 × 10−14, combined score = 365.31; Figure 5A) and upregulated genes (p‐adj = 2.02 × 10−11, combined score = 339.49; Figure 5A). On day 4, the upregulated genes were weakly related to the p53 pathway and apoptosis (p‐adj = 3.50 × 10−5 and 7.91 × 10−5, combined score = 32.6 and 26.3, respectively; Figure 5B). On the other hand, the downregulated genes were very strongly associated with pathways regulating DNA replication and cell cycle (p‐adj = 1.52 × 10−41 and 9.88 × 10−42, combined score = 1191 and 694, respectively; Figure 5B). Overall, Myb knockdown caused downregulation of cyclins and other proteins that promote cell cycle progression, while simultaneously upregulating prominent inhibitors of cell cycle progression, such as Cdkn1a (p21) and Rbl2 (p130; Figure 5C). Furthermore, Gene Set Enrichment Analysis 64 showed an association between the gene signature following Myb knockdown at day 4 and the gene sets corresponding to targets of the DREAM complex, which has been previously described as a master coordinator of cell cycle‐dependent gene expression, 65 , 66 and with genes that are upregulated upon loss of RB1 (Figure 5D), a well‐known inhibitor of DNA replication and cell cycle progression. 67
Figure 5.
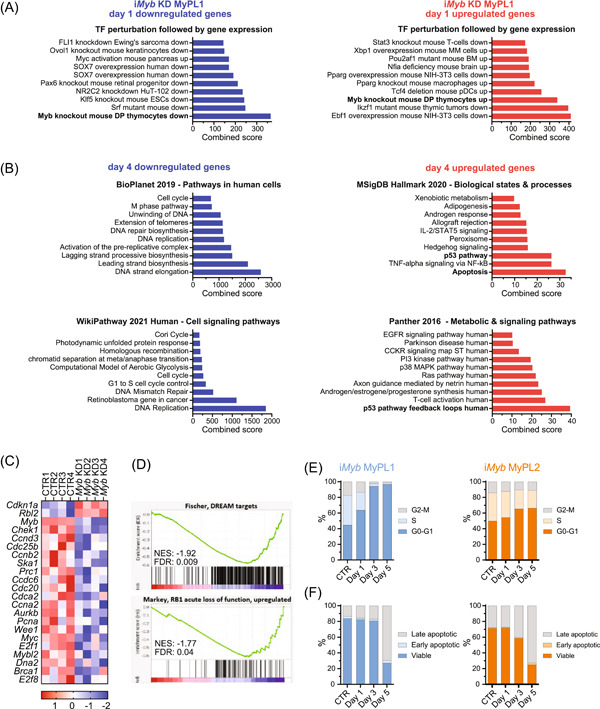
Loss of Myb halts cell cycle progression and induces cell death in murine T‐ALL. (A, B) Bar graphs of enrichment analysis using a gene expression signature of iMyb MyPL1 cells that suffered from Myb knockdown, or not, on experimental day 1 (A) and day 4 (B). Enrichment analysis showed that the gene signature of Myb knockdown in iMyb MyPL1 matches significantly the genes that are affected by Myb knockout in mouse thymocytes (A), both for the downregulated genes (in blue), and the upregulated genes (in red). After 4 days without MYB (B), iMyb MyPL1 cells showed very significant downregulation of genes involved in DNA replication and cell cycle progression (in blue), and an upregulation of genes implicated in the p53 pathway, and in apoptosis (in red). (C) Heatmap of messenger RNA expression of genes that are involved in cell cycle regulation after 4 days in untreated (CTR1‐4) and doxycycline‐treated iMyb MyPL1 cells (Myb KD1‐4). (D) Gene set enrichment analysis of targets of the DREAM complex and upregulated genes upon loss of RB1. (E) Stacked bar chart of the percentage of cells per phase of the cell cycle in untreated (CTR) iMyb MyPL1 (in blue) or iMyb MyPL2 (in orange) cells or in doxycycline‐treated cells on experimental days 1, 3, and 5. Myb knockdown led to a decreased number of cycling cells, as shown by the accumulation of cells in G0/G1, and the reduced proportion of cells at the S and G2/M phases of the cell cycle. A representative result from two independent experiments is shown. (F) Stacked bar chart of the percentage of viable, early apoptotic, or late apoptotic cells (as determined by Annexin/PI staining) in either untreated (CTR), or doxycycline‐treated iMyb MyPL1 or iMyb MyPL2 cells on experimental days 1, 3, and 5. A representative result from two independent experiments is shown. FDRq, false discovery rate q‐value; NES, normalized enrichment score.
Since these molecular analyses strongly point to MYB being a key regulator of cell cycle progression in T‐ALL, we performed cell cycle analysis on the iMyb MyPL1 cell line using propidium iodide (PI) staining. The absence of MYB induced a strong cell cycle arrest over time, with a nearly complete accumulation of cells in G0/G1, and a proportional decrease in the S and G2/M fractions by day 4 (Figure 5E). Because MYB has been described to play an anti‐apoptotic role in multiple cancers, 36 , 49 , 68 we also performed annexin V/propidium iodide apoptosis assay on iMyb MyPL1 cells. Loss of Myb resulted in a progressive accumulation of apoptotic cells, with over two‐thirds of the cells undergoing apoptosis by day 5 (Figure 5F). On the other hand, the loss of MYB in iMyb MyPL2 cells only caused a mild cell cycle block in G0/G1, but still triggered a strong induction of apoptosis (Figure 5E,F). Unlike what has been previously reported for human T‐ALL cell lines with MYB‐duplication, 28 flow cytometry analysis did not reveal differentiation of iMyb MyPL1 or MyPL2 cells upon Myb knockdown (Supporting Information S1: Figure S5), although mRNA expression of Cd4, Cd8a, Cd8b1, and Cd3e was mildly upregulated in iMyb MyPL1 cells (Cd4, 0.43‐fold, p‐adj. = 0.026; Cd8a, 0.52‐fold, p‐adj. = 1.6 × 10−13; Cd8b1, 0.56‐fold, p‐adj. = 2.1 × 10−5; Cd3e, 0.77‐fold, 4.4 × 10−12). In conclusion, the loss of MYB inhibited cell cycle progression and triggered apoptosis of murine T‐ALL cells.
MYB is essential for cellular division and survival in human T‐ALL
Since Myb knockdown triggered a robust cell cycle arrest and apoptosis in our T‐ALL mouse model, we decided to further investigate the consequences of loss of MYB expression in human T‐ALL. We performed MYB knockdown in two human T‐ALL cell lines with different immunophenotypes and MYB duplication statuses. Jurkat is a mature single positive CD4 (CD3+CD4+CD8−) TAL‐LMO cell line with PTEN frameshift and missense mutations and is wild type for MYB. Conversely, RPMI‐8402 is an immature double‐negative (CD3−CD4−CD8−) TAL‐LMO cell line with a SIL‐TAL1 translocation, PTEN missense and frameshift mutations, and MYB duplication. Both cell lines were lentivirally transduced with the abovementioned lentiviral vector for inducible shRNA MYB knockdown (iMYB). 49 Following the induction of shRNA expression, a strong reduction in the levels of MYB protein was observed in both cell lines (Figure 6A). We then monitored the effect of sustained MYB loss for 7 days and observed that, despite the differences in differentiation stage and MYB CNG status, a similar effect occurred in both cell lines, with a gradual drop in cell counts starting from day 3 (Figure 6B). This effect coincided with a cell cycle block, as shown by an accumulation of cells in G0/G1 that went up from 81% to 91% by day 7 (Figure 6C). Furthermore, a simultaneous increase in cell death occurred, with 59%–92% of cells undergoing apoptosis by the 7th day (Figure 6D). Similar to Myb KD in murine T‐ALLs, MYB KD in Jurkat also resulted in a reduction of total H3K27ac levels (Supporting Information S1: Figure S6A,B). Moreover, we analyzed TAL1 levels upon MYB KD in Jurkat, since these cells harbor a microinsertion, upstream of TAL1, that creates a de novo MYB binding site and recruits a super‐enhancer complex that drives TAL1 overexpression. 31 In line with this notion, MYB KD in Jurkat also caused a decrease in TAL1 expression (Supporting Information S1: Figure S6C). To validate this antileukemic effect in vivo, we transplanted iMYB Jurkat cells into immunocompromised NXG mice, which were randomly separated into two groups (n = 11/group) on experimental day 4, and given either a conventional diet or a doxycycline‐containing diet. At experimental day 42, leukemia progression was compared between both groups. To this end, five randomly selected mice from each group were killed, and the leukemic burden in their bone marrow was analyzed by determining the percentage of human CD45+/GFP+ cells by flow cytometry (Figure 6E). At this intermediate time point, leukemic infiltration was detected in the bone marrow of all control animals, but no Jurkat cells were found in mice treated with doxycycline (Figure 6F,G). The remaining six mice in each cohort were used for survival analysis (Figure 6H). Control mice had a median survival of 50.5 days, while all doxycycline‐fed mice survived until the end of the experiment, at day 130 (Figure 6I), with no signs of Jurkat cells in their bone marrow (data not shown). In conclusion, MYB was required for the expansion and survival of human T‐ALL cells, both in vitro and in vivo.
Figure 6.
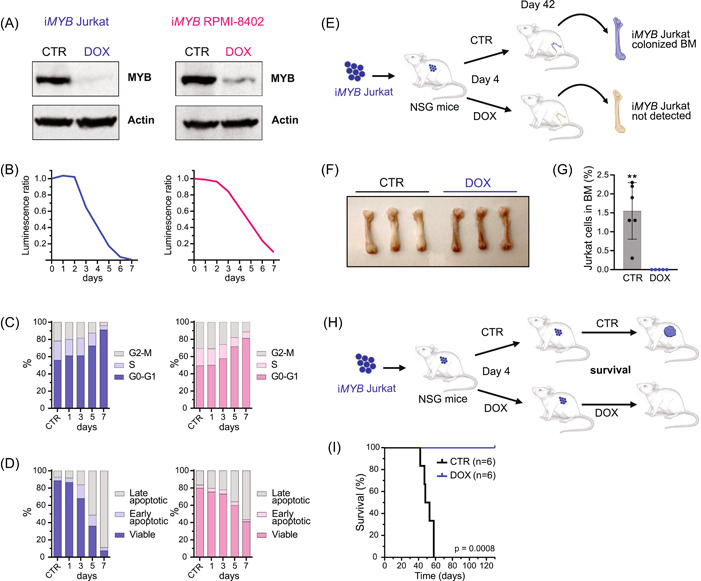
Loss of MYB blocks cell cycle progression and causes cell death in human T‐ALL. (A) Immunoblotting analysis of MYB protein levels in Jurkat and RPMI‐8402 T‐ALL cell lines that have been lentivirally transduced with a tetracycline‐inducible vector to express an shRNA targeting MYB (iMYB). Following 2 days of doxycycline treatment, the transduced cells showed a strong reduction in MYB protein levels. The control condition (CTR) had no tetracyclines in the medium. The treated condition had 1 µg/mL of doxycycline hyclate (DOX) added to the culture medium. Actin was used as the loading control. (B–D) iMYB Jurkat (in blue) and iMYB RPMI‐8402 (in pink) were treated with 1 µg/mL of doxycycline hyclate in the culture medium for 7 consecutive days (D1–D7), or not (CTR), and were analyzed for cell viability (B), cell cycle (C), and apoptosis (D). A representative result from two independent experiments is shown. (B) Line chart of the ratio of luminescence detected with CellTiter‐Glo normalized against the control condition. (C) Stacked bar charts showing the percentage of cells per phase of the cell cycle and (D) the percentage of viable, early apoptotic, or late apoptotic cells per day. (E) Schematic representation of the in vivo experiment with iMYB Jurkat cells. iMYB Jurkat cells (9 × 105/mouse) were transplanted intravenously into immunodeficient NXG mice (n = 5/group), which were given four days post‐transplantation either a normal diet (CTR mice) or a diet containing 200 ppm of doxycycline (DOX mice). On day 42, the percentage of human CD45‐positive (hCD45+) Jurkat cells in the bone marrow was analyzed. (F) Photograph of femurs from CTR and DOX mice at day 42. Pallor is observed in femurs of CTR mice, as a consequence of the replacement of bone marrow cells with hCD45+ Jurkat cells. This pallor was not seen in DOX mice. (G) Bar chart representing the percentage of hCD45+/eGFP+ Jurkat cells detected in the bone marrow of the CTR and DOX mice. **p = 0.0043. (H) Schematic representation of the in vivo survival experiment with iMYB Jurkat cells showing that mice that were treated with DOX from day 4 until the end of the experiment had significantly increased survival. (I) Kaplan–Meier survival curve of NXG mice that were transplanted with iMYB Jurkat cells and were treated as described in E (n = 6/group). A log‐rank Mantel–Cox test showed that the survival of DOX mice was significantly higher than that of control mice (median survival of CTR mice was 50.5 days, and DOX mice did not show any sign of disease up to day 130).
Targeting the transcriptional activity of MYB in murine and human T‐ALL
Since our data showed that MYB is a dependency factor in both mouse and human T‐ALL, the inhibition of its transcriptional activity might be an attractive strategy for T‐ALL therapy. The transcriptional activity of MYB involves DNA binding and the recruitment of coactivators, such as CBP or its paralogue p300, via the transactivation domain of MYB. In particular, the MYB residue E308 is essential for this interaction, as was elegantly shown in the Booreana mouse strain, which has a naturally occurring Myb E308G mutation and is resistant to induction of myeloid leukemia. 69 Here, we blocked the interaction between MYB and its transcriptional coactivators CBP/p300 using MYBMIM, a cell‐penetrant peptidomimetic inhibitor. 36 As a control peptide, we used an inactive version of MYBMIM, named TG3, in which three key MYB residues for this CBP/p300 interaction were mutated. Both the mouse cell lines derived from the Myb/Pten Lck mice and the human T‐ALL cells were sensitive to the treatment with MYBMIM, and no significant effects were seen upon treatment with TG3 in the concentration range used (Figure 7A,B). Probably due to being more immediate than the shRNA‐mediated knockdown of Myb, MYBMIM achieved a faster antileukemic effect and caused over 50% viability reduction of all T‐ALL cell lines after 48 h with concentrations ranging from 10 to 40 µM. To assess whether blocking the interaction between MYB and CBP/p300 is comparable to the antileukemic effect of MYB loss by shRNA, we treated Jurkat and KOPT‐K1 cells with a single concentration of MYBMIM and TG3 for 48 h and performed viability and apoptosis assays, as well as cell cycle analysis. We found that besides preventing the expansion of T‐ALL cells, MYBMIM also caused a reduction in their viability (Figure 7C). Furthermore, our results show that blocking the interaction between MYB and CBP/p300 caused a mild cell cycle arrest in G0/G1 while simultaneously triggering cell death in T‐ALL cell lines (Figure 7D,E).
Figure 7.
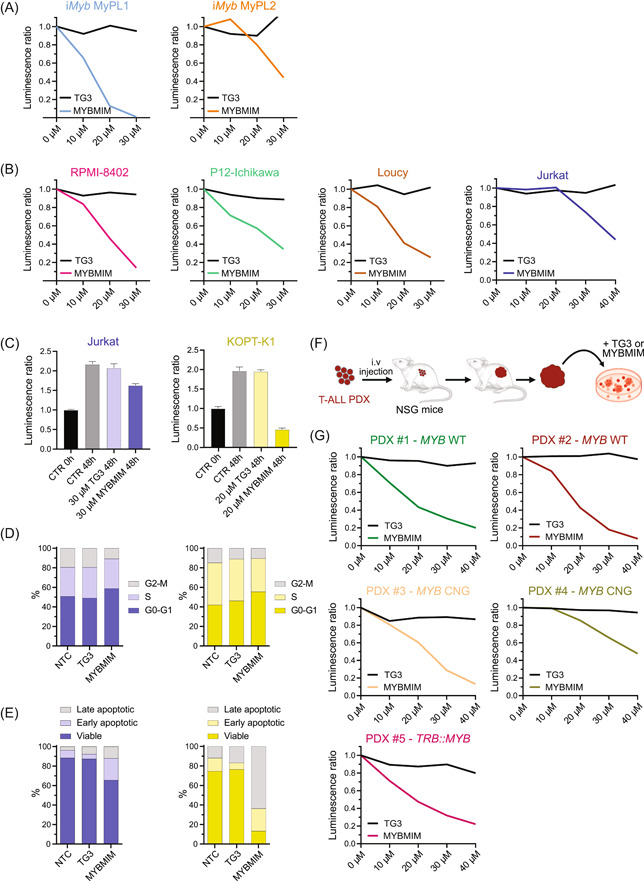
Peptidomimetic blockade of MYB triggers cell death in T‐ALL. (A, B) Line chart of the ratio of luminescence detected using CellTiter‐Glo cell viability assay (hereafter called relative viability) upon treatment of cell lines with 10–40 µM of MYBMIM (colored line) or its inactive version, named TG3 (black line), normalized to the untreated condition. (A) MyPL1 and MyPL2 are murine R26‐Myb tg/tg ; Pten fl/fl ; Lck‐Cre tg/+ T‐ALL cell lines. (B) RPMI‐8402, P12‐Ichikawa, Loucy, KOPT‐K1, and Jurkat are human T‐ALL cell lines. (C) Bar chart showing the relative viability of Jurkat and KOPT‐K1 cells at the beginning of the experiment (0 h), and 48 h later, when treated with either 30 µM (for Jurkat) or 20 µM (for KOPT‐K1) of MYBMIM, TG3, or untreated (CTR 48 h). (D) Stacked bar charts showing the percentage of cells per phase of the cell cycle and (E) the percentage of viable, early apoptotic, or late apoptotic cells at 48 h. A representative result from two independent experiments is shown. (F) Scheme of generation of T‐ALL patient‐derived xenografts (PDXs) through intravenous injections, followed by their ex vivo treatment with either TG3 or MYBMIM. (G) Line chart of the relative viability of T‐ALL PDX with either chromosomal translocation (TRB::MYB), genomic gain of MYB (MYB CNG), or wild‐type MYB (MYB WT), upon treatment with 10–40 µM of MYBMIM (colored line) or TG3 (black line), normalized to the untreated condition.
Next, using MYBMIM ex vivo, we treated five T‐ALL PDXs with either chromosomal translocation, genomic duplication, or wild‐type MYB. Similar to the effects seen in the cell lines, all PDXs had their viability reduced by over 50% after 48 h with MYBMIM concentrations ranging from 10 to 40 µM, but no significant differences were seen when TG3 was used in the same concentration range (Figure 7F,G).
Taken together, our findings suggest that the CBP/p300‐dependent functions of MYB are essential for maintaining an oncogenic program in T‐ALL, which might be associated with the generation of super‐enhancers by acetylating H3K27, opening chromatin, and recruiting transcription factors that promote an oncogenic T‐ALL core regulatory circuit.
DISCUSSION
MYB is highly expressed in ALL, and a subset of T‐ALL cases harbor genomic aberrations that further potentiate the expression or activity of this proto‐oncogene. Previous studies have associated MYB with a malignant role in the induction, pathogenesis, and maintenance of several cancers. 22 , 23 , 24 , 25 , 26 , 27 However, its role in T‐ALL is still relatively poorly understood.
In this study, we used the PeCan database and found a negative association between increased copy number of MYB, which leads to higher MYB levels, and the event‐free survival of pediatric T‐ALL patients. However, we could not validate the prognostic value of MYB CNG in an independent validation T‐ALL cohort. Using our previously described approach, 70 we generated a mouse with conditional Myb overexpression 23 and used it to assess the effects of Myb overexpression in two different spontaneous T‐ALL mouse models. We demonstrated that the gain of Myb synergizes with Pten deletion to accelerate the initiation of T‐cell malignancies. The T‐ALL mouse model driven by the loss of Pten resembles human TAL‐LMO T‐ALL, which often displays a more mature immunophenotype, 6 , 8 , 9 , 10 along with frequent mutations affecting the PI3K pathway, 56 , 57 and where the consistently high MYB levels have been reported to sustain the TAL1‐driven oncogenic feed‐forward circuit. 60 Our Myb/Pten Lck model added an extra layer of complexity to the original Pten‐deletion model, 39 and hinted that Myb overexpression may cooperate with Pten loss, particularly during the events associated with the initiation of the malignant transformation. During T‐cell transformation, Pten‐null thymocytes accumulate multiple oncogenic hits, including reciprocal t(14:15) chromosomal translocations involving the T‐cell receptor alpha/delta locus and Myc. 58 , 59 We have confirmed the presence of these Myc translocations in Pten‐null T‐ALLs. More research will be necessary to determine whether the MYC mutational status plays an essential role in the observed accelerated leukemia initiation upon MYB gain in the Pten Lck model. On the other hand, Myb overexpression did not influence the initiation of leukemia in the Lmo2‐driven T‐ALL model. Overexpression of LMO2 hinders T‐cell differentiation, both in mice and humans, and promotes an accumulation of self‐renewing immature cells associated with the development of early T‐cell precursor ALL (ETP‐ALL), 40 , 47 , 71 , 72 , 73 , 74 which is often characterized by high levels of LYL1. 1 , 75 In AML, MYB is known to generate oncogenic super‐enhancers and recruit an aberrant complex of transcriptional coactivators that includes LMO2 and LYL1. 76 In the context of T‐ALL, MYB also forms super‐enhancers to activate the transcription of oncogenes and promote a core regulatory circuit. 31 , 60 For example, somatic noncoding mutations that create de novo binding motifs for MYB in TAL1, LMO1, or LMO2 genes have been demonstrated to result in the recruitment of transcriptional machinery and cause aberrant overexpression of those oncogenes. 31 , 32 , 77 The TAL1 transcriptional complex recruited by MYB in the super‐enhancers also includes and requires the involvement of LMO2, 31 thus suggesting that these two proteins can cooperate in T‐ALL. However, the role of MYB is highly context‐dependent, 78 and even though LMO2 seems to promote a positive regulatory network in ETP‐ALL, 72 MYB has yet to be implicated in the regulation of this LMO2‐LYL1 oncogenic axis. Furthermore, LMO2‐LYL1 cases display relatively low levels of MYB, compared to other T‐ALL subgroups (Figure 1B). 8 Taken together, these considerations seem to indicate that LMO2‐LYL1 T‐ALLs might not require sustained MYB overexpression for malignant transformation and maintenance.
Using a validated doxycycline‐inducible shRNA‐mediated knockdown approach, 25 , 49 we observed that MYB sustained MyPL cell lines by promoting cell cycle progression and inhibiting apoptosis. This finding is concordant with previous reports about the role of MYB in hematopoietic malignancies. 25 , 79 Nonetheless, Lahortiga and collaborators have reported that the loss of MYB had only a minor impact on viability, proliferation, and cell cycle in human T‐ALL cell lines. 28 Our results showed that the consequences of Myb loss were progressive (Figure 4H), and it took several days for the cell cycle arrest and apoptosis to become evident (Figure 6B–D). This might indicate that T‐ALL cells can temporarily tolerate lower MYB levels down to a certain threshold before succumbing. It is possible that the time point chosen by Lahortiga and colleagues was too early for the detection of significant cell cycle arrest and apoptosis.
Although the in vivo Myb knockdown in MyPL1 cells improved mouse survival, the NSG mice still developed GFP‐negative and knockdown‐resistant tumors. A similar outcome was reported by De Dominici and collaborators, using the same inducible shRNA on Philadelphia chromosome‐positive (Ph+) leukemia cells. 25 In their in vivo study, inducible MYB KD in Ph+ leukemia cells increased the survival of the recipient mice, which ultimately still developed tumors that regained MYB expression. Those tumors displayed a lower GFP intensity, possibly associated with a reduced expression of MYB shRNA. In our study, it is unclear whether the GFP‐negative MyPL1 tumors resulted from silencing of the retroviral insert or simply arose due to clonal selection of nontransduced cells. Regardless, these findings seem to reiterate the importance of MYB during T‐ALL maintenance.
Our results suggest that MYB may be a good therapeutic target for T‐ALL, since both lentiviral knockdown and pharmacological inhibition of MYB resulted in antileukemic effects in all examined T‐ALL cell lines and PDXs, irrespective of their immunophenotype, T‐ALL subgroup, and even MYB duplication status. Transcription factors are generally regarded as difficult targets for therapy due to their short half‐life, 80 and up to now, no clinical‐grade MYB‐targeting strategy exists. 35 , 81 For this reason, researchers frequently attempt to identify druggable downstream targets rather than targeting the transcription factor directly. De Dominici and colleagues identified several downstream targets of MYB that are critical for its oncogenic function in Ph+ leukemia, and they were able to partially mitigate the antileukemic consequences of MYB silencing by ectopically expressing CDK6, cyclin D3, BCL2, and silencing p21. Afterward, they combined the CDK4/6 inhibitor palbociclib with a BCL2 antagonist compound and observed a synergistic antileukemic effect in vivo. 25 We found a similar upregulation of the p21 gene, Cdkn1a, and downregulation of Ccnd3 in MyPL1 cells following Myb knockdown, along with many other genes involved in the regulation of the cell cycle (Figure 4G). However, we could not validate Cdk6 and Bcl2 as targets of MYB in MyPL1 cells.
Other groups have successfully demonstrated the therapeutic potential of MYB targeting in vivo for other hematopoietic cancers while showing that transient MYB targeting is compatible with normal hematopoiesis. 36 , 37 , 82 In this study, we tried a peptidomimetic approach 36 based on blocking the interaction between MYB and its coactivators, CBP and p300, aimed at disrupting the oncogenic super‐enhancer and transcription factor complexes, and we validated its antileukemic potential in T‐ALL.
In conclusion, our data indicate that MYB overexpression plays an oncogenic role in cooperation with the loss of Pten to promote the formation of T‐ALL. Furthermore, the tools used in this study allowed us to validate both the direct inhibition of MYB as well as the disruption of the MYB‐CBP/p300 interaction as promising alternative therapeutic approaches for patients with chemotherapy‐resistant or relapsed T‐ALL.
AUTHOR CONTRIBUTIONS
André Almeida, Luca Pagliaro, Steven Goossens, Pieter Van Vlierberghe and Tim Pieters: Conceived research. André Almeida, Steven Goossens, Tim Pieters, and Luca Pagliaro: Wrote the paper. André Almeida, Sara T'Sas, Tim Pieters, Luca Pagliaro, Igor Fijalkowski, Wouter Sleeckx, Hannah Van Steenberge, Raffaella Zamponi, Béatrice Lintermans, Wouter Van Loocke, Bruno Palhais, Lisa Demoen, Lindy Reunes, Dieter Deforce, Filip Van Nieuwerburgh, and Nadine Van Roy: Data collection and analysis. Roberta La Starza, Cristina Mecucci, Alexandra Reekmans, and Barbara De Moerloose: Provided clinical data. Alex Kentsis, Panagiotis Ntziachristos, and Giovanni Roti: Contributed new reagents, models, and analytic tools.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
FUNDING
This work was supported by the following funding agencies: Research Foundation – Flanders (FWO) and the Baillet Latour Funds. The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation ‐ Flanders (FWO). AK is a Scholar of the Leukemia & Lymphoma Society and is supported by the US National Institute of Health grants P30 CA08748 and RO1 CA204396. The Ntziachristos laboratory is supported by the Research Foundation Flanders (FWO, G0F4721N and G0A8B24N), start‐up funds from the Department of Biomolecular Medicine, Ghent University, a Flanders interuniversity consortium grant (BOF.IBO.2023.0006.02), and a Cancer Research Institute Ghent (CRIG) partnership grant.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank Drs T. Gonda and M. De Dominici for generating and kindly providing the pLVTSH‐MYB shRNA vector.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/, reference number GSE253616.
REFERENCES
- 1. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T‐ALL. Blood. 2017;129(9):1113‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Teachey DT, O'Connor D. How I treat newly diagnosed T‐cell acute lymphoblastic leukemia and T‐cell lymphoblastic lymphoma in children. Blood. 2020;135(3):159‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patel AA, Thomas J, Rojek AE, Stock W. Biology and treatment paradigms in T cell acute lymphoblastic leukemia in older adolescents and adults. Curr Treat Options Oncol. 2020;21(7):57. [DOI] [PubMed] [Google Scholar]
- 4. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371(9617):1030‐1043. [DOI] [PubMed] [Google Scholar]
- 5. Marks DI, Rowntree C. Management of adults with T‐cell lymphoblastic leukemia. Blood. 2017;129(9):1134‐11421142. [DOI] [PubMed] [Google Scholar]
- 6. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brady SW, Roberts KG, Gu Z, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54(9):1376‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen B, Jiang L, Zhong ML, et al. Identification of fusion genes and characterization of transcriptome features in T‐cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2018;115(2):373‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai YT, Zhang F, Fang H, et al. Transcriptome‐wide subtyping of pediatric and adult T cell acute lymphoblastic leukemia in an international study of 707 cases. Proc Natl Acad Sci USA. 2022;119(15):e2120787119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Homminga I, Pieters R, Langerak AW, et al. Integrated transcript and genome analyses reveal NKX2‐1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19(4):484‐497. [DOI] [PubMed] [Google Scholar]
- 11. Lieu YK, Reddy EP. Conditional c‐myb knockout in adult hematopoietic stem cells leads to loss of self‐renewal due to impaired proliferation and accelerated differentiation. Proc Natl Acad Sciences USA. 2009;106(51):21689‐21694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ess KC, Witte DP, Bascomb CP, Aronow BJ. Diverse developing mouse lineages exhibit high‐level c‐Myb expression in immature cells and loss of expression upon differentiation. Oncogene. 1999;18(4):1103‐1111. [DOI] [PubMed] [Google Scholar]
- 13. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8(7):523‐534. [DOI] [PubMed] [Google Scholar]
- 14. Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c‐Myb. J Mol Biol. 2004;337(3):521‐534. [DOI] [PubMed] [Google Scholar]
- 15. Mucenski ML, McLain K, Kier AB, et al. A functional c‐myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65(4):677‐689. [DOI] [PubMed] [Google Scholar]
- 16. Bender TP, Kremer CS, Kraus M, Buch T, Rajewsky K. Critical functions for c‐Myb at three checkpoints during thymocyte development. Nat Immunol. 2004;5(7):721‐729. [DOI] [PubMed] [Google Scholar]
- 17. Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia. 2013;27(2):269‐277. [DOI] [PubMed] [Google Scholar]
- 18. Ganter B, Lipsick JS. Myb and oncogenesis. Adv Cancer Res. 1999;76:21‐60. [DOI] [PubMed] [Google Scholar]
- 19. Mukai HY, Motohashi H, Ohneda O, Suzuki N, Nagano M, Yamamoto M. Transgene insertion in proximity to the c‐myb gene disrupts erythroid‐megakaryocytic lineage bifurcation. Mol Cell Biol. 2006;26(21):7953‐7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beug H, von Kirchbach A, Döderlein G, Conscience JF, Graf T. Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinct phenotypes of differentiation. Cell. 1979;18(2):375‐390. [DOI] [PubMed] [Google Scholar]
- 21. Shen‐Ong GLC, Potter M, Mushinski JF, Lavu S, Reddy EP. Activation of the c‐myb locus by viral insertional mutagenesis in plasmacytoid lymphosarcomas. Science. 1984;226(4678):1077‐1080. [DOI] [PubMed] [Google Scholar]
- 22. Badiani PA, Kioussis D, Swirsky DM, Lampert IA, Weston K. T‐cell lymphomas in v‐Myb transgenic mice. Oncogene. 1996;13(10):2205‐2212. [PubMed] [Google Scholar]
- 23. Pieters T, Almeida A, T'Sas S, et al. Myb drives B‐cell neoplasms and myeloid malignancies in vivo. Blood Adv. 2022;6(10):2987‐2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zuber J, Rappaport AR, Luo W, et al. An integrated approach to dissecting oncogene addiction implicates a Myb‐coordinated self‐renewal program as essential for leukemia maintenance. Genes Dev. 2011;25(15):1628‐1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Dominici M, Orazzi P, Oliera AR, et al. Targeting CDK6 and BCL2 exploits the “MYB Addiction” of Ph+ acute lymphoblastic leukemia. Cancer Res. 2018;8:097‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanghvi VR, Mavrakis KJ, Van der Meulen J, et al. Characterization of a set of tumor suppressor microRNAs in T cell acute lymphoblastic leukemia. Sci Signal. 2014;7(352):ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen N, Vishwakarma BA, Oakley K, et al. Myb expression is critical for myeloid leukemia development induced by Setbp1 activation. Oncotarget. 2016;7(52):86300‐86312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lahortiga I, De Keersmaecker K, Van Vlierberghe P, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39(5):593‐595. [DOI] [PubMed] [Google Scholar]
- 29. Clappier E, Cuccuini W, Kalota A, et al. The C‐MYB locus is involved in chromosomal translocation and genomic duplications in human T‐cell acute leukemia (T‐ALL), the translocation defining a new T‐ALL subtype in very young children. Blood. 2007;110(4):1251‐1261. [DOI] [PubMed] [Google Scholar]
- 30. Noronha EP, Marques LVC, Andrade FG, Thuler LCS, Terra‐Granado E, Pombo‐de‐Oliveira MS. The profile of immunophenotype and genotype aberrations in subsets of pediatric T‐cell acute lymphoblastic leukemia. Front Oncol. 2019;9:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mansour MR, Abraham BJ, Anders L, et al. An oncogenic super‐enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346(6215):1373‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rahman S, Magnussen M, León TE, et al. Activation of the LMO2 oncogene through a somatically acquired neomorphic promoter in T‐cell acute lymphoblastic leukemia. Blood. 2017;129(24):3221‐3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu S, Qian M, Zhang H, et al. Whole‐genome noncoding sequence analysis in T‐cell acute lymphoblastic leukemia identifies oncogene enhancer mutations. Blood. 2017;129(24):3264‐32683268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uttarkar S, Dassé E, Coulibaly A, et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood. 2016;127(9):1173‐1182. [DOI] [PubMed] [Google Scholar]
- 35. Walf‐Vorderwülbecke V, Pearce K, Brooks T, et al. Targeting acute myeloid leukemia by drug‐induced c‐MYB degradation. Leukemia. 2018;32(4):882‐889. [DOI] [PubMed] [Google Scholar]
- 36. Ramaswamy K, Forbes L, Minuesa G, et al. Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat Commun. 2018;9(1):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith C, Touzart A, Simonin M, et al. Harnessing the MYB‐dependent TAL1 5'super‐enhancer for targeted therapy in T‐ALL. Mol Cancer. 2023;22(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarvaiya PJ, Schwartz JR, Hernandez CP, Rodriguez PC, Vedeckis WV. Role of c‐Myb in the survival of pre B‐cell acute lymphoblastic leukemia and leukemogenesis. Am J Hematol. 2012;87(10):969‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suzuki A, Yamaguchi MT, Ohteki T, et al. T cell‐specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14(5):523‐534. [DOI] [PubMed] [Google Scholar]
- 40. McCormack MP, Young LF, Vasudevan S, et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self‐renewal. Science. 2010;327(5967):879‐883. [DOI] [PubMed] [Google Scholar]
- 41. Zhou X, Wang J, Patel J, et al. Exploration of coding and non‐coding variants in cancer using GenomePaint. Cancer Cell. 2021;39(1):83‐95 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McLeod C, Gout AM, Zhou X, et al. St. Jude cloud: a pediatric cancer genomic data‐sharing ecosystem. Cancer Disc. 2021;11(5):1082‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun V, Sharpley M, Kaczor‐Urbanowicz KE, et al. The metabolic landscape of thymic T cell development in vivo and in vitro. Front Immunol. 2021;12:716661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verboom K, Van Loocke W, Volders PJ, et al. A comprehensive inventory of TLX1 controlled long non‐coding RNAs in T‐cell acute lymphoblastic leukemia through polyA+ and total RNA sequencing. Haematologica. 2018;103(12):e585‐e589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dankort D, Curley DP, Cartlidge RA, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature Genet. 2009;41(5):544‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hennet T, Hagen FK, Tabak LA, Marth JD. T‐cell‐specific deletion of a polypeptide N‐acetylgalactosaminyl‐transferase gene by site‐directed recombination. Proc Natl Acad Sci USA. 1995;92(26):12070‐12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith S, Tripathi R, Goodings C, et al. LIM domain only‐2 (LMO2) induces T‐cell leukemia by two distinct pathways. PLoS One. 2014;9(1):e85883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Roy N, Laureys G, Cheng NC, et al. I;17 translocations and other chromosome 17 rearrangements in human primary neuroblastoma tumors and cell lines. Genes Chromosom Cancer. 1994;10(2):103‐114. [DOI] [PubMed] [Google Scholar]
- 49. Drabsch Y, Hugo H, Zhang R, et al. Mechanism of and requirement for estrogen‐regulated MYB expression in estrogen‐receptor‐positive breast cancer cells. Proc Natl Acad Sci USA. 2007;104(34):13762‐13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mets E, Van der Meulen J, Van Peer G, et al. MicroRNA‐193b‐3p acts as a tumor suppressor by targeting the MYB oncogene in T‐cell acute lymphoblastic leukemia. Leukemia. 2015;29(4):798‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bardelli V, Arniani S, Pierini V, et al. MYB rearrangements and over‐expression in T‐cell acute lymphoblastic leukemia. Genes Chromosom Cancer. 2021;60(7):482‐488. [DOI] [PubMed] [Google Scholar]
- 55. La Starza R, Pierini V, Pierini T, et al. Design of a comprehensive fluorescence in situ hybridization assay for genetic classification of T‐cell acute lymphoblastic leukemia. J Mol Diagn. 2020;22(5):629‐639. [DOI] [PubMed] [Google Scholar]
- 56. Zuurbier L, Petricoin EF 3rd, Vuerhard MJ, et al. The significance of PTEN and AKT aberrations in pediatric T‐cell acute lymphoblastic leukemia. Haematologica. 2012;97(9):1405‐1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mendes RD, Sarmento LM, Canté‐Barrett K, et al. PTEN microdeletions in T‐cell acute lymphoblastic leukemia are caused by illegitimate RAG‐mediated recombination events. Blood. 2014;124(4):567‐578. [DOI] [PubMed] [Google Scholar]
- 58. Guo W, Lasky JL, Chang CJ, et al. Multi‐genetic events collaboratively contribute to Pten‐null leukaemia stem‐cell formation. Nature. 2008;453(7194):529‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X, Karnell JL, Yin B, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J Clin Invest. 2010;120(7):2497‐2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):209‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fuglerud BM, Ledsaak M, Rogne M, Eskeland R, Gabrielsen OS. The pioneer factor activity of c‐Myb involves recruitment of p300 and induction of histone acetylation followed by acetylation‐induced chromatin dissociation. Epigenetics Chromatin. 2018;11(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90‐W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25(1):114‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fischer M, Schade AE, Branigan TB, Müller GA, DeCaprio JA. Coordinating gene expression during the cell cycle. Trends Biochem Sci. 2022;47(12):1009‐1022. [DOI] [PubMed] [Google Scholar]
- 67. Engeland K. Cell cycle regulation: p53‐p21‐RB signaling. Cell Death Differ. 2022;29(5):946‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Srivastava SK, Bhardwaj A, Arora S, et al. MYB is a novel regulator of pancreatic tumour growth and metastasis. Br J Cancer. 2015;113(12):1694‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pattabiraman DR, McGirr C, Shakhbazov K, et al. Interaction of c‐Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. 2014;123(17):2682‐2690. [DOI] [PubMed] [Google Scholar]
- 70. Pieters T, T'Sas S, Demoen L, et al. Novel strategy for rapid functional in vivo validation of oncogenic drivers in haematological malignancies. Sci Rep. 2019;9(1):10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cleveland SM, Smith S, Tripathi R, et al. Lmo2 induces hematopoietic stem cell‐like features in T‐cell progenitor cells prior to leukemia. Stem Cells. 2013;31(5):882‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McCormack MP, Shields BJ, Jackson JT, et al. Requirement for Lyl1 in a model of Lmo2‐driven early T‐cell precursor ALL. Blood. 2013;122(12):2093‐2103. [DOI] [PubMed] [Google Scholar]
- 73. Wiekmeijer AS, Pike‐Overzet K, Brugman MH, et al. Overexpression of LMO2 causes aberrant human T‐cell development in vivo by three potentially distinct cellular mechanisms. Exp Hematol. 2016;44(9):838‐849 e9. [DOI] [PubMed] [Google Scholar]
- 74. Pike‐Overzet K, de Ridder D, Weerkamp F, et al. Ectopic retroviral expression of LMO2, but not IL2Rγ, blocks human T‐cell development from CD34+ cells: implications for leukemogenesis in gene therapy. Leukemia. 2007;21(4):754‐76363. [DOI] [PubMed] [Google Scholar]
- 75. Genesca E, la Starza R. Early T‐cell precursor all and beyond: immature and ambiguous lineage T‐ALL subsets. Cancers. 2022;14(8):1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Takao S, Forbes L, Uni M, et al. Convergent organization of aberrant MYB complex controls oncogenic gene expression in acute myeloid leukemia. eLife. 2021;10:e65905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Z, Abraham BJ, Berezovskaya A, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T‐ALL. Leukemia. 2017;31(10):2057‐2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. George O, Ness S. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers. 2014;6(4):2049‐2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ye P, Zhao L, Gonda TJ. The MYB oncogene can suppress apoptosis in acute myeloid leukemia cells by transcriptional repression of DRAK2 expression. Leuk Res. 2013;37(5):595‐601. [DOI] [PubMed] [Google Scholar]
- 80. Henley MJ, Koehler AN. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat Rev Drug Discovery. 2021;20(9):669‐688. [DOI] [PubMed] [Google Scholar]
- 81. Uttarkar S, Frampton J, Klempnauer KH. Targeting the transcription factor Myb by small‐molecule inhibitors. Exp Hematol. 2017;47:31‐35. [DOI] [PubMed] [Google Scholar]
- 82. Calabretta B, Sims RB, Valtieri M, et al. Normal and leukemic hematopoietic cells manifest differential sensitivity to inhibitory effects of c‐myb antisense oligodeoxynucleotides: an in vitro study relevant to bone marrow purging. Proc Natl Acad Sci USA. 1991;88(6):2351‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are openly available in Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/, reference number GSE253616.