

Abstract
Mentha is a commonly used spice worldwide, which possesses medicinal properties and fragrance. These characteristics are conferred, at least partially, by essential oils such as menthol. In this study, a gap-free assembly with a genome size of 414.3 Mb and 31,251 coding genes was obtained for Mentha suaveolens ‘Variegata’. Based on its high heterozygosity (1.5%), two complete haplotypic assemblies were resolved, with genome sizes of 401.9 and 405.7 Mb, respectively. The telomeres and centromeres of each haplotype were almost fully annotated. In addition, we detected a total of 41,135 structural variations. Enrichment analysis demonstrated that genes involved in terpenoid biosynthesis were affected by these structural variations. Analysis of volatile metabolites showed that M. suaveolens mainly produces piperitenone oxide rather than menthol. We identified three genes in the M. suaveolens genome which encode isopiperitenone reductase (ISPR), a key rate-limiting enzyme in menthol biosynthesis. However, the transcription levels of ISPR were low. Given that other terpenoid biosynthesis genes were expressed, M. suaveolens ISPRs may account for the accumulation of piperitenone oxide in this species. The findings of this study may provide a valuable resource for improving the detection rate and accuracy of genetic variants, thereby enhancing our understanding of their impact on gene function and expression. Moreover, our haplotype-resolved gap-free genome assembly offers novel insights into molecular marker-assisted breeding of Mentha.
Introduction
The genus Mentha, commonly known as mint, comprises several strongly scented herb species of the Labiatae family. This herb is cultivated worldwide owing to its distinct aroma and commercial value [1]. This versatile plant contains a diverse array of components, such as essential oils and non-essential compounds, rendering it suitable for a wide range of potential applications [2]. Mentha essential oil has a long history of medicinal use as a digestive aid and analgesic [3]. Research showed that the Mentha essential oil possesses various biological activities (e.g. antioxidant, antibacterial, antiradiation, anticancer, and hypotensive) [4]. Understanding the Mentha genome offers valuable insights into its genetic traits and aids in identifying specific genes responsible for the aforementioned biological activities. The Mentha genome consists of large chromosomes and numerous small chromosomes, resulting in a wide range of chromosome numbers, typically ranging from 24 to 120. Pineapple mint (M. suaveolens) is the cultivated variegated form of apple mint [5]. M. suaveolens is a diploid (2n = 2x = 24) species that grows as a wild plant worldwide, and is widely used in the medical field owing to its numerous therapeutic properties [6].
Decoding complete genome sequence information can assist in detecting gene variation in genomes [7]. The telomere-to-telomere (T2T) genome assembly has been regarded as the ultimate goal of genome assembly [8]. However, due to the considerable diversity in chromosome numbers, obvious polyploidization phenomenon, and high heterozygosity [9], limited progress has been achieved in obtaining a high-quality genome assembly for Mentha. For highly heterozygous species, a haplotype-resolved genome could be combined with a T2T genome assembly to construct a superior reference genome (i.e. haplotype-resolved T2T genome) [10]. The first chromosome-level genome assemblies were generated in Mentha longifolia (M. longifolia) (2n = 2x = 24) [11]. However, a number of low-quality regions or undetectable sequence gaps in M. longifolia genome remained due to the technological limitations at the time. The completion of a haplotype-resolved genome assembly in kiwifruit [12], tea [13], and apple [14] provides a reference for assembling a high-quality M. suaveolens genome and lays the foundation for downstream analysis and precision breeding.
High-quality genome assembly facilitated the identification of genes encoding key enzymes in the secondary metabolite synthesis pathway [15]. Additionally, the haplotype-resolved genome is valuable in revealing the impact of genomic variation on gene function and expression [16], thus further helping us to understand the regulatory mechanisms of secondary metabolite biosynthesis [17]. Numerous recent studies focused on the biosynthesis of monoterpene compounds in Mentha [18]. Essential oils in Mentha are mainly monoterpenes, such as menthol, carvol, and pulegone. Menthol is the most abundant compound in the essential oils of most Mentha plants (e.g. Mentha piperita [19], Mentha arvensis [20], and Mentha canadensis [21]). However, a large number of studies have shown that piperitenone oxide (PO), rather than menthol, is the main volatile chemical component of M. suaveolens [22]. PO and menthol share the steps of isopiperitenone biosynthesis. Subsequently, isopiperitenone is converted by unknown reactions to produce PO rather than being converted by isopiperitenone reductase (ISPR) to produce menthol [11]. ISPR may be the key factor inducing diversity in essential oils.
In this study, we assembled the first haplotype-resolved and gap-free genome of M. suaveolens by integrating data from the MGI sequencing platform, Oxford Nanopore Technologies (ONT) ultralong reads, PacBio High-Fidelity (HiFi) reads, and Hi-C sequencing technologies. We further analyzed the features and mechanisms of structural variations (SVs). We also investigated the expression of terpenoid biosynthesis genes to decipher the genetic aspects that influence the accumulation of volatile terpenoids in Mentha. This study lays a foundation for Mentha genomics and provides a scientific basis for subsequent germplasm innovation and fine variety selection.
Results
Gap-free reference genome assembly and phased diploid genome
The K-mer analysis showed that the estimated genome size of M. suaveolens was 420.48 Mb, accompanied by a high level of heterozygosity (1.50%) and repetitive sequences (53.83%) (Fig. 1a, Fig. S1). We incorporated different sequencing platforms to develop a gap-free genome assembly for M. suaveolens using 29.73 Gb ONT ultralong sequencing data with N50 being 54.51 kb; 31.27 Gb HiFi sequencing data using the PacBio sequel II platform; and 71.2 Gb Hi-C sequencing data (Table S1). The matrix generated from the Hi-C short reading library based on the relationship between contig interaction intensity and position showed that 12 chromosomes were reasonably assembled (Fig. S2). The final genome size was 414.3 Mb with a contig N50 of 32.9 Mb, containing 12 chromosomes with 12 contigs (Fig. S3). The final genome size was in accordance with the estimated genome size according to the K-mer analysis. Based on HiFi reads, we generated two fully resolved haplotypes (termed hapA and hapB) using Hi-C and ONT ultralong reads for assisted haplotyping (Fig. 1b). The assembled haplotypes contained 12 pseudomolecules with a total length of 401.9 and 405.7 Mb, respectively. Moreover, the contig N50 of hapA was 35.1 Mb, while that of hapB was 36.3 Mb (Table 1). The final genome size was similar to that estimated by the K-mer analysis. Finally, according to the characteristic repetitive base sequence of the telomere region (CCCTAAA/TTTAGGG), we identified 45 telomeres and 24 centromeres in 24 pseudochromosomes of the haplotype-resolved gap-free genome of M. suaveolens; three telomeres remain to be determined (Fig. 1c, Table S2). Compared with the 3266 gaps that remained in the M. longifolia genome, the absence of gaps in all chromosomes represented the continuity of the M. suaveolens genome assembly (Fig. S4).
Figure 1.

Overview of the genomic features of M. suaveolens. a Image of M. suaveolens. b Circos plot of M. suaveolens haplotype-resolved gap-free genomic features. I: Chromosome length. II: LTR/Copia coverage. III: LTR/Gypsy elements. IV: Gene density. V: Repeat sequence density. VI: GC content. The innermost part of the plot represents the collinear relationship between the M. suaveolens haplotype-resolved genomes. c Location of the predicted centromere region and identified telomere sequences in M. suaveolens. Abbreviations: GC, guanine-cytosine; LTR, long terminal repeat; Mlon, Mentha longifolia; Msua, Mentha suaveolens.
Table 1.
Summary statistics of M. suaveolens genome assembly
| Feature | M. longifolia V3 [11] | Gap-free genome (M. suaveolens) | HapA (M. suaveolens) | HapB (M. suaveolens) |
|---|---|---|---|---|
| Assembly total length (Mb) | 469.1 | 414.9 | 401.9 | 405.7 |
| Protein-coding genes | 42 107 | 31 251 | 31 688 | 32 011 |
| Contig number | 3586 | 12 | 12 | 12 |
| Contig N50 (bp) | 394 381 | 36 297 221 | 35 112 294 | 36 357 466 |
| Number of gaps in chromosomes | 3266 | 0 | 0 | 0 |
| Number of telomeres | 0 | 22 | 23 | 22 |
| Number of definite centromeres | 0 | 12 | 12 | 12 |
| Genome BUSCOs (%) | 91.5 | 99.2 | 99.1 | 99.1 |
Using the hapA genome as a reference, >99.1% of the core conserved genes (i.e. 1600 of 1614 Benchmarking Universal Single-Copy Orthologs (BUSCOs) were completely assembled (Table 1). We mapped the reads to evaluate genome consistency, resulting in 97.17% mapping rate and 99.96% coverage. The assembly accuracy (estimated as qualification value) was 46.46, indicating the construction of the haplotype-resolved and gap-free genome assembly of M. suaveolens with high integrity and accuracy (Table S3). Through this process, we completed the first high-quality haplotype-resolved gap-free genome assembly for Mentha.
Repetitive sequences accounted for 61.8% of the M. suaveolens genome (total length: 249,298,078 bp), which were mainly composed of tandem repeats and dispersed repeats. Long terminal repeats (LTRs) accounted for 34.08%, including Gypsy (19.7%) and Copia (10.02%), with a total length of 137,470,926 bp (Table S4). We further assessed genome completeness by LTR Assembly Index (LAI) LAI evaluation and, obtained 20.29 LAI value. We predicted 31,688 coding genes in the genome, with an average messenger RNA length of 3957 bp, an average coding sequence length of 1164 bp, and an average number of 5.12 exons per gene (Table S5, S6). Approximately 99% of the core conserved genes (1598 of 1614 BUSCOs) were complete in the M. suaveolens genome annotation (Table S7). Functional annotation of genes was performed by tagging gene functions and associated metabolic pathways involved based on various databases, including predictions of motifs, domains, protein functions, and the metabolic pathways in which they were involved. The results showed that 92.74% (29,388) of the genes were annotated in at least one database (Table S8). Moreover, we identified a total of 162 microRNAs (miRNAs), 534 transfer RNAs (tRNAs), 333 ribosomal RNAs (rRNAs), and 2135 small nuclear RNAs (snRNAs) (Table S9).
Phylogenetic and whole genome duplication analysis
We sought to explore the phylogenetic position of M. suaveolens through comparative genomic analysis. Therefore, we downloaded the protein sequences of 14 species (i.e. Vitis vinifera, Artemisia carvifolia, Catharanthus roseus, Lycopersicon esculentum, Sesamum indicum, Scutellaria baicalensis, Schizonepeta tenuifolia, Thymus quinquecostatus, M. longifolia, Salvia splendens, Salvia miltiorrhiza, Arabidopsis thaliana, and Oryza sativa). The results revealed a total of 56,244 homologous gene families comprising 457,838 genes across all species; of note, 5810 gene families were shared by all species. The M. suaveolens genome had 1334 unique gene families with 1609 genes (Fig. S5; Table S10). The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that a significant proportion of the unique gene families were mainly enriched in the spliceosome and phenylpropanoid biosynthesis (Fig. S6). We further focused on the Labiatae species, which are representative of essential oil-containing plants (i.e. M. suaveolens, M. longifolia, S. tenuifolia, and T. quinquecostatus). There were 1422 unique gene families in M. suaveolens genome, while 8564 genes families were shared by all four species (Fig. S7). The shared gene families were mainly enriched in endocytosis and glycolysis, and participated in monoterpenoid and terpenoid backbone biosynthesis (Fig. S8). The unique gene families of M. suaveolens were mainly enriched in spliceosome and phenylpropanoid biosynthesis (Fig. S9).
By utilizing >100 single-copy orthologous genes of M. suaveolens and the 14 species to construct a phylogenetic tree, we estimated that the divergence time between Labiatae species and Sesamum indicium (a Pedaliaceae species) was 73.2 million years ago (Mya). The estimated divergence time between Mentha and T. quinquecostatus, and between M. suaveolens and M. longifolia was 24 and 9.1 Mya, respectively. (Fig. 2a).
Figure 2.

Phylogenetic analysis and identification of WGD events. a The phylogenetic tree constructed based on 178 single-copy genes.b The numbers of expanded or contracted gene families among 14 species. c WGD signatures in Labiatae (Rate-adjusted mixed Ks distribution for M. suaveolens). Gray: the anchor pair Ks distribution of this M. suaveolens, the vertical dashed lines labeled ‘a’ indicated WGD age estimates based on Ks. d Phylogram of M. suaveolens, Mentha longifolia, Schizonepeta tenuifolia, Thymus quinquecostatus, and Vitis vinifera by Ks rates, with branch lengths. Abbreviations: Acar, Artemisia carvifolia; Atha, Arabidopsis thaliana; Cros, Catharanthus roseus; Ks, synonymous substitutions per synonymous site; Lesc, Lycopersicon esculentum; Mlon, M. longifolia; M. suaveolens, Mentha suaveolens; Osat, Oryza sativa; Sbai, Scutellaria baicalensis; Sind, Sesamum indicum; Smil, Salvia miltiorrhiza; Sspl, Salvia splendens; Sten, Schizonepeta tenuifolia; Tqui, Thymus quinquecostatus; Vvin, V. vinifera; WGD, whole genome duplication.
Furthermore, we identified 383 expanded and 393 contracted gene families in the M. suaveolens genome (Fig. 2b). The KEGG analysis showed that the contracted gene families mainly participated in the mitogen-activated protein kinase (MAPK) signaling pathway and phenylpropanoid biosynthesis, while the expanded gene families were mainly enriched in ubiquitin-mediated proteolysis, phenylalanine metabolism, and terpenoid backbone biosynthesis pathways (Fig. S10). Gene ontology (GO) analysis demonstrated that the contracting gene families were mainly enriched in polysaccharide binding and the apoplast, as well as in the terpenoid biosynthetic process; the expanded gene families were mainly enriched in FDA binding (Fig. S11).
The number of synonymous substitutions per synonymous site (Ks) analysis depicted that M. suaveolens had experienced a whole genome duplication (WGD) event at Ks ~ 1.13, which was shared with Labiatae (Fig. 2c). The Ks plot for the paralogous genes did not show any signs of M. suaveolens-specific WGD. In addition, the dot-plot analysis between M. suaveolens with M. Longifolia, T. quinquecostatus and S. tenuifolia showed a 1:1 pattern, which was further confirmed M. suaveolens had experienced a WGD event (Fig. S12). By constructing a phylogram of M. suaveolens, M. longifolia, S. tenuifolia, and T. quinquecostatus using the Ks rates, we were able to put the estimated values of the same event more closely together. Moreover, correct phylogenetic localization of WGD was achieved in this lineage (Fig. 2d).
SV in M. suaveolens may influence terpenoid biosynthesis
The resolution of two complete haplotypes enabled a genome variation analysis using Synteny and Rearrangement Identifier (SyRI) (Fig. 3a). A total of 3,508,508 variants were identified, including single nucleotide polymorphisms, indels, and SVs (Table S11). A total of 41,135 SVs were discovered, including 6519 deletions (15.8%) (length distribution: 31–40 bp); 6277 insertions (15.3%) ranging from 1 to 5 bp (with 1-bp insertions being the most abundant type); 22,774 duplications (55.4%) and 5352 translocations (13%) (with lengths generally <1000 bp); and 213 inversion (0.5%) (with lengths generally <5000 bp) (Fig. 3a, b). Large SVs, such as duplications and inversions, were observed in the M. suaveolens genome (Fig. S13). Overlapping of SVs and gene annotation revealed that 8874 SVs were located within 2 kb upstream of genes (19.0%), 2653 SVs were located within the coding region of genes (5.7%), 7212 SVs were located within introns (15.4%), 8349 SVs were located within 2 kb downstream of genes (17.9%), and 19 605 SVs were located within intergenic regions (42%).
Figure 3.
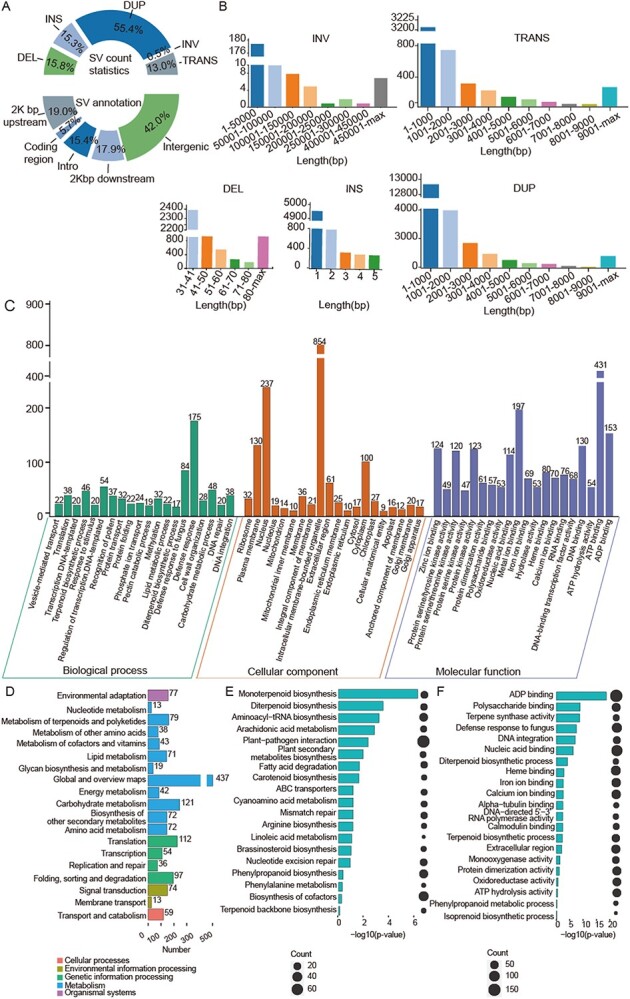
Analysis of SVs and related genes. a Quantity statistics and annotation of SV. b SV length statistics for INV, DEL, TRANS, INS, and DUP. c GO annotation of SV-affected genes. d KEGG annotation statistics of SV-affected genes. e KEGG enrichment analysis of SV-affected genes. f GO enrichment analysis of SV-affected genes. Abbreviations: DEL, deletion; DUP, duplication; GO, Gene Ontology; INS, insertion; INV, inversion; KEGG, Kyoto Encyclopedia of Genes and Genomes; SV, structural variation; TRANS, translocation.
GO and KEGG annotations, followed by enrichment analysis of SV-affected genes, were performed to link functions and metabolic pathways with these genes. The results of the GO analysis showed that a large proportion of genes affected by SVs were involved in the defense response and terpenoid biosynthesis pathway (Fig. 3c). According to the KEGG analysis, numerous genes affected by SVs were involved in the global and overview maps, as well as the metabolism of terpenoids and polyketides (Fig. 3d). Based on the GO and KEGG enrichment analyses, several terpene biosynthesis-related terms were identified among the top enriched groups. For example, GO terms monoterpenoid biosynthesis and diterpenoid biosynthesis, and KEGG term terpene synthase activity ranked first, second, and third respectively. These results implied that SV plays important roles in the synthesis of aromatic compounds and production of volatile terpenoids (Fig. 3e, f).
Piperitenone oxide is the main volatile compound in M. suaveolens
The analysis of volatile metabolites revealed that terpenoids accounted for the largest proportion (26.65%) (Fig. 4a). The downstream product of isopiperitenone, PO, was predominant among all terpenoid compounds (14.77%) (Fig. 4b). The mass spectra of major volatile metabolites are shown in Fig. 4c. Subsequently, samples obtained from different stages of development were analyzed. The results showed that the primary components of M. suaveolens volatile oils were consistent with previous findings. Furthermore, menthol was almost undetectable during the growth and development process (Fig. S14). This discovery highlights the need for further exploration of the metabolic pathways and products of M. suaveolens to gain a more comprehensive understanding of its growth and medicinal value.
Figure 4.

Analysis of volatile metabolites in M. suaveolens.a Classification of all volatile metabolites. b Classification of terpenoid compounds in M. suaveolens.c The position of menthol and piperitenone oxide peaks in the total ion flow diagram (TIC diagram) of the QC sample mass spectrum. d The main biosynthetic pathway of monoterpene biosynthesis in Mentha. The dotted line shows the unidentified enzyme. The expression (log2(FPKM+1)) of each gene is shown as a heatmap in leaves (L); 1, 2, and 3 represent three biological replicates. e Relative proportion of five metabolites in the proposed monoterpene biosynthetic pathway. Abbreviations: FPKM, fragments per kilobase of exon model per million mapped fragments; IPGI, isopulegone isomerase; ISPD, isopiperitenone dehydrogenase; ISPR, isopiperitenone reductase; L3H, limonene-3-hydroxylase; LS, 4-hydroxy-3-methylbut2-en-1-yl diphosphate reductase; M. suaveolens/Msua, Mentha suaveolens; PR, pulegone reductase; Sten, Schizonepeta tenuifolia; TIC, total ion chromatogram; QC, quality control.
Monoterpene biosynthesis pathway in Mentha
Previous research showed that Mentha plants use the methylerythritol phosphate pathway as the main source of C5 raw material for essential oil production and other monoterpene biosynthesis [23, 24]. Geranyl diphosphate produces menthol and pulegone via two pathways catalyzed by a series of enzymes [25] (Fig. 4d). We identified genes that may participate in monoterpene biosynthesis through Basic Local Alignment Search Tool (BLAST) search of orthologous genes with known functions in the M. suaveolens genomes (Table S12). Regarding genes involved in monoterpenoid biosynthesis (Table S13), four limonene synthase (LS) genes, three limonene-3-hydroxylase (L3H) genes, two isopiperitenone dehydrogenase (ISPD) genes, three ISPR genes, three menthol reductase (MR) genes, and five pulegone reductase (PR) genes exhibiting high homology with previously reported genes were identified in the M. suaveolens genomes (Fig. S15, S16).
We analyzed the monoterpene biosynthesis pathway in Mentha by combining transcriptome and metabolome data. After trans-isopiperitenone, the Mentha monoterpene biosynthetic pathway diverges into two branches; one branch leads to the formation of PO, while the other leads to the formation of menthol. The first step of the menthol biosynthetic branch, i.e. from isopiperitenone to isopulegone, is catalyzed by ISPR. Thus, the expression and function of ISPR in M. suaveolens may be the main reason responsible for the competitional formation of PO over menthol. A possible explanation for the lack of menthol biosynthesis in M. suaveolens is the loss of ISPR genes in the M. suaveolens genome. Nevertheless, three ISPR genes were identified, while their expression was consistently limited, e.g. MsISPR2, and MsISPR3. The fragments per kilobase of exon model per million mapped fragments (FPKM) values were zero in all tested tissues (leaf, root, stem) and growth stages (Fig. S17). However, the FPKM values of three MR genes encoding the enzyme catalyzing two reactions in downstream branches, were relatively high. Consequently, we hypothesized that these MRs participate in the PO biosynthetic branch. In S. tenuifolia, pulegone and menthone were identified as the intermediates of menthol biosynthesis, accounting for the largest proportions of monoterpenes (Fig. 4e). Two ISPRs were identified in the S. tenuifolia genome, and had high FPKM values compared with three ISPRs detected in the M. suaveolens genome.
Putative enzyme catalyzing isopiperitenone into PO
At least two enzymes were required to produce PO from isopiperitenone, namely an unknown isopulegone isomerase (IPGI) and a terpene epoxidase. Isopiperitenone is converted to piperonone via a reaction catalyzed by IPGI, and piperitenone subsequently produces PO via a reaction catalyzed by terpene epoxidase. It was reported that Δ5-3-ketosteroid isomerase, a member of the nuclear transport factor 2 (NTF2) family [26], from Pseudomonas putida possesses IPGI activity [27]. In addition, CYP71D is a type of terpene epoxidase that catalyzes the conversion of piperonone to PO [28, 29]. The key enzymes in Mentha remain to be determined. Eight NTF2 and eight CYP71D genes were found in the M. suaveolens genome using homologous sequence alignment and co-expression analysis (Fig. 5a, b). The expression of two established monoterpene biosynthesis genes (i.e. L3H2 and ISPD2) was positively correlated with the accumulation of PO and compounds in its biosynthesis pathway (i.e. limonene, and trans-isopiperitenol). The highest expression and greatest accumulation of these gene and compounds were detected in the leaf, followed by the stem and root. According to the co-expression analysis of eight NTF2 and eight CYP71D with two known genes (L3H2 and ISPD2) and metabolites (PO, limonene, trans-isopiperidol) involved in monoterpene biosynthesis, two NTF2 genes (MsuaChr6AG00168710 and MsuaChr12AG00311870) and five CYP71D genes (MsuaChr5AG00141030, MsuaChr5AG00140150, MsuaChr5AG00141070, MsuaChr2AG00115530, MsuaChr2AG00106980) exhibited strong positive correlations, and were identified as candidates genes (Fig. 5c, d). These genes may participate in the biosynthesis of monoterpenes or related precursors and play a crucial role in converting isopiperitenone into PO.
Figure 5.

Search for candidate piperitenone oxide biosynthesis genes. The phylogenetic relationship of the M. suaveolens hapA genome. NTF2 (a) or CYP71D (c) and their orthologous genes were constructed. The expression (log2(FPKM+1)) of each gene is shown as a heatmap. L: leaf, R: root, S: stem. Correlation of candidate NTF2 (b) or CYP71D (d) genes with the intermediate genes and metabolites of the monoterpene biosynthesis pathway in M. suaveolens. A CYP71D network was constructed with r > 0.8, p < 0.05, and FPKM >1. A NTF network was constructed with r > 0.6, p < 0.05, and FPKM >1. Abbreviations: FPKM, fragments per kilobase of exon model per million mapped fragments.
Discussion
Mentha has been cultivated for >400 years in China, and breeding of Mentha has been highly valued [30]. A high-quality genome plays an important role in explaining the origin, evolution, and spread of plants, effectively protecting germplasm resources [31] and assisting in the exploration of new genes and efficient germplasm innovation [32]. For Mentha, two genome versions of M. longifolia have been released and updated since 2017 [11, 33]. High-quality genomes of other Mentha species have not been released thus far. Regarding other Labiatae species, the high-quality genome of Japanese catnip (S. tenuifolia) (a species closely related to Mentha) was recently released [34]. In addition, high-quality genome assembly was generated in Salvia splendens (scarlet sage) [35], Salvia miltiorrhiza [36], Salvia officinalis (sage) [37], Perilla frutescens [38], and Thymus mongolicu [39]. Thus far, although genomes of multiple Labiatae plants have been published, a T2T genome assembly for the family has not been established thus far. In this study, the first haplotype-resolved gap-free genome of Mentha was generated.
WGD events occurred occasionally in Labiatae. A very recent polyploidization occurred in Perilla frutescens within 10 000 years [38]. Two WGD events and a WGT event were reported in Salvia splendens genome at Ks = ~0.08, ~0.2, and 0.6–0.8, respectively [35]. Two WGD events were reported in the T. quinquecostatus genome at Ks = ~0.07 and ~ 1.22, respectively [39]. Two large-scale gene duplications were reported in the S. tenuifolia at Ks = 0.1–0.2 and ~ 1, respectively [34]. The Ks plot for the paralogues of M. suaveolens revealed that its genome was subjected to a WGD event, which was shared with Labiatae. However, the dot-plot analysis between M. suaveolens with M. Longifolia, T. quinquecostatus, and S. tenuifolia showed a 1:1 pattern (Fig. S18). The peaks observed at Ks = ~0.07 in T. quinquecostatus and at Ks = 0.1–0.2 in S. tenuifolia were identified as suspicious peaks. Subsequently, we extracted the corresponding collinear regions of S. tenuifolia and T. quinquecostatus at Ks = ~0.07 (Fig. S19). The results showed that the corresponding peaks are more likely to be caused by tandem repeats, replication, and insertion of certain genomic regions, rather than a WGD event.
Several reports demonstrated the chemical diversification of Mentha species [9]. Gain of enzyme gene copies usually accounts for the accumulation of specific metabolites, while loss of enzyme genes may result in paucity. For example, Liao et al. found that amorpha-4,11-diene synthase (ADS) gene expansion was significantly related to the levels of artemisinin [40]. Wang et al. revealed that loss of 12 exons of dammarenediol synthase (DDS) in Aralia elata may lead to low levels of dammarane-type saponin in A. elata [41]. The lack of artemisinin production in Artemisia argyi may be attributed to the partial deletion of the ADS gene and loss of function of the ADS homologue [42]. Consequently, we speculated that ISPR gene loss may cause the low menthol production in M. suaveolens. Surprisingly, three ISPR copies were identified, with zero and very low FPKM values for two and one ISPR copies, respectively. In addition to differences in the expression of key enzyme genes, we observed other factors that may be involved in the intra- and inter-species chemical diversity of terpenes in Mentha. Wang et al. discovered that SV had a great impact on gene function (e.g. terpene compound synthase genes) [43]. The total number of genes in M. suaveolens is 31 688; of those, 5493 genes (17.3%) were affected by SVs. Based on the GO and KEGG enrichment analyses, we suspected that SVs impact the synthesis and metabolism of volatile terpenoids in M. suaveolens. For example, SVs affected 11 genes (HDS: 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase, four LS, three ISPR, and three MR) in the monoterpenoid pathway of M. suaveolens (Table S14). These variations can possibly affect the chemical composition and biological activity of plants. These findings provide valuable insights into the genetic basis of terpenoid biosynthesis and highlight the potential regulatory effects of SVs on this important pathway.
In conclusion, this work presents the first haplotype-resolved and gap-free genome of Mentha, thereby providing a solid scientific basis for investigating its origins. Additionally, it provides valuable resources for precise gene annotation and functional studies in the future. Using the chemical diversity of Mentha as the starting point, we explored the impact of SVs on the synthesis of terpenoid compounds, which ultimately affects the mint aroma characteristics. In addition, this study putatively identified two previously unknown key enzymes in the biosynthetic pathway of Mentha monoterpenes. Thus, this research may provide a ‘gold standard’ reference genome of Mentha and facilitate molecular marker-assisted breeding.
Materials and methods
Plant materials and sequencing
Diploid (2n = 2x = 24) M. suaveolens plants were obtained from the Wangcheng District, Changsha City, Hunan Province, China. Fresh leaves of M. suaveolens were collected for ONT ultralong, PacBio HiFi, and Hi-C sequencing. The MGI DNBSEQ-T7 platform was used to generate next-generation sequencing data. The ONT PromethION sequencer was used to generate ONT ultralong reads. A PCR-free SMRTBell library was constructed using high-quality purified long reading DNA for PacBio HiFi sequencing. Hi-C libraries were constructed and sequenced using BGI platform. Stems, leaves, and roots of M. suaveolens were frozen in liquid nitrogen and stored at −80°C for transcriptome and metabolome analyses.
Genome assembly and pseudochromosome construction
The genome size and heterozygosity of M. suaveolens were estimated using K-mer analysis (K = 19) [44]. ONT ultralong sequencing data were assembled using nextDenovo (option: read_cutoff =1 k, blocksize = 1 g, nextgraph_options = −a 1; https://github.com/Nextomics/NextDenovo), Canu, and Flye (option: -I 3 -m 10 000). The PacBio HiFi data were assembled using Hifiasm [45]. Hi-C interaction is used to determine the strength of associations between different contigs in valid data and cluster contigs. Contigs were clustered, ordered, and oriented to chromosomes using ALLHiC [46]. Next, the ordered and oriented contigs were manually sequenced and oriented with the Juicebox software [47]. The haplotype-resolved, chromosome-level genome was obtained, remained six gaps. The corrected ONT and PacBio HiFi reads were used to align the chromosome-level genome using Winnowmap (v1.11, option: k = 15, –MD) [48]. The sequence with the best alignment was used to replace the corresponding sequence in the gap region. Finally, we obtained the haplotype-resolved gap-free genome of M. suaveolens. We use LTR Finder and LTR Retriever calculates LAI to evaluate repeat elements [49].
Identification of telomeres and prediction of centromere regions
The sequence of CCCTAAA was used to identify telomeres by medaka_Consu (option: -m r941_min_high_g360; https://github.com/nanoporetech/medaka). The centromeres of M. suaveolens were predicted as previously reported [10]. We calculated the coverage of Tandem Repeats Finder (TRF; short tandem repeat sequences) and gene coverage in the filamentous region, with 100 k as the window size using bedtools [50]. The continuous high TRF coverage and very low gene coverage of each chromosome could be used to predict the centromere position.
Genome annotation
We predicted the LTR sequences using RepeatModeler (option: BuildDatabase -name mydb; RepeatModeler -database mydb -pa 10) and LTR_FINDER (option: -threads 16 -harvest_out -size 1 000 000 -time 300) software. Redundant LTR sequences were removed using LTR_ retriever (option: - threads 16) [51]. The repeat sequences were masked using RepeatProteinMask (option: - noLowSimple - pvalue 0.0001). The gene structures of M. suaveolens were predicted combined with integrating ab initio gene prediction, homologous proteins, and transcriptome annotation. Functional annotation of genes was identified based on sequence and motif similarity. The tRNA sequences in the genome were identified by tRNAscan SE software [52]. The rRNA and noncoding RNA sequences were predicted based on the rRNA and Rfam database, respectively.
Comparative genomic analysis
Gene family clustering was performed using Orthofinder (option: -M msa) [53] based on the protein sequences of 14 species. Common single-copy genes were used to construct a maximum likelihood phylogenetic tree using Randomized Axelerated Maximum Likelihood (RAxML) software (model: PROTGAMMAWAG) [54]. The divergence times of the selected species were estimated using the MCMCtree subroutine of Phylogenetic Analysis by Maximum Likelihood (PAML; option: nsample: 3000000, burn: 8000000, seqtype: 0, model: 4) [55]. The Computational Analysis of gene Family Evolution (CAFE) was used to predict the contraction and expansion of gene families. MCScanX (option: -a -e 1e−5 -s [5]) [56] was used to analyze the genome collinearity. The ratio of the number of nonsynonymous substitutions per nonsynonymous site/Ks (Ka/Ks) values of the collinear gene pairs were calculated using the yn00 module in PAML.
SV detection
The genome was aligned using MUMmer (version 4.0.0rc) [57] with the haplotype A used as reference. Subsequently, SyRI (version 1.5) [58] was used to detect and identify variations in the haplotype genome.
Key enzymes in monoterpenoid biosynthesis
To identify the genes involved in monoterpenoid biosynthesis, a series of functional protein sequences were retrieved from the National Center for Biotechnology Information (Table S12). The related genes involved in monoterpene biosynthesis were identified in the M. suaveolens genome using Protein BLAST (BLASTP; option: e-value 1e−10). Subsequently, the phylogenetic trees of monoterpenoid biosynthetic genes were constructed and visualized using MEGA-X64 software with 1000 bootstrap replicates.
RNA sequencing and volatile metabolite analysis by gas chromatography–mass spectrometry
Tissues of roots, leaves, stems of plants grown for 30, 120, 240 days were collected and frozen in liquid nitrogen for transcriptome and metabolome analyses. Thereafter, we extracted and isolated total RNA from those tissues. The qualified samples were used to build a library using the MGIEasy RNA Library Prep Kit for BGI®. An Agilent 2100 Bioanalyzer was used to detect the quality of the library, and DNBSEQ was used for RNA sequencing. Low-quality, irrelevant sequences were filtered using fastq [59]. The Spliced Transcripts Alignment to a Reference (STAR) was used to align clean data [60], and StringTie [61] was used to assemble the transcripts. The expression levels of each gene in terms of FPKM were generated using RNA-Sequencing by Expectation–Maximization (RSEM) [62].
A gas chromatograph (8890; Agilent) and a mass spectrometer (7000D; Agilent) were used for gas chromatography–mass spectrometry analysis. DB-5MS (5% phenyl-polydimethylsiloxane) capillary column (Agilent) was used for the separation of VOCs, the line speed was 1.2 mL/min. The injector temperature was kept at 250°C, the detector at 280°C, and the solvent delay was 3.5 minutes. The oven temperature was programmed from 40°C (3.5 minutes), increasing at 10°C/min to 100°C, at 7°C/min to 180°C, at 25°C/min to 280°C, and held for 5 min. The mass spectrometer was operated under the following conditions: electron impact mode, 70 eV, four-stage rod temperature, ion source and mass spectrometry interface set at 150°C, 230°C, and 280°C. Accurate scanning of qualitative and quantitative ions was performed in the ion detection mode (selected ion monitoring), and MassHunter was used for data analysis.
Co-expression network construction
An Hidden Markov Model (HMM) search (NTF2, PF02136 and CYP450, PF00067) combined with BLASTP was performed to identify candidate IPGI and CYP71D genes in the M. suaveolens genome. The protein data of IPGI and CYP71D genes with known function are listed in Tables S15 and S16. The phylogenetic trees of IPGI and CYP71D genes were constructed and visualized using MEGA-X64 software with 1000 bootstrap replicates. The heatmap was generated using expression data to visualize the levels of NTF2 and CYP71D genes in different tissues. Correlation analysis was used to evaluate NTF2 and CYP71D genes exhibiting high correlations with monoterpene biosynthesis genes and metabolites. The co-expression network was visualized using Cytoscape. The cutoff values were set as follows: FPKM values of the tested gene >1, p-value of the correlation test <0.05, and Pearson correlation coefficient (r) >0.8. Of note, the NTF2 protein sequence used in this study was derived from bacteria; hence, the parameter settings were adjusted (i.e. r > 0.6).
Acknowledgements
This work was supported by introduces the talented person scientific research start funds subsidization project of Chengdu University of Traditional Chinese Medicine (030040015, 030040017) and Hubei science and technology planning project (2020BCB038).
Author Contributions
H.Y., C.S., L.L., C.W., and S.C conceived and supervised the study. H.Y., and G.Z. prepared the materials. Z.W., L.Y., H.T., G. C., and X.K. performed genome assembly and annotation. T.H., H.Z., X.T., and Z.L. analyzed the data. X.T., H.Y., Y.W., and Y.Z. drew the figures. H.Y., S.Z., C.S., L.L., and C.W wrote the manuscript. All authors read and approved the final manuscript.
Data availability statement
The genome data of M. suaveolens were uploaded in 1 K Medicinal Plant Genome Database (http://www.herbgenome.com) [63]. RNA-seq data of different tissues were available in NCBI under Biological Project ID (PRJNA938973, https://dataview.ncbi.nlm.nih.gov/object/PRJNA938973?reviewer=mp56u3u4kcflnb7g3q0gum9qmg).
Conflict of interests
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Hanting Yang, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Pharmacy College, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Can Wang, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Guanru Zhou, Hubei University of Chinese Medicine, Wuhan 430065, China.
Yuxuan Zhang, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Pharmacy College, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Tianxing He, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Pharmacy College, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Lulu Yang, Wuhan Benagen Technology Co., Ltd, Wuhan 430000, China.
Ya Wu, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Pharmacy College, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Zhengnan Wang, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China.
Xin Tang, Chongqing Academy of Chinese Materia Medica, Chongqing College of Traditional Chinese Medicine, Chongqing, China.
Gang Chen, Wuhan Benagen Technology Co., Ltd, Wuhan 430000, China.
Zhaoyu Liu, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Huanyu Tang, Wuhan Benagen Technology Co., Ltd, Wuhan 430000, China.
Hanlin Zhou, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Xumei Kang, Wuhan Benagen Technology Co., Ltd, Wuhan 430000, China.
Sanyin Zhang, Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Liang Leng, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Shilin Chen, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Hubei University of Chinese Medicine, Wuhan 430065, China.
Chi Song, Institute of Herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China; Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
References
- 1. Anwar F, Abbas A, Mehmood T. et al. Mentha: a genus rich in vital nutra-pharmaceuticals—a review. PTR. 2019;33:2548–70 [DOI] [PubMed] [Google Scholar]
- 2. Zhao H, Ren S, Yang H. et al. Peppermint essential oil: its phytochemistry, biological activity, pharmacological effect and application. Biomed Pharmacother. 2022;154:113559 [DOI] [PubMed] [Google Scholar]
- 3. Baek JP, Park KW, Craker LE. et al. Changes in growth and quality of three mint cultivars at different harvesting periods. Hortic Environ Biotechnol. 2016;57:207–12 [Google Scholar]
- 4. Oumzil H, Ghoulami S, Rhajaoui M. et al. Antibacterial and antifungal activity of essential oils of Mentha suaveolens. Phytother Res. 2002;16:727–31 [DOI] [PubMed] [Google Scholar]
- 5. Lawrence BM. Mint: The Genus Mentha[M]. CRC press; 2006: [Google Scholar]
- 6. Kasrati A, Jamali CA, Bekkouche K. et al. Essential oil composition and antimicrobial activity of wild and cultivated mint timija (Mentha suaveolens subsp. timija (Briq.) Harley), an endemic and threatened medicinal species in Morocco. Nat Prod Res. 2013;27:1119–22 [DOI] [PubMed] [Google Scholar]
- 7. Zhang L, Liang J, Chen H. et al. A near-complete genome assembly of Brassica rapa provides new insights into the evolution of centromeres. Plant Biotechnol J. 2023;21:1022–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nurk S, Koren S, Rhie A. et al. The complete sequence of a human genome. Science. 2022;376:44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tucker AO III. Genetics and breeding of the genus Mentha: a model for other polyploid species with secondary constituents. Journal of Medicinally Active Plants. 2012;1:19–29 [Google Scholar]
- 10. Guk JY, Jang MJ, Choi JW. et al. De novo phasing resolves haplotype sequences in complex plant genomes. Plant Biotechnol J. 2022;20:1031–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vining KJ, Pandelova I, Lange I. et al. Chromosome-level genome assembly of Mentha longifolia L. reveals gene organization underlying disease resistance and essential oil traits. G three. 2022;12:jkac112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han X, Zhang Q, Ma N. et al. Two haplotype-resolved, gap-free genome assemblies for Actinidia latifolia and Actinidia chinensis shed light on the regulatory mechanisms of vitamin C and sucrose metabolism in kiwifruit. Mol Plant. 2023;16:452–70 [DOI] [PubMed] [Google Scholar]
- 13. Zhang X, Chen S, Shi L. et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat Genet. 2021;53:1250–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun X, Jiao C, Schwaninger H. et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat Genet. 2020;52:1423–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi X, Cao S, Wang X. et al. The complete reference genome for grapevine (Vitis vinifera L.) genetics and breeding. Hortic Res. 2023;10:uhad061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ebert P, Audano PA, Zhu Q. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372:eabf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu X, Simpson SA, Youngblood RC. et al. Two haplotype-resolved genomes reveal important flower traits in bigleaf hydrangea (hydrangea macrophylla) and insights into Asterid evolution. Hortic Res. 2023;10:uhad217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ribeiro B, Shapira P. Anticipating governance challenges in synthetic biology: insights from biosynthetic menthol. Technol Forecast Soc Change. 2019;139:311–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kennedy D, Okello E, Chazot P. et al. Volatile terpenes and brain function: investigation of the cognitive and mood effects of mentha × piperita l. essential oil with in vitro properties relevant to central nervous system function. Nutrients. 2018;10:1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Makkar MK, Sharma S, Kaur H. Evaluation of Mentha arvensis essential oil and its major constituents for fungitoxicity. J Food Sci Technol. 2018;55:3840–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamatou GPP, Vermaak I, Viljoen AM. et al. Menthol: a simple monoterpene with remarkable biological properties. Phytochemistry. 2013;96:15–25 [DOI] [PubMed] [Google Scholar]
- 22. Moreno L, Bello R, Primo-Yúfera E. et al. Pharmacological properties of the methanol extract from Mentha suaveolens Ehrh. Phytothérapie. 2002;16:10–3 [DOI] [PubMed] [Google Scholar]
- 23. Chen Q, Fan D, Wang G. Heteromeric geranyl(geranyl) diphosphate synthase is involved in monoterpene biosynthesis in Arabidopsis flowers. Mol Plant. 2015;8:1434–7 [DOI] [PubMed] [Google Scholar]
- 24. Tomlinson ML, Zhao M, Barclay EJ. et al. Diterpenoids from Scutellaria barbata induce tumour-selective cytotoxicity by taking the brakes off apoptosis. Med Plant Biol. 2022;1:1–16 [Google Scholar]
- 25. Shou C, Zheng YC, Zhan JR. et al. Removing the obstacle to (−)-menthol biosynthesis by building a microbial cell factory of (+)-cis-isopulegone from (−)-limonene. ChemSusChem. 2022;15:e202101741 [DOI] [PubMed] [Google Scholar]
- 26. Lu S, Wang J, Chitsaz F. et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Currin A, Dunstan MS, Johannissen LO. et al. Engineering the “missing link” in biosynthetic (−)-menthol production: bacterial isopulegone isomerase. ACS Catal. 2018;8:2012–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lange BM, Srividya N. Enzymology of monoterpene functionalization in glandular trichomes. J Exp Bot. 2019;70:1095–108 [DOI] [PubMed] [Google Scholar]
- 29. Wüst M, Little DB, Schalk M. et al. Hydroxylation of limonene enantiomers and analogs by recombinant (−)-limonene 3- and 6-hydroxylases from mint (Mentha) species: evidence for catalysis within sterically constrained active sites. Arch Biochem Biophys. 2001;387:125–36 [DOI] [PubMed] [Google Scholar]
- 30. Božović M, Pirolli A, Ragno R. Mentha suaveolens Ehrh. (Lamiaceae) essential oil and its main constituent piperitenone oxide: biological activities and chemistry. Molecules. 2015;20:8605–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu J, Guo S, Yin X. et al. Genomic, transcriptomic, and epigenomic analysis of a medicinal snake, Bungarus multicinctus, to provides insights into the origin of Elapidae neurotoxins. Acta Pharm Sin B. 2023;13:2234–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen S, Li Z, Zhang S. et al. Emerging biotechnology applications in natural product and synthetic pharmaceutical analyses. Acta Pharm Sin B. 2022;12:4075–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vining KJ, Johnson SR, Ahkami A. et al. Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement. Mol Plant. 2017;10:323–39 [DOI] [PubMed] [Google Scholar]
- 34. Liu C, Smit SJ, Dang J. et al. A chromosome-level genome assembly reveals that a bipartite gene cluster formed via an inverted duplication controls monoterpenoid biosynthesis in Schizonepeta tenuifolia. Mol Plant. 2023;16:533–48 [DOI] [PubMed] [Google Scholar]
- 35. Dong AX, Xin HB, Li ZJ. et al. High-quality assembly of the reference genome for scarlet sage, Salvia splendens, an economically important ornamental plant. Gigascience. 2018;7:giy068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song Z, Lin C, Xing P. et al. A high-quality reference genome sequence of salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome. 2020;13:e20041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li CY, Yang L, Liu Y. et al. The sage genome provides insight into the evolutionary dynamics of diterpene biosynthesis gene cluster in plants. Cell Rep. 2022;40:111236 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y, Shen Q, Leng L. et al. Incipient diploidization of the medicinal plant Perilla within 10,000 years. Nat Commun. 2021;12:5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun M, Zhang Y, Zhu L. et al. Chromosome-level assembly and analysis of the thymus genome provide insights into glandular secretory trichome formation and monoterpenoid biosynthesis in thyme. Plant Commun. 2022;3:100413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao B, Shen X, Xiang L. et al. Allele-aware chromosome-level genome assembly of Artemisia annua reveals the correlation between ADS expansion and artemisinin yield. Mol Plant. 2022;15:1310–28 [DOI] [PubMed] [Google Scholar]
- 41. Wang Y, Zhang H, Ri HC. et al. Deletion and tandem duplications of biosynthetic genes drive the diversity of triterpenoids in Aralia elata. Nat Commun. 2022;13:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen H, Guo M, Dong S. et al. A chromosome-scale genome assembly of Artemisia argyi reveals unbiased subgenome evolution and key contributions of gene duplication to volatile terpenoid diversity. Plant Commun. 2023;4:100516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang P, Yu J, Jin S. et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Hortic Res. 2021;8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k -mers. Bioinformatics. 2011;27:764–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cheng H, Concepcion GT, Feng X. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18:170–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, Zhang S, Zhao Q. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on hi-C data. Nat Plants. 2019;5:833–45 [DOI] [PubMed] [Google Scholar]
- 47. Durand NC, Robinson JT, Shamim MS. et al. Juicebox provides a visualization system for hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jain C, Rhie A, Zhang H. et al. Weighted minimizer sampling improves long read mapping. Bioinformatics. 2020;36:i111–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ou S, Chen J, Jiang N. Assessing genome assembly quality using the LTR assembly index (LAI). Nucleic Acids Res. 2018;46:e126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quinlan AR. BEDTools: the swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11.12.1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Flynn JM, Hubley R, Goubert C. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020;117:9451–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan PP, Lin BY, Mak AJ. et al. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91 [DOI] [PubMed] [Google Scholar]
- 56. Wang Y, Tang H, DeBarry JD. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marçais G, Delcher AL, Phillippy AM. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018;14:e1005944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goel M, Sun H, Jiao WB. et al. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 2019;20:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen S, Zhou Y, Chen Y. et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dobin A, Davis CA, Schlesinger F. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pertea M, Pertea GM, Antonescu CM. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Su X, Yang L, Wang D. et al. 1 K medicinal plant genome database: an integrated database combining genomes and metabolites of medicinal plants. Hortic Res. 2022;9:uhac075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome data of M. suaveolens were uploaded in 1 K Medicinal Plant Genome Database (http://www.herbgenome.com) [63]. RNA-seq data of different tissues were available in NCBI under Biological Project ID (PRJNA938973, https://dataview.ncbi.nlm.nih.gov/object/PRJNA938973?reviewer=mp56u3u4kcflnb7g3q0gum9qmg).