

Abstract
Restlessness is a core symptom underlying restless legs syndrome (RLS), neuroleptic-induced akathisia, and opioid withdrawal. These three conditions also share other clinical components suggesting some overlap in their pathophysiology. Recent prospective studies demonstrate the frequent incidence of RLS-like symptoms during opioid withdrawal and supervised prescription opioid tapering. Based on the therapeutic role of µ-opioid receptor (MOR) agonists in the three clinical conditions and recent preclinical experimental data in rodents, we provide a coherent and unifying neurobiological basis for the restlessness observed in these three clinical syndromes and propose a heuristic hypothesis of a key role of the specific striatal neurons that express MORs in akathisia/restlessness.
Keywords: restless legs syndrome, akathisia, neuroleptics, opioid withdrawal
Graphical Abstract
Graphical Abstract.
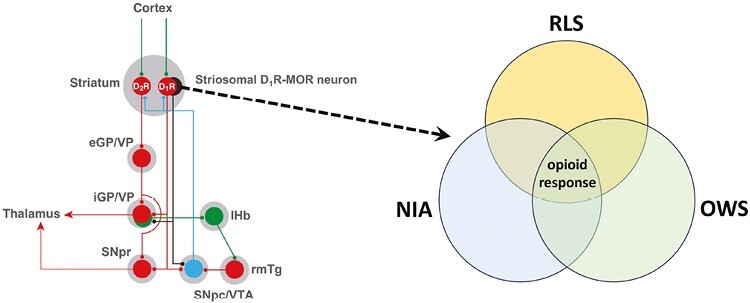
Statement of Significance.
We analyze the significant overlap between the symptomatology and pharmacology of the restlessness of restless legs syndrome, neuroleptic-induced akathisia, and opioid withdrawal. Their significant common dependence on opioid mechanisms allowed us to establish a unifying neurobiological hypothesis and postulate a key role of the specific striatal neurons that express µ-opioid receptors in the restlessness associated with the three clinical conditions.
Introduction
Restless legs syndrome (RLS) is a common neurological disorder affecting between 2.5% and 15% of the general population [1]. Four key clinical symptoms have been described as necessary characteristics for RLS diagnosis. These include an uncontrollable need or urge to move the legs, often associated with an uncomfortable or unpleasant sensation in the legs (essential feature 1), that is induced by rest or decreased arousal (essential feature 2) in a circadian-dependent manner (decreased symptomatology in the morning, and increased symptoms in the evening and first half of the sleep cycle; essential feature 3), and that is substantially relieved by movement, standing, walking, or an increase in attention or arousal (essential feature 4) [2]. “Idiopathic” RLS (henceforth denoted as RLS) includes the four basic clinical criteria but also excludes confounding conditions: leg cramps, positional discomfort, local leg injury, arthritis, leg edema, venous stasis, peripheral neuropathy, radiculopathy, habitual foot tapping, myalgia, anxiety, and drug-induced akathisia [2]. The first known recorded medical description of RLS appears to have been by Sir Thomas Willis in 1672 [3]. Karl Ekbom, in 1944 [4], is credited with defining the four basic criteria of RLS and, in later publications, further elaborating on broader clinical features [5, 6].
The term akathisia (from Greek, “inability to sit”) was coined by Ladislav Haskovec in 1901 [7] to describe the symptoms in two patients who were unable to sit and who compulsively needed to move to relieve a sensation of “jumping.” Although the description of the two patients by Haskovec could today potentially fit the diagnosis of RLS, the term “akathisia” has become synonymous with neuroleptic-induced akathisia (NIA), which was recognized in the early 1960s to be one of the most common and distressing side effects of neuroleptic medication (for a historical overview, see [8]). There are clinical similarities between the restlessness observed in an RLS patient and that observed in those with NIA (described below), and several cases of RLS have been diagnosed as secondary to neuroleptic treatment, with symptoms subsiding upon drug withdrawal (reviewed in [9]).
“Restlessness” is also a common symptom of opioid withdrawal and is an essential domain in a variety of clinical opioid-withdrawal scales (CINA, COWS, OOWS, and SOWS) [10–12]. Several recent case reports and clinical studies have described this opioid-withdrawal “restlessness” as “RLS symptoms,” as the patients fulfilled the four essential diagnostic criteria for RLS during their opioid withdrawal or taper. In a retrospective analysis of 120 patients with opioid use disorder (OUD), Scherbaum et al. found 15 patients who spontaneously reported transient symptoms of RLS during detoxification [13]. This prevalence is most likely an underestimate; however, a systematic interview of all patients had not been undertaken. In a prospective study of 124 patients with OUD during detoxification withdrawal, 50% fulfilled the four essential clinical criteria of RLS [14]. Similar results were later reported in another study with 19 patients with OUD under detoxification withdrawal [15]. A more recent prospective study of 97 patients with chronic pain who underwent supervised prescription opioid tapering found that 35% of the patients developed de novo symptoms of RLS [16].
Here we analyze the brief literature that addresses similarities, as well as differences between RLS, NIA, and opioid-withdrawal restlessness, and their pharmacological approaches, stressing the significant therapeutic effect of µ-opioid receptor (MOR) agonists in the three clinical conditions. We then posit that their similar features stem from common opioid-moderated neuronal mechanisms and propose a heuristic hypothesis of a key role of the specific striatal neurons that express MORs in akathisia/restlessness.
Phenotypic and Pharmacological Comparison of RLS, NIA, and Opioid-Withdrawal Restlessness
In an early description of three cases, Blom and Ekbom concluded that NIA described in the literature, as well as that seen in their patients, may be “extremely like RLS,” but with some differences, such as restlessness that is not constrained to the legs, and less circadian dependence [17] (Figure 1). Even periodic leg movements during sleep (PLMS), an RLS clinical feature (“the motor sign of RLS”) that contribute to sleep disruption in this syndrome [1], can be observed in some patients with NIA, although much less frequently and (when present) with many less movements than in patients with RLS [18].
Figure 1.
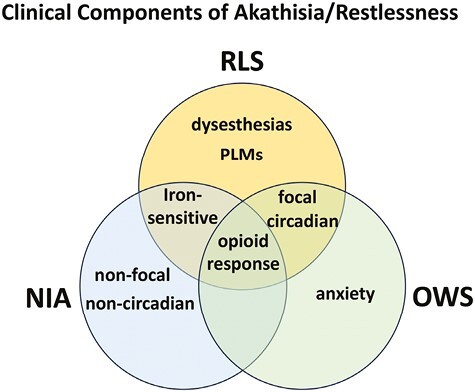
Putative representation of the distinctive and shared clinical components of akathisia/restlessness of RLS, NIA, and the opioid-withdrawal syndrome (OWS). In RLS the restlessness is generally localized in the legs (focal), while in NIA and OWS is usually more generalized (non-focal) and does not show dysesthesias. RLS has a circadian component, shared with OWS, and sensitivity to low peripheral iron levels, shared by NIA. PLMs are predominant in RLS, and an anxiety component is present in OWS. Finally, the three distinctive neuropsychiatric conditions share as a common component the therapeutic response to opioids.
Iron provides another convergence between RLS and NIA (Figure 1): low iron levels or frank iron deficiency is one of the major environmental factors associated with triggering or worsening RLS symptoms [19]. A recent meta-analysis has shown a relationship between low iron and risk of developing NIA [20]. On the other hand, RLS and NIA have distinct pharmacological responsiveness. For instance, propranolol, which has no clinical benefits in RLS, is commonly used as a first-line treatment for NIA. Mirtazapine (a serotonin 5-HT2A receptor antagonist), which may aggravate RLS, has been shown to be of value in treating NIA, but at higher doses, can worsen NIA symptoms [21].
Even though a significant proportion of patients with opioid-withdrawal restlessness fulfill the main diagnostic criteria of RLS [14, 16], to the best of our knowledge, no attempts have been made to characterize in detail the phenotypic similarities and differences between these types of restlessness. Nevertheless, a need to move that occurs when at rest, improves with movement, and is worse at night appears to be present in both conditions, while a component of anxiety is commonly present in the restlessness of opioid withdrawal (Figure 1) [10–12]. Furthermore, there are common effective treatments for opioid-withdrawal restlessness and RLS, which is integral to gaining an understanding of the two conditions’ potential neurobiological overlap. RLS is effectively treated with agonists of dopamine D2-like receptors (which include D2, D3, and D4 receptor subtypes, here abbreviated D2R, D3R, and D4R) and α2δ ligands, as well as MOR agonists [22]. The D2-like receptor agonist pramipexole and the α2δ ligand gabapentin have also been reported to be effective in opioid-withdrawal-induced restlessness [23, 24]. The effectiveness of opioids in treating RLS and brain autopsy studies have led to the proposal that intrinsic deficits in the opioid system underlie RLS [25]. Finally, the opioid codeine has been reported to be effective in reducing NIA [26], altogether supporting shared neuronal mechanisms for NIA, RLS, and opioid-withdrawal restlessness, involving opioid-moderated brain circuits (Figure 1). Although other forms of NIA (e.g. tardive akathisia or akathisia with drug withdrawal) do occur, the clinical entity for which a comparison appears feasible is acute drug-induced akathisia. In fact, a second analysis of the study with codeine suggested that the least responsive patients with tardive akathisia [27], in which symptoms appear upon prolonged treatment with neuroleptics and, different from acute NIA, persist after the neuroleptic treatment is withdrawn.
Aiming at a Common Neuronal Mechanism of Akathisia/Restlessness in RLS, NIA, and Opioid-Withdrawal Restlessness
The striatum is the main input structure of the basal ganglia, which can be subdivided into two major functional compartments: the dorsal striatum (caudate and putamen), innervated by the dopaminergic neurons of the substantia nigra pars compacta (SNpc), and the ventral striatum (nucleus accumbens), innervated by the ventral tegmental area (VTA) [28, 29]. There are two types of striatal GABAergic efferent neurons, which express different subtypes of dopamine receptors and project to different basal ganglia nuclei. Those are the striatopallidal neuron, which expresses D2Rs, and the striatonigral neuron, which expresses D1Rs (Figure 2). In the dorsal striatum, the striatopallidal neuron projects to the external segment of the globus pallidus (eGP; globus pallidus in rodents), and in the ventral striatum, it projects to the ventral pallidum (VP) [28, 29]. The striatonigral neuron of the dorsal striatum projects to the internal segment of the globus pallidus (iGP; entopeduncular nucleus in rodents) and to the SNpc or the substantia nigra pars reticulata, and it also projects to the VP and the VTA in the ventral striatum (Figure 2) [28, 29].
Figure 2.
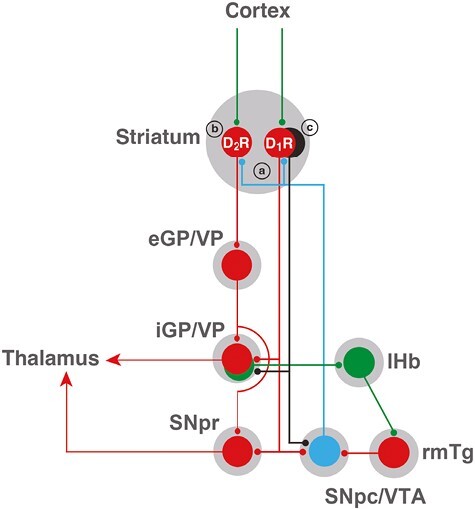
Scheme of cortical-striatal-pallidal-thalamic and cortical-striatal-pallidal-habenular basal ganglia circuits. Glutamatergic, GABAergic, and dopaminergic neurons are represented in green, red, and blue, respectively. D1R and D2R represent two main subtypes of striatal GABAergic neurons, the striatonigral and the striatopallidal neurons, expressing dopamine D1 receptors (D1Rs) and dopamine D2 receptors (D2Rs), respectively. The GABAergic striosomal D1R-MOR neurons are represented in black. It is postulated that an imbalance in the normal dopaminergic receptor activation of the D1R and D2R neurons and the specific increased activation of the striosomal D1R-MOR neurons, leading to inactivation of the pallidal-habenular neurons, mediates the akathisia/restlessness in RLS, NIA, and opioid withdrawal (see text). In NIA, this is dependent on an increased dopamine release (a), secondary to autoreceptor blockade, and a concomitant blockade of postsynaptic D2Rs (b). In RLS, there is also an increased dopamine release (a), secondary to glutamate release, a downregulation of D2Rs (b), and a concomitant increase in D1R sensitivity (c). With opioid withdrawal, there is no presynaptic component, but a significant increase in the sensitivity of the striosomal D1R-MOR neurons (c). Abbreviations: eGP/VP, external segment of the globus pallidus/ventral pallidum; iGP, internal segment of the globus pallidus; lHb, lateral habenula; rmTg, rostromedial tegmental nucleus; SNpc, substantia nigra pars compacta; SNpr, substantia nigra pars reticulata; VTA, ventral tegmental area. For simplification, the indirect connection of the GPe to the GPi and SNpr through the subthalamic nucleus is not included. Also, only the cortex is shown as an input to the striatum, but it also receives inputs from the midline and intralaminar thalamic nuclei, and the ventral striatum also receives inputs from the hippocampus and amygdala.
Postmortem studies and results from CSF analysis and imaging studies (positron emission tomography and single-photon emission computerized tomography) indicate the existence of a presynaptic hyperdopaminergic state in RLS, with an increased synthesis of dopamine and synaptic dopamine by the dopaminergic cells that innervate the caudate and putamen (dorsal striatum), with a possible secondary downregulation of dorsal striatal D2Rs [30–32]. This may create an imbalance in the normal dopaminergic receptor activation of the striatonigral and striatopallidal neurons, with increases in presynaptic synthesis and release of dopamine promoting a preferential activation of postsynaptic D1Rs versus D2Rs. The presynaptic hyperdopaminergic state has been reproduced in the iron deficiency rodent model of RLS, which also showed striatal D2R downregulation and, in addition, a significant downregulation of adenosine A1 receptors (A1Rs) in the brain [33]. Importantly, A1Rs do not only mediate the brake that adenosine imposes on striatal glutamate release and on dopamine synthesis and release (which is dependent on glutamate release), but also on D1R signaling (through A1R-D1R complexes or heteromers). A1R downregulation therefore provides a putative mechanism for the presynaptic hyperdopaminergic state and for an increased dopaminergic sensitivity of the striatal GABAergic neurons that express D1Rs [34, 35]. It has then been proposed that the akathisia of RLS is mostly mediated by presynaptic and postsynaptic mechanisms that promote an increased activation of the striatal GABAergic neurons that express D1Rs [36] (Figure 2).
To understand the paradox of the efficacy of low doses of D2-like receptor agonists (pramipexole, ropinirole) in RLS, as well as the paradox of the restlessness of NIA, which is generally accepted to be mediated by D2R receptor blockade and should therefore be expected to promote motor depression, we need to turn to the dopamine autoreceptors. These are D2-like receptors (mostly of the short isoform of the D2R, or D2SR, and the D3R subtypes) that are localized at the somatodendritic level and in the nerve terminals of dopaminergic cells and exert a strong inhibitory control on their ability to synthesize and release dopamine [37–40]. Dopamine autoreceptors mediate the classically described decreased locomotor activity in rodents induced by low doses of nonselective D2-like receptor agonists, as well as the locomotor activation induced by low doses of D2R antagonists [41, 42]. Low doses of D2-like receptor agonists preferentially activate dopamine autoreceptors versus postsynaptic D2Rs (mostly of the long isoform of the D2R, or D2LR [37]), counteracting the presynaptic hyperdopaminergic state of RLS [36, 42, 43]. On the other hand, their blockade by neuroleptics promotes dopamine synthesis and release [40–44], which should preferentially activate postsynaptic D1Rs, since a significant proportion of postsynaptic D2R are concomitantly blocked. Therefore, the same as for the akathisia of RLS, it has been proposed that NIA depends on presynaptic and postsynaptic mechanisms that promote an increased activation of the striatal GABAergic neurons that express D1Rs [36] (Figure 2).
There is still another level of complexity in the striatal architecture, the matrix, and striosomes (or patches) sub-compartments [29, 45]. A classical distinction of the striosomes is their high density of MORs, which are practically absent from the matrix [46]. Both sub-compartments contain the two subtypes of GABAergic efferent neurons, but MORs are mostly expressed by the striatonigral neuron of the striosomes, the striosomal D1R-MOR neurons [47, 48]. Another important distinction between the matrix and striosomes sub-compartments is the differential projections of the striatonigral neuron. The matrix striatonigral neuron projects to the GABAergic neurons of the substantia nigra pars reticulata and the GPi, while the striosomal D1R-MOR neuron projects specifically to glutamatergic neurons of the iGP or VP and to the dopaminergic neurons of the SNpc and VTA (Figure 2) [47–50].
Akpinar (1987) was the first to propose an opioid-dopaminergic interaction in RLS: “. . . disturbance in the dopamine system is primary whereas dysfunction in the system of opioid peptides may be secondary . . .” [51]. We hypothesize that the striosomal D1R-MOR neurons that project to glutamatergic neurons of the GPi (and possibly the VP), which are known to be involved in arrest and withdrawal from aversive stimuli [49, 50], represent a key neuronal target of the opioid-dopaminergic interactions that underlie akathisia/restlessness. The iGP/VP glutamatergic neurons project to glutamatergic neurons of the lateral habenula, which innervate GABAergic neurons of the rostromedial tegmental nucleus, which finally innervate and tonically inhibit the dopaminergic cells of the SNpc/VTA (Figure 2). Activation of the pallidal-habenular neurons leads, therefore, to inhibition of the dopaminergic neurons, which leads to arrest behavior. A recent study in mice [48] demonstrated that acute activation of MORs in the nerve terminals of the striosomal D1R-MOR neurons of the dorsal striatum inhibits their GABA release in the iGP, while the withdrawal from chronic opioid treatment leads to sensitization of the striosomal D1R-MOR neurons and increased iGP GABA transmission, leading to inactivation of the pallidal-habenular neurons, which we postulate underlies akathisia/restlessness. With the present hypothesis, nevertheless, we need to assume that an increased activation or sensitivity of the striosomal D1R-MOR neurons should provide a predominant excitatory effect on SNpc (and possibly VTA) dopaminergic neuronal activity mediated by the inactivation of pallidal-habenular neurons, over their potential inhibitory effect mediated by their direct innervation of the dopaminergic neurons (Figure 2).
Conclusion and Future Directions
The three clinical conditions, RLS, NIA, and opioid-withdrawal-induced restlessness, have phenotypic similarities but also distinctions. We propose that their similar features stem from common neuronal mechanisms. Based on converging clinical data indicating a significant role of opioids in the treatment of RLS and NIA and on recently reported RLS-like symptoms during opioid withdrawal and supervised prescription opioid tapering [14–16], and recent preclinical experimental data in rodents [48], we propose the heuristic hypothesis of a key role of the striosomal D1R-MOR neuron in akathisia/restlessness. This hypothesis can be tested in animal models of RLS [43, 52] and opioid-withdrawal syndrome [48] as well as in animals acutely treated with D2R antagonists. These studies could then provide a more coherent and unifying neurobiological basis for the restlessness observed in these three clinical syndromes. Figure 1 shows a still putative representation of the distinctive and shared clinical components of akathisia/restlessness of RLS, NIA, and opioid withdrawal. Future studies should consider more careful, comparative phenotyping, as well as comparative pharmacologic treatment responses, to better clarify those distinctions and similarities [53–55]. Finally, objective measures used in RLS, such as the suggested immobilization test or PLMS, may provide further understanding of these phenotypes [53].
Contributor Information
Sergi Ferré, Integrative Neurobiology Section, National Institute on Drug Abuse, Intramural Research Program, National Institutes of Health, Baltimore, MD, USA.
John W Winkelman, Departments of Psychiatry and Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Diego García-Borreguero, Sleep Research Institute, Madrid, Spain.
Annabelle M Belcher, Division of Addiction, Research, and Treatment, Department of Psychiatry, School of Medicine, University of Maryland, Baltimore, MD, USA.
Joy H Chang, Substance Abuse Consultation Service, Department of Psychiatry, School of Medicine, University of Maryland, Baltimore, MD, USA.
Christopher J Earley, Department of Neurology and Sleep Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Funding
S.F. is funded with the intramural funds of the National Institute on Drug Abuse (ZIA DA000493). J.W.W. is funded by the National Institute of Drug Abuse, the RLS Foundation, and the Baszucki Brain Research Fund.
Disclosure Statement
(a) J.W.W. receives consultation fees from Emalex, Noctrix, and Disc Medicine; (b) the authors declare no conflicts of interest.
References
- 1. Earley CJ. Clinical practice restless legs syndrome. N Engl J Med. 2003;348(21):2103–2109. doi: 10.1056/NEJMcp021288 [DOI] [PubMed] [Google Scholar]
- 2. Allen RP, Picchietti DL, Garcia-Borreguero D, et al.; International Restless Legs Syndrome Study Group. Restless legs syndrome/Willis-Ekbom disease diagnostic criteria: updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria--history, rationale, description, and significance. Sleep Med. 2014;15(8):860–873. doi: 10.1016/j.sleep.2014.03.025 [DOI] [PubMed] [Google Scholar]
- 3. Willis T. De Animae Brutorum. Well and Scott; 1672. [Google Scholar]
- 4. Ekbom. Asthenia crurum paraesthetica (irritable legs). Acta Medica Scandinavica. 1944;118:197. [Google Scholar]
- 5. Ekbom K. Restless legs syndrome. Neurology. 1960;19(9):868–873. [DOI] [PubMed] [Google Scholar]
- 6. Ekbom KA. Resless lewgs, a report of 70 new cases. Acta Medica Scandinavica. 1950;246:64. [PubMed] [Google Scholar]
- 7. Haskovec L. L’akathisie. Rev Neurol (Paris). 1901;9:1107–1109. [Google Scholar]
- 8. Sachdev P. The development of the concept of akathisia: a historical overview. Schizophr Res. 1995;16(1):33–45. doi: 10.1016/0920-9964(94)00058-g [DOI] [PubMed] [Google Scholar]
- 9. Patatanian E, Claborn MK.. Drug-induced restless legs syndrome. Ann Pharmacother. 2018;52(7):662–672. doi: 10.1177/1060028018760296 [DOI] [PubMed] [Google Scholar]
- 10. Handelsman L, Cochrane KJ, Aronson MJ, Ness R, Rubinstein KJ, Kanof PD.. Two new rating scales for opiate withdrawal. Am J Drug Alcohol Abuse. 1987;13(3):293–308. doi: 10.3109/00952998709001515 [DOI] [PubMed] [Google Scholar]
- 11. Peachey JE, Lei H.. Assessment of opioid dependence with naloxone. Br J Addict. 1988;83(2):193–201. doi: 10.1111/j.1360-0443.1988.tb03981.x [DOI] [PubMed] [Google Scholar]
- 12. Wesson DR, Ling W.. The Clinical Opiate Withdrawal Scale (COWS). J Psychoactive Drugs. 2003;35(2):253–259. doi: 10.1080/02791072.2003.10400007 [DOI] [PubMed] [Google Scholar]
- 13. Scherbaum N, Stüper B, Bonnet U, Gastpar M.. Transient restless legs-like syndrome as a complication of opiate withrawal. Pharmacopsychiatry. 2003;36(2):70–72. doi: 10.1055/s-2003-39047 [DOI] [PubMed] [Google Scholar]
- 14. Mackie SE, McHugh RK, McDermott K, Griffin ML, Winkelman JW, Weiss RD.. Prevalence of restless legs syndrome during detoxification from alcohol and opioids. J Subst Abuse Treat. 2017;73:35–39. doi: 10.1016/j.jsat.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gupta R, Ali R, Ray R.. Willis-Ekbom disease/restless legs syndrome in patients with opioid withdrawal. Sleep Med. 2018;45:39–43. doi: 10.1016/j.sleep.2017.09.028 [DOI] [PubMed] [Google Scholar]
- 16. McCarter SJ, Labott JR, Mazumder MK, et al. Emergence of restless legs syndrome during opioid discontinuation. J Clin Sleep Med. 2023;19(4):741–748. doi: 10.5664/jcsm.10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blom S, Ekbom KA.. Comparison between akathisia developing on treatment with phenothiazine derivatives and the restless legs syndrome. Acta Med Scand. 1961;170:689–694. doi: 10.1111/j.0954-6820.1961.tb00286.x [DOI] [PubMed] [Google Scholar]
- 18. Walters AS, Hening W, Rubinstein M, Chokroverty S.. A clinical and polysomnographic comparison of neuroleptic-induced akathisia and the idiopathic restless legs syndrome. Sleep. 1991;14(4):339–345. [PubMed] [Google Scholar]
- 19. Allen RP, Picchietti DL, Auerbach M, et al.; International Restless Legs Syndrome Study Group (IRLSSG). Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/Willis-Ekbom disease in adults and children: an IRLSSG task force report. Sleep Med. 2018;41:27–44. doi: 10.1016/j.sleep.2017.11.1126 [DOI] [PubMed] [Google Scholar]
- 20. Schoretsanitis G, Nikolakopoulou A, Guinart D, Correll CU, Kane JM.. Iron homeostasis alterations and risk for akathisia in patients treated with antipsychotics: a systematic review and meta-analysis of cross-sectional studies. Eur Neuropsychopharmacol. 2020;35:1–11. doi: 10.1016/j.euroneuro.2020.04.001 [DOI] [PubMed] [Google Scholar]
- 21. Zareifopoulos N, Katsaraki M, Stratos P, et al. Pathophysiology and management of Akathisia 70 years after the introduction of the chlorpromazine, the first antipsychotic. Eur Rev Med Pharmacol Sci. 2021;25(14):4746–4756. doi: 10.26355/eurrev_202101_26386 [DOI] [PubMed] [Google Scholar]
- 22. Silber MH, Buchfuhrer MJ, Earley CJ, Koo BB, Manconi M, Winkelman JW; Scientific and Medical Advisory Board of the Restless Legs Syndrome Foundation. The management of restless legs syndrome: an updated algorithm. Mayo Clin Proc. 2021;96(7):1921–1937. doi: 10.1016/j.mayocp.2020.12.026 [DOI] [PubMed] [Google Scholar]
- 23. Freye E, Levy JV, Partecke L.. Use of gabapentin for attenuation of symptoms following rapid opiate detoxification (ROD)--correlation with neurophysiological parameters--. Neurophysiol Clin. 2004;34(2):81–89. doi: 10.1016/j.neucli.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 24. Makhinson M, Gomez-Makhinson J.. A successful treatment of buprenorphine withdrawal with the dopamine receptor agonist pramipexole. Am J Addict. 2014;23(5):475–477. doi: 10.1111/j.1521-0391.2014.12133.x [DOI] [PubMed] [Google Scholar]
- 25. Walters AS, Ondo WG, Zhu W, Le W.. Does the endogenous opiate system play a role in the Restless Legs Syndrome? A pilot post-mortem study. J Neurol Sci. 2009;279(1-2):62–65. doi: 10.1016/j.jns.2008.12.022 [DOI] [PubMed] [Google Scholar]
- 26. Walters A, Hening W, Chokroverty S, Fahn S.. Opioid responsiveness in patients with neuroleptic-induced akathisia. Mov Disord. 1986;1(2):119–127. doi: 10.1002/mds.870010206 [DOI] [PubMed] [Google Scholar]
- 27. Walters AS, Hening A.. Opioids a better treatment for acute than tardive akathisia: possible role for the endogenous opiate system in neuroleptic-induced akathisia. Med Hypotheses. 1989;28(1):1–2. doi: 10.1016/0306-9877(89)90145-x [DOI] [PubMed] [Google Scholar]
- 28. Haber SN, Behrens TEJ.. The neural network underlying incentive-based learning: implications for interpreting circuit disruptions in psychiatric disorders. Neuron. 2014;83(5):1019–1039. doi: 10.1016/j.neuron.2014.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerfen CR. Basal ganglia. In: Paxinos G (ed.), The Rat Nervous System. San Diego, CA: Academic Press; 2004: 445–508. [Google Scholar]
- 30. Earley CJ, Kuwabara H, Wong DF, et al. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep. 2013;36(1):51–57. doi: 10.5665/sleep.2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Connor JR, Wang XS, Allen RP, et al. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain. 2009;132(Pt 9):2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allen RP, Connor JR, Hyland K, Earley CJ.. Abnormally increased CSF 3-Ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med. 2009;10(1):123–128. doi: 10.1016/j.sleep.2007.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quiroz C, Gulyani S, Ruiqian W, et al. Adenosine receptors as markers of brain iron deficiency: implications for restless legs syndrome. Neuropharmacology. 2016;111:160–168. doi: 10.1016/j.neuropharm.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferre S, Garcia-Borreguero D, Allen RP, Earley CJ.. New insights into the neurobiology of restless legs syndrome. Neuroscientist. 2019;25(2):113–125. doi: 10.1177/1073858418791763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferre S, Quiroz C, Guitart X, et al. Pivotal role of adenosine neurotransmission in restless legs syndrome. Front Neurosci. 2017;11:722. doi: 10.3389/fnins.2017.00722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferre S, Guitart X, Quiroz C, et al. Akathisia and restless legs syndrome: solving the dopaminergic paradox. Sleep Med Clin. 2021;16(2):249–267. doi: 10.1016/j.jsmc.2021.02.012 [DOI] [PubMed] [Google Scholar]
- 37. Radl D, Chiacchiaretta M, Lewis RG, Brami-Cherrier K, Arcuri L, Borrelli E.. Differential regulation of striatal motor behavior and related cellular responses by dopamine D2L and D2S isoforms. Proc Natl Acad Sci U S A. 2018;115(1):198–203. doi: 10.1073/pnas.1717194115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bello EP, Mateo Y, Gelman DM, et al. Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci. 2011;14(8):1033–1038. doi: 10.1038/nn.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zapata A, Shippenberg TS.. D(3) receptor ligands modulate extracellular dopamine clearance in the nucleus accumbens. J Neurochem. 2002;81(5):1035–1042. doi: 10.1046/j.1471-4159.2002.00893.x [DOI] [PubMed] [Google Scholar]
- 40. Kehr W, Carlsson A, Lindqvist M, Magnusson T, Atack C.. Evidence for a receptor-mediated feedback control of striatal tyrosine hydroxylase activity. J Pharm Pharmacol. 1972;24(9):744–747. doi: 10.1111/j.2042-7158.1972.tb09104.x [DOI] [PubMed] [Google Scholar]
- 41. Jackson DM, Ross SB, Larsson LG.. Dopamine D-2 receptor agonist-induced behavioural depression: critical dependence upon postsynaptic dopamine D-1 function A behavioural and biochemical study. Naunyn Schmiedebergs Arch Pharmacol. 1989;340(4):355–365. doi: 10.1007/BF00167035 [DOI] [PubMed] [Google Scholar]
- 42. Strömbom U. Antagonism by haloperidol of locomotor depression induced by small doses of apomorphine. J Neural Transm. 1977;40(3):191–194. doi: 10.1007/BF01300133 [DOI] [PubMed] [Google Scholar]
- 43. Salminen AV, Clemens S, García-Borreguero D, et al.; International Restless Legs Syndrome Study Group (IRLSSG). Consensus guidelines on the construct validity of rodent models of restless legs syndrome. Dis Model Mech. 2022;15(8):dmm049615. doi: 10.1242/dmm.049615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moghaddam B, Bunney BS.. Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem. 1990;54(5):1755–1760. doi: 10.1111/j.1471-4159.1990.tb01230.x [DOI] [PubMed] [Google Scholar]
- 45. Brimblecombe KR, Cragg SJ.. The Striosome and matrix compartments of the striatum: a path through the labyrinth from neurochemistry toward function. ACS Chem Neurosci. 2017;8(2):235–242. doi: 10.1021/acschemneuro.6b00333 [DOI] [PubMed] [Google Scholar]
- 46. Dilts RP, Kalivas PW.. Autoradiographic localization of mu-opioid and neurotensin receptors within the mesolimbic dopamine system. Brain Res. 1989;488(1-2):311–327. doi: 10.1016/0006-8993(89)90723-3 [DOI] [PubMed] [Google Scholar]
- 47. Stephenson-Jones M, Bravo-Rivera C, Ahrens S, et al. Opposing contributions of GABAergic and glutamatergic ventral pallidal neurons to motivational behaviors. Neuron. 2020;105(5):921–933.e5. doi: 10.1016/j.neuron.2019.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang W, Xie X, Zhuang X, et al. Striatal μ-opioid receptor activation triggers direct-pathway GABAergic plasticity and induces negative affect. Cell Rep. 2023;42:112089. doi: 10.1016/j.celrep.2023.112089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R.. Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron. 2012;74(3):475–481. doi: 10.1016/j.neuron.2012.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Faget L, Zell V, Souter E, et al. Opponent control of behavioral reinforcement by inhibitory and excitatory projections from the ventral pallidum. Nat Commun. 2018;9(1):849. doi: 10.1038/s41467-018-03125-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Akpinar S. Restless legs syndrome treatment with dopaminergic drugs. Clin Neuropharmacol. 1987;10(1):69–79. doi: 10.1097/00002826-198702000-00007 [DOI] [PubMed] [Google Scholar]
- 52. Earley CJ, Jones BC, Ferre S.. Brain-iron deficiency models of restless legs syndrome. Exp Neurol. 2022;356:114158. doi: 10.1016/j.expneurol.2022.114158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kohnen R, Allen RP, Benes H, et al. Assessment of restless legs syndrome--methodological approaches for use in practice and clinical trials. Mov Disord. 2007;22(suppl 18):S485–S494. doi: 10.1002/mds.21588 [DOI] [PubMed] [Google Scholar]
- 54. Fulda S, Allen RP, Earley CJ, et al. We need to do better: a systematic review and meta-analysis of diagnostic test accuracy of restless legs syndrome screening instruments. Sleep Med Rev. 2021;58:101461. doi: 10.1016/j.smrv.2021.101461 [DOI] [PubMed] [Google Scholar]
- 55. Winkelman JW. Opioids and restless legs syndrome: a double-edged sword. J Clin Sleep Med. 2023;19(4):641–642. doi: 10.5664/jcsm.10524 [DOI] [PMC free article] [PubMed] [Google Scholar]