

Abstract
The ability to label proteins by fusion with genetically encoded fluorescent proteins is a powerful tool for understanding dynamic biological processes. However, current approaches for expressing fluorescent protein fusions possess drawbacks, especially at the whole organism level. Expression by transgenesis risks potential overexpression artifacts while fluorescent protein insertion at endogenous loci is technically difficult and, more importantly, does not allow for tissue-specific study of broadly expressed proteins. To overcome these limitations, we have adopted the split fluorescent protein system mNeonGreen21–10/11 (split-mNG2) to achieve tissue-specific and endogenous protein labeling in zebrafish. In our approach, mNG21–10 is expressed under a tissue-specific promoter using standard transgenesis while mNG211 is inserted into protein-coding genes of interest using CRISPR/Cas-directed gene editing. Each mNG2 fragment on its own is not fluorescent, but when co-expressed the fragments self-assemble into a fluorescent complex. Here, we report successful use of split-mNG2 to achieve differential labeling of the cytoskeleton genes tubb4b and krt8 in various tissues. We also demonstrate that by anchoring the mNG21–10 component to specific cellular compartments, the split-mNG2 system can be used to manipulate protein function. Our approach should be broadly useful for a wide range of applications.
Keywords: Zebrafish, CRISPR-Cas, protein tagging, split fluorescent protein
Introduction
Protein labeling by fusion with genetically encoded fluorescent proteins has been a powerful tool for studying biological processes, allowing scientists to visualize and track proteins of interest in live cells. Fluorescent protein labeling has been especially useful for investigating the dynamic processes that occur during embryonic development. However, traditional methods for generating and expressing fluorescent fusion proteins, especially in multicellular organisms, have several drawbacks. In zebrafish and other model organisms, expression of fusion proteins can be achieved by injection of in vitro transcribed mRNA (Rosen et al., 2009), which is ubiquitous, or by transgenesis, which utilizes gene regulatory elements to drive spatiotemporal restricted expression (Clark et al., 2011). These approaches, however, run the risk of producing overexpression artifacts, in which proteins may not function or localize correctly when expressed at higher than wild-type levels (Simiczyjew et al., 2014). An alternative approach is to knock in fluorescent protein coding sequences into the genetic locus of that protein of interest (Albadri et al., 2017; Auer and Del Bene, 2014; Kimura et al., 2014). Although this approach has the advantage of preserving endogenous regulation of that protein’s expression, many proteins are expressed broadly; issues arise when there is a need to study a broadly expressed protein in a specific tissue. Thus, there is a need for tissue-specific and endogenous tagging of proteins.
Split fluorescent proteins (split-FPs) are self-complementing protein fragments that only fluoresce when bound together. Split-FPs have been successfully used to visualize and quantify cell-cell interactions (Feinberg et al., 2008), signaling pathway activation (Harvey and Smith, 2009), and subcellular protein localization (Cho et al., 2022). One commonly used split-FP system is based on the yellow-green fluorescent protein monomeric NeonGreen2 (mNG2) in which strands 1–10 of the mNG2 beta-barrel (mNG21–10) and strand 11 (mNG211) are expressed as independent protein fragments (Feng et al., 2017). On their own, the fragments are nonfluorescent, but when present in the same cell, they will self-assemble into a bimolecular complex with similar spectral properties to the intact, full-length fluorescent protein. The split-mNG2 system has been demonstrated to function in several different organisms and cell types (Cho et al., 2022; Kesavan et al., 2021; O’Hagan et al., 2021). Here, we adapt it for use in zebrafish to achieve tissue-specific and endogenous protein labeling. In our approach, mNG21–10 is expressed under the control of a tissue-specific promoter using standard zebrafish transgenesis techniques. Because the mNG211 fragment is only 16 amino acids long, its short sequence can be easily inserted into endogenous genetic loci by CRISPR/Cas-directed gene editing. In this way, the mNG211-tagged protein will continue to be expressed at endogenous levels, but fluorescent signal will only be detected in tissues in which mNG21–10 is co-expressed (Fig. 1A).
Figure 1. Split fluorescent protein fragments are functional in zebrafish embryos.
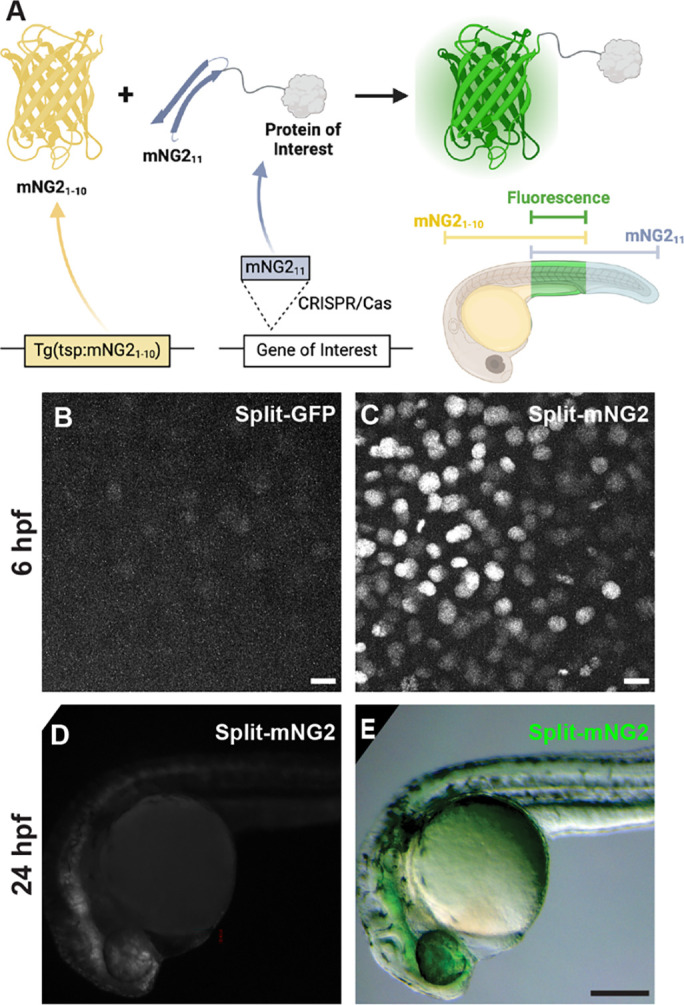
A. Schematic illustrating our protein labeling strategy using a split fluorescent protein. Transgenic (Tg) mNG21–10 is expressed under the control of a tissue-specific promoter (tsp) while mNG211 is inserted into protein-coding genes by CRISPR/Cas-directed gene editing. Fluorescence (green) is only generated in tissues co-expressing mNG21–10 and the mNG211-tagged protein of interest. B–E. Embryos were injected with GFP1–10 and GFP11-H2B (split-GFP, B) or mNG21–10 and mNG211-H2B (split-mNG2, C–E) mRNAs then imaged at 6 hours post-fertilization (hpf) on a confocal microscope (B, C) or at 24 hpf on a fluorescence stereomicroscope (D, E). Scale bars in B and C, 50 μm. Scale bar in E, 200 μm.
Results
mNG21–10 and mNG211 can assemble fluorescent complexes in zebrafish embryos
To assess the viability of our protein labeling strategy, we first determined if split-FP fragments could self-assemble in zebrafish embryos to form functional fluorescent complexes (Fig. 1B–D). We tested two different FP1–10/11-type systems, split-GFP (Kamiyama et al., 2016) and split-mNG2 (Feng et al., 2017). We injected mRNAs encoding GFP1–10 and GFP11-H2B (GFP11 fused to histone 2B) or mNG21–10 and mNG211-H2B (mNG211 fused to histone 2B) into zebrafish embryos. For both systems, expression of the FP1–10 or FP11 fragments alone did not produce fluorescence. However, when both fragments were co-expressed, we could detect nuclear-localized fluorescent signals by 6 hours post-fertilization (hpf) using confocal fluorescence microscopy (Fig. 1B–C). We observed that embryos expressing split-mNG2 fragments (Fig. 1C) were brighter than those expressing the split-GFP fragments (Fig. 1B), consistent with previous studies showing that split-mNG2 exhibits improved signal-to-background ratios compared to split-GFP (Feng et al., 2017). This difference in brightness persisted over time so that by 24 hpf split-mNG2 fluorescence was bright enough to be detected by a fluorescence stereomicroscope (Fig. 1D–E). Based on these observations, we only used the split-mNG2 system for further experiments.
Generating mNG21–10 transgenic lines
We next determined whether transgene-driven expression of mNG21–10 could be used to spatially restrict fluorescence (Fig. 2). We generated multiple transgenic zebrafish lines that express mNG21–10 under control of various promoters representing a broad range of tissue types including fezf2 (brain and eye) (Berberoglu et al., 2009), myl7 (myocardium) (Huang et al., 2003), and ubb (ubiquitous expression) (Mosimann et al., 2011). To verify that these transgenic lines were functional, we injected transgenic embryos with mNG211-H2B mRNA, which would be distributed ubiquitously, and qualitatively assessed fluorescence patterns at 24 or 48 hpf. We found that uninjected mNG21–10 transgenic embryos exhibited no detectable fluorescence (Supplemental Fig. 1). In contrast, transgenic embryos injected with mNG211-H2B mRNA exhibited fluorescence in spatially restricted patterns consistent with the promoter used to drive mNG21–10 expression (Fig. 2A–F). Moreover, these fluorescence patterns were qualitatively similar to those in embryos expressing full-length, intact GFP under control of the same tissue-specific promoters (Fig. 2G–L).
Figure 2. Split fluorescent protein labeling can be spatially restricted by transgenic expression of mNG21–10.

A–F. Transgenic embryos expressing mNG21–10 under control of the fezf2 (A–B), myl7 (C–D), or ubb (E–F) promoters and injected with mNG211-H2B mRNA. D–F. Transgenic embryos expressing GFP under control of the fez1 (G–H), myl7 (I–J), or ubb (K–L) promoters. Images were acquired at 24 hours post-fertilization. Scale bars in B, F, H, and L, 200 μm. Scale bars in D, J, 50 μm.
mNG211 tagging by CRISPR/Cas-directed gene editing
We next determined whether proteins of interest could be tagged with mNG211 at their endogenous genetic loci by CRISPR/Cas-guided homology directed repair (Fig. 3). Previous reports have suggested that split-FP tagging works best for highly expressed genes (Goudeau et al., 2021; O’Hagan et al., 2021). Therefore, we targeted three genes that are highly expressed with relatively broad patterns — tubb4b, which codes for Beta-tubulin 4b; krt8, which codes for Keratin 8; and h2az2b, which codes for histone H2A. We designed guide RNAs (gRNAs) targeting each gene just downstream of the start (tubb4b) or upstreak of the stop (krt8, h2aza2b) codon to generate, respectively, N- or C-terminal mNG211 tags. We injected gRNAs together with Cas9 mRNA and a repair template that contained the coding sequence for mNG211 and a short linker (Fig. 3A); the repair template consisted of double-stranded DNA with single-stranded homology arms of p30 bp at each end (Liang et al., 2017). To verify that the knock-in was successful, we pooled injected embryos and performed insert-specific PCR that amplified the mNG211 insertion but not the unedited wild-type (Fig. 3B).
Figure 3. mNG211 tagging by CRISPR/Cas-directed gene editing.

A. Schematic of CRISPR/Cas-directed mNG211 insertion into target genes. Purple, endogenous exon sequence. Yellow, linker (LK). Green, mNG211. ATG, start codon. Arrows denote primers used in B. B. mNG211 insertion was assessed by PCR. The primers used correspond to the arrows shown in A. C. Amino acid sequences of wild-type, predicted mNG211 fusions, and recovered alleles for Tubb4b and Krt8. Mismatches between the predicted and recovered sequences are highlighted in red. Asterisks, stop codons. D–I. Representative images of mNG211-tubb4b (D-F) and krt8-mNG211 (G–I) embryos injected with mNG21–10 mRNA (D–E, G–H) or uninjected (F, I). Images were acquired at 24 hours post-fertilization. Images in F and I have been overexposed to emphasize lack of fluorescence. Scale bars, 50 μm.
For tubb4b, we estimated the knock-in efficiency using quantitative PCR. To determine mNG211 prevalence, we pooled and extracted DNA from 30 injected F0 embryos at 24 hpf. We amplified mNG211 using insert-specific primers and amplified the untargeted, single-copy gene prox1a for comparison; we obtained a ∆Ct of 5 cycles between the two. As zebrafish are diploid, prox1a is present in two copies per cell, but each mNG211 knock-in likely occurred only in one tubb4b allele per cell. We thus estimated that roughly 1 in every 16 cells in our pooled sample carried the knock-in allele, corresponding to a knock-in efficiency of about 6%, although not necessarily in-frame nor equally distributed among embryos. This knock-in efficiency is on par with other reports of CRISPR-guided knock-in in zebrafish (Auer and Del Bene, 2014; Zhang et al., 2023).
To establish stable, germline-transmitted lines for the mNG211 insertions, we raised injected F0 fish to adulthood and identified several founders representing multiple alleles for each gene. Some alleles contained indel mutations at the insertion junctions or within the insertion itself. For example, both alleles recovered for h2az2b contained mutations within the mNG211 sequence and produced very dim fluorescence (Supplemental Fig. 2). Therefore, we chose to propagate only alleles with precise integration of the mNG211 sequence, resulting in establishment of one line each for tubb4b (tubb4bucm131, referred to here as mNG211-tubb4b) and krt8 (krt8ucm132, referred to here as krt8-mNG211).
To confirm that the mNG211 tag is functional and does not alter endogenous expression patterns, we injected embryos with mNG21–10 mRNA and qualitatively assessed fluorescence. For mNG211-tubb4b, we observed strong fluorescence at 24 hpf that was especially prominent in the eye and brain (Fig. 3D) and along the neural tube (Fig. 3E). For krt8-mNG211, fluorescence appeared restricted to the skin epidermis at 24 hpf (Fig. 3G, H). These fluorescence patterns are consistent with the reported expression patterns for both tubb4b (Thisse and Thisse, 2008; Zhuo et al., 2012) and krt8 (Fischer et al., 2014; Thisse and Thisse, 2008). At the subcellular level, we observed that fluorescence for both genes was enriched at the cell periphery and excluded from the nucleus, which would be expected for cytoskeletal filaments. For both genes, we observed no fluorescence in uninjected embryos (Fig. 3F, I)
Combinatorial expression of tissue-specific mNG21–10 and mNG211-tagged proteins
After successfully generating mNG21–10 transgenic lines and mNG211 insertions, we next determined whether these lines could be combined to achieve tissue-specific protein labeling (Fig. 4A). We crossed each of our mNG211-tagged lines — mNG211-tubb4b and krt8-mNG211 — with each of our mNG21–10 transgenic lines — fezf2:mNG21–10, myl7:mNG21–10, and ubb:mNG21-10. For mNG211-tubb4b, crossing to ubb:mNG21–10 produced fluorescence broadly throughout the head (Fig. 4B), similar to mNG21–10 mRNA injection and to the reported expression pattern for tubb4b (Thisse and Thisse, 2008; Zhuo et al., 2012). In contrast, crossing to fezf2:mNG21–10 resulted in fluorescence restricted to the brain and eye (Fig. 4C), consistent with the known expression pattern for fezf2 (Jeong et al., 2006). Finally, crossing to myl7:mNG21–10 resulted in no observable fluorescence (Fig. 4D). This result is consistent with the reported expression pattern for tubb4b, which has not been reported to be expressed in the heart.
Figure 4. Combinatorial expression of tissue-specific mNG21–10 and mNG211-tagged proteins.
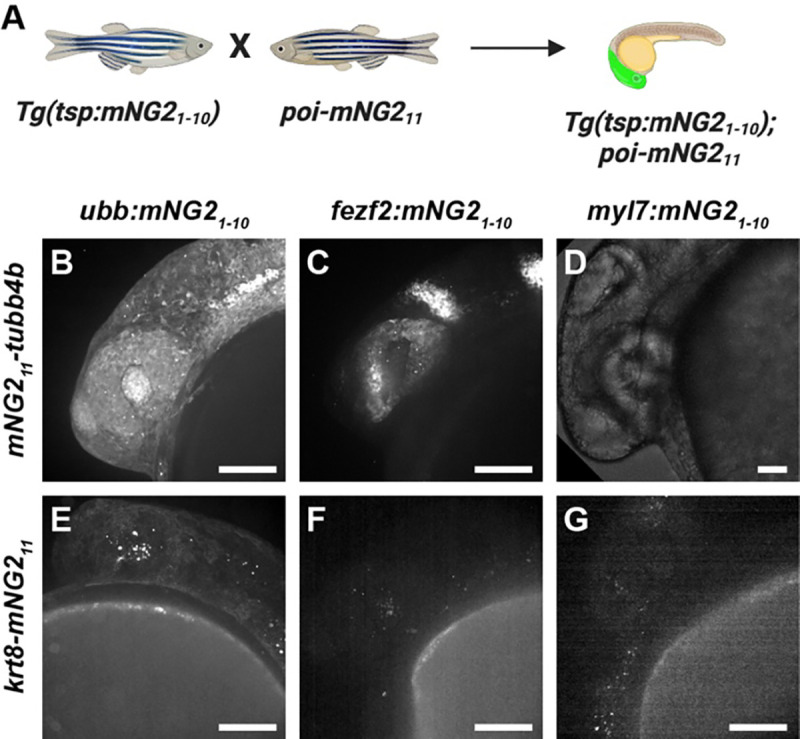
A. Schematic of crossing strategy. Tsp, tissue-specific promoter. poi, protein of interest. B-G. Representative images of embryos resulting from crosses between mNG211-tubb4b (B–D) or krt8-mNG211 (E–G) and ubb:mNG21–10 (B, E), fez2f:mNG2110 (C, F), or myl7:mNG21–10 (D, G). Images were acquired at 24 hours post-fertilization. Image in D has been overexposed to emphasize lack of fluorescence. Scale bars, 100 μm.
The results we obtained for krt8-mNG211 similarly demonstrated retention of endogenous expression patterns. Crossing to ubb:mNG21–10 resulted in fluorescence primarily in the skin (Fig. 4E), similar to mNG21–10 mRNA injection and to the reported expression pattern for krt8 (Fischer et al., 2014; Thisse and Thisse, 2008). And, crossing to both fezf2:mNG21–10 or myl7:mNG21–10 resulted in no observable fluorescence (Fig. 4F–G), which is expected as krt8 has not been reported to be expressed in either cardiac or neural tissues.
Altogether, our results show that combining transgenic mNG21–10 expression and mNG211 tagging can achieve tissue-specific fluorescent protein labeling that preserves endogenous expression patterns.
Directing protein localization with split-mNG2
Given that split-mNG2 fragments self-assemble, it may be possible to use mNG21–10 as a “bait” to direct mNG211-tagged proteins to specific subcellular locations. To determine the feasibility of this application, we fused mNG21–10 to a localization signal for the outer mitochondrial membrane (mito-mNG21–10) (Bear et al., 2000) (Fig. 5A). We then injected mRNA for mito-mNG21–10 into krt8-mNG211 embryos. Compared to control embryos injected with untagged mNG21–10 (Fig. 5B–D), embryos injected with mito-mNG21–10 exhibited qualitatively different fluorescence localization patterns that co-localized with the mitochondrial dye MitoTracker (Fig. 5E–F). These results suggested that mito-mNG21–10 was indeed directing mNG211-tagged Keratin 8 to the mitochondria. Interestingly, krt8-mNG211 embryos injected with mito-mNG21–10 exhibited a higher frequency of defects consistent with mislocalization of Keratin 8 protein including skin blistering (Fig. 5I) and collapsed fin folds (Fig. 5J). These defects were observed in 17.3% of embryos injected with mito-mNG21–10 (n=248 embryos) compared with 1.1% of uninjected embryos (n=90 embryos). Together, these results demonstrate that by anchoring mNG21–10 to specific cellular compartments, the split-mNG2 system can be used to direct protein localization and perturb protein function.
Figure 5. Directing protein localization with split-mNG2.

A. Schematic illustrating use of the split-mNG2 system to sequester proteins of interest on mitochondria. B–G. Representative images of krt8-mNG211 embryos injected with mNG21–10 (B–D) or mito-mNG21–10 (E–G) mRNA. Images were acquired from the tail fin epidermis at 48 hours post-fertilization (hpf). Mitochondria were labeled with MitoTracker dye. Scale bars, 50 μm. H–J. Representative images of the tails of uninjected control embryos (H) krt8-mNG211 embryos injected with mito-mNG21–10 mRNA (I, J). Images were acquired at 48 hpf. Arrows denote blisters. Scale bars, 200 μm.
Discussion
In this study, we describe using the mNG21–10/11 split fluorescent protein system to achieve tissue-specific fluorescent labeling of endogenous proteins in zebrafish embryos. We further demonstrate that the split-mNG2 system can be used to control protein localization by anchoring the mNG21–10 fragment to specific cellular compartments.
Similar FP1–10/11 systems are now commonly used as endogenous protein labeling tools in cell lines (Cho et al., 2022; Feng et al., 2017; Kamiyama et al., 2016; Leonetti et al., 2016). The popularity of these systems is primarily due to the ease with which the short FP11 sequences can be inserted into gene loci. The general utility of split-FP systems for protein labeling has also been demonstrated in multicellular organisms including zebrafish (Kesavan et al., 2021) and mouse embryos (O’Hagan et al., 2021), but in these studies the corresponding FP1–10 fragment was delivered constitutively. Tissue specificity has been achieved in C. elegans (Goudeau et al., 2021; He et al., 2019; Hefel and Smolikove, 2019; Noma et al., 2017) and Drosophila (Kamiyama et al., 2021) and now in zebrafish (this study).
Control of protein localization is a novel application of the split-mNG2 system. As we demonstrate by tethering Keratin 8 to mitochondria, this approach can be used for manipulating protein function by sequestering proteins away from their normal site of function. But the same approach could also be used to force constitutive localization of a protein to its site of activity or to direct ectopic localization. Reconstituted fluorescent proteins tagged with specific localization sequences retain their fluorescence, thus, successful mislocalization can easily be confirmed. When combined with transgenic expression of mNG21–10, these perturbations can be applied to specific tissues of interest for even broadly expressed proteins.
Previous reports have suggested that not all proteins can be easily labeled with the split-mNG2 system (Cho et al., 2022; Leonetti et al., 2016; O’Hagan et al., 2021). Fluorescent labeling may fail because the target protein does not tolerate mNG211 tagging. Thus, mNG211 fusion proteins should be designed using the same considerations as with any epitope tag. Even if tagging is tolerated, some proteins may not be expressed at high enough levels to produce a detectable fluorescent signal (Leonetti et al., 2016; O’Hagan et al., 2021). This challenge could be overcome by inserting multiple repeats of the mNG211 sequence to increase fluorescent signal as has been demonstrated for split-GFP (He et al., 2019; Hefel and Smolikove, 2019; Kamiyama et al., 2016, 2021; Noma et al., 2017). Additionally, a third generation split-mNG system was recently developed and reported to have improved spectral properties (Zhou et al., 2020), which may extend the use of split-FP labeling to low or moderately expressed proteins.
In this study, we focused on the use of split-mNG2 as a protein labeling tool. However, the ability to control expression of of these protein fragments independently, paired with their ability to self-assemble, could be leveraged for other applications. For example, they could be used as coincidence detectors to monitor cell states or signaling pathway activation. And because fluorescence is only produced when the two fragments bind, they could be used to visualize interactions at multiple length scales, i.e., between proteins, organelles, cells, or adjacent tissues.
In summary, we have demonstrated that the split-mNG2 system can function in zebrafish to endogenously label proteins in a tissue-specific manner, with other potential applications that make it broadly useful to many areas of investigation.
Materials and Methods
Zebrafish strains
Adult Danio rerio zebrafish were maintained under standard laboratory conditions. Zebrafish in an outbred AB, TL, or EKW background were used as wild-type strains. Strains generated in this study are: Tg(fezf2:mNG21–10)ucm120; Tg(myl7:mNG21–10)ucm121; Tg(ubb:mNG21–10)ucm117; krt8ucm132; and tubb4bucm132. This study was performed with the approval of the Institutional Animal Care and Use Committee (IACUC) of the University of California Merced (Protocol #2023–1144).
mRNA expression
All expression plasmids for in vitro mRNA synthesis were generated in a pCS2 backbone. To generate pCS2-GFP1–10, GFP1–10 was PCR amplified from pACUH- GFP1–10 (Bo Huang, University of California San Francisco) and cloned into pCS2 by enzymatic assembly (Gibson et al., 2009). To generate pCS2-mNG21–10, mNG21–10 was PCR amplified from pSFFV-mNG21–10 (Bo Huang, University of California San Francisco) and cloned into pCS2 by enzymatic assembly. To generate pCS2-GFP11-H2B and pCS2-mNG211-H2B, GFP11 (5ʹ-CGTGACCACATGGTCCTTCATGAGTATGTAAATGCTGCTGGGATTACA-3ʹ) and mNG211 (5ʹ-ACCGAGCTCAACTTCAAGGAGTGGCAAAAGGCCTTTACCGATATGATG-3ʹ) were directly synthesized by Integrated DNA Technologies and H2B was PCR amplified from GFP-H2B (Hesselson et al., 2009); fragments were fused and cloned into pCS2 by enzymatic assembly. To generate pCS2-mito-mNG21–10, the outer mitochondrial membrane signal sequence was PCR amplified from pMSCV-FPPPP-mito (Bear et al., 2000) and cloned into pCS2-mNG21–10 by enzymatic assembly. Capped messenger RNA was synthesized using the mMESSAGE mMACHINE kit (Ambion), and 500 pg of each mRNA was injected at the one- or two-cell stage.
Generation of mNG21–10 transgenic lines
All transgene plasmids were generated in a pμTol2 backbone (LaBelle et al., 2021). mNG21–10 and promoter sequences for fezf2 (Berberoglu et al., 2009), myl7 (Huang et al., 2003), or ubb (Mosimann et al., 2011) were PCR amplified then fused and cloned into pμTol2 by enzymatic assembly to generate pμTol2-fez:mNG21–10, pμTol2-myl7:mNG21–10, and pμTol2-ubb:mNG21–10, respectively. The constructs were used to generate Tg(fez:mNG21–10)ucm120; Tg(myl7:mNG21–10)ucm121; Tg(ubb:mNG21–10)ucm117 using standard transgenesis protocols (Clark et al., 2011; Kawakami, 2004).
CRISPR/Cas-directed insertion of mNG211
Guide RNAs (gRNAs) were designed using CRISPRscan (Moreno-Mateos et al., 2015) and synthesized as previously described (Varshney et al., 2016). The double-stranded DNA template for homology directed repair was assembled from two oligomers synthesized by Integrated DNA Technologies. Each oligomer contained the sequence for mNG211, a 10-amino acids-encoding linker sequence (5’- GGAGCTGGTGCAGGCGCTGGAGCCGGTGCC-3’), and a homology arm. Oligomers were hybridized to obtain a double-stranded template with single-stranded, 30 bp-long homology arms at each end (Liang et al., 2017). gRNAs, donor DNA, and Cas9 mRNA were injected at the one-cell stage as previously described (Gagnon et al., 2014).
To verify insertion, we pooled 40 injected embryos at 24 hpf, isolated genomic DNA, and performed PCR using two sets of primer pairs per gene covering the 5’ and 3’ insertion sites. The same primer sets were used for quantitative PCR (qPCR) to estimate knock-in efficiency. Each qPCR reaction contained 2X PerfeCTa® SYBR Green FastMix (Quantabio), five-fold diluted genomic DNA, and 325 nM of each primer. Reactions were carried out on a QuantStudio3 (Applied Biosystems) real-time PCR machine using the following program: initial activation at 95°C for 10 min, followed by 40 cycles of 30 s at 95°C, 30 s at 60°C, and 1 min at 72°C. Once the PCR was completed, a melt curve analysis was performed to determine reaction specificity. The gene prox1a was used as a reference. Primers used in this study (presented 5’–3’):
5’ h2az2b-mNG211 forward: TTGTGTGTTTGTGCGTCCGC
5’ h2az2b-mNG211 reverse: GCCACTCCTTGAAGTTGAGC 3’ h2az2b-mNG211 forward: GCTCAACTTCAAGGAGTGGC
3’ h2az2b-mNG211 reverse: ACGAAGCCCCGAAAGCACAC
5’ mNG211-krt8 forward: ATACAGCGGCGGATACAGCG
5’ mNG211-krt8 reverse: GCCACTCCTTGAAGTTGAGC
3’ mNG211-krt8 forward: GCTCAACTTCAAGGAGTGGC
3’ mNG211-krt8 reverse: AAGGCACGACAAGAGCGGTG
5’ mNG211-tubb4b forward: CACATCTCGAATTACGACCTCA
5’ mNG211-tubb4b reverse: GCCTTTTGCCACTCCTTGAAG
3’ mNG211-tubb4b forward: GCTCAACTTCAAGGAGTGGC
3’ mNG211-tubb4b reverse: AAAACAAGCAAGGATTAGCGTC
prox1a forward: TGTCATTTGCGCTCGCGCTG
prox1a reverse: ACCGCAACCCGAAGACAGTG
To verify germline transmission and establish stable lines, injected F0 embryos were raised to adulthood then outcrossed to wild-type zebrafish. We pooled 40 of the resulting F1 embryos at 24 hpf, isolated genomic DNA, and performed PCR using the same primer sets as above. PCR fragments were cloned in pGEM-T (Promega), and the inserts were sequenced by Sanger sequencing (University of California Berkeley DNA Sequencing Facility). Only clutches containing precise insertion of the mNG211 plus linker sequence were kept for propagation. At adulthood, individual F1 zebrafish were genotyped by fin clipping using the same primer sets as described above. Only animals containing precise insertion of the mNG211 sequence were kept for line propagation.
Microscopy and image processing
Fluorescence and brightfield images were acquired on an Olympus SZ51 stereomicroscope equipped with a DP23 monochrome camera and cellSens software (Evident) or with an Olympus IX83 microscope equipped with a 10x/0.4NA or 30x/1.05 NA objective (Evident), a spinning-disk confocal unit (CSU-W1; Andor), a scientific complementary metal–oxide–semiconductor (sCMOS) camera (Prime 95b; Teledyne Photometrics), and MicroManager software (Edelstein et al., 2014). Dechorionated embryos or larvae were embedded in 1.5% low-melting agarose (ISC BioExpress) containing 0.01% tricaine (Sigma-Aldrich) within glass-bottom Petri dishes (MatTek Corporation). For mitochondria labeling, embryos were incubated in 50 nM MitoTracker Red CMXRos (Invitrogen) for 30 minutes prior to agarose embedding. Standard filter settings were applied and brightfield and fluorescence images were merged after acquisition. Identical exposure settings for fluorescence images were used for all embryos from the same set of experiments. Images were processed in Fiji (Schindelin et al., 2012) as follows: denoised using the Non-local Means Denoise plugin (Buades et al., 2005), maximum intensity Z-projection, brightness and contrast levels adjusted, converted to 8-bit depth, and cropped. Illustrations were created with BioRender (https://biorender.com).
Supplementary Material
Acknowledgements
We thank Bo Huang (University of California San Francisco) for providing the split-GFP and split-mNG2 plasmids and advice, Anne Pipathsouk (University of California San Francisco) and Manuel Leonetti (Chan Zuckerberg Biohub) for helpful discussions, the Department of Animal Research Services (University of California Merced) for excellent fish care, and members of the Woo and Materna labs for helpful comments on the manuscript. This work was supported by grants from the Society for Developmental Biology, the National Institutes of Health (NIH R15HD102829), and the National Science Foundation (NSF-IOS-2238304) to S.W. J.L. was supported by NIH training grant T32GM141862. K.S. received support from the NSF-CREST: Center for Cellular and Biomolecular Machines at the University of California, Merced (NSF-HRD-1547848).
References
- Albadri S, Del Bene F, Revenu C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods San Diego Calif 2017;121–122:77–85. [DOI] [PubMed] [Google Scholar]
- Auer TO, Del Bene F. CRISPR/Cas9 and TALEN-mediated knock-in approaches in zebrafish. Methods San Diego Calif 2014;69:142–50. [DOI] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fässler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell 2000;101:717–28. 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Berberoglu MA, Dong Z, Mueller T, Guo S. fezf2 expression delineates cells with proliferative potential and expressing markers of neural stem cells in the adult zebrafish brain. Gene Expr Patterns 2009;9:411–22. 10.1016/j.gep.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buades A, Coll B, Morel J-M. A Non-Local Algorithm for Image Denoising. 2005 IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recognit. CVPR05, vol. 2, San Diego, CA, USA: IEEE; 2005, p. 60–5. 10.1109/CVPR.2005.38. [DOI] [Google Scholar]
- Cho NH, Cheveralls KC, Brunner A-D, Kim K, Michaelis AC, Raghavan P, et al. OpenCell: Endogenous tagging for the cartography of human cellular organization. Science 2022;375:eabi6983. 10.1126/science.abi6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KJ, Urban MD, Skuster KJ, Ekker SC. Transgenic zebrafish using transposable elements. Methods Cell Biol 2011;104:137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using μManager software. J Biol Methods 2014;1:e10. ht 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, et al. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 2008;57:353–63. [DOI] [PubMed] [Google Scholar]
- Feng S, Sekine S, Pessino V, Li H, Leonetti MD, Huang B. Improved split fluorescent proteins for endogenous protein labeling. 2017;8:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Metzger M, Richardson R, Knyphausen P, Ramezani T, Franzen R, et al. p53 and TAp63 promote keratinocyte proliferation and differentiation in breeding tubercles of the zebrafish. PLoS Genet 2014;10:e1004048. 10.1371/journal.pgen.1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PloS One 2014;9:e98186. 10.1371/journal.pone.0098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009;6:343–5. [DOI] [PubMed] [Google Scholar]
- Goudeau J, Sharp CS, Paw J, Savy L, Leonetti MD, York AG, et al. Split-wrmScarlet and split-sfGFP: tools for faster, easier fluorescent labeling of endogenous proteins in Caenorhabditis elegans. Genetics 2021;217:iyab014. 10.1093/genetics/iyab014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SA, Smith JC. Visualisation and quantification of morphogen gradient formation in the zebrafish. PLoS Biol 2009;7:e1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Cuentas-Condori A, Miller DM. NATF (Native and Tissue-Specific Fluorescence): A Strategy for Bright, Tissue-Specific GFP Labeling of Native Proteins in Caenorhabditis elegans. Genetics 2019;212:387–95. 10.1534/genetics.119.302063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefel A, Smolikove S. Tissue-Specific Split sfGFP System for Streamlined Expression of GFP Tagged Proteins in the Caenorhabditis elegans Germline. G3 Bethesda Md 2019;9:1933–43. 10.1534/g3.119.400162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselson D, Anderson RM, Beinat M, Stainier DYR. Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc Natl Acad Sci U S A 2009;106:14896–901. 10.1073/pnas.0906348106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-J, Tu C-T, Hsiao C-D, Hsieh F-J, Tsai H-J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn Off Publ Am Assoc Anat 2003;228:30–40. [DOI] [PubMed] [Google Scholar]
- Jeong J-Y, Einhorn Z, Mercurio S, Lee S, Lau B, Mione M, et al. Neurogenin1 is a determinant of zebrafish basal forebrain dopaminergic neurons and is regulated by the conserved zinc finger protein Tof/Fezl. Proc Natl Acad Sci U S A 2006;103:5143–8. 10.1073/pnas.0600337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, et al. Versatile protein tagging in cells with split fluorescent protein. Nat Commun 2016;7:11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama R, Banzai K, Liu P, Marar A, Tamura R, Jiang F, et al. Cell-type-specific, multicolor labeling of endogenous proteins with split fluorescent protein tags in Drosophila. Proc Natl Acad Sci U S A 2021;118:e2024690118. 10.1073/pnas.2024690118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol 2004;77:201–22. [DOI] [PubMed] [Google Scholar]
- Kesavan G, Machate A, Brand M. CRISPR/Cas9-Based Split Fluorescent Protein Tagging. Zebrafish 2021;18:369–73. 10.1089/zeb.2021.0031. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Hisano Y, Kawahara A, Higashijima S-I. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci Rep 2014;4:6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBelle J, Ramos-Martinez A, Shen K, Motta-Mena LB, Gardner KH, Materna SC, et al. TAEL 2.0: An Improved Optogenetic Expression System for Zebrafish. Zebrafish 2021;18:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A 2016;113:E3501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Potter J, Kumar S, Ravinder N, Chesnut JD. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol 2017;241:136–46. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK, Khokha MK, et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 2015;12:982–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Dev Camb Engl 2011;138:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Goncharov A, Ellisman MH, Jin Y. Microtubule-dependent ribosome localization in C. elegans neurons. eLife 2017;6:e26376. 10.7554/eLife.26376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan D, Kruger RE, Gu B, Ralston A. Efficient generation of endogenous protein reporters for mouse development. Dev Camb Engl 2021;148:dev197418. 10.1242/dev.197418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen JN, Sweeney MF, Mably JD. Microinjection of zebrafish embryos to analyze gene function. J Vis Exp JoVE 2009:1115. 10.3791/1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simiczyjew A, Mazur AJ, Popow-Woźniak A, Malicka-B\laszkiewicz M, Nowak D. Effect of overexpression of β- and γ-actin isoforms on actin cytoskeleton organization and migration of human colon cancer cells. Histochem Cell Biol 2014;142:307–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 2008;3:59–69. [DOI] [PubMed] [Google Scholar]
- Varshney GK, Carrington B, Pei W, Bishop K, Chen Z, Fan C, et al. A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish. Nat Protoc 2016;11:2357–75. 10.1038/nprot.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Marshall-Phelps K, De Almeida RG. Fast, precise and cloning-free knock-in of reporter sequences in vivo with high efficiency. Development 2023;150:dev201323. 10.1242/dev.201323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Feng S, Brown D, Huang B. Improved yellow-green split fluorescent proteins for protein labeling and signal amplification. PloS One 2020;15:e0242592. 10.1371/journal.pone.0242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo H-Q, Huang L, Huang H-Q, Cai Z. Effects of chronic tramadol exposure on the zebrafish brain: a proteomic study. J Proteomics 2012;75:3351–64. 10.1016/j.jprot.2012.03.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.