

Abstract
The development of bioactive small molecules as probes or drug candidates requires discovery platforms that enable access to chemical diversity and can quickly reveal new ligands for a target of interest. Within the past 15 years, DNA-encoded library (DEL) technology has matured into a widely used platform for small-molecule discovery, yielding a wide variety of bioactive ligands for many therapeutically relevant targets. DELs offer many advantages compared to traditional screening methods, including efficiency of screening, easily multiplexed targets and library selections, minimized resources needed to evaluate an entire DEL, and large library sizes. This review provides accounts of recently described small molecules discovered from DELs, including their initial identification, optimization, and validation of biological properties including suitability for clinical applications.
Introduction
DNA-encoded library (DEL) technology is a powerful small-molecule discovery method first described in the 1990s1 and further developed2,3 through advances in library syntheses4–11, selection methodology8,11–13, data workup8,11,13, and ligand discovery7–9,13. Over the course of the past decade, DEL technology has been increasingly applied to the discovery of chemical probes and novel clinical candidates. Indeed, since the discovery of the first DNA-encoded small molecules ~12–15 years ago7–9,14, the number of studies reporting new libraries, selection schemes, and ligands with biological activity has grown rapidly. DEL technology is widely used in academia and industry, facilitating the widespread application of small-molecule libraries. Indeed, more than 70 unique DELs have been reported since initial studies in 200415.
The widespread adoption of DEL technology is the result of its accessibility, versatility, efficiency, and cost-effectiveness, especially when compared to traditional synthetic library screening-based technologies. High-throughput screening (HTS) technologies are both resource and time intensive, often relying on dedicated automated cores that require extensive liquid handling systems and plate manipulation, assaying, and storage capabilities15. HTS requires large amounts of consumables, including individual library members, targets of interest, buffers, plasticware, and other reagents15. Furthermore, because HTS necessitates the analysis of individual members one at a time, these experiments often take days to weeks, scaling with the size of the library and the number of targets co-screened. The overall experimental time is further lengthened by the need to validate a functional assay for each new target type.
In contrast, DEL selections typically can be conducted over a few days, requiring only tens of micrograms of a typical protein target, picomole quantities of DEL, and a next-generation DNA sequencing (NGS) kit. Because DELs are created by covalently attaching each library member to an identifying DNA barcode (Fig. 1a), entire libraries can be pooled together and analyzed for target binding in a single test tube. DEL-enabled small-molecule discovery typically uses an in vitro selection for binding immobilized protein. Following incubation of the entire pooled DEL with the target, active ligands are captured by the immobilized target, and their covalently attached DNA barcodes are amplified by PCR and sequenced to identify target-binding ligands (Fig. 1b). This pooled library selection format, coupled with the ability of DNA tags to be amplified by PCR from sub-attomoles of material, drastically decreases the physical scale of screening. The reduced scale of a DEL selection, compared to HTS screens, allows libraries as large as 105–1012 members to be assessed for binding to many targets in parallel with minimal extra time and materials per added target. Finally, DEL selections are read out through quantification of bound library members by NGS, bypassing the need to validate and implement a new functional screening assay for each target type.
Fig. 1 |. DNA-encoded libraries as a small molecule discovery platform.

a, Architecture of a DNA-encoded library member. Individual building blocks (green, yellow, and purple shapes) that comprise the entire molecular structure are encoded by regions of DNA (green, yellow, and purple DNA, respectively) on a covalently attached DNA oligonucleotide. PCR binding sites on the appended DNA allow the amplification of the entire DNA barcode by PCR for downstream identification and quantification. b, General cycle of a DNA-encoded library selection campaign. The target of interest is prepared in a form suitable for in vitro selection (1) and incubated with the entire pooled DNA-encoded library (2). Library members with target affinity are selectively retained, resulting in their enrichment over non-binding library members (3). Target-bound library members are isolated (4). Surviving library members can then either be reintroduced back into the selection for further rounds of selection (5a) or amplified by PCR and sequenced (5b) to identify and quantify these hit candidates.
The practical advantages of DELs have also made it possible for researchers to access small-molecule libraries through the services offered by CROs. DEL selection services now make it possible for many academic and industry researchers with a target of interest to use a powerful small-molecule discovery platform. Still, others opt to develop their own DELs, given the increasingly capable and streamlined methods now available for their synthesis. DEL campaigns often yield new bioactive small molecules that can be used as chemical probes or clinical candidates, making them well-suited for drug discovery efforts. Indeed, almost all major pharmaceutical companies use DELs, to some degree, for their drug discovery programs15. Notably, over half of the DEL-mediated novel ligands discovered to date were reported in the past six years alone (Fig 2a) and have spanned a range of target types (Fig. 2b).
Fig. 2 |. Publications reporting novel small molecules discovered from DNA-encoded libraries.

a, Publications are binned by year and are only included if the study reports new chemical matter with in vitro activity against the target of interest. b, Number of novel DNA-encoded library discovered ligands per target type. Examples are binned by target type and are only included if the study reports new chemical matter with in vitro activity against the target of interest. One representative ligand for each compound series with a target is counted.
This review will summarize examples of DEL-mediated discovery and development of novel chemical matter. DEL-compatible chemistry16–19 (Box 1), DEL library synthesis and development17–23 (Box 2), or DEL selection methods20,22–27 (Box 3) will not be reviewed, as recent reviews have extensively covered these areas. Examples will be restricted to the past six years and discuss select case studies of DEL-mediated small-molecule discovery will be discussed. These cases exemplify in vitro or biological characterization of DEL-developed small molecules discovered using prototypical immobilization workflows, creative in vitro selection strategies, or selection platforms on living systems. Notably, many of these small molecules exhibit target engagement in biological models, and in such cases, their specific activities will be highlighted. Collectively, these examples illustrate that DEL technology has developed compatibility with targets spanning a wide range of biochemistry, protein families, localization, and biological roles, making it a powerful and broadly applicable small-molecule discovery platform.
Box 1 |. DEL-compatible chemical reactions used to synthesize DNA-encoded libraries.
DELs are unique in that they blend elements of both genetically encoded (the DNA tag) and synthetic combinatorial libraries (the small molecule warhead)19. In principle, one can synthesize a small molecule, covalently attach a unique DNA-tag, and pool all molecules into one pot. However, such an approach requires individual synthesis of each library member, negating some of the throughput advantages that DELs offer over high-throughput screens. Rather, reaction sequences must be carefully designed in a pooled format with a DNA-barcode already present. As such, DNA-compatible chemical reactions for creating DELs are guided by the following criteria19,22:
Reagents are physically compatible with DNA properties (i.e. mild conditions, water or polar solvent).
Reaction conditions maintain integrity of DNA functional groups (sugars, phosphodiesters, anilines, etc.)
Reactions must be high yielding or have an effective method to remove side-products in a pooled format.
Reactions must have high substrate scope to input chemical diversity.
Despite these requirements, several reactions have seen use throughout the field to create customized DELs. These include (but are not limited to) amide coupling, reductive amination, palladium cross-coupling reactions, click reactions, Wittig olefination, Diels-Alder cycloaddition, nucleophilic substitution, Michael addition, nucleophilic capping reactions (acylation, sufonylation, isocyanates, isothiocyanates), nucleophilic aromatic substitution, and various heterocycle syntheses17,18. An ever-expanding list of new DNA-compatible reactions are developed each year, steadily expanding the scope of DEL synthesis capabilities and accessible structures.
Box 2 |. Methods to synthesize DELs de novo.
Synthesizing DELs deviates from traditional small-molecule library synthesis, even though their chemical composition may be very similar. Researchers typically seek access to DELs that contain very large numbers (for example, millions to biliions) of compounds for selections against targets of interest. Only pooled library synthesis routes offer the ability to practically access such large libraries. Multiple methods have emerged over the last two and a half decades to efficiently synthesize DELs while effectively maintaining their genotype-structure linkage23. Pioneering work in this field include DNA-templated synthesis (DTS)143, encoded self-assembling chemical libraries (ESAC)9, yoctoreactor6, DNA routing10,144, and split and pool based methodologies (or DNA-recorded synthesis)8,11,13. The split and pool techniques have since become the most popular DEL synthesis method145. This technique takes a pool of existing chemotypes, each with a unique covalently attached DNA-barcode, and splits them into separate reaction pools to add unique chemical building blocks. Each new building block is then encoded by a unique DNA-barcode that is ligated onto the DNA-tag already present on each library member. The separate reaction sequences are then re-pooled, and the cycle is repeated for each additional building block pool. This technique is easily implemented and has supported the construction of libraries eclipsing 109 members.
These techniques have used a wide variety of DEL architectures, which can vary in both small-molecule diversity and in the architecture of the DNA tag (Fig. 1b). dsDNA architectures provide a robust architecture for most DELs. DTS, DNA routing, ESAC, and related methods require ssDNA regions to support DNA hybridization. Further efforts have also used these strategies to create solid-phase DEL libraries146, where DNA-barcodes and their encoded small molecules are each covalently attached to a solid-support in a one-bead-one-compound format. This type of DEL allows alternative assay manipulation options during selections, including release of compound for analysis of DELs in microfluidic devices129,130,147 or flow cytometers106,127,148.
Box 3 |. Techniques to evaluate DELs against targets of interest.
Small-molecule discovery from DELs typically involves a binding-based assay on an immobilized target (Fig. 1b). First, library members are pooled into a single solution and incubated with a target of interest containing an affinity tag immobilized on beads. A small subset of library members binds to the target and washing removes non-binding library members. Putative binders are isolated by eluting target-bound library members. If needed, surviving library members can be subjected to another cycle of selection to further enrich bona fide binders. The identities of library members that pass selection are revealed by PCR amplification of surviving DNA tags, followed by NGS and analysis of DNA sequences to infer the structure of active library members. Researchers have developed several data analysis platforms to process the many unique sequences isolated from DEL selections. The enrichment of individual members is often plotted compared to their abundance in the pre-selection library. Unusually enriched barcodes are correlated with their encoded chemical building blocks, and compounds of interest are resynthesized and assayed in a DNA-free form. Subsequent medicinal chemistry optimization and further development can advance the most promising compounds for research or clinical applications.
Separate from immobilization methods, other selection workflows have also been developed to address target engagement in solution, bypassing target immobilization restrictions. These methods include interaction determination using unpurified proteins (IDUP)149, binder trap enrichment150, capillary electrophoresis151,152, crosslinking strategies153–157, DNA-encoded dynamic libraries (DEDL)158–160 , and microfluidics129,130,147. Further efforts have advanced the use of DELs on proteins in biological environments, rather than as isolated targets. Examples include selections against overexpressed targets on cell surface (mimicking protein immobilization workflows)103,161, native cell surface targets labeled with DNA-programmed affinity labeling108, cell penetrating peptide libraries for intracellular protein assessement140, intracellular injection of DELs139, and phenotype-based selections104.
Compounds from simple immobilization selections
Applying DELs for small molecule discovery most commonly uses binding-based selections on single immobilized targets (Fig.1b, Box 3). This simple workflow can quickly identify new ligands with affinity for a target of interest as a starting point for optimization, characterization, further development, and clinical trials. Below, studies that exemplify this process are highlighted. In many cases, DEL libraries were also synthesized in the same study, highlighting the facile translation between library synthesis, target evaluation, and hit characterization.
In vitro probes
In contrast to non-pooled library formats of the same size, the pooled format of DELs enables the rapid synthesis and implementation of novel, large libraries (millions to billions of members) with researcher-defined properties, accelerating the discovery of new chemical matter. Indeed, over the past few years, several groups have quickly identified novel in vitro probes using DELs (Table 1). For example, parts of the thrombin inhibitor argatroban were used as a scaffold to synthesize a protease-focused DEL of up to 9.8 million members28. A selection with immobilized, biotinylated thrombin enabled the discovery of a potent inhibitor of thrombin protease activity (#1). This study illustrates the utility of structure-focused DELs and that DELs can facilitate the rapid discovery of novel, clinically relevant in vitro hits.
Table 1 |. Examples of small molecules discovered from simple immobilization selections.
Studies are grouped by ligands with characterized in vitro activity on target, or ligands with verified bioactivity. Compounds that fall under both ‘Top library hit(s)’ and ‘Optimized compound(s)’ column headings were lead compounds directly isolated from the DEL selection that were not optimized further.
| Target | Library | Selection conditions | Top library hits | Optimized compounds | Unique properties | Crystal Structure(s) |
|---|---|---|---|---|---|---|
| Ligands with in vitro activity | ||||||
| Thrombin28 | Protease focused DEL Size: 9,837,660 |
|
#1 (Compound 5) IC50=1 nM - inhibition |
|
N/A | |
| IL229 | AG-DEL Size: 669,240 |
|
#2 (Compound 1) Kd= 0.25 μM - FP |
#3 (Compound 18) Kd= 0.34 μM - FP IC50= 34 μM - competition with anti-IL2 Fab |
|
N/A |
| LC3B31 | 21 pooled DELs Size: 7,000,000,000 |
|
#6 (Compound 2) Kd= 0.58 μM - SPR |
|
N/A | |
| Ligands with characterized bioactivity | ||||||
| CypD, CypE33 | DNA-templated macrocycle library Size: 256,000 |
|
#7 (JOMBt) IC50= 17 μM - inhibition |
#8 (B52) IC50= 10 nM – CypD inhibition #9 (C3A) IC50= 13 nM – CypE inhibition |
|
JOMBt with CypD (PDB ID 7TGS) B52 with CypD (PDB ID 7THD) |
| TEAD435 | tiDEL Size: 8,112 |
|
#10 (Compound 9) IC50=0.41 μM - competition with FITC palmitic acid IC50=6.75 μM - competition with FITC-YAP |
|
N/A | |
| Mcl-136 | (Size and structure not disclosed) |
|
#11 (Compound 1) IC50= 1.49 μM – Bim peptide competition |
#12 (Compound 26) IC50<3 nM - Bim peptide competition EC50=3.78 μM - apoptosis |
|
Compound 1 with Mcl-1 (PDB ID 5KU9) |
| Compound 29 (Compound 26 derivative) with Mcl-1 (PDB ID 5MES) | ||||||
| BRD4-BD137 | Benzimidazole library (hit sub-library) Size: 117,000,000 |
|
#13 (Compound 10) plC50= 6.6 μM - competition with I-BET762 |
#14 (I-BET469) plC50= 7.9 - competitive binding plC50= 8.3 - cell BRET |
|
Compound 10 with BRD4-BD1 (PDB ID 6TPX) |
| DEL34–39 (total) Size: not disclosed |
I-BET469 with BRD4-BD1 (PDB ID 6TPZ) | |||||
| ATAD238 | 11 total DELs, Size: 65,000,000,000 |
|
#15 Name and potency not disclosed |
#16 (BAY-850) IC50= 22nM - tetra-acetylated H4 N-terminal peptide competition |
|
N/A |
| PARP1, PAPR4, PARP10, PARP12, PARP14, PARP15, SRIT640 | NADEL Size: 58,302 |
|
#19 (A65-(CONHMe)-B101) IC50= 170 nM - PARP1 inhibition #20 (A82-(CONHMe)-B354) IC50= 6 μM - PARP10 inhibition #21 (A101-(CONH2)-B322) IC50= 200 nM - PARP15 inhibition #22 (A127-(CONHPr)-B178) IC50= 6.7 μM - SIRT6 inhibition |
|
N/A | |
| Antibiotic targets for: S. aureus,A. baumannii, M. tuberculosis42–43 | 100 total DELs, Size: >1,000,000,000 |
|
#23 (Compound 1) S. aureus methionyl-tRNA synthetase (MRS) IC50=0.83 nM - inhibition MIC= 0.5 μg/mL (S. aureus) #24 (Compound 5) A. baumannii Undecaprenyl pyrophosphate synthase(UppS) IC50=0.043 μM - inhibition MIC>128 μg/mL (A. baumannii) #25 (Compound 8) M. tuberculosis Dihydrofolate reductase (DHFR) IC50= 0.61 μM – inhibition MIC= 2.5 μg/mL (M. tuberculosis) #26 (Compound 1) S. aureus Undecaprenyl pyrophosphate synthase (UppS) IC50= 190 nM - inhibition MIC=8 μg/mL (S. aureus) |
|
Compound 1 with S. aureus UppS (PDB ID 5KH5) | |
| Mpro (SARS-CoV-2)44 | Size: 3,987,000,000 |
|
#27 (CDD-1713) IC50= 45 nM - inhibition IC50= 5.19 μM - rescue of cell viability |
#28 (CDD-1976) IC50= 37 nM inhibition IC50= 2.5 μM - rescue of cell viability |
|
CDD-1713 with MPro (PDB ID 7LTN) |
A similar approach was applied in the discovery of novel IL2 binders, designed to abrogate the interaction between IL2 and the alpha subunit of the IL2 receptor (IL2Rα), as the alpha subunit is preferentially expressed in immunosuppressive Treg cells29. A 669,240-member DEL (AG-DEL) was constructed and applied in a selection with an immobilized biotinylated L19-IL2 fusion protein (an IL2 phase 3 clinical trial candidate) as L19-IL2 is easier to formulate and store than recombinant IL2. This yielded fluorescein-labeled #2, which upon further optimization resulted in #3, which bound strongly to IL2 and displayed attenuated binding to non-target plasma chaperone proteins. These properties made #3 a promising starting point for the discovery of potent and selective IL2 binders.
A third example of DEL-mediated in vitro probe discovery is the identification of non-mechanism-based inhibitors of the β-lactamase OXA-48, a clinically resistant carbapenemase (Fig. 3a)30. Following construction of a 3-building block, triazine core DEL consisting of up to 162 million library members, three rounds of selection with immobilized His6-OXA-48 yielded inhibitory compound #4, later truncated to #5. A co-crystal structure of #5 bound to OXA-48 (PDB ID 6UVK) indeed showed active-site and non-covalent binding to OXA-48. #5 was also found to inhibit other OXA enzymes with potencies directly proportional to the sequence homology of each enzyme with OXA-48. This study highlights the utility of DEL-mediated identification of novel chemotypes for antibiotic discovery.
Fig. 3 |. Highlighted studies that use the typical immobilization DEL selection workflow to discover novel ligands.

a, The discovery and development of non-covalent, non-β-lactam based OXA-48 inhibitors. b, First example of small molecules that inhibited polymerization of Z α1-antitrypsin. Compound #18 (GSK716) selectively bound to Z mutant over wild-type in a novel pocket containing the E342K mutant residue and increased monomeric Z α1-antitrypsin in mice.
Collectively, these three studies illustrate the rapid progression between DEL library synthesis, target selection, and hit validation.
Finally, when combined with other discovery platforms, DELs can facilitate tailored ligand discovery. For example, a comprehensive screening campaign identified ligands of LC3, a member in the autophagy-related (Atg8) proteins with understudied mechanistic roles and few known ligands31. Fragment-based strategies yielded only modestly potent fragments that preferentially bound to the HP2 pocket of LC3 and could not be extended into the HP1 pocket to improve target affinity. To discover larger and more potent ligands, a DEL of up to 7 billion compounds was used in a selection against immobilized, His-tagged LC3B and LC3C. From this selection, the reversibly covalent compound #6 was discovered, with predicted binding at or near the HP1 or HP2 pockets. A future DEL selection scheme that leverages the knowledge gained from these DEL and fragment-based platforms might help identify ligands that selectively interact with the HP1 pocket. Alternatively, the use of bivalent DEL libraries might yield ligands that occupy both pockets. Indeed, DEL conditions can be customized to preferentially bias specific ligand binding modes which, in principle, enable tailored selection pressures. We detail this capability of DELs in a later section of this review.
Chemical probes with biological activity
Over the past six years, DELs have mediated the discovery of several bioactive compounds spanning multiple target classes (Table 1). As DEL selections rely on a binding-based assay, large DELs enable the discovery of new ligands that bind to shallow protein-binding surfaces, a historically challenging task for small-molecule discovery32. A clear example of this is the identification of novel subtype-selective inhibitors of cyclophilin D (CypD), a prolyl-isomerase and a key regulator of the mitochondrial permeability transition pore (mPTP)33. Key structural features of CypD are two shallow and solvent-exposed binding pockets (active pocket and S2 pocket). A selection of a macrocycle DEL with up to 256,000 members34, using immobilized His6-CypD, found a novel active site CypD inhibitor (#7) with no selectivity for CypD. Using structure guided analysis and structure-activity relationships (SAR), a dicarboxylate derivative, #8 (PDB ID 7THD) showed potent and selective inhibition for CypD via contacts made in the S2 pocket. Derivatives of #8 similarly inhibited CypD-mPTP in isolated mitochondria. To further validate this selective binding mode, a first-in-class CypE selective inhibitor was designed (#9) which formed a reversible covalent interaction with CypE’s S2 pocket K217 residue. We anticipate that a greater number of cyclophilin ligands can be discovered by using clever DEL counter-selection schemes to enrich for subtype selective ligands.
Another challenge facing small-molecule discovery efforts is the wide, yet shallow, binding interfaces of protein-protein interactions (PPIs)32. To address this challenge and identify ligands that bind to PPI interfaces, many groups have leveraged the binding-based selections of DELs. For example, a focused DEL of up to 8,112 members (tiDEL) based around tryptophan motifs, an enriched residue in PPI interactions, was synthesized, with the aim of discovering ligands that ablated the PPI between transcriptional enhancer factor-1 domains (TEAD1–4) and co-transcription factor Yes-associated protein (YAP)35. Discovery of the PPI inhibitor compound #10 resulted from a selection conducted on His-tagged, immobilized YAP-interaction domain of hTEAD4. As the formation of the TEAD-YAP complex in the nucleus is responsible for CTGF gene expression, #10 significantly reduced CTGF transcript levels. This study illustrates that focused DELs of even modest library sizes with appropriate structural motifs can quickly find bioactive ligands for traditionally intractable classes of targets. Furthermore, this study shows that DELs can support transcription factor ligand discovery despite the presence of DNA tags.
DELs have similarly been used to enable the rapid discovery of selective, bioactive PPI inhibitors in apoptotic pathways. Novel inhibitors of Mcl-1, an apoptotic suppressor overexpressed in multiple cancer types, were discovered using various discovery strategies, including a series of DELs36. Selections from DEL platforms resulted in the isolation of binding compound #11, which bound to the BH3 binding groove of Mcl-1 (PDB ID 5KU9). Structure optimization led to macrocyclic compound #12, and a closely-related derivative (PDB ID 5MES) revealed enhanced binding to the hydrophobic pocket of Mcl-1. Compound #12 also exhibited >1000-fold selectivity for Mcl-1 over similar proteins Bcl-2 and Bcl-xL and induced apoptosis in Mcl-1-dependent leukemia cells.
In addition, selective bioactive PPI inhibitors for an epigenetic protein, the histone acetylysine reader BET BRD4-BD1, have been identified37 by using a series of DELs for an affinity selection against the His6-BRD4 dual domain (both BD1 and BD2 domains). Compound #13 was identified as an initial ligand (PDB ID 6TPX) from the selection, which following substantial medicinal chemistry efforts, resulted in compound #14, which adopted an optimized binding mode (PDB ID 6TPZ) and exhibited reduced off-target activity. Compound #14 showed increased activity in cells and increased selectivity for the BD1 domains of the BET family. In a model of endotoxic shock, #14 suppressed IL-6 proinflammatory cytokine levels in mice challenged with lipopolysaccharide (LPS). #14 also reduced IgG1 levels in mice challenged with 2,4,6-trinitrophenyl keyhole limpet hemocyanin (TNP-KLH) antigen, illustrating its potential ability to suppress auto-immune responses.
In a final example of DEL-mediated PPI-inhibitor discovery, DELs were leveraged to discover bromodomain inhibitors of ATPase family AAA-domain containing protein 2 (ATAD2)38. Using DELs containing up to 65 billion compounds for a selection with immobilized GST-ATAD2 bromodomain, an original binding library hit (#15) was optimized to ATAD2 selective compound #16. This compound had a unique binding mode that dimerized two equivalents of ATAD2 protein, a property offered through an unbiased binding-based DEL selection. #16 also displaced labeled ATAD2 from chromatin in MCF7 cells but did not affect the expression of ATAD2 target genes. It was suggested that a proteolysis-targeting chimera (PROTAC) derivative of #16 could explore ATAD2 oncology, a task for which DELs are particularly well-suited (discussed later in this review).
Together, these four examples illustrate how DELs enable the rapid discovery of bioactive ligands that modulate protein surfaces, including those that mediate PPIs. DELs can also sample chemotypes for PPI inhibition across a variety of target types and for which the lead molecules can impart a downstream phenotype. Furthermore, the ability to quickly synthesize some types of DELs enables researchers to create small libraries with user-defined chemotypes. For example, in this section two cases are noted in which researchers leverage small, focused DELs for improved binding at intractable protein surfaces, including those that include macrocyclic scaffolds or tryptophan motifs.
DELs have also been used to solve complex and therapeutically relevant target-binding problems that have challenged small molecule discovery efforts. For example, α1-antitrypsin is a critical protein for healthy kidney functioning which, when harboring the E342K mutant (known as Z-α1-antitrypsin), is prone to polymerize and results in liver cirrhosis, hepatocellular carcinoma, and emphysema. Despite its clear clinical relevance, several factors made it challenging to identify Z-α1-antitrypsin ligands using traditional discovery methods. The protein is conformationally dynamic, its chaperone-binding modes are unknown, and chaperone-mimetic ligands are rare, necessitating the rapid sampling of large libraries. Recognizing that DELs would be well suited for overcoming these challenges, a DEL was used to perform a selection and identify ligands that stabilized and prevented the polymerization of Z-α1-antitrypsin (Fig. 3b)39. Using a library of up to 2 trillion members, an affinity selection identified a potent in vitro inhibitor of Z α1-antitrypsin polymerization (#17). Subsequent optimizations led to compound #18, which also refolded denatured Z α1-antitrypsin in vitro. The co-crystal structure of Z α1-antitrypsin with #18 (PDB ID 7AEL) revealed an interaction with a novel binding site at the E342K mutation, not present Z α1-antitrypsin apo structures. In iPSC-derived human hepatocytes, #18 inhibited polymerization, increased secretion of Z α1-antitrypsin polymers, and increased circulating monomeric Z α1-antitrypsin in mouse models. An HTS screen of 1,700,000 compounds was also conducted, but compounds could not be advanced beyond early lead optimization. Indeed, the unbiased, binding-focused nature of DEL selections with an enormous library enabled the development of #18. Altogether, this study highlights the capacity of DELs to mediate the discovery of ligands with complex targets and with unique, previously undefined binding modes.
A major advantage of DELs is that they enable selections to be conducted concurrently against multiple targets within a single workflow. For example, when carefully designed, moderately sized DELs that mimic a particular bioactive small molecule (focused DELs) can enable rapid ligand discovery across multiple targets. This strategy was recently used to create an NAD+ mimicking DEL (NADEL) of up to 58,302 members to facilitate NAD+-dependent enzyme ligand discovery40. Using NADEL, novel inhibitors for various NAD+-dependent enzymes were discovered, validating the potential for structurally focused DELs to identify ligands for multiple target types (Table 1). Affinity selections with a series of immobilized biotinylated poly- and mono-(ADP-ribose) polymerases (PARPs) resulted in the successful discovery of many validated ligands for multiple PARPs (#19-#21)40,41. A further selection against the mono-ADP-ribosyltransferase SIRT6 identified the selective inhibitor #22. In HUVECs, inhibition by #22 showed bioactive SIRT6 engagement through elevated DNA-damage biomarkers, consistent with the role of SIRT6 in telomere maintenance. Compound #22 also decreased extracellular release of LPS-stimulated TNF-α in HP-1 monocytes, a SIRT6 dependent pathway. This study illustrates that DEL selections can be multiplexed when ligands for a family of targets are sought, even from a modestly sized library.
The ability to rapidly conduct DEL selections against a large volume of disparate therapeutic targets has also been demonstrated in a search focused on finding novel antibiotic compounds against 39 S. aureus, 70 A. baumannii, and 42 M. Tuberculosis targets42. From various tagged and immobilized protein targets and a DEL containing up to several billion compounds, potent target binders or inhibitors for each target panel (14 for S. aureus, 52 for A. baumannii, and 27 for M. Tuberculosis) were identified. Off-DNA synthesis for each target panel found 7 S. aureus binders to have activity, two of which had confirmed MoAs, 17 A. baumannii binders to exhibit activity, three of which had confirmed MoAs, and at least four M. Tuberculosis binders to have confirmed activity.
As the target and compound space for this effort is vast, a short list of exemplary compounds discovered from the study is included in Table 1 (#23–26). One key example is the identification of hits developed from S. aureus Undecaprenyl pyrophosphate synthase (UppS)43. This protein is a cis-prenyltransferase, and its downstream pathway product, undecaprenylphosphate (UP), is essential for cell viability in E. coli, P. aeruginosa, and S.pneumoniae. FLAG and strep-tagged UppS from S.aureus was immobilized and used in a selection with the DELs mentioned above. Isolated compound #26 inhibited S.aureus UppS, exhibited a MIC of 8 μg/mL against methicillin-resistant S. aureus (MRSA), and bound to S. pneumoniae UppS (PDB ID 5KH5) near the base of its substrate binding site. The success rate of these studies was similar to those obtained from HTS-based discovery efforts, but with vastly improved timelines and fewer resources - 3–4 months from isolated protein to analysis of DEL selections and 2–3 months of subsequent chemical synthesis and testing.
Another illustration of the short timeframe of DEL campaigns from selection to hit characterization and development is a study developing SARS-CoV-2 protease inhibitors44. An affinity selection (1–2 weeks) on immobilized His-tagged Mpro (cloning and expression in 1–2 weeks) with a collection of DELs totaling up to 4 billion compounds was performed. This DEL campaign resulted in the identification of inhibitory library member compound #27 and a synthesized derivative #28 (1–2 weeks for compound characterization), which both exhibited a covalent binding mode to a coronavirus main protease-conserved S1 pocket (PDB ID 7LTN). Notably, both compounds rescued cell death in VERO E6 cells treated with live strain SARS-CoV-2 (1–2 weeks for antiviral testing). This study exemplifies the power of DELs to enable rapid (<3 months) screening of a time-sensitive therapeutic target. An analogous HTS well-based approach would have enabled the screening of no more than ~100,000 small molecules44.
Collectively these three studies illustrate that libraries of various sizes can be used quickly against a large series of targets in parallel, enabling rapid characterization and implementation of new bioactive ligands.
Clinical candidates
Traditional DEL selection workflows, which use single immobilized proteins, have enabled the identification of multiple clinical candidates. To date, three clinical candidates have been assessed or approved in clinical trials (Fig. 4a–c).
Figure 4 |. Clinical candidates discovered from DNA-encoded libraries.

a, A lead sEH inhibitor that has been used in Phase I and Phase II trials. b, A series of RIP1 kinase inhibitors used in Phase I and Phase II trials, encompassing a variety of drug formulations, patient cohorts, and disease types. c, An autotaxin inhibitor that attenuated lung fibrosis in vivo and has been approved for Phase I clinical trials.
The first DEL-enabled clinical candidate was a soluble epoxide hydrolase (sEH) inhibitor (Fig. 4a), developed and optimized from an original library hit (#29)18,20,45–51, into the clinical candidate GSK2256294 (#30)48. sEH converts epoxyeicosatrienoic acids (EETs) to dihydroxyepoxyeicosatrienoic acids (DHETs), the former of which is a more potent anti-inflammatory metabolite. Inhibition of this enzyme, therefore, can potentially treat conditions associated with over-inflammation. Phase I clinical trials on safety and tolerability of GSK2256294 were successfully completed (NCT01762774, NCT02262689)52,53. Additional Phase I trial data (NCT01762774) from 2017 revealed that treatment with GSK2256294 successfully increased EET-mediated vasodilation in COPD patients or overweight smokers with impaired EET-mediated endothelial function and bradykinin vasodilation54. Two Phase II clinical trials were also recently reported for GSK2256294: one for the potential treatment of aneurysmal subarachnoid hemorrhage (NCT03318783)55 and the other for potentially altering insulin sensitivity in obese or pre-diabetic patients (NCT03486223)56. The first trial (NCT03318783) reported a large increase in the EET/DHET ratio in the serum of patients treated with GSK2256294 compared to the placebo. At clinical endpoints, patients treated with GSK2256294 had shortened hospital stays, resulting in a quicker transition back to home compared to placebo-treated patients, albeit with a small sample size (10 for GSK2256294, 9 for placebo). Although the second trial (NCT03486223)56 is still ongoing, GSK2256294-dependent decreases of biomarkers for oxidative stress in diabetic, obese, and smoking patients have been reported. The many clinical trials conducted with GSK2256294 demonstrate that a DEL discovery platform can translate candidate ligands into clinical settings.
DELs have also facilitated the development of a receptor-interacting serine/threonine-protein 1 (RIP1) kinase inhibitor—a key regulator of inflammation—that entered clinical trials (Fig. 4b). An initial library hit, GSK’48157,58 (#31), was discovered from an immobilized selection with RIP1, exhibited potent and specific RIP1 inhibition, and was subsequently optimized into GSK2982772 (#32). After validating the candidate in cell culture (in vitro) and in mouse models of TNF-induced acute lethal shock (in vivo), GSK2982772 was moved into Phase I clinical trials with healthy individuals (NCT02302404, NCT03305419, NCT03590613)59,60 and tested with alternate formulations (NCT03266172, NCT03649412)61,62. In all cases, the candidate was well tolerated. Phase II trials for three conditions (psoriasis, arthritis, and ulcerative colitis) resulted in reduced epidermal thickness, infiltration of inflammatory immune cells in patients with mild-to-moderate plaque-type psoriasis (NCT02776033)63; attenuated bone erosion in patients with moderate-to-severe rheumatoid arthritis (NCT02858492)64; and no therapeutic benefit for patients with ulcerative colitis (NCT02903966)65. Other studies involving GSK2982772 derivatives have also been conducted, revealing a range of capabilities from activity in cell models of necrosis66, to blood-brain barrier penetration67, to increasing the immune response in pancreatic ductal adenocarcinoma models as a separate clinical candidate, GSK3145095 (#33) (NCT02903966)68,69. That so many potential therapeutic applications have resulted from this clinical candidate highlights the biomedical relevance of DEL discovery platforms from in vitro screen to clinical application.
The most recent example of a clinical candidate discovered by DELs is the discovery of a potent autotaxin inhibitor (Fig. 4c)70. A DEL with a three-building-block peptidomimetic-like structure of up to 225 million compounds was used in a selection with immobilized FLAG-tag autotaxin, a secreted phosphodiesterase associated with lung fibrosis and idiopathic pulmonary fibrosis70 and the primary source of blood-borne lysophosphatidic acid (LPA). This selection revealed compound #34 as a potent autotaxin inhibitor, optimized to substrate competitive X-165 (#35) (PDB ID 6W35), which reduced mice plasma LPA levels and exhibited antifibrotic activity. The FDA has since cleared X-165 for Phase 1 clinical trials.
Collectively, these three examples illustrate that DEL technologies can advance new small molecules into clinic trials, despite their relatively recent development as a ligand discovery strategy. We anticipate that many more DEL-mediated clinical candidates will be discovered either through this immobilized single-target workflow or more complex selections (described in the following sections).
Engineering DEL selection conditions
Researchers often require ligands with specific binding properties or ligands for targets not amenable to traditional DEL workflows. Therefore, researchers have sampled various iterations of the traditional DEL immobilization workflow. To address ligand design, researchers often take advantage of the multiplexable nature of DEL selections, in which single or multiple targets can be co-incubated with varied conditions to select for user-defined binding modes. Additionally, the small scale and ease with which DELs can be re-synthesized enables additional rounds of selection, optimized hits, and improved binders. Various groups have also addressed targets not stable to traditional immobilization techniques and have developed methods for alternatively immobilized or non-immobilized in vitro selections. Examples of novel small molecule discovery using these techniques are highlighted in this section.
Tuning selection conditions
Individual selection conditions or entire selection campaigns, using the immobilization workflows described previously, can be tuned to identify ligands with unique or user-defined properties (Table 2).
Table 2 |. Examples of DEL discovery campaigns that use engineered selection conditions to improve selection outcomes.
Compounds that fall under both ‘Top library hit(s)’ and ‘Optimized compound(s)’ column headings were lead compounds directly isolated from the DEL selection that were not optimized further.
| Target | Library | Selection conditions | Top library hit(s) | Optimized compound(s) | Unique properties | Crystal structure(s) |
|---|---|---|---|---|---|---|
| DDR171 | 2 pools of DELs Size: 83,000,000,000 and 85,000,000,000 |
|
#36 (Compound 2a) IC50= 1.4 μM - competition binding |
#37 (Compound 2.45) IC50= 0.029 μM - competition binding |
|
Compound 2a with DDR1 (PDB ID 6FEW) |
| Compound 2.45 with DDR1 (PDB ID 6FEX) | ||||||
| BRD4-BD272 | Glycine based (Hit sub-library) Size: 1,290,000 |
|
#38 (Compound 8) plC50= 6.6 - competition binding |
#39 (Compound 60) plC50= 8.3- competition binding |
|
Compound 8 with BRD4-BD2 (PDB ID 70E0) |
| Pooled DEL34-DEL97 Size: total not disclosed | Compound 60 with BRD4-BD2 (PDB ID 70ET) | |||||
| TAK174 | Hit sub-library size: 3,760,000 |
|
#43 (Compound 7) IC50= 1.3 μM - inhibition |
#44 (Compound 22) IC50= 0.6 μM - inhibition Kd= 52 nM - KdELECT |
|
Compound 22 with TAK1 (PDB ID 7NTI) |
| 21 pooled DELs Size: total not disclosed | ||||||
|
#45 (Compound 54) IC50= 2 nM - inhibition | ||||||
| HAO176 | DEL C (Hit sub-library) size: 740,00048 total DELs |
|
#48 (Compound 5) IC50= 37 nM - inhibition EC50=17 μM - rescue HAO1 toxicity |
#49 (Compound 31) IC50= 2.9 μM - oxidase inhibition |
|
Compound 5 with HAO1 (PDB ID 6W45) |
| Size: total not disclosed | ||||||
| LpxA Pseudomonas aeruginosa77 | Hit sub-library size: 3,600,000 |
|
#50 (Compound 1) IC50= 400 nM - P. aeruginosa LpxA inhibition IC50>50 μM - E. coli LpxA inhibition |
#51 (Compound 50) IC50= 3 nM -P. aeruginosa LpxA inhibition MIC= 4 μg/mL - P. aeruginosa LESb58 |
|
Compound 1 with E.coli LpxA (PDB ID 70JP) |
| 11 total DELs Size: not disclosed | Compound 1 with P. aeruginosa LpxA (PDB ID 70J6) | |||||
| Naa5078 | Hit sub-library (DEL951) Size: 69,000,000 |
|
#52 (Compound 4a) Kd=27 nM - SPR with AcCoA IC50=7 nM - inhibition |
|
4a with Naa50 and CoA (PDB ID 6WFK) or AcCoA (PDB ID 6WFN) | |
| Total pooled DELs Size: 2,200,000,000 (total) | ||||||
| PLAP81 | NF-DEL Size: 670,752 |
|
#53 (Compound 16) IC50= 32 nM - inhibition |
#54 (Compound 18) |
|
N/A |
| GB-DEL Size: 366,600 | ||||||
| BTK82–83 | Size: 110,261,100 |
|
#55 (Compound 1) IC50= 2.5 μM - competition binding #56 (Compound 3) IC50= 0.55 nM - competition binding IC50= 3.8/3.0 nM - reporter displacement assay for wild-type/C481S mutant IC50= 30 nM - Ramos B-cell stimulation |
|
Compound 3 with BTK (PDB ID 5U9D) | |
| Mer, Axl84,85 | 46 total DELs Size: >90,000,000,000 |
|
#57 (Compound 12) plC50= 7.6/8.0 - Mer/Axl kinase inhibition #58 (Compound 13) plC50= 8.3/7.8 – Mer/Axl kinase inhibition #59 (Compound 14) plC50= 8.3/6.4 Mer/Axl kinase inhibition |
#60 (AZ14145845) plC50= 7.8/7.0 - cellular ELISA for Mer/Axl phosphorylated products plC50= 7.6 - efferocytosis inhibition, CD14+ monocytes |
|
Compound 12 with Mer (PDB ID 7AW3) |
| Compound 13 with Mer (PDB ID 7AW4) | ||||||
| Compound 14 with Mer (PDB ID 7AW2) | ||||||
| AZ14145845 with Mer (PDB ID 70LX) | ||||||
| β2AR92–96 | For antagonist: 3 DELs Size: 190,000,000 Structure not disclosed | Antagonist discovery
|
#64 (Compound 15) (antagonist) Kd= 1.7 μM – ITC IC50= 1.9 μM - competition binding with isoproterenol agonist EC50= 0.48 μM - cooperative binding with [3H]-ICI-118,551 inverse agonist |
|
Compound 15 with β2AR and carazolol (PDB ID 5X7 D) | |
| For agonist: 4 DELs Size: 500,000,000 Structure not disclosed | ||||||
Agonist discovery
| ||||||
| Compound 6 with β2AR and BI-167107 (PDB ID 6N48) | ||||||
|
#65 (Compound 6) (agonist) Kd= 5.2 μM – ITC EC50=1.32 μM - increase in 3H-Fen agonist binding | ||||||
Conditions to enrich selective or multi-target binders
In the most straightforward implementation, hit output can be enriched for subtype selective ligands using workflows that rely on in-parallel counter-selections against related family members. This method was used to find the first selective small-molecule modulators of Discoidin Domain Receptor 1 (DDR1), a receptor tyrosine kinase that autophosphorylates and activates downstream inflammatory responses71. Using two pooled libraries (up to 83 billion and 85 billion members), a selection against immobilized GST-tagged DDR1 and DDR2 was conducted. By computationally excluding binders that enriched for both DDR1 and DDR2, compound #36 was identified to selectively bind to DDR1 over DDR2. A significant medicinal chemistry effort resulted in compound #37 with >1,400 kinome selectivity for DDR1 and >14-fold selectivity over DDR2, made possible through a unique kinase binding mode (PDB ID 6FEW, 6FEX). This compound also showed therapeutic benefit in a mouse model of Alport syndrome. This study illustrates that a simple counter selection can be conducted alongside the main target when subtype selectivity is desired.
A similar counter-selection method was used to identify BET BRD4-BD2 domain-selective ligands72. Selections with 6-His-BRD4 (1–477, Y390A) to identify BRD4-BD1 ligands, 6-His-BRD4 (1–477, Y97A) to identify BRD4-BD2 ligands, and then finally against BRD4 dual domain (1–477), were conducted using a series of DELs. Isolated compound #38 enriched in the Y97A selection and the dual domain BRD4 constructs, but not for the Y390A mutant, which suggested BD2 selectivity. Indeed, #38 exhibited near equipotent binding to BD2 domains of BRD4, BRD3, and BRD2, while showing >30-fold better potency compared to each of their respective BD1 domains. A lengthy structure-guided medicinal chemistry campaign on #38 (PDB ID 7OEO) led to compound #39, a potent BD2 binder with >5,000-fold selectivity over BD1 and an optimized BD2 binding mode (PDB 7OET). Both these examples demonstrate that the addition of a few more selection conditions can enable the identification of subtype selective ligands straight from the library, an experimental change requiring minimal extra time and resources in DEL selection workflows.
Conversely, concurrent parallel selections can also rapidly identify ligands with multi-target potency, a capability that is particularly useful for rapidly mutating targets in oncology or infectious diseases. For example, multiple novel binders to estrogen receptor α (ERα), a nuclear hormone receptor and transcription factor overexpressed in ~70% of all breast cancers with many clinically identified gain-of-function mutants (D538G, S463P, and Y537S), were discovered through a DEL affinity selection (Fig. 5a)73. 44 DELs totaling up to 120 billion compounds were used against immobilized ligand binding domains of His-tagged ERα WT and these gain-of-function mutants73. Multiple hits—including compound #40—enriched for ERα WT and the three ERα mutants. #40 exhibited binding in vitro to ERα WT and to all three ERα mutants, but showed an agonistic phenotype in an MCF7 antiproliferation assay. A short SAR campaign to convert #40 to an ERα antagonist led to #41, which exhibited improved in vitro potency, targeted all three ERα mutants, and inhibited MCF7 growth. These results demonstrate that multiplexed selections can enable the discovery of ligands capable of inhibiting multiple clinically relevant protein subtypes directly from library selection.
Fig. 5 |. Highlighted studies showing successful implementation of concurrent selection conditions to develop tailored ligands.

a) Discovery of ERα wild-type and mutant inhibitors. A VHL-based PROTAC, #42 (Compound 21) was also developed from lead compounds, which inhibited proliferation of many ER positive cancer cell lines and exhibited in vivo tumor suppression. b) BRDT-BD2 inhibitors that were selective for BET family BD2 domains. c) GPCR allosteric agonists, (#61 (AZ2429)) and allosteric antagonists (#62 (Compound 2) and #63 (AZ3451)). #63 (AZ3451) exhibited bioactivity in cellular and in vivo inflammatory disease models.
Conditions for defined ligand binding modes
Another way to tailor selection output is to introduce parallel selection conditions that allow certain desired binding modes of library hits to be inferred. To accomplish this, libraries can be co-incubated with both protein targets and other target ligands or substrates that have known binding modes. For example, by introducing a parallel condition to nominate ligands with an active site binding mode, novel inhibitors for growth factor β-activated kinase 1 (TAK1, MAP3K7) were found74. Using 21 pooled DELs, a selection was conducted on immobilized, His-tagged TAK1-TAB1 fusion which was co-incubated with or without active site inhibitor 5Z-7-oxozeaenol. Hits from a sub-library that did not enrich in conditions with 5Z-7-oxozeaenol resulted in inhibitory compound #43 with a putatively active site binding mode. Medicinal chemistry and structure-guided optimization led to intermediary compound #44 and optimized compound #45, which exhibited kinome selectivity for TAK1. A co-crystal structure of #45 bound to TAK1 (PDB ID 7NTH) revealed a unique type-I binding mode at the hinge region that enabled kinome selectivity, confirming the competitive binding observed with 5Z-7-oxozeaenol from the DEL selection. This study demonstrates the utility of running a parallel condition to direct selection output for a particular ligand binding mode.
To prioritize ligands with selective binding modes, parallel ligand co-incubations can be combined with multiple proteins. For example, by pooling >50 DELs totaling up to 4.5-billion compounds for a selection with immobilized His-tagged BRDT-BD2 and BRDT-BD1 (and running parallel conditions of each protein co-incubated with promiscuous bromodomain binder JQ1), potent and specific inhibitors for the BD2 domain over the BD1 domain of BET bromodomain testis (BRDT), a male contraceptive target, were rapidly discovered (Fig. 5b)75. From these selections, #46 was identified as a BD2 selective binder, which enriched with the BD2 domain but did not enrich with either the BD1 domain or BD2 domain co-incubated with JQ-1. Optimization led to #47, which exclusively interacted with BET family BD2 domains. Co-crystal structures of both #46 and #47 (PDB IDs 7L9A, 7L99, respectively) revealed BD2-domain selectivity was contingent on interacting residues (P352 and H355) not conserved with the BD1 domain (K110 and D113), an area JQ1 does not contact. This study further highlights the ability of parallel DEL selections to accelerate the rate of selective ligand discovery.
Similarly, selection schemes were used to find inhibitors of hydroxy acid oxidase 1 (HAO1), that avoided inhibition of off-target protein lactate dehydrogenase B (LDHB), to treat primary hyperoxaluria (PH), a liver disorder that overproduces oxalate76. Using a collection of DELs in a selection against C-tag-HAO1 and LDHB, immobilized with an Anti-C-tag matrix, a novel chemical series of HAO1 inhibitors was identified. To gain further information on binding modes from hit candidates, a HAO1 condition co-incubated with a previously reported active-site inhibitor was also included. Hits from a well-enriched sub-library resulted in inhibitory compound #48. This compound and series did not enrich in conditions co-incubated with the previously reported inhibitor or with LDHB off-target, suggesting a HAO1-specific, active site binding mode. A co-crystal structure of #48 bound to HAO1 (PDB ID 6W45) proved active site binding occupying the glycolate/glyoxylate binding site. #48 also rescued cell viability in CHO cells overexpressing HAO1. Compound #49, a carboxylate bioisostere of #48 containing a benzotriazole with improved physiochemical properties yet reduced in vitro potency was also developed.
To discover clinically useful binding modes on unverified targets, multiple selections can be designed and performed in parallel under different conditions. For example, in a search for the first inhibitors of UDP-N-acetylglucosamine acyltransferase (LpxA) from Pseudomonas aeruginosa, eleven distinct DELs were pooled and incubated against E. coli and P. aeruginosa LpxA with a variety of substrate, product, and inhibitor co-incubations (see Table 2)77. Enriched hits for P. aeruginosa LpxA led to inhibitory compound #50, which showed reduced enrichment in the selection condition with its enzymatic product. A co-crystal structure of #50 bound to both P. aeruginosa LpxA (PDB. ID 7OJP) and E.coli LpxA (PDB ID 7OJ6), showing binding near the substrate binding and catalytic regions of the LpxA, consistent with selection outcomes. Next, a structure-guided approach was used to successfully design a large series of selective LpxA inhibitors with varying potencies in vitro, in bacterial cultures, and physiochemical properties, such as #51. This compound also inhibited a series of clinically relevant isolates of P. aeruginosa growth, demonstrating the clinical utility of targeting this protein with this type of binding mode. This study demonstrates that multiple parallel conditions can enable clinically relevant hit discovery on a target with an enzymatically unique binding mode.
Multiple DEL selections can also be performed to enrich ligands with a pre-defined binding mode. For example, a complex selection scheme was conducted to identify N-α-acetyltransferase 50 (Naa50) ligands that are acetyl-CoA (AcCoA) cooperative78. A series of DELs totaling up to 2.2-billion members for a selection against immobilized His-tagged Naa50 was used, along with a parallel set of selections co-incubated with either AcCoA, or a previously reported active-site inhibitor. One sub-library (from the Naa50+AcCoA selection) yielded compounds that were less strongly enriched when AcCoA was absent and not enriched in the presence of the Naa50+active site inhibitor. These results suggested that this family of compounds were AcCoA-cooperative active site inhibitors. Inhibitory compound #52 from the series selectively stabilized Naa50 with AcCoA. Potent binding was also observed in the presence of AcCoA or CoA. Additionally, co-crystal structures of #52 with either CoA or AcCoA bound to Naa50 (PDB ID 6WFK, 6WFN) were solved, which verified that the compound bound to the Naa50 active site adjacent to the CoA-binding pocket without displacing either cofactor, suggesting uncompetitive binding with the AcCoA cofactor and competitive binding with peptide substrates.
Collectively, these studies illustrate the use of DEL selections that incorporate counter-selections against closely related targets, together with conditions to exclude ligands that engage binding sites. Using this strategy, binding information about putative library hits can be acquired efficiently and effectively. Prioritized hits with desired properties can then be resynthesized and optimized for downstream efforts.
Bifunctional compounds
DEL selections inherently evaluate bifunctional compounds containing a target-binding ligand and a covalently linked DNA barcode. Typically, the DNA barcode is solvent-exposed and does not substantially contribute to target binding, allowing subsequent replacement during compound optimization or characterization. This inherent structural feature of DEL members—a modifiable linker at the site of DNA attachment—can be leveraged to develop bifunctional molecules from selection data. For example, the researchers who developed ERα WT and mutant inhibitors also developed a proteolysis targeting chimera (PROTAC) derivative of compound #41 (Fig. 5a)73. By conjugating the modifiable linker of #41 to a von Hippel-Lindau protein (VHL) E3 ligase-recruiting ligand, compound #42 was developed (Fig. 5a)73. Compound #42 retained binding to ERα WT and all three mutants, while exhibiting a strong cellular ERα degradation profile. This compound also had potent antiproliferation effects on MCF7 cells and multiple ER-positive breast cancer cell lines, and reduced ERα levels and tumor volume in an MCF7 xenograft mouse model.
DELs selection schemes were also exploited to develop bifunctional cell-recruiting molecules. Two DELs were used in parallel - GB-DEL (up to 366,600 members)79, NF-DEL (up to 670,752 members)80 — in a selection with immobilized placental alkaline phosphatase (PLAP), a protein expressed on the surfaces of tumors in the female reproductive tract81. Isolated compound #53 from this selection inhibited PLAP phosphatase activity. Next, compound #54 was synthesized, effectively a bifunctional molecule with #53 and fluorescein, and its tumor-targeting capabilities were subsequently tested. #54 bound selectively to PLAP-positive tumors both in cell lines and in xenograft mice, confirming its ability to target tumors of the female reproductive tract. #54 was also used as a bispecific bridge between UniCAR T-cells and PLAP-positive tumors, where anti-fluorescein antibodies are expressed on T-cell surfaces. Substantial cell lysis was observed only when CAR-T cells, HeLa cells, and varying doses of #54 were combined.
Together, these studies illustrate that the architecture of DEL small molecules can facilitate the rapid development of clinically relevant bifunctional ligands.
Multiplexed conditions to enrich multiple ligand types
In cases when various ligand types are needed for a single target, multiple DEL selection conditions can be used to span a variety of potentially useful binding modes. For example, because inhibitors of Burton’s tyrosine kinase (BTK) were starting to develop clinical resistance82, new inhibitors were sought. A selection with a DEL encompassing up to 110,261,100-members was conducted using immobilized 6-His BTK, 6-His BTK with saturating amounts of ATP, or 6-His BTK with saturating amounts of dasatinib, a known BTK inhibitor. Isolated inhibitory compound #55 showed reduced enrichment in the ATP or dasatinib selections. Another inhibitory hit, compound #56 also enriched against BTK but maintained enrichment in the ATP conditions, suggesting a different binding mode than #55. Further in vitro mechanism of action studies revealed that #55 was ATP-competitive, consistent with selection results. In contrast, #56 was either a non-competitive binder with ATP, or ATP competitive yet so potent that ATP could not displace it. A co-crystal structure of #56 bound to BTK (PDB ID 5U9D) revealed critical interactions with the ATP binding pocket and a conformational shift that blocks access to the selectivity pocket, suggesting very tight binding near the ATP binding pocket. In a follow-up study using a different in vitro assay, #56 was shown to inhibit both wild-type BTK and a clinically relevant BTK C481S mutant, a cysteine mutant that conferred resistance to previous inhibitors83. This study illustrates that multiple concurrent DEL selections can be used to discover a series of ligands with various binding properties for a single target.
The utility of running parallel DELs to discover multiple ligand types is also illustrated in a search for Mer and Axl receptor tyrosine kinase inhibitors84. Using various Mer and Axl constructs (see Table 2), and selection conditions for immobilized affinity-based selections with 46 combined DELs totaling > 90 billion compounds, inhibitory compound #57 was identified. This compound exhibited an ATP-competitive type I binding mode on Mer. Another DEL series that enriched in both the +/− ATP conditions resulted in the identification of inhibitory compound #58. Co-crystal structure analysis (PDB ID 7AW4) confirmed a type-II, non-ATP competitive binding mode. The series of final hits led to the identification of inhibitory compound #59, which exhibited a type I½ binding mode (ATP competitive binding to inactive kinase confirmation) by co-crystal structure (PDB ID 7AW2) and selectivity over other paneled kinases. #59 was optimized in a follow-up study, leading to #6085, which showed complete selectivity for Mer and Axl over a panel of kinases and inhibited efferocytosis in CD14+ moncytes with a similar binding mode (PDB ID 7OLX) to #59. #60 showed antiproliferation in both Mer and Axl-dependent Ba/F3 cell lines and mouse xenograft models. In an MC38 tumor mouse model, #60, in combination with ionizing radiation (IR) and an anti-PD1 antibody, showed improved overall survival compared to IR alone.
Together these 2 studies demonstrate that the malleability and multiplexed capability of DEL selections enable the enrichment of hits that span a variety of different, biologically useful binding modes to a target of interest. Indeed, the highlighted DEL selection examples above enabled the development of multiple pre-clinical candidate ligands.
Selections on membrane-bound proteins
DEL selections owe many of their advantages to their use of in vitro, immobilized targets. However, this feature raises concerns about conducting selections on membrane proteins and other hard-to-isolate targets, such as G protein-coupled receptors (GPCRs). Furthermore, because putative GPCR binders can have either agonist or antagonistic downstream effects, and in vitro selections cannot select for this information, analysis is further complicated. Despite this concern, two recent studies have successfully used DELs on GPCRs for agonist and antagonist ligand discovery.
First, allosteric ligands of the protease-activated receptor 2 (PAR2), a putative GPCR target for pain or inflammatory-related diseases, have been identified from DELs (Fig. 5c)86. To retain a properly folded membrane protein during the selection process, a thermostabilized GPCR (StaRs) construct in the apo form, pre-complexed with a stabilizing antagonist AZ6343, or either of those two conditions with competitor antagonists AZ8838 or AZ7188, was used. Using a collection of 20 DELs, compound #61 was identified87. This compound was enriched in the apo and AZ6343 stabilized forms, but not in the presence of competitors AZ8838 or AZ7188, and exhibited PAR2 agonist activity. Compound #62 was also identified as a PAR2 antagonist, enriched with AZ6343 stabilized PAR2 but attenuated enrichment with AZ8838 or AZ7188. Optimization of #62 yielded a more potent compound #63, revealed as an allosteric binder (PDB ID 5NDZ)88. A follow-up study determined that antagonist #63 inhibited peptide activation of PAR2 and reduced downstream biomarkers, while agonist #61 exhibited the opposite phenotypes87. Both #61 and #63 strongly bound to PAR2 and concurrently bound to their respective PAR2 allosteric sites. #63 also attenuated immune responses in rat models of PAR2 agonist-induced inflammation. Subsequent papers have used antagonist #63 to further probe PAR2 biology89, and as a potential therapeutic for both osteoarthritis90 and cardiovascular endothelial dysfunction91.
A second study that illustrates the use of DELs for agonist/antagonist discovery sought to find positive and negative allosteric modulators of the β2-adrenoceptor (β2AR). Although non-allosteric antagonists of β2AR (known as β-blockers) are already used as cardiovascular disease therapeutics, new allosteric modulators could potentially confer newer selectivity or phenotypes. Initially, novel ‘β-blocker’ type antagonist compound #64 of β2AR was discovered from DELs totaling up to 190 million members against detergent solubilized, immobilized Flag-tagged β2AR92. #64 was a negative allosteric modulator of β2AR, reduced downstream β2AR biomarkers in cells, exhibited allosteric binding at the cytoplasmic surface of the receptor (PDB ID 5X7D)93, and stabilized β2AR’s inactive conformation. A collection of DELs of up to 500,000,000 members was also used for agonist discovery, using biotinylated β2AR reconstituted in detergent-free immobilized HDL particles94. A co-incubation with active conformation stabilizer BI167107 was included. From this selection, the first positive allosteric modulator of β2AR, (#65), was isolated, which potentiated the binding of orthosteric agonists to β2AR, elevated downstream biomarkers, and stabilized the agonist-induced active conformation of β2AR (PDB ID 6N48)95. In a separate study, #65 was reported to also enhance agonist activity on β1AR96.
Both of these DEL campaigns resulted in a highly comprehensive set of structural, biochemical, and biological studies for new ligands exhibiting agonist and antagonist binding properties. These studies highlight the inherent advantages offered by binding-based DEL selections, including specific binding modes (such as allosteric modulation) or selectivity over a series of proteins. Additionally, these studies illustrate how different selection conditions can be leveraged to dictate protein conformations, ligand output, and downstream biological activity. Finally, these studies use valuable methods for stabilizing difficult-to-isolate membrane targets with DEL selections.
Non-immobilized selections
Selections on isolated, immobilized proteins have dominated DEL-mediated ligand discovery due to their straightforward implementation and their applicability to most targets. While feasible for most targets, including difficult-to-isolate proteins such as GPCRs, the immobilization process may not be compatible with all targets, may occlude ligand binding sites, or may promote non-specific interactions with the solid-phase resin. Alternatively, solution-phase selections are an evolving technology within DEL platforms that can assess protein targets in more relevant contexts, access other binding modes, and avoid potential protein- or assay-dependent complications from the immobilization process. Furthermore, unique ligand properties can be selected for by using solution-based selections not possible with immobilization techniques. Although these techniques typically require more laborious selection workflows, or significant modification of library architecture, novel ligands using these techniques have begun to emerge in the DEL field.
For example, a DNA-encoded dynamic library (DEDL) in-solution approach has been used to identify ligands for sirtuins (Table 3)97. DEDLs are similar to encoded self-assembling chemical (ESAC) libraries9, which use two fragments bound to each strand of a DNA-duplex reminiscent of fragment-based screening. However, DEDLs use a DNA strand with short hybridization sites (6–7 bases), where transient pairs of DNA fragments exist in a solution that provides ever-changing combinatorial pairs that only becomes stabilized upon concurrent interaction with a target. The fragments are then UV cross-linked and amplified by PCR for NGS to identify fragment pairs. The study used a DEDL of up to 10,000-members against SIRT1, SIRT2, and SIRT5 in solution; isolated crosslinked DNA duplexes by PAGE electrophoresis; amplified the surviving fragments by PCR, and sequenced DNA barcodes by NGS. Enriched fragments were synthesized and screened with various linkers and inhibitory compounds #66 (SIRT1), #67 (SIRT2) and #68 (SIRT5) were isolated. This study nicely highlights how in-solution DEL technology can be fused with strategies of other verified ligand discovery platforms, such as fragment screening.
Table 3 |. DEL selections that:
employ in-solution techniques, library retooling, or resynthesis to enhance ligand discovery or selection platforms on living cells or organisms.
| Target | Library | Selection conditions | Top library hit(s) | Unique properties |
|---|---|---|---|---|
| In-solution selections | ||||
| SIRT1, SIRT2, SIRT597 | DNA-encoded dynamic library (DEDL) Size: 10,000 |
|
#66 (Compound 79-L3–66) IC50= 78 μM - SIRT1-p53 deacetylation inhibition |
|
|
#67 (Compound 82-L5–21) IC50= 7 μM - SIRT2-p53 deacetylation inhibition | ||||
|
#68 (Compound 84-L6–39) IC50= 183 μM - SIRT5- desuccinylation inhibition Kd= 2.47 μM - SPR | ||||
| Library retooling or resynthesis | ||||
| c-myc promotor G-quarter99 | 33 DELs Size: not disclosed |
|
# 70 (Compound 2) Kd= 328 nM - SPR |
|
| BTK83 | Library 1 Size: 26,183,954 Acrylamide electrophile |
|
#71 (Compound 3) IC50= 4.8 μM - reporter displacement assay |
|
| Library 2 Size: 157,103,670 Haloalkane and epoxide electrophiles |
#72 (Compound 6) IC50= 58 nM - reporter displacement assay |
|||
| Selections on cells | ||||
| B. subtilis and E. coli104 | Solid phase DEL Size: 7,488 |
|
#76 (Compound 1) MIC= 32 μg/mL - E.coli MIC= 1 μg/mL - B. subtilis |
|
| IgG105 | Solid phase DEL Size: 448,000 |
|
#77 (Compound 2-B) |
|
Another de novo ligand discovery effort using in-solution selections aimed to discover c-Src kinase inhibitors that bind to the rarely-targeted kinase-substrate site (Fig. 6a)98. An in-solution DEL selection was designed that could enrich for c-Src enzyme substrates from a newly synthesized DEL of up to 550,000 members. DEL molecules were treated with active c-Src and ATP, followed by enrichment of phosphorylated molecules by pulldown with a broadly specific anti-phosphotyrosine antibody, enabling the isolation of compound #69, exhibiting both c-Src substrate and c-Src inhibition properties. A methyl ester version of #69 reduced c-Src-dependent STAT3 phosphorylation in a cell culture model of EGFR-transformed mammary epithelial (NME) cells. These two examples show that in-solution-based selections can produce novel ligands with unique properties. Solution-phase selections allow greater variability in experimental setup compared to immobilization, consequently enabling the identification of more nuanced binding or activity modes.
Fig. 6 |. Studies that incorporate non-immobilized selections or library retooling to enhance ligand discovery efforts.

a, Employing non-immobilized selection conditions to discover ligands that act as c-Src substrates. Methyl ester version of lead compound, #69 (SrcDEL10), showed bioactivity by reducing STAT3 phosphorylation in cellular models. b, Leveraging library resynthesis to discover and iteratively improve CBX chromodomain ligands. Lead CBX8 inhibitor, #74 (SW2_110A) was selective for CBX8 over other PRC1 CBX chromodomains, decreased transcription of CBX8 dependent genes, and inhibited proliferation of CBX8 dependent cell lines. CBX2 inhibitor, #75 (SW2_152F), was selective for CBX2 over other PRC1 CBX chromodomains, displaced CBX2 from chromatin, and inhibited cell proliferation in NED prostate cancer models synergistically with androgen receptor antagonists.
Selections using library retooling
DELs can be quickly synthesized and selection hits can be characterized shortly thereafter. As a result, DELs can be re-synthesized or re-tooled for a particular chemotype, target type, or biochemical function. Due to both the pooled nature of DEL synthesis and the small library needed in a selection, researchers can practically implement this procedure to enhance discovery of active ligands. By contrast, whole-library resynthesis for traditional libraries and screening platforms is often impractical (Table 3).
The most straightforward way to retool a library is to modify it to serve as a control. For example, DELs have been used to identify nucleic acid binders, highlighting the broad utility of a binding-based selection99. More than 30 different DELs were used in selections against immobilized biotin-tagged c-mycG4, which are DNA G-quartets found in the promotor region of the c-myc gene that control 80–90% of its expression. A portion of the libraries was modified by removing the small molecule (leaving only the DNA barcode) and this truncated library was used in a parallel selection with the intact library. This truncated, no-small-molecule control library was included to filter out any false positive DNA-DNA hybridization events that might occur with a nucleic acid-based target. Compound #70 was identified from this selection as a binder of c-mycG4 and a variety of other G4-like sequences, which inhibited PCR amplification of c-mycG4. These experiments verified target engagement and validated the goal of using DELs for novel nucleic acid binders, expanding the target scope of DELs to nucleic acids. Other studies using non-ligand control libraries might similarly enable new ligands for nucleic acid epitopes.
The main advantage of library re-tooling is that it further diversifies library members, expands library size, and provides new properties on currently used DELs to enhance discovery efforts. To extend the BTK campaign highlighted above82, a new library that mimicked the DEL architecture of discovered non-covalent BTK inhibitors was modified with acrylamide electrophiles (up to 26,183,954 compounds) to find novel covalent inhibitors of BTK83. An optimized selection protocol was used with immobilized His6-BTK at multiple time points, with and without kinase inhibitor staurosporine. Parallel selections with 17 off-target proteins were conducted to eliminate non-specific interactions and compound #71 was enriched with increasing incubation time. Additionally, #71 exhibited kinome-selective BTK binding in vitro, but did not inhibit BTK C481S, suggesting a covalent inhibition. The covalent complex was verified by mass spectroscopy and a co-crystal structure. An expanded electrophile DEL with six different electrophiles (up to 157,103,670 members) was also synthesized and a similar selection conducted. Epoxide-containing compound #72 exhibited improved potency, showed inactivity over BTK C481S mutant, and formed a covalent adduct on MS. The results of this study allow for future DEL selection schemes for covalent inhibitors. The optimized protocol reported should be emulated for DEL synthesis and selection of putative covalent binders.
Finally, re-tooling libraries is a valuable strategy from a medicinal chemistry perspective, as increasingly focused libraries can be developed to improve ligand potency. An example of this is the discovery of novel CBX chromodomain (ChD) ligands (Fig. 6b)100. In this study, selection conditions for finding ChD selective inhibitors were initially optimized and a positional scanning library (PSL1), which contains a trimethyllysine incorporated penta-peptide with broad/moderate binding affinity for CBX domains, was synthesized100. On-DNA based medicinal chemistry was conducted through selection data by making the parental peptide variable at each of the four amino acid positions outside of trimethyllysine. To find CBX8 ligands, selections against all 5 His-tagged ChDs (CBX2, CBX4, CBX5, CBX7, CBX8) using PSL1 were performed, isolating compound #73 as a weak CBX8 binder with no cellular CBX8 inhibition101. Next, two libraries were generated based around #73, PSL2A, and PSL2B and all four positions following trimethyllysine were varied with new chemotypes. Isolated compound #74 as a fluorescein conjugate was a potent binder and selective for CBX8 over other CBXs. #74 displaced CBX8 from chromatin, reduced transcription of CBX8-dependent genes in a leukemia model, and reduced cell viability of CBX8-dependent leukemia cells. From this same selection campaign, compound #75 was identified as a CBX2 binder102. #75 reduced both CBX2 and CBX8 binding to chromatin, inhibited cell proliferation, and increased expression of AR and AR target genes in a prostate cancer cell-line model independent of the androgen receptor (AR). Co-treatment with an AR antagonist further reduced cell viability, suggesting cells can be re-sensitized to AR inhibition through CBX2 inhibition.
These three examples demonstrate how library re-tooling and additional selections can serve as an alternative method to traditional medicinal-chemistry-based derivatization. Indeed, library retooling potentially enables the characterization of more potent and selective lead compounds straight out of libraries.
Collectively, the examples highlighted in the previous subsections illustrate the variety of conditions that DEL selections can use to discover unique and active ligands. The advantages inherent to DELs, such as multiplexed selection conditions, ease of resynthesis and retooling, and the utility of non-immobilized conditions, have resulted in the identification of many new small molecules. These library traits make DEL technology a unique and powerful tool for finding ligands with particular in vitro properties that can be preemptively defined.
Selections on living cells
Some targets are not amenable to any isolated protein or in vitro selection due to protein instability or a lack of well-established isolation protocols. Furthermore, as with any target-based binding screen, the qualities of ligand binding, biochemical modulation of the target, and downstream phenotype are disconnected. Selections using biological models instead of isolated proteins to find novel ligands can expand usable selection environments. Indeed, such a strategy has precedent outside the timeframe of this review103, and more recent examples are highlighted in the following section (Table 3). Additionally, functional or phenotype-based assays (as opposed to binding selections), have been substantially developed and reported. However, these assays have only recently been used to find new small molecules.
In a creative example of DEL-based phenotypic screens, a solid phase on-bead DEL was used to screen for novel antibiotics against E. coli and B. subtilis104. A one-bead one-compound solid phase DEL of up to 7,488-members was used in a screen on bacteria agar cultures. UV irradiation released the active component off-beads, and active antibiotics formed growth inhibition zones (GIZ) around its DEL bead. Next, beads were isolated, DNA barcodes amplified by PCR and sequenced, and compounds assessed for their MIC. These efforts resulted in identification of the novel hit compound #76. Library members that were derivatives of ciprofloxacin, previously reported to have antibiotic activity, were also identified. Future phenotype-based screens can use this technology with larger libraries to interrogate larger, novel chemical spaces.
Another study highlighting a unique application of DELs used a solid phase peptoid DEL containing up to 448,000-members, to search for diagnostic epitope surrogates of IgGs that circulate during non-infectious, latent state (LTB), and active infectious states (ATB) of mycobacterium tuberculosis (Mtb) infection105. Host immunological states might better diagnose between the three epitope surrogates, given that current Mtb antigens do not offer appropriate specificity or sensitivity. A selection was conducted using fluorescent-activated cell sorting (FACS) with a solid phase, on-bead DEL, incubated with either a mixture or individual samples of sera acquired from ATB, LTB, and non-Mtb-infected individuals. Beads were incubated with separate secondary fluorescence anti-IgG to label and distinguish serum IgG binding hit compounds and hits were verified by resynthesis on beads. Samples were analyzed for serum IgG binding and epitope detection by flow cytometry106 and an unexpected library side-product compound #77 showed ATB binding, attenuated binding to LTB serum, and little-to-no binding in non-infected serum. A secreted protein Ag85B, a diacylglycerol acetyltransferase, was identified as the antigen #77 mimic, as it competed strongly with #77 for serum IgG.
These two studies reverse the phase of typical DEL selections, which traditionally rely on immobilized protein and soluble DEL. A solid-phase DEL allows selections against more complex targets and offers more options for selection assays outside a typical DEL binding assay.
Living cells can also serve as target-based selection platforms. On-cell target selections are valuable tools for DELs, as this condition is an endogenously relevant environment and enables ligand discovery for traditionally hard-to-isolate membrane proteins. By deploying DNA-programmed affinity labeling (DPAL) technology107 on endogenous cell surface targets, ligands to folate receptor (FR) and epidermal growth factor receptor (EGFR) were recently discovered (Fig. 7). The technique required no overexpression on cell surfaces and allowed for DEL selections on native protein substrates on cell membranes. A cell surface protein of interest was labeled with a DNA tag through photocrosslinking with a known binding epitope or ligand, followed by library incubation on cells. If library members bound to the target protein, an avidity effect ensued between the library DNA-tag and the DNA-tag on the target protein, which promoted hybridization with short complementary regions. Non-binders formed unstable duplexes by comparison and were washed away. Cell-bound library members were then heat eluted and isolated for PCR and NGS. This selection procedure was used on a DEL of up to 30,420,000-members with HeLa cells for FR and A431 cells for EGFR108. From the FR selection, isolated compound #78 exhibited strong binding affinity. FAM-labeled #78 also stained the cell surfaces of HeLa cells but not those with low FR expression. For EGFR, isolated compound #79 exhibited modest binding. This method is one of the only reported means to access cell-based selections against endogenous targets. As many cell surface protein targets already have developed antibodies, one can adapt this approach to discover novel small molecule binders of cell surface targets, especially membrane proteins that suffer from poor recombinant expression and stability.
Fig. 7 |. On-cell DEL-based small molecule discovery.
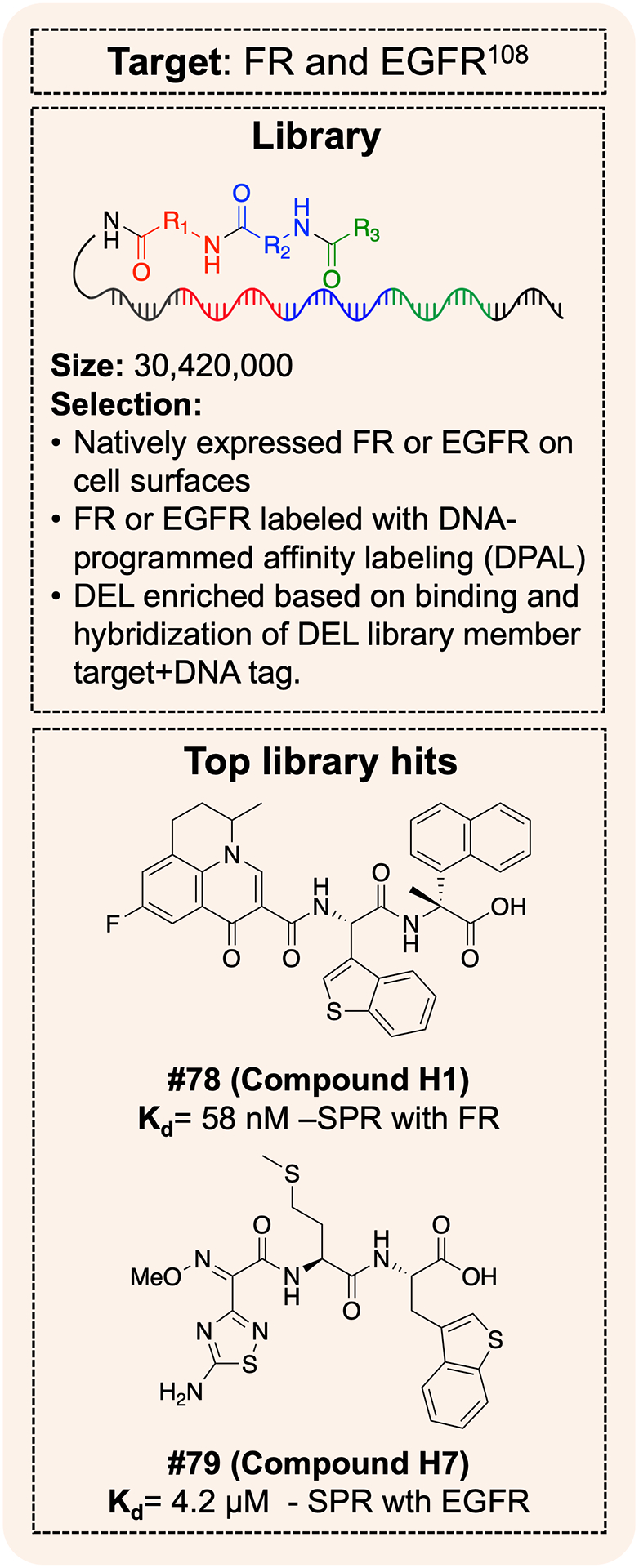
Novel ligand discovery was mediated through selections with natively expressed FR and EGFR on whole cells, without overexpression of target needed.
DEL selections are increasingly being conducted on complex biological systems and have recently played a bigger role in facilitating novel ligand discovery. We anticipate that these selection platforms will allow DELs to sample targets in more biological contexts, an area in which HTS has traditionally enjoyed a profound advantage. Future discovery efforts within these models will enable DELs to transition seamlessly between in vitro validation and biological activity.
Future Perspectives
DNA-encoded libraries (DELs) have emerged as a popular platform for discovering novel chemical matter. Various DEL selection setups can be tuned to the target and desired ligand type. This review has highlighted multiple on-bead, co-incubation, or parallel selection conditions to help researchers identify or even select for ligands with desired properties. DEL architectures have become structurally diverse despite constraints on the chemistries that can be used to prepare them. While large libraries (>109) can enhance discovery efforts, there are many examples of focused, small DELs that have yielded highly bioactive small molecules. Indeed, DELs of multiple sizes can include important chemotypes used in the current small-molecule discovery landscape. For example, macrocyclic compounds can offer protein-binding capabilities normally associated with larger biomolecules, but with biophysical properties more similar to small molecules, and several macrocyclic DELs have been developed109. Covalent DELs have also been developed83,110,111 and can now offer a unique approach to targeting nucleophilic residues that may enhance potency and selectivity on key targets. Fragment-based screening has also been used in DEL architectures, allowing for rapid screening of binding fragments that can later be synthesized into active ligands. As a result of these advantages, studies reporting DEL-mediated discovery of new ligands have increased over the last 20 years (Fig. 2a). Thus far, the biological space DELs have accessed is broad and we anticipate that the use of DEL libraries and their corresponding small molecules will continue to increase. The breadth of targets accessible to DELs ranges from traditional enzymes, to cell surface receptors, protein-protein interactions, and folding chaperones (Fig. 2b).
Given the recent breakthroughs in targeted protein degradation112,113,122,123,114–121, DELs may be especially valuable for researchers seeking to discover bifunctional molecules with new degradation activities. DEL library members are inherently bifunctional molecules with a linker between the small molecule and the DNA-tag. As the DNA-tag does not typically engage in interactions with the target of interest and is solvent exposed, the tag’s point of conjugation with the small molecule is usually replaceable with a wide variety of other groups, as already demonstrated with PROTACs73, and cell recruiters81. Since PROTACs capable of recruiting enzymes, cellular cargo, or whole cells, are becoming increasingly prevalent115, DEL-mediated discovery of small molecules for targets of interest are well-poised to take advantage of this modularity. DEL-mediated discovery of recruiting ligands for effector proteins, such as E3 ligases, also benefit from this site of modularity. This property can facilitate conjugation to a target’s ligand, enabling new tools for targeted protein degradation. The field currently uses a limited set of ligands for a small number of well-characterized E3 ligases, yet hundreds more E3 ligases exist in the human genome115. DELs have been already utilized to find ligands for new E3 ligases124 and may expand the capabilities of PROTAC systems.
Molecular glues116,119 are a broader class of ligands that bring together two targets of interest (Target A and Target B) and can have unique downstream phenotypes. Indeed, the targeted degradation field often relies on these types of ligands. DELs have already been used to discover molecular glues, including BRD4-VHL125 and BRD4-Cereblon(CRBN)126 dimerizers that promote target protein degradation. These studies used libraries based on ligands for individual proteins (VHL and CRBN) and included additional diversity elements and linking elements for binding to the second protein target. In both studies, the facile synthesis of target-focused DELs and the implementation of parallel selections facilitated the identification of compounds that form ternary complexes with both protein targets. Other key advantages of DEL technology that can be leveraged for molecular glue discovery include: the large size of many DELs (up to 1012 library members) to discover statistically infrequent molecular glue activities; the ability to synthesize focused libraries based on SAR from initial DEL selection efforts to improve discovery rates; and the capacity to use multiple protein constructs or ligand co-incubations to select for molecular glues with research-specified binding modes to each target. Based on the many advantages DEL technologies offer, additional molecular glues will likely be discovered through this platform.
DEL-mediated functional selections are also valuable as they can be used to identify library members that bind and impart a biochemical effect or phenotype. Some groups have pioneered solid-phase DELs in FACS-based selections for protein activity127–130, illustrating the use of DELs in modulating biochemical activity or fluorescent based binding selections131. As highlighted in this review, solid-phase DELs can also be used for phenotypic screens to discover antibiotics104. Given that most enzymatic functional assays use a fluorescence readout, these solid-phase DELs can be expanded to include more library members. Such an expansion would enable further exploration of classes of enzyme targets. Likewise, solid-phase DEL phenotypic screens for antibiotics can also be expanded to diversify DEL library applications. For example, DEL-mediated discovery of ligand agonists, such as for GPCRs, already uses creative immobilized selection conditions to isolate agonist-like binding modes86–96. However, more functional-based selections would enable better diagnostic identification of agonist ligands that can be characterized in the selection rather than off-DNA in a separate assay.
Target types are not limited to proteins, as DELs have also been used to find ligands for nucleic acid targets. For example, as summarized above, efforts were made to find G-quartet promoter ligands of the c-myc gene99. Furthermore, DELs have recently been used to find RNA-binding ligands, including FMN riboswitch132 and primary microRNA-27a131. As DELs inherently have DNA-tags that may promiscuously bind to nucleic acid targets in a non-ligand dependent manner, these studies have developed methods to overcome this limitation to find bioactive nucleic acid ligands. These examples diversify the target depth that DELs can sample for therapeutic benefit.
DEL technology also offers a new way to quickly sample known chemical space using currently approved drugs in the clinic, natural products, or well-characterized chemical probes. Natural-product-based or drug-based DELs are particularly useful for repurposing known molecules against new targets in new contexts. By creating DNA-tagged versions of these molecules and conducting traditional or modified DEL selections, other targets can be quickly identified using libraries of previously-reported compounds133–136. Repurposing ligands can also result in campaigns to increase their activity, as affinity maturation libraries and campaigns have been reported, with a recent maturation of acetazolamide coinciding with a campaign to generate a cancer imaging agent137,138. Furthermore, focused libraries based around either a known ligand or putative chemotypes for a specific protein are more tractable with DELs, as their synthesis and screening are on a much smaller scale compared to HTS.
Future targets also include proteins that are not easy to isolate or proteins that are not stable outside of native contexts. Indeed, DELs have been successfully used in serum105, bacteria culture plates104, and against cell-surface expressed protein targets in either overexpressed103 or native conditions107,108. Recent studies have described the adaptation of DELs to access intracellular-based targets139,140, though such efforts have not yet led to any well-developed small molecules.
Finally, as the chemical size of DNA-encoded libraries is far greater than any other type of combinatorial non-genetically encoded library and selection output is primarily next-generation sequencing, researchers can use various computational methods to sift through dense selection data. Machine learning or virtual combinatorial library platforms, that can predict ways of improving ligands based on sequencing input from a DEL selection, could enable rapid compound optimization. This technology remains a valuable and expandable area within the DEL community41,141,142.
Supplementary Material
Outlook.
We anticipate that DELs will continue to serve as a powerful tool for finding new modulators of biological activity. As a platform, DELs offer a combination of desirable properties that are often mutually exclusive using other means of small-molecule discovery. DELs are chemically diverse, easy to screen, and use a low quantity of consumable and chemical materials. Given its broad utility and demonstrated impact, DEL technology will likely continue to play an important role facilitating the discovery of new chemical probes for use in basic research or clinical settings.
Acknowledgements
We would like to thank Dr. Anahita Vieira for her valuable assistance and input. We thank Dr. Mary O’Reilly for contributing DEL architecture and selection figures. This work was supported by NIH R35 GM118062 and the Howard Hughes Medical Institute.
Footnotes
Competing Interests
A.A.P. and D.R.L. are co-inventors on patent applications on DNA-encoded libraries and their applications. D.R.L. is a consultant and co-founder of Exo Therapeutics, a company that uses DNA-encoded libraries.
Supplementary Information
Structures of all compounds highlighted in this manuscript as well as references tallied in Fig. 2a may be found in the Supplementary File.
References
- 1.Brenner S & Lerner RA Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. U. S. A 89, 5381–5383 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark MA Selecting chemicals: the emerging utility of DNA-encoded libraries. Curr. Opin. Chem. Biol 14, 396–403 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Kleiner RE, Dumelin CE & Liu DR Small-molecule discovery from DNA-encoded chemical libraries. Chem. Soc. Rev 40, 5707–5717 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gartner ZJ & Liu DR The generality of DNA-templated synthesis as a basis for evolving non-natural small molecules. J. Am. Chem. Soc 123, 6961–6963 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tse BN, Snyder TM, Shen Y & Liu DR Translation of DNA into a library of 13 000 synthetic small-molecule macrocycles suitable for in vitro selection. J. Am. Chem. Soc 130, 15611–15626 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen MH et al. A yoctoliter-scale DNA reactor for small-molecule evolution. J. Am. Chem. Soc 131, 1322–1327 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Wrenn SJ, Weisinger RM, Halpin DR & Harbury PB Synthetic ligands discovered by in vitro selection. J. Am. Chem. Soc 129, 13137–13143 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark MA et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol 5, 647–654 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Melkko S, Scheuermann J, Dumelin CE & Neri D Encoded self-assembling chemical libraries. Nat. Biotechnol 22, 568–574 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Halpin DR & Harbury PB DNA display II. Genetic manipulation of combinatorial chemistry libraries for small-molecule evolution. PLoS Biol. 2, (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buller F et al. Design and synthesis of a novel DNA-encoded chemical library using Diels-Alder cycloadditions. Bioorganic Med. Chem. Lett 18, 5926–5931 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Doyon JB, Snyder TM & Liu DR Highly sensitive in vitro selections for DNA-linked synthetic small molecules with protein binding affinity and specificity. J. Am. Chem. Soc 125, 12372–12373 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Mannocci L et al. High-throughput sequencing allows for the identification of binding molecules from DNA-encoded chemical libraries. Proc. Natl. Acad. Sci 105, 17670–17675 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleiner RE, Dumelin CE, Tiu GC, Sakurai K & Liu DR In vitro selection of a DNA-templated small-molecule library reveals a class of macrocyclic kinase inhibitors. J. Am. Chem. Soc 132, 11779–11791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunkari YK, Siripuram VK, Nguyen TL & Flajolet M High-power screening (HPS) empowered by DNA-encoded libraries. Trends Pharmacol. Sci 43, 4–15 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Castan IFSF, Graham JS, Salvini CLA, Stanway-Gordon HA & Waring MJ On the design of lead-like DNA-encoded chemical libraries. Bioorganic Med. Chem 43, 116273 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Fitzgerald PR & Paegel BM DNA-Encoded Chemistry: Drug Discovery from a Few Good Reactions. Chem. Rev 121, 7155–7177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song M & Hwang GT DNA-Encoded Library Screening as Core Platform Technology in Drug Discovery: Its Synthetic Method Development and Applications in DEL Synthesis. J. Med. Chem 63, 6578–6599 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Dickson P & Kodadek T Chemical composition of DNA-encoded libraries, past present and future. Org. Biomol. Chem 17, 4676–4688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gironda-Martínez A, Donckele EJ, Samain F & Neri D DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges. ACS Pharmacol. Transl. Sci 4, 1265–1279 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunig VBK, Potowski M, Klika Škopić M & Brunschweiger A Scanning Protein Surfaces with DNA-Encoded Libraries. ChemMedChem 16, 1048–1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao G, Huang Y, Zhou Y, Li Y & Li X Future challenges with DNA-encoded chemical libraries in the drug discovery domain. Expert Opin. Drug Discov 14, 735–753 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Neri D & Lerner RA DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem 87, 479–502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Li Y & Li X Strategies for developing DNA encoded libraries beyond binding assays. Nat. Chem 14, 129–140 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Song Y & Li X Evolution of the Selection Methods of DNA-Encoded Chemical Libraries. Acc. Chem. Res 54, 3491–3503 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Huang Y & Li X Recent Advances on the Selection Methods of DNA-Encoded Libraries. ChemBioChem 22, 2384–2397 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Kodadek T, Paciaroni NG, Balzarini M & Dickson P Beyond protein binding: Recent advances in screening DNA-encoded libraries. Chem. Commun 55, 13330–13341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawadi S et al. Discovery of potent thrombin inhibitors from a protease-focused DNA-encoded chemical library. Proc. Natl. Acad. Sci. U. S. A 117, 16782–16789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gironda-Martínez A et al. Identification and Validation of New Interleukin-2 Ligands Using DNA-Encoded Libraries. J. Med. Chem 64, 17496–17510 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Taylor DM et al. Identifying Oxacillinase-48 Carbapenemase Inhibitors Using DNA-Encoded Chemical Libraries. ACS Infect. Dis 6, 1214–1227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steffek M et al. A Multifaceted Hit-Finding Approach Reveals Novel LC3 Family Ligands. Biochemistry (2022). doi: 10.1021/acs.biochem.1c00682 [DOI] [PubMed] [Google Scholar]
- 32.Scott DE, Bayly AR, Abell C & Skidmore J Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov 15, 533–550 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Peterson AA et al. Discovery and molecular basis of subtype-selective cyclophilin inhibitors. Nat. Chem. Biol 18, 1184–1195 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Usanov DL, Chan AI, Maianti JP & Liu DR Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem 10, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunig VBK et al. TEAD–YAP Interaction Inhibitors and MDM2 Binders from DNA-Encoded Indole-Focused Ugi Peptidomimetics. Angew. Chemie - Int. Ed 59, 20338–20342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannes JW et al. Structure Based Design of Non-Natural Peptidic Macrocyclic Mcl-1 Inhibitors. ACS Med. Chem. Lett 8, 239–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wellaway CR et al. Discovery of a Bromodomain and Extraterminal Inhibitor with a Low Predicted Human Dose through Synergistic Use of Encoded Library Technology and Fragment Screening. J. Med. Chem 63, 714–746 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Fernández-Montalván AE et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem. Biol 12, 2730–2736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lomas DA et al. Development of a small molecule that corrects misfolding and increases secretion of Z α 1 -antitrypsin . EMBO Mol. Med 13, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuen LH et al. A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD+-Dependent Enzymes. J. Am. Chem. Soc 141, 5169–5181 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Lemke M et al. Integrating DNA-encoded chemical libraries with virtual combinatorial library screening: Optimizing a PARP10 inhibitor. Bioorganic Med. Chem. Lett 30, 127464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacHutta CA et al. Prioritizing multiple therapeutic targets in parallel using automated DNA-encoded library screening. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Concha N et al. Discovery and Characterization of a Class of Pyrazole Inhibitors of Bacterial Undecaprenyl Pyrophosphate Synthase. J. Med. Chem 59, 7299–7304 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Chamakuri S et al. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 Mpro inhibitors. Proc. Natl. Acad. Sci. U. S. A 118, 8–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Podolin PL et al. In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediat. 104–105, 25–31 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Belyanskaya SL, Ding Y, Callahan JF, Lazaar AL & Israel DI Discovering Drugs with DNA-Encoded Library Technology: From Concept to Clinic with an Inhibitor of Soluble Epoxide Hydrolase. ChemBioChem 18, 837–842 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Ding Y et al. Discovery of soluble epoxide hydrolase inhibitors through DNA-encoded library technology (ELT). Bioorganic Med. Chem 41, 116216 (2021). [DOI] [PubMed] [Google Scholar]
- 48.DING Y, THALJI RK & MARINO JPJ NOVEL sEH INHIBITORS AND THEIR USE FIELD. (2009).
- 49.Ottl J, Leder L, Schaefer JV & Dumelin CE Encoded library technologies as integrated lead finding platforms for drug discovery. Molecules 24, 1–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satz AL What Do You Get from DNA-Encoded Libraries? ACS Med. Chem. Lett 9, 408–410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arico-Muendel CC From haystack to needle: Finding value with DNA encoded library technology at GSK. Medchemcomm 7, 1898–1909 (2016). [Google Scholar]
- 52.Lazaar AL, Baines A, Ahmed M, Boardley R & Hussaini A Inhibition of Soluble Epoxide Hydrolase Does Not Augment Hypoxic Pulmonary Vasoconstriction in Healthy Subjects. 1 (2016). [Google Scholar]
- 53.Lazaar AL et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol 81, 971–979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang L et al. Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers. Chest 151, 555–563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martini RP et al. A Double-Blind, Randomized, Placebo-Controlled Trial of Soluble Epoxide Hydrolase Inhibition in Patients with Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care (2021). doi: 10.1007/s12028-021-01398-8 [DOI] [PubMed] [Google Scholar]
- 56.Luther JM et al. GSK2256294 Decreases sEH (Soluble Epoxide Hydrolase) Activity in Plasma, Muscle, and Adipose and Reduces F2-Isoprostanes but Does Not Alter Insulin Sensitivity in Humans. Hypertension 1092–1102 (2021). doi: 10.1161/HYPERTENSIONAHA.121.17659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris PA et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem 59, 2163–2178 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Harris PA et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem 60, 1247–1261 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Weisel K et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect 5, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tompson DJ et al. Comparison of the Pharmacokinetics of RIPK1 Inhibitor GSK2982772 in Healthy Western and Japanese Subjects. Eur. J. Drug Metab. Pharmacokinet 46, 71–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tompson DJ et al. Development of a Prototype, Once-Daily, Modified-Release Formulation for the Short Half-Life RIPK1 Inhibitor GSK2982772. Pharm. Res 38, 1235–1245 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tompson D et al. Development of a Once-Daily Modified-Release Formulation for the Short Half-Life RIPK1 Inhibitor GSK2982772 using DiffCORE Technology. Pharm. Res 153–165 (2022). doi: 10.1007/s11095-021-03124-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weisel K et al. Response to Inhibition of Receptor-Interacting Protein Kinase 1 (RIPK1) in Active Plaque Psoriasis: A Randomized Placebo-Controlled Study. Clin. Pharmacol. Ther 108, 808–816 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weisel K et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther 23, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weisel K et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 8, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xia C et al. Structure-based bioisosterism design of thio-benzoxazepinones as novel necroptosis inhibitors. Eur. J. Med. Chem 220, 113484 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Yoshikawa M et al. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H -pyrazolo[3,4- c] pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. J. Med. Chem 61, 2384–2409 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Wang W et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 34, 757–774.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harris PA et al. Identification of a RIP1 Kinase Inhibitor Clinical Candidate (GSK3145095) for the Treatment of Pancreatic Cancer. ACS Med. Chem. Lett 10, 857–862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cuozzo JW et al. Novel Autotaxin Inhibitor for the Treatment of Idiopathic Pulmonary Fibrosis: A Clinical Candidate Discovered Using DNA-Encoded Chemistry. J. Med. Chem 63, 7840–7856 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Richter H et al. DNA-Encoded Library-Derived DDR1 Inhibitor Prevents Fibrosis and Renal Function Loss in a Genetic Mouse Model of Alport Syndrome. ACS Chem. Biol 14, 37–49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rianjongdee F et al. Discovery of a Highly Selective BET BD2 Inhibitor from a DNA-Encoded Library Technology Screening Hit. J. Med. Chem 64, 10806–10833 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Disch JS et al. Bispecific Estrogen Receptor α Degraders Incorporating Novel Binders Identified Using DNA-Encoded Chemical Library Screening. J. Med. Chem 64, 5049–5066 (2021). [DOI] [PubMed] [Google Scholar]
- 74.Veerman JJN et al. Discovery of 2,4–1 H-Imidazole Carboxamides as Potent and Selective TAK1 Inhibitors. ACS Med. Chem. Lett 12, 555–562 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu Z et al. Discovery and characterization of bromodomain 2-specific inhibitors of BRDT. Proc. Natl. Acad. Sci. U. S. A 118, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee ECY et al. Discovery of Novel, Potent Inhibitors of Hydroxy Acid Oxidase 1 (HAO1) Using DNA-Encoded Chemical Library Screening. J. Med. Chem 64, 6730–6744 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Ryan MD et al. Discovery of Novel UDP- N-Acetylglucosamine Acyltransferase (LpxA) Inhibitors with Activity against Pseudomonas aeruginosa. J. Med. Chem 64, 14377–14425 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Kung PP et al. Characterization of Specific N-α-Acetyltransferase 50 (Naa50) Inhibitors Identified Using a DNA Encoded Library. ACS Med. Chem. Lett 11, 1175–1184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bassi G et al. A Single-Stranded DNA-Encoded Chemical Library Based on a Stereoisomeric Scaffold Enables Ligand Discovery by Modular Assembly of Building Blocks. Adv. Sci 7, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Favalli N et al. Stereo- and regiodefined DNA-encoded chemical libraries enable efficient tumour-targeting applications. Nat. Chem 13, 540–548 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bassi G et al. Specific Inhibitor of Placental Alkaline Phosphatase Isolated from a DNA-Encoded Chemical Library Targets Tumor of the Female Reproductive Tract. J. Med. Chem 64, 15799–15809 (2021). [DOI] [PubMed] [Google Scholar]
- 82.Cuozzo JW et al. Discovery of a Potent BTK Inhibitor with a Novel Binding Mode by Using Parallel Selections with a DNA-Encoded Chemical Library. ChemBioChem 18, 864–871 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Guilinger JP et al. Novel irreversible covalent BTK inhibitors discovered using DNA-encoded chemistry. Bioorganic Med. Chem 42, (2021). [DOI] [PubMed] [Google Scholar]
- 84.Nissink JWM et al. Generating Selective Leads for Mer Kinase Inhibitors-Example of a Comprehensive Lead-Generation Strategy. J. Med. Chem 64, 3165–3184 (2021). [DOI] [PubMed] [Google Scholar]
- 85.McCoull W et al. Optimization of an Imidazo[1,2-a]pyridine Series to Afford Highly Selective Type I1/2 Dual Mer/Axl Kinase Inhibitors within Vivo Efficacy. J. Med. Chem 64, 13524–13539 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Brown DG et al. Agonists and Antagonists of Protease-Activated Receptor 2 Discovered within a DNA-Encoded Chemical Library Using Mutational Stabilization of the Target. SLAS Discov. 23, 429–436 (2018). [DOI] [PubMed] [Google Scholar]
- 87.Kennedy AJ et al. Protease-activated receptor-2 ligands reveal orthosteric and allosteric mechanisms of receptor inhibition. Commun. Biol 3, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheng RKY et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545, 112–115 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Carey RM et al. Polarization of protease-activated receptor 2 (PAR-2) signaling is altered during airway epithelial remodeling and deciliation. J. Biol. Chem 295, 6721–6740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang X et al. Protease-activated receptor 2 (PAR-2) antagonist AZ3451 as a novel therapeutic agent for osteoarthritis. Aging (Albany. NY). 11, 12532–12545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun L et al. Protease-Activated Receptor 2 (PAR-2) Antagonist AZ3451 Mitigates Oxidized Low-Density Lipoprotein (Ox-LDL)-Induced Damage and Endothelial Inflammation. Chem. Res. Toxicol 34, 2202–2208 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Ahn S et al. Allosteric ‘beta-blocker’ isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. U. S. A 114, 1708–1713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X et al. Mechanism of intracellular allosteric β 2 AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ahn S et al. Small-molecule positive allosteric modulators of the β2-adrenoceptor isolated from DNA-encoded libraries. Mol. Pharmacol 94, 850–861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu X et al. Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science (80-. ). 364, 1283–1287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J et al. β-Arrestin–Biased Allosteric Modulator Potentiates Carvedilol-Stimulated β Adrenergic Receptor Cardioprotection. Mol. Pharmacol 100, 568–579 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou Y, Shen W, Peng J, Deng Y & Li X Identification of isoform/domain-selective fragments from the selection of DNA-encoded dynamic library. Bioorganic Med. Chem 45, 116328 (2021). [DOI] [PubMed] [Google Scholar]
- 98.Kim D et al. Application of a Substrate-Mediated Selection with c-Src Tyrosine Kinase to a DNA-Encoded Chemical Library. Molecules 24, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Litovchick A et al. Novel nucleic acid binding small molecules discovered using DNA-encoded chemistry. Molecules 24, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Denton KE et al. Robustness of In Vitro Selection Assays of DNA-Encoded Peptidomimetic Ligands to CBX7 and CBX8. SLAS Discov. 23, 417–428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang S et al. Optimization of Ligands Using Focused DNA-Encoded Libraries to Develop a Selective, Cell-Permeable CBX8 Chromodomain Inhibitor. ACS Chem. Biol 15, 112–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang S et al. A Potent, Selective CBX2 Chromodomain Ligand and Its Cellular Activity During Prostate Cancer Neuroendocrine Differentiation. ChemBioChem 22, 2335–2344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu Z et al. Cell-Based Selection Expands the Utility of DNA-Encoded Small-Molecule Library Technology to Cell Surface Drug Targets: Identification of Novel Antagonists of the NK3 Tachykinin Receptor. ACS Comb. Sci 17, 722–731 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Cochrane WG, Fitzgerald PR & Paegel BM Antibacterial Discovery via Phenotypic DNA-Encoded Library Screening. ACS Chem. Biol 16, 2752–2756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mendes KR et al. High-throughput identification of DNA-encoded IgG ligands that distinguish active and latent mycobacterium tuberculosis infections. ACS Chem. Biol 12, 234–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Doran TM & Kodadek T A liquid array platform for the multiplexed analysis of synthetic molecule-protein interactions. ACS Chem. Biol 9, 339–346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang Y, Deng Y, Zhang J, Meng L & Li X Direct ligand screening against membrane proteins on live cells enabled by DNA-programmed affinity labelling. Chem. Commun 57, 3769–3772 (2021). [DOI] [PubMed] [Google Scholar]
- 108.Huang Y et al. Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells. Nat. Chem 13, 77–88 (2021). [DOI] [PubMed] [Google Scholar]
- 109.Plais L & Scheuermann J Macrocyclic DNA-encoded chemical libraries: a historical perspective. RSC Chem. Biol 3, 7–17 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ge R et al. Discovery of SARS-CoV-2 main protease covalent inhibitors from a DNA-encoded library selection. SLAS Discov. Adv. life Sci. R D 27, 79–85 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li L et al. Triazine-Based Covalent DNA-Encoded Libraries for Discovery of Covalent Inhibitors of Target Proteins. ACS Med. Chem. Lett 13, 1574–1581 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lu J et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 22, 755–763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhong Y et al. Emerging targeted protein degradation tools for innovative drug discovery: From classical PROTACs to the novel and beyond. Eur. J. Med. Chem 231, 114142 (2022). [DOI] [PubMed] [Google Scholar]
- 115.Békés M, Langley DR & Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 21, 181–200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Modell AE, Lai S, Nguyen TM & Choudhary A Bifunctional modalities for repurposing protein function. Cell Chem. Biol 28, 1081–1089 (2021). [DOI] [PubMed] [Google Scholar]
- 117.Belcher BP, Ward CC & Nomura DK Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry (2021). doi: 10.1021/acs.biochem.1c00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Faust TB, Donovan KA, Yue H, Chamberlain PP & Fischer ES Small-Molecule Approaches to Targeted Protein Degradation. Annu. Rev. Cancer Biol 5, 181–201 (2020). [Google Scholar]
- 119.Gerry CJ & Schreiber SL Unifying principles of bifunctional, proximity-inducing small molecules. Nat. Chem. Biol 16, 369–378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (80-. ). 348, 1376–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lai AC et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chemie - Int. Ed 55, 807–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cowan AD & Ciulli A Driving E3 Ligase Substrate Specificity for Targeted Protein Degradation: Lessons from Nature and the Laboratory. Annu. Rev. Biochem 91, 295–319 (2022). [DOI] [PubMed] [Google Scholar]
- 123.Kramer LT & Zhang X Expanding the landscape of E3 ligases for targeted protein degradation. Curr. Res. Chem. Biol 2, 100020 (2022). [Google Scholar]
- 124.Chana CK et al. Discovery and Structural Characterization of Small Molecule Binders of the Human CTLH E3 Ligase Subunit GID4. J. Med. Chem (2022). doi: 10.1021/acs.jmedchem.2c00509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mason JW et al. DNA-encoded library (DEL)-enabled discovery of proximity-inducing small molecules. bioRxiv 1–29 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen Q et al. Optimization of PROTAC Ternary Complex Using DNA Encoded Library Approach. ACS Chem. Biol 18, 25–33 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shin MH, Lee KJ & Lim HS DNA-Encoded Combinatorial Library of Macrocyclic Peptoids. Bioconjug. Chem 30, 2931–2938 (2019). [DOI] [PubMed] [Google Scholar]
- 128.Lee KJ, Bang G, Kim YW, Shin MH & Lim HS Design and synthesis of a DNA-encoded combinatorial library of bicyclic peptoids. Bioorganic Med. Chem 48, 116423 (2021). [DOI] [PubMed] [Google Scholar]
- 129.Cochrane WG et al. Activity-Based DNA-Encoded Library Screening. ACS Comb. Sci 21, 425–435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.MacConnell AB, Price AK & Paegel BM An Integrated Microfluidic Processor for DNA-Encoded Combinatorial Library Functional Screening. ACS Comb. Sci 19, 181–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Benhamou RI et al. DNA-encoded library versus RNA-encoded library selection enables design of an oncogenic noncoding RNA inhibitor. Proc. Natl. Acad. Sci. U. S. A 119, 1–8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen Q et al. Expanding the DNA-encoded library toolbox: Identifying small molecules targeting RNA. Nucleic Acids Res. 50, E67 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chan AI, McGregor LM, Jain T & Liu DR Discovery of a Covalent Kinase Inhibitor from a DNA-Encoded Small-Molecule Library × Protein Library Selection. J. Am. Chem. Soc 139, 10192–10195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang DY et al. Target Identification of Kinase Inhibitor Alisertib (MLN8237) by Using DNA-Programmed Affinity Labeling. Chem. - A Eur. J 23, 10906–10914 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Xie J et al. Selection of Small Molecules that Bind to and Activate the Insulin Receptor from a DNA-Encoded Library of Natural Products. iScience 23, 101197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li J et al. A DNA-encoded library for the identification of natural product binders that modulate poly (ADP-ribose) polymerase 1, a validated anti-cancer target. Biochem. Biophys. Res. Commun 533, 241–248 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Wichert M et al. Dual-display of small molecules enables the discovery of ligand pairs and facilitates affinity maturation. Nat. Chem 7, 241–249 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Kulterer OC et al. A Microdosing Study with 99mTc-PHC-102 for the SPECT/CT Imaging of Primary and Metastatic Lesions in Renal Cell Carcinoma Patients. J. Nucl. Med 62, 360–365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Petersen LK et al. Screening of DNA-encoded small molecule libraries inside a living cell. J. Am. Chem. Soc 143, 2751–2756 (2021). [DOI] [PubMed] [Google Scholar]
- 140.Cai B et al. Selection of DNA-Encoded Libraries to Protein Targets within and on Living Cells. J. Am. Chem. Soc 141, 17057–17061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McCloskey K et al. Machine learning on DNA-encoded libraries: A new paradigm for hit finding. J. Med. Chem 63, 8857–8866 (2020). [DOI] [PubMed] [Google Scholar]
- 142.Xiong F et al. Discovery of TIGIT inhibitors based on DEL and machine learning. Front. Chem 10, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gartner ZJ et al. DNA-templated organic synthesis and selection of a library of macrocycles. Science (80-. ). 305, 1601–1605 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Halpin DR & Harbury PB DNA display I. Sequence-encoded routing of DNA populations. PLoS Biol. 2, 1015–1021 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.DNA-encoded chemical libraries. Nat. Rev. Methods Prim 2, 43586 (2022). [Google Scholar]
- 146.MacConnell AB, McEnaney PJ, Cavett VJ & Paegel BM DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci 17, 518–534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hackler AL, Fitzgerald FG, Dang VQ, Satz AL & Paegel BM Off-DNA DNA-Encoded Library Affinity Screening. ACS Comb. Sci 22, 25–34 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Roy A, Koesema E & Kodadek T High-Throughput Quality Control Assay for the Solid-Phase Synthesis of DNA-Encoded Libraries of Macrocycles**. Angew. Chemie - Int. Ed 60, 11983–11990 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mcgregor LM, Jain T & Liu DR Identification of Ligand – Target Pairs from Combined Libraries of Small Molecules and Unpuri fi ed Protein Targets in Cell Lysates. J. Am. Chem. Soc 136, 3264–3270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Blakskjaer P, Heitner T & Hansen NJV Fidelity by design: Yoctoreactor and binder trap enrichment for small-molecule DNA-encoded libraries and drug discovery. Curr. Opin. Chem. Biol 26, 62–71 (2015). [DOI] [PubMed] [Google Scholar]
- 151.Kochmann S, Le ATH, Hili R & Krylov SN Predicting efficiency of NECEEM-based partitioning of protein binders from nonbinders in DNA-encoded libraries. Electrophoresis 39, 2991–2996 (2018). [DOI] [PubMed] [Google Scholar]
- 152.Bao J et al. Predicting Electrophoretic Mobility of Protein–Ligand Complexes for Ligands from DNA-Encoded Libraries of Small Molecules. Anal. Chem 88, 5498–5506 (2016). [DOI] [PubMed] [Google Scholar]
- 153.Zhao P et al. Selection of DNA-encoded small molecule libraries against unmodified and non-immobilized protein targets. Angew. Chemie - Int. Ed 53, 10056–10059 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Shi B, Deng Y, Zhao P & Li X Selecting a DNA-Encoded Chemical Library against Non-immobilized Proteins Using a ‘ligate-Cross-Link-Purify’ Strategy. Bioconjug. Chem 28, 2293–2301 (2017). [DOI] [PubMed] [Google Scholar]
- 155.Sannino A et al. Critical Evaluation of Photo-cross-linking Parameters for the Implementation of Efficient DNA-Encoded Chemical Library Selections. ACS Comb. Sci 22, 204–212 (2020). [DOI] [PubMed] [Google Scholar]
- 156.Denton KE & Krusemark CJ Crosslinking of DNA-linked ligands to target proteins for enrichment from DNA-encoded libraries. Medchemcomm 7, 2020–2027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ma H et al. PAC-FragmentDEL - photoactivated covalent capture of DNA-encoded fragments for hit discovery. RSC Med. Chem 13, 1341–1349 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhou Y et al. DNA-Encoded Dynamic Chemical Library and Its Applications in Ligand Discovery. J. Am. Chem. Soc 140, 15859–15867 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Reddavide FV, Lin W, Lehnert S & Zhang Y DNA-Encoded Dynamic Combinatorial Chemical Libraries. Angew. Chemie - Int. Ed 54, 7924–7928 (2015). [DOI] [PubMed] [Google Scholar]
- 160.Deng Y et al. Selection of DNA-Encoded Dynamic Chemical Libraries for Direct Inhibitor Discovery. Angew. Chemie - Int. Ed 59, 14965–14972 (2020). [DOI] [PubMed] [Google Scholar]
- 161.Oehler S et al. Affinity Selections of DNA-Encoded Chemical Libraries on Carbonic Anhydrase IX-Expressing Tumor Cells Reveal a Dependence on Ligand Valence. Chem. - A Eur. J 27, 8985–8993 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.