

SUMMARY
A limitation of conventional bulk-tissue proteome studies in amyotrophic lateral sclerosis (ALS) is the confounding of motor neuron (MN) signals by admixed non-MN proteins. Here, we leverage laser capture microdissection and nanoPOTS single-cell mass spectrometry-based proteomics to query changes in protein expression in single MNs from postmortem ALS and control tissues. In a follow-up analysis, we examine the impact of stratification of MNs based on cytoplasmic transactive response DNA-binding protein 43 (TDP-43)+ inclusion pathology on the profiles of 2,238 proteins. We report extensive overlap in differentially abundant proteins identified in ALS MNs with or without overt TDP-43 pathology, suggesting early and sustained dysregulation of cellular respiration, mRNA splicing, translation, and vesicular transport in ALS. Together, these data provide insights into proteome-level changes associated with TDP-43 proteinopathy and begin to demonstrate the utility of pathology-stratified trace sample proteomics for understanding single-cell protein dynamics in human neurologic diseases.
In Brief
Guise and Misal et al. report the unbiased single-cell proteomic analysis of ALS motor neurons directly captured from human tissues to explore disease-associated protein dynamics across TDP43-pathological strata. This study highlights both important technical challenges accompanying single-cell omics studies and emerging avenues for exploring human disease biology with nanoPOTS.
Graphical Abstract
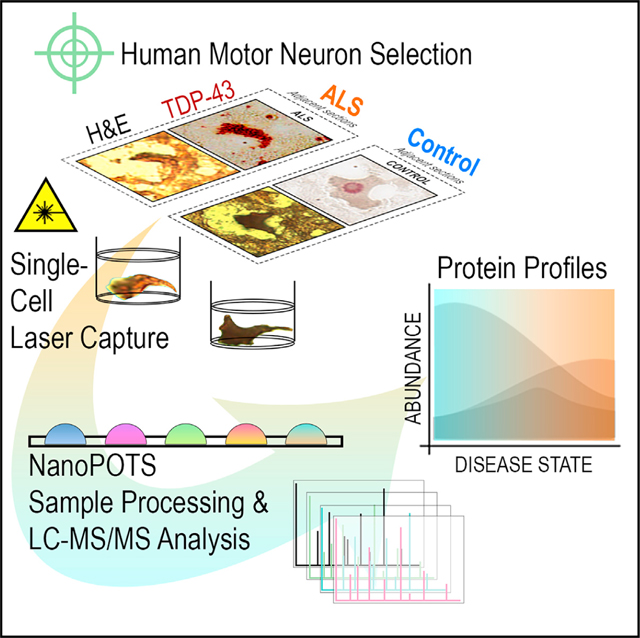
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease (NDD) commonly characterized by loss of spinalcord somatic motor neurons (MN) and motor function. Approved pharmacologic treatments for ALS impart either only modest effects on disease progression1–3 (riluzole, edaravone, sodium phenylbutyrate/taurursodiol) or are limited to specific genetic subpopulations4 (tofersen). Furthermore, there is a lack of biomarkers of specific MN pathologies, causing a significant hindrance to the clinical development of investigational drugs. New molecular insights are essential to these critical unmet needs.
Substantial evidence implicates transactive response DNA-binding protein 43 (TDP-43) dysregulation in MN disease pathogenesis. Neuronal cytoplasmic inclusions (NCIs) of ubiquitinated, phosphorylated C-terminal TDP-43 fragments are a neuropathologic hallmark of a majority of ALS subtypes,5–7 and the TARDBP gene itself harbors mutations in ~3% of ALS.8 Prior studies link TDP-43 perturbations with widespread alterations in mRNA and cryptic exon (CE) expression9–14; however, the contributions of individual CE-inclusion events to ALS pathophysiology and the protein-level impacts of TDP-43 loss of function (LOF) in MNs remain to be elucidated. Increased knowledge of protein-level dysregulation in MNs will augment understanding of disease biology with potential to uncover new and cell-type-specific ALS biomarker and/or therapeutic candidates.
To date, proteome studies have primarily focused on bulk tissue/pooled cell populations in ALS15–18 where measurements of MN-relevant targets are readily obscured by cellular heterogeneity.19 While single-cell genomic and transcriptomic analyses benefit from signal amplification,20 protein-level characterization has remained challenging. Nonetheless, recent advances in trace sample proteomics have begun to address this gap, enabling quantitative protein abundance measurements in single MNs.21,22
Here, we present an unbiased survey of single-cell protein expression profiles in human spinal MNs directly sampled from ALS tissues via laser-capture microdissection and analyzed via nanoPOTS19 coupled with ultrasensitive mass spectrometry-based detection. A pilot study of single-MN proteomes readily revealed disease-state differentiability and deficiencies in proteins with roles in oxidative phosphorylation, mRNA splicing, translation, and vesicular transport. In a follow-up experiment incorporating TDP-43-focused stratification of single-MN proteomes, we found conserved reductions in metabolic, RNA regulatory, and endolysosomal trafficking proteins in MNs with and without TDP-43+ NCIs. Our single-cell datasets support losses in STMN2 consistent with existing literature11,13 and prominent deficiencies in endolysosomal trafficking components (retromer, ESCRT-III [endosomal sorting complexes required for transport III], GARP/EARP [Golgi- and endosome-associated retrograde protein]) in ALS MNs prior to appearance of detectable TDP-43+ NCIs. Against a backdrop of rapid advancements in single-cell proteomics technologies, this report demonstrates the evolving potential for single-cell proteomics to augment our understanding of cell-type-specific mechanisms and pathologies underlying ALS and other NDDs.
RESULTS AND DISCUSSION
Pilot study: Characterization of single-MN proteomes in human ALS
We compared protein abundances in somatic MNs individually dissected from postmortem thoraco-lumbar ventral spinal horns of ALS or control (CTL; no history of NDD) donors (Table 1). Individual MNs identified by morphology and the presence of Nissl substance were laser excised prior to nanoPOTS liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis,19,22–24 yielding the quantification of, on average, 890 proteins (5% false discovery rate [FDR] ∩ ≥ 1 unique peptide) per single MN (Figure 1A; Table S1). ALS and CTL MNs were readily differentiated by dimensionality reduction of the top 500 most-variable proteins (Figure 1B). The MN-associated proteins acetylcholinesterase and peripherin were abundantly present in 67% and 100% cells, respectively, while choline O-acetyltransferase (ChAT) was detected infrequently in 22% cells (Table S1). In a subsequent experiment where cells were selected based on histologic ChAT positivity in adjacent sections, ChAT was detected by MS/MS in only 64% (CTL) and 36% (ALS) MNs (Table S2), suggesting the possibility of diminished ChAT expression in ALS MNs and poor flyability of ChAT peptides in general.
Table 1.
Patient characteristics
| ALS (n = 8) | CTL (n = 6) | Overall (n = 14) | p value | |
|---|---|---|---|---|
|
| ||||
| PMI, h | ||||
|
| ||||
| Mean (SD) | 15.3 (10.2) | 15.7 (3.27) | 15.5 (7.79) | 0.937 |
| Median [min, max] | 10.7 [5.00, 30.7] | 14.5 [12.9, 21.0] | 13.5 [5.00, 30.7] | — |
|
| ||||
| Age, years | ||||
|
| ||||
| Mean (SD) | 71.3 (8.01) | 55.5 (6.38) | 64.5 (10.8) | 0.00192 |
| Median [min, max] | 71.0 [59.0, 81.0] | 57.0 [46.0, 63.0] | 63.5 [46.0, 81.0] | — |
|
| ||||
| Sex | ||||
|
| ||||
| F | 4 (50.0%) | 1 (16.7%) | 5 (35.7%) | 0.469 |
| M | 4 (50.0%) | 5 (83.3%) | 9 (64.3%) | — |
Donor demographic data and characteristics are shown. Statistical comparisons were determined using Student’s t test (numerical data) or chisquared test (categorical data). F, female; M, male.
Figure 1. Ultrasensitive single-cell proteomic mapping of ALS motor neurons.

(A) Proteins (high-confidence master proteins, HCMPs; 5% global FDR) identified across single MNs (n = 6/donor) laser excised from ALS or CTL donor tissues (n = 3/diagnosis).
(B) Principal-component analysis (PCA) dimensionality reduction of the top 500 most-variable proteins measured across ALS and CTL MNs.
(C) Differential protein abundance in ALS vs. CTL MNs with correction for donor origin. Significant DAPs (|log2(ALS/CTL)| ≥ 1.5 ∩ p-adj < 0.05) indicated in purple.
(D) (Left) Age and PMI distributions for ALS and CTL donors; median ± range. (Right) Differential protein abundance with correction for additional covariates (age, sex, PMI). Significant DAPs indicated in purple.
(E) PPI network of significant DAPs. Node color indicates fold change in normalized protein abundance; edges denote high-confidence PPIs.
Oxidative phosphorylation, mRNA splicing, translation, and retromer-mediated vesicular transport protein abundances are significantly reduced in ALS MNs
We identified significantly differentially abundant proteins (DAPs) in ALS MNs using a linear mixed model accounting for the within-donor correlation structure. This model identified 298 high-confidence DAPs (|log2(ALS/CTL)| ≥ 1.5 ∩ adjusted p value [p-adj] < 0.05) in ALS vs. CTL MNs (Figure 1C; Table S1B). We further explored the impact of incorporating additional potential covariates (donor age, sex, postmortem interval [PMI]) into the model (Figure 1D), resulting in retention of 7 DAPs supporting pathway-level dysregulation of cellular respiration (CYCS) and mRNA splicing (SNRPD1, SNRPD3), transport (CAPRIN1), and translation (EEF1A1) (Figure 1E).
To examine the functional relationships between DAPs, we constructed a protein-protein interaction (PPI) network (Figure 1E) and report significant deficiencies in protein complexes with critical roles in cellular metabolism, RNA processing, and proteostasis, including diminished ribosomal protein and elongation factor abundances (Figures 1E and S1A). While consistent with previous observations of ALS-associated translational dysregulation,25,26 it is important to note that downregulation of translation and losses in mitochondrial respiration are also associated with cellular response to hypoxic conditions.27,28 While several studies have suggested that ischemia and hypoxia are involved in the pathogenesis of ALS,29–33 the possibility remains that changes in protein abundance are influenced by perimortem hypoxic/ischemic tissue insults or antemortem interventions that may elevate levels of HIF1α and other hypoxia-induced proteins.34,35 Interestingly, while we did not identify HIF1α in our dataset, we observed lower abundance of the hypoxia-upregulated protein 1 (HYOU1)36 in ALS relative to CTL MNs (Tables S1 and S4).
Evaluating the broad dynamic range of protein detection in single MNs and targeted investigation of proteins below the limit of detection
Comparison of median MS1 protein intensities measured in single MNs revealed proteins ranging from high-abundance structural neurofilament proteins (NfPs; NEFH, NEFM, NEFL) to lower-abundance membrane-associated synaptic proteins (LIN7C) (Figure 2A). Interestingly, GFAP was the most readily detected protein in both ALS and CTL MNs (Figure 2A) despite being a known astrocytic protein.37 Several scenarios could account for this observation, including the following: (1) astroglial projections captured along with MN cell bodies upon excision, either from the xy-plane cell periphery or greater z depths, or (2) protein contamination from tissue shearing during cryogenic sectioning. Cellular material carried along the blade as it traverses the tissue block may be deposited atop cells of interest and become inseparable from underlying tissue during fixation. In both scenarios, GFAP mimics behavior of “common contaminant” proteins often found in abundance in proteomic samples (e.g., keratins, albumins) due to sample handling or culture conditions.38 It is, however, important to note that while both scenarios explain detection of GFAP in single MNs, neither accounts for its elevated abundance in ALS MNs; instead, increased GFAP abundance more likely reflects a true tissue-level difference in which the local environment surrounding an ALS MN may be more densely populated with astrocytes than that surrounding a healthy MN.
Figure 2. Absence of STMN2 protein detection in human ALS MNs parallels decreased abundance of Stathmin-2 (STMN2) RNA and absence of functional TDP-43 interaction partners.

(A) Rank-ordered median protein intensities in single CTL or ALS MNs.
(B) Uniquely identified and overlapping proteins detected in ALS or CTL single MNs.
(C) PPI network of proteins uniquely detected in CTL (left) or ALS (right) single MNs; TDP-43 node added manually. Node border: magenta, BioGRID-reported direct or indirect TDP-43 PPIs; purple, downstream TDP-43 splice target STMN2; edges indicate STRING-annotated physical PPIs.
(D) Prevalence of TDP-43 interactors among proteins uniquely detected in CTL MNs, proteins uniquely detected in ALS MNs, or detected in the background human proteome (two-sided Fisher’s exact test).
(E) Dual expression of STMN2 RNA (red) and TDP-43 protein (teal) in ALS and CTL human spinal MNs.
(F) Quantitation of STMN2 RNA abundance in ALS (n = 5) and CTL (n = 10) donors; violin plots show median (dashed line), interquartile range (IQR; dotted lines), range, and Mann-Whitney U test p values.
(G) Cryptic and canonical STMN2 transcript visualization in adjacent ALS MN cross-sections.
Though downshifted in MS1 intensity, the relative protein rank orders in ALS MNs largely paralleled their counterparts in CTLs, highlighting proteins at the lower end of the abundance range in CTL samples that fall below the limit of detection in ALS samples, such as Stathmin-2 (STMN2) (Figure 2A). Presence-absence comparison of single-cell proteomes identified 127 proteins uniquely present in CTL and 140 proteins uniquely present in ALS MNs (Figures 2B and 2C). Gene Ontology (GO) and pathway analyses of condition-unique protein sets identified significant enrichment in RNA-processing-/splicing-associated proteins in CTL and innate immune signaling components in ALS (Figures S1B–S1C). Consistent with this observation, TDP-43 PPIs were significantly over-represented within the unique-to-CTL population relative to their frequency both in the human proteome39 and on the edge of significance (p = 0.06) for the unique-to-ALS protein set (Figures 2C and 2D).
TDP-43 is an RNA-binding protein whose cytoplasmic inclusions in neurons (NCIs) represent a distinguishing pathological feature of ALS.40 An intriguing possibility is that sequestration of interactors alongside TDP-43 within insoluble NCIs limits their accessibility for trypsin digestion—and subsequent detection—reflecting their functional unavailability in ALS MNs. These interactors include proteins with critical roles in mRNA processing/splicing (SNRNP200, NHP2L1, SRSF4, PRPF4, SART3, EIF4G3), splice target protein products (STMN2), and stress-granule-associated proteins (LSM12, LSM14A, FUS, and G3BP1). G3BP1 was recently shown to co-localize with cytoplasmic, mutant TDP-43 in ribonucleoprotein condensates in a cellular ALS model.41
A recently identified target of TDP-43, STMN2 is aberrantly spliced in human ALS MNs, resulting in the suppression of canonical STMN2 RNA and protein and inhibited axonal regeneration following injury.11,13 To evaluate whether STMN2 expression and splicing might be altered in our cohort of ALS MNs, we detected STMN2 RNA via in situ hybridization (Figure 2E) and report significantly decreased STMN2 RNA levels in ALS vs. CTL MNs (Figure 2F), consistent with less frequent MS-based detection of STMN2 protein in ALS MNs (Figure 2A). We further observed that expression of cryptic STMN2 is concomitant with the sparse detection of canonical STMN2 expression (and vice versa) bisected single MNs analyzed (Figure 2F), suggesting that STMN2 protein losses observed by single-cell proteomic analysis are likely the result of STMN2 mis-splicing events. Interestingly, we observed loss of canonical STMN2 RNA expression in MNs lacking apparent TDP-43+ NCIs (Figure 2D, ALS#7, bottom), implicating TDP-43 LOF in STMN2 mis-splicing and suggesting functional deficiency prior to TDP-43-mislocalization and formation of histologically evident NCIs.
MN stratification by TDP-43+ NCIs to enable pseudolon-gitudinal assessment of protein dynamics in postmortem tissues
As postmortem CNS tissues often represent patients with end-stage ALS, we hypothesized that MNs representative of advanced NDD could be overrepresented in our pilot experiment. To test the hypothesis that MNs at earlier vs. advanced stages of neurodegeneration might show different proteomic profiles, we generated a second, larger single-cell dataset consisting of MNs stratified by TDP-43+ NCI burden (Figures 3A and S3A). Using TDP-43 immunohistochemistry in adjacent histologic sections (Figure 3A), we classified MNs using a qualitative staging system, CTL: control MNs with normal appearing TDP-43; NON: ALS MNs with no overt TDP-43+ NCIs; MLD: ALS MNs with mild TDP-43+ NCIs; MOD: ALS MNs with moderate TDP-43+ NCIs; and SEV: ALS MNs with severe TDP-43+ NCIs (Figures 3A and S2A), using an in-house-developed antibody targeting the C-terminal domain of TDP-43 that robustly detects NCIs in human ALS and TDP-43-mutant mouse (TAR4/4) CNS tissues (Figure S2B). It is important to recognize several limitations of this approach, including (1) subjectivity in manual assignment of TDP-43 strata, (2) the assumption that larger or more prominent TDP-43+ NCIs imply a more advanced disease state, and (3) inclusion formation follows a simple and non-reversible trajectory.
Figure 3. Significant disruption of proteostasis, mitochondrial dysfunction, and induction of pro-apoptotic signaling are apparent prior to overt TDP-43 aggregation.

(A) Schematic of single-MN selection for laser-capture microdissection by dual detection of TDP-43 and ChAT in immediately adjacent tissue sections. Captured MNs were stratified based on TDP-43+ NCI status.
(B) Frequency of STMN2 protein detection in single MNs across TDP-43 strata.
(C) Differential protein abundances identified across TDP-43+ NCI strata (NON/MLD/MOD/SEV vs. CTL). Significant DAPs (|log2(ALS/CTL)| ≥ 1.5 ∩ p-adj < 0.05) shown in color.
(D and E) Shared or unique proteins across TDP-43 strata with significantly (D) decreased or (E) increased abundance in ALS MNs.
(F) PPI network of significant DAPs (|log2(ALS/CTL)| ≥ 1.5 ∩ p-adj < 0.05) common to all TDP-43 stages. Node color indicates fold change in protein abundance; edges denote high-confidence PPIs.
Target sample sizes were determined based on post hoc analyses of pilot dataset protein variability measurements (Figure S3) and modeled impacts on effect sizes for single-cell data.42 We captured 25 MNs from each ALS donor (n = 3), yielding between 13 and 29 MNs per stratified TDP-43 class (Figure S4A). It is important to note per-donor differences in the distributions of MNs across TDP-43 strata (Figure S4A); ALS#1 and ALS#3 appear more closely matched, while ALS#4 likely has greater influence in MLD group comparisons. In line with the TDP-43-agnostic pilot data, we observed decreased frequency of STMN2 detection in ALS MNs, including in normal-appearing ALS MNs lacking TDP-43+ NCIs (Figure 3B). Together, these data may suggest that mis-splicing and diminished STMN2 translation are early events relative to the accumulation of TDP-43+ NCIs.
Beyond STMN2, we observed robust, significant protein abundance differences in ALS MNs lacking TDP-43+ NCIs relative to healthy CTL MNs (Figure S4Bi) and trends toward more subtle abundance alterations between NON and MLD ALS MNs (Figure S4Bii), though the latter did not meet pre-specified significance thresholds. Interestingly, stratum-to-stratum comparisons identified no significant DAPs across MLD, MOD, and SEV ALS MN groups (Figure S4B; Tables S4, S5, and S6). We observed substantial overlap in DAPs identified in the TDP-43 agnostic pilot (Figures 1C–1E; Table S1) with those identified in normal-appearing ALS vs. CTL MNs (Figures 3C–3G; Table S2). Additional analyses revealed 485 significant DAPs common across individual TDP-43 strata vs. CTL comparisons (Figures 3D and 3E), suggesting that deficiencies in proteins associated with oxidative phosphorylation, mRNA splicing, translation, and endolysosomal transport observed in normal-appearing ALS MNs persist across TDP-43 inclusion strata (Figures 1E and 3C–3F).
Endolysosomal sorting complex component abundances exhibit strong inverse correlations with increasing presence of cytoplasmic TDP-43 inclusions
Using TDP-43+ NCI stratification assignments, pseudotemporal profiles for individual proteins were determined both using the combined dataset and individually per donor based on Spearman correlation scores (Rs) (Table S6), identifying a subset of proteins whose abundance values were significantly (p-adj < 0.05) correlated or anti-correlated (Figure 4A; Tables S4C and S4D) with increasing TDP-43+ NCI status.
Figure 4. Neurofilament protein and retromer complex component abundances track inversely with increasing TDP-43 aggregation in postmortem human MNs.

(A) Volcano plot of Spearman rank correlations (Rs) for abundance of individual proteins vs. corresponding TDP-43 stratum; purple highlight, significantly (p-adj < 0.05, Bonferroni) correlated proteins; green dashed lines indicate NfP correlations.
(B) Distribution of NEFH, NEFM, and NEFL protein abundances in individual MNs across TDP-43+ NCI strata.
(C) (i) Over-represented (blue highlight; p-adj < 0.05, p-adj < 0.01, Benjamini-Hochberg) KEGG pathways and (ii) physical protein complex assignments for the top 5% negative TDP-43-correlators; median ± IQR; whiskers indicate range.
(D) TDP-43-stratified protein abundances trajectories for retromer (blue), GARP/EARP (mauve), HOPS/CORVET (teal), and ESCRT-III (gold); median ± IQR; whiskers indicate range.
(E) Glial:neuronal abundance ratios for proteins with significant, positive RS.
(F) TDP-43-stratified protein abundance trajectories for SYNM and CNTNAP2; median ± IQR; whiskers indicate range.
Included in the significantly anti-correlated subset are NfPs (Figures 4B and S4C), following a pattern consistent with increased neuroaxonal stress/degeneration with greater TDP-43 burden. Release of cytoskeletal NfPs in response to axonal damage has been suggested across a broad range of NDDs, including multiple sclerosis, Alzheimer’s disease (AD), frontotemporal dementia, Lewy body disease, traumatic brain injury, Parkinson’s disease (PD), and Huntington’s disease.43 Multiple cross-sectional NDD studies have demonstrated elevation of NEFL in peripheral biofluids of affected individuals and correlation with measures of cognitive decline, brain volume, and survival.44–47 Interestingly, while NfPs were significantly anti-correlated with TDP-43+ NCI burden, they were not the strongest correlators. In addition to NDD-associated pathway enrichments driven by metabolic and proteasomal proteins, we observed significant over-representation of endocytic, endolysosomal sorting, and retrograde trafficking proteins (Figure 4C) among strong negative correlators (≥95th percentile; RS ≥ 0.62).
Separation of strong negative correlators based on physical PPIs revealed core components of the retromer (VPS26B/SNX6/SNX12), ESCRT-III (VPS4B, CHMP4B), and GARP/EARP (VPS52) complexes (Figure 4Cii). Remaining members of the heteropentameric retromer complex (VPS26A, VPS29, and VPS35) and components of the CORVET/HOPS (class C core vacuole/endosome tethering/homotypic fusion and protein sorting) tethering and sorting complexes (VPS18) and GARP/EARP transport complexes (VPS51, VPS52) also showed strong inverse correlations with TDP-43+ NCI burden, with conservation of correlation significance across all ALS donors profiled (Figure 4D; Table S6). To evaluate the possibility that correlations are driven by inclusion of the CTL group, we performed the correlation analysis across only ALS MNs (Table S6). Notably, we observed that SNX6, SNX12, VPS4B, and VPS18 retain negative correlation with TDP43+ NCI burden when CTL datapoints are excluded (Table S6).
The retromer complex is responsible for endosomal recycling of transmembrane protein cargoes to the plasma membrane, trans-Golgi network, or lysosomes,48 and dysregulation of VPS35 has been linked to AD49–52 and PD53–55 pathogenesis. Consistent with our observation of declining retromer abundance with increasing TDP43+ NCI burden, VPS35 mRNA deficiency has been shown to track with stereotyped neurodegeneration across AD brain regions, with increased VPS35 downregulation in the comparatively vulnerable entorhinal cortex relative to the more resistant dentate gyrus.56 VPS35 LOF was recently shown to induce insoluble pTDP-43 accumulation in murine nervous tissues,57 and several reports have linked retromer and/or endosome dysfunction to human ALS51,58 and disruption of TDP-43 processing/NCI formation.59–61 VPS35 mRNA deficiencies were further documented in human cortical tissues from a cohort of frontotemporal lobar degeneration (FTLD) donors harboring progranulin mutations. FTLD shares many pathological hallmarks with ALS, including profound TDP-43 pathology and NCI accumulation,5,7 and it has been suggested that ALS and FTLD may represent two different manifestations of TDP-43-accumulation-driven NDD.62
The retromer core has been nominated as a potential therapeutic target in multiple NDD contexts,52,55 and adeno-associated virus (AAV)-mediated replacement of VPS35 was sufficient to reduce accumulation of C-terminal APP fragments in mouse models.51 Virally encoded VPS35 was capable of binding VPS26 and VPS29, indicating that exogenous VPS35 replacement can restore retromer core assembly.51 Consequently, therapies designed to restore retromer component functions have the potential to ameliorate the proteotoxic effects of protein accumulation in vulnerable neuronal populations51 across a spectrum of NDD proteinopathies, including ALS.
Stress granule protein abundances correlate inversely with TDP-43+ NCI burden
The stress granule and aggresome proteins G3BP1, USP10, and ATXN2 were also inversely correlated with increasing TDP-43+ NCI burden (Figure 4Cii; Table S6). The ubiquitin-specific protease USP10 has previously been shown to promote TDP-43+ stress granule clearance and TDP-43+ aggresome formation following proteasome inhibition, while depletion of USP10 increased insoluble cytoplasmic TDP-43 cleavage products.63 The repeat-expansion-containing protein ATXN2 (Ataxin 2) is decreased in abundance and co-localizes with pTDP-43 in NCIs and with dystrophic neurites in FTLD-TDP hippocampi,64 and intermediate-length ATXN2 repeat expansions are associated with increased ALS risk.65,66
Neuronal and astrocyte proteins increased in MNs with TDP-43+ NCIs
Fifteen DAPs increased in ALS MNs demonstrated significant positive correlations with the extent of TDP-43+ NCIs (Figures 4A, S4D, and S4E), including GFAP and SYNM. We determined glial:neuronal abundance ratios using reported cell-type-specific proteomic measurements67 to identify proteins consistent with astrocyte co-capture with MNs68 and which may indicate increased astrocyte activity/reactivity in degenerating MNs (Figures 4E and 4F). CNTNAP2 (contactin-associated protein 2) was the most neuron-enriched protein positively correlated with increasing presence of TDP-43+ NCIs. CNTNAP2 is a transmembrane neurexin localized in channel-rich axonal juxtaparanodes with important roles in mediating cell:cell interactions69,70 and axonal excitability.71 Notably, CNTNAP2 extracellular domain peptides have also been detected in human cerebrospinal fluid (CSF),72 including in CSF from patients with ALS and FTLD.73
Comparison to ALS proteomic and transcriptomic studies
Mitochondrial dysfunction and impaired translation have been previously implicated in ALS disease by bulk proteomic studies of CNS tissues from ALS/FTLD donors.15–17,74,75 Reminiscent of these findings, reduced stability of transcripts encoding many oxidative phosphorylation and ribosomal proteins has been reported in ALS patient-derived fibroblasts, iPSCs, and CNS tissues.76 Transcriptome data from human FTLD cortical cells lacking nuclear TDP-43 identified numerous differentially expressed transcripts, including TDP-43 splice targets12,14; we identified proteins corresponding to 24% (16/66) of these TDP-43 targets, with 75% (12/16) showing significantly altered abundance (reduced: CADPS, CAMK2B, CYFIP2, EIF4G3, EPB41L1, HP1BP3, IMMT, KIF3A, PRUNE2, STXBP1, SYNE1, UQCRC2; increased: ARHGEF11) in ALS MNs lacking TDP-43+ NCIs. Interestingly, transcriptomic studies of laser-captured lumbar MNs in bulbar-onset ALS—which could reflect earlier stages of NDD relative to limb onset—reported widespread increases in mRNA abundance in ALS MNs relative to CTL MNs.68,77 The utility of correlating RNA with protein abundance measurements has been the topic of significant interest and debate in the field,78–82 ultimately suggesting their complementarity and that RNA:protein data must be interpreted on a case-by-case basis to shed light on the mechanistic regulation of a given locus.
Limitations of this study
As a nascent field, we expect single-cell proteomic approaches to render even greater insights into disease biology as technologies mature. In this initial application of single-cell proteomics to ALS MNs, we encountered several challenges. Notably, MN proteome coverage was low at ~2,500 proteins, an estimated ~20%–25% of the human proteome.83 Although we measured clear, consistent, and biologically relevant abundance differences in ~500 proteins in ALS vs. CTL MNs, these challenges clearly limit the discovery of low-abundance proteins potentially altered in NDD. Furthermore, due to our methodology’s low throughput and technical reliance on manual cell selection, only 135 MNs from 6 ALS donors were analyzed in in total. While per-group sample sizes were significantly increased in our TDP-43-focused experiment relative to the pilot based on measured (Figure S3) and modeled42 protein variability in CTL cells, we found comparisons across MNs with more prominent TDP-43+ NCIs insufficiently powered. It is not difficult to imagine that widespread alterations in protein abundances early in NDD cascades could exacerbate proteome variability at later stages, even among cells appearing similar with respect to histologically detected TDP-43.
We sought to stratify MNs based on TDP-43+ NCIs and consensus assignment to individual strata, and it is critical to acknowledge the qualitative/subjective nature of this method when interpreting these data. We operated under an assumption that TDP-43 staining observed in one-half of an MN is representative of total TDP-43 burden, which may not always be the case. Recent advancements in antibody-stained tissue processing methods will provide an improved means of capturing the exact material used for phenotypic stratification, especially in conjunction with data-driven cell selection and classification algorithms to further limit potential biases.
In addition to stratification via TDP-43 immunoreactivity, future studies may benefit from incorporation of MS-based measures of C-terminal, phosphorylated TDP-43 species as additional indicators of TDP-43 LOF. While we did detect unmodified TDP-43 peptides (Tables S1 and S2), evidence was insufficient to conclude that there were differences in TDP-43 between ALS and CTL MNs, which may be attributable to several factors: (1) the hyperphosphorylated C terminus of TDP-43, relatively lacking in tryptic cleavage sites (lysine, arginine), may be more amenable to digestion with cyanogen bromide84 (cleaving at methionine) than trypsin, which is widely used in bottom-up proteomics workflows for its consistent, efficient digestion of a majority of protein species and safety of use; (2) our study focused on unmodified proteoforms rather than phosphopeptides; and (3) conceivably, TDP-43+ protein inclusions/aggregates may not be fully accessible to enzymatic digestion. Future studies enriching for phosphorylations and/or employing complementary digestion or targeted detection methods represent alternative methodologies for cell stratification in the context of proteinopathies.
Autopsy tissues provide a cross-sectional sample of cells at an advanced stage of human disease. Like other deep molecular phenotyping technologies, single-cell proteomic trajectory analyses may soon be capable of delineating molecular stages of disease progression without true longitudinal tissue sampling. We employed immunohistochemical assays in adjacent frozen sections to stratify MNs across four phenotypic stages based on the presence of TDP-43+ NCIs, operating under the assumption that greater histologically evident inclusion burden indicates more advanced NDD. With our approach and sampling depth, we were unable to delineate cell trajectories de novo using the CellTrails85 algorithm (data not shown), which may be related to the small number of MNs and relatively few associated features in our single-cell proteomics dataset compared to single-cell RNA sequencing (RNA-seq) studies.86 Alternatively, we identified DAPs with the strongest Spearman correlations with TDP-43+ NCI strata as candidates that may play important mechanistic roles in ALS. However, our assumption of the direct relationship between an MN’s neurodegenerative progression and observable TDP-43+ NCIs does not account for the effects of oligomer toxicity or the potential neuroprotective roles of large aggregates.87,88
A further limitation stems from our study’s reliance on banked human autopsy specimens, which imparts additional variability in sample handling outside our control. Tissue banking protocols vary site to site, and while we ensured that ALS and CTL tissues originated from the same site and tried to match samples based on available demographic information, we were limited by sample availability and noted PMI variation in ALS vs. CTL donors (Figure 1D; Table S7). In postmortem tissue studies, it is also not possible to fully deconvolute the effects of perimortem insults including tissue hypoxia89,90 from those of disease. In an ideal situation with greater sample sizes, incorporating correction for donor characteristics is undeniably the preferred method for high-confidence detection of DAPs in a dataset consisting of multiple samples from multiple individuals.
To derive as much information as possible from rare and precious human tissues where knowledge of donor case histories and comorbidities is incomplete, we elected to perform analyses by (1) correcting for the within-donor correlations between MNs from the same individual and (2) with additional correction for donor-specific age, sex, and PMI data91 (Figures 1C and 1D). Not surprisingly, we observed a striking reduction in DAPs identified with full covariate inclusion (Figures 1C and 1D). While only 7 proteins survived this stringent correction, these high-confidence DAPs confer additional support for pathway-level dysregulation of cellular respiration, RNA splicing, and protein translation in ALS MNs (Figures 1C–1E). We further show conservation of protein abundance correlations in TDP-43-stratified MNs at the level of individual donors and upon exclusion of CTL samples (Figures S4C–S4E; Table S6) in addition to pooled donor analyses (Figure 4). Ultimately, collection of a larger dataset of single-cell measurements from additional subjects will be necessary for increased confidence in DAPs reported in this present study and is the focus of ongoing efforts to build on this demonstration of single-cell proteomics applied to human disease tissues.
Conclusion
Herein, we have presented unbiased proteomic studies of single somatic MNs from postmortem ALS donor spinal cords. Trace sample protein processing using nanoPOTS (1) rendered identification of >2,700 proteins and quantitative comparison of >1,300 proteins in our pilot study, (2) enabled disease-state differentiation of single MNs, and (3) revealed reductions in protein abundances suggesting impairments in cell energetics, protein translation, proteostasis, and trafficking mechanisms, including an emphasis on Golgi-lysosome trafficking. In a follow-up experiment where MNs were stratified by TDP-43 neuropathology, we observed similar complements of DAPs in MNs lacking TDP-43+ NCIs, suggesting that impairments in cellular energetic and proteostatic mechanisms occur early with respect to inclusion formation. Specifically, we report declining expression of retromer components accompanying increasing presence of TDP-43+ NCIs and propose retromer-complex-mediated endolysosomal sorting as a potential point of future mechanistic research in the formation or clearance of TDP-43 inclusions in ALS MNs (Figure S5). We further confirmed loss of canonical STMN2 transcripts and protein in ALS MNs concurrent with early TDP-43 pathology, indicative of loss of TDP-43-mediated splicing functions prior to formation of large TDP-43+ NCIs. Our study begins to show the potential of single-cell proteomics to augment the study of human neurologic diseases and provide insights into temporal dynamics of disease progression directly from human tissues.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Edward D. Plowey (ed.plowey@biogen.com).
Materials availability
Custom BaseScope probes targeting Stathmin 2 that were generated for this study are available through the Advanced Cellular Diagnostics catalog.
Data and code availability
Data reported in this paper are available upon request form the lead contact. Mass spectrometry data generated in this study have been deposited to ProteomeXchange (PXD042799) and MassIVE (MSV000092119).
Custom code is available on GitHub (https://doi.org/10.5281/zenodo.10268532).
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Postmortem human tissue specimens were provided by the University of Miami Brain Bank under requisition ID B2AE76. Subject details are summarized in Table 1. Human tissue samples sourced from the University of Miami Brain Endowment Bank were collected under informed consent outlined in Brain Endowment Bank IRB Protocol Number 19920358 (CR0001775) approved by the University of Miami Institutional Review Board.
METHOD DETAILS
Case selection
ALS and CTL cases were selected from a cohort of frozen thoraco-lumbar spinal cord tissue samples from the University of Miami Brain Bank (Table S7). Spinal cord tissues were divided into ~3mm sub-blocks. Two to seven sub-blocks were generated per sample, depending on initial tissue dimensions. Sub-blocks were generated by allowing the tissue to thaw slightly to avoid fracturing, then sliced into sub-blocks using a razor blade. Sub-blocks were immediately transferred to histology cassettes on dry ice and stored at −80°C. One ~3mm sub-block per donor was embedded in pre-chilled 2.5% carboxymethylcellulose solution (2.5% (w/v) carboxymethylcellulose (Sigma, Cat. No. 419273–100G, Lot. No. MKCG5725) in dH2O) in cryomolds on dry ice and allowed to freeze fully at −80°C. Subsequently, 5um sections were collected on positively-charged histology slides (Fisher Superfrost) and post-fixed by immersion in 10% Neutral Buffered Formalin (Fisher Scientific) for 15 min followed by two 10-min washes in 1X phosphate buffered saline (PBS) (Abcam, Ref. #ab128983). All sections were collected using a Leica CM1520 cryostat (CMMS ID XX-99339) using a Leica high-profile microtome blade at an internal cryostat temperature of −23C. Fixed slides were allowed to air dry following PBS rinse and stored at room temperature prior to staining. For tissue evaluation and case selection, haematoxylin and eosin (H&E) staining was performed on a single frozen section per donor using an automated Leica Spectra staining protocol (H&E no oven) and coverslipped using an automated coverslipper (TissueTek). Brightfield images of stained tissues were collected of all tissues at 20× on 3DHIS-TECH Panoramic slide scanner. In addition to evaluation of tissue quality, samples were selected to minimize differences in age at time of autopsy and postmortem interval (PMI) across sample groups.
Tissue sectioning and laser capture microdissection
Zeiss PEN membrane slides (Zeiss, Ref # 415190–9041-000, Lot # 000671–19) and Superfrost PlusGold slides (Fisher, Ref # 15–188-48, Lot # 19906–665158) were exposed to UV light for 30 min in a laminar flow hood to promote tissue adhesion. Spinal cord blocks were transferred from the −80°C freezer to the cryostat (−23°C) and allowed to equilibrate in temperature for 20 min 12-μm-thick sections were collected by cryosectioning in the following order: (1) one 12 μm section on a Superfrost PlusGold slide; (2) ten adjacent 12 μm sections (two per slide) on five PEN membrane slides; (3) one 12 μm section on a Superfrost PlusGold slide.
Slides were pre-chilled on the interior surface of the cryostat prior to section collection. Single sections were collected on PlusGold slides, adhered to the slide by warming the reverse side of the slide with a finger, and then immediately refrozen on the inner cryostat surface. Two sections were collected at the same time onto each PEN membrane slide, melted onto the slide by warming the reverse side of the slide with a finger, and then were immediately refrozen on the inner cryostat surface. Once sections were frozen and all sections (A-C) for a sample were collected, tissue sections were fixed in 70% ethanol for 15 min at room temperature and then transferred immediately to storage at −80°C in slide boxes with desiccant packs (Humidity Sponge).
For TDP-43-agnostic MN captures (ALS Pilot), all sections were collected over the course of four days, ensuring that equal numbers of ALS and non-disease control cases were collected in individual sectioning sessions using the same cryostat. All sections were stored at −80°C prior to H&E staining, ethanol dehydration, and vacuum desiccation. Stained tissue sections were scanned on a Zeiss PALM MicroBeam system at 40× resolution. Motor neurons were selected for single cell proteomics based on morphology and presence of Nissl substance and if the same cell was confidently identified across two or more adjacent sections to allow capture of two 12um-thick excised cell cross-sections per sample. Six individual MNs from each case (3 ALS and 3 CTL) were selected for subsequent analysis. Specifically, individual MNs matched across two adjacent sections were selected from laminae VII/IX of the ventral horn, excised by laser capture microdissection, and collected in individual nanoPOTS wells prefilled with DMSO to aid in sample collection using the Slide Collector 48 adapter. 10MN boost samples were generated for each donor by capturing and combining twenty 12um-thick excised cell cross-sections to approximate the protein contents of ten cells (two cross-sections per cell). Nanowells were imaged at 10× resolution to confirm collection of each excised cell. Following collection, nanoPOTS chips were sealed to avoid the evaporation of the solution from the nanowells.
For TDP-43-pathology-guided MN captures (TDP-43 Pseudotime), 20um-thick frozen sections were collected onto Zeiss PEN membrane slides, as described above, and adjacent 10um-thick frozen sections were collected onto Plus Gold slides for subsequent chromogenic detection of TDP-43 and ChAT protein by immunohistochemistry (IHC) staining. Frozen human spinal cord cross sections were fixed in 10% NBF, dried, and stored at room temperature prior to immunostaining. Staining was conducted on a Ventana Ultra platform using standard chromogenic methods. For antigen retrieval (HIER) and permeabilization, slides were heated in a pH9 EDTA-based buffer for 10 m at 94°C, followed by incubation with a mouse monoclonal antibody against TDP-43 (L95A-42, Biogen) at 1:40,000 and a rabbit monoclonal antibody targeting ChAT (Clone: EPR16590, Abcam, Ref. ab178850) at 1:3,000. Bound anti-TDP-43 and anti-ChAT primary antibodies were detected using an AP-conjugated-anti-mouse and HRP-conjugated-anti-rabbit secondary polymers with chromogenic visualization with Ventana DISCOVERY Red and Ventana DISCOVERY Yellow, respectively. A subset of slides was counterstained with haematoxylin to visualize nuclei. Stained slides were imaged at 203. Images from adjacent H&E-stained 20um- and TDP-43-ChAT-stained 10um-sections were manually aligned for identification of single MNs spanning both sections for laser capture microdissection (as described above) and classification of TDP-43 pathology, respectively. TDP-43 strata were defined along a semi-quantitative 4-point scale based on quantity and morphology of cytoplasmic TDP-43 protein inclusions.
NanoPOTS sample processing
Following cell capture, remaining DMSO was allowed to evaporate prior to adding additional processing reagents. Samples were further processed using the nanoPOTS workflow as described previously.19,23,92 Briefly, proteins were extracted with 0.1% dodecyl-β-d-maltopyranoside (DDM) and reduced with 5 mM dithiothreitol (DTT) followed by alkylation with 10 mM iodoacetamide (IAA). The two-step enzyme digestion was performed with LysC (0.25 ng) for 4 h followed by trypsin (0.25 ng) for an additional 16 h at 37°C. Digestion reactions were quenched with 0.1% formic acid (FA) and digested peptide samples were collected in 200-μm-i.d. fused silica capillaries using a robotic liquid handling system. The samples were stored individually at −20°C prior to LC-MS/MS analysis. Additional “boost” samples (10 MN equivalents) were prepared using pooled MNs from each case to facilitate feature identification and matching across single-cell analyses of the same case. Prior to injection and MS analysis, samples were block-randomized to minimize batch effects and impact of instrument drift during data acquisition.
nanoLC-MS/MS analysis
Samples were equilibrated to 4°C prior to analysis and were positioned for in-line loading on to an in-house-packed SPE column (5 cm x 75-μm-i.d.). Samples were loaded onto the column over 10 min using 100% Mobile Phase A (0.1% FA in water) at a flow rate of 0.5 μL/min using an UltiMate 3000 RSLCnano pump (Thermo Fisher) to ensure complete desalting. Peptide separation was performed by connecting the SPE column to an in-house-packed analytical SPE column (50 cm x 30-μm-i.d.) connected to a nanospray emitter by a zero-dead-volume union (Valco, Houston, TX). Peptides were separated by a 100-min linear gradient (8–25% mobile phase B (0.1% FA in acetonitrile) followed by an additional 20-min linear gradient (25–45% B) to elute hydrophobic peptides. For column washing, mobile phase B was increased to 90% over 5 min and held constant for 5 min to wash the column, then was reduced to 2% over 5 min and held constant for 15 min to re-equilibrate the column. Post-split (50 cm x 75-μm-i.d. split column) mobile phase flow rates were 20 nL/min; 250 nL/min programmed flow was provided by the UltiMate 3000 RSLCnano pump.
Peptides were injected into a Thermo Orbitrap Exploris 480 mass spectrometer by electrospray using in-house-pulled nanospray emitters (20-μm-i.d.). MS and MS/MS data were acquired by employing an electrospray potential of 2000 V at the source for ionization. The ion transfer tube temperature was 200°C for desolvation and the ion funnel RF level was 40. Full MS scans were acquired at 375–1800 m/z with an orbitrap resolution of 120,000 (m/z 200). The AGC target and maximum injection time were set to 1E6/200 ms. Data-dependent MS/MS spectrum acquisitions were triggered for precursor ions with intensities > 5E3 and charge states of +2 to +7. The scan range was defined from first mass to 100 m/z with a cycle time of 3 s. Monoisotopic precursor ion peaks were fragmented by higher energy collision-induced dissociation (HCD) with a normalized collision energy of 28% and with AGC target and maximum injection time set to 1E5/500 ms. Fragment ions were detected in the orbitrap at 30,000 resolution (m/z 200). MS/MS isolation windows were 1.6 Da with a mass tolerance of ±10 ppm and dynamic exclusion time was set to 90s.
Protein identification and quantitation
Raw MS data were searched against a protein database consisting of reviewed human proteins (20,353 reviewed protein sequences, UniProtKB, downloaded: July 20th, 2020) appended with common contaminants using a two-step database search was done with Sequest HT and Sequest HT INFERYS rescoring algorithms in Proteome Discoverer (version 2.5, ThermoFisher Scientific, San Jose, CA) specifying fully tryptic enzymatic digestion (7–30 amino acids, 2 missed cleavages). Fixed carbamidomethylation (C) and variable oxidation (M), deamidation (N,Q) and modification of protein N-termini (acetyl, Met-loss, pGlu) were included in the search parameters as modifications. Precursor and fragment mass tolerances were set to 10 ppm and 0.02 Da, respectively. The peak matching feature detection option was enabled to allow a maximum chromatographic retention time shift to 10 min with a mass tolerance of 10 ppm. Peptide identifications were refined using a target-decoy approach followed by percolation based on q-values and imposing a strict FDR cut-off of 0.01 and a relaxed FDR cut-off of 0.05 at the PSM and peptide level. Protein abundances were determined using the precursor ions quantifier node based on the top three distinct peptides (unique and razor) from each protein and normalized to total peptide signal per single cell sample (“boost” samples were included in the search to facilitate protein identification via feature matching but were excluded in normalization and quantitative comparison steps).
In situ hybridization and immunohistochemistry staining
FFPE tissue sections from human spinal cord samples were evaluated for RNA quality using positive (BA-Hs-POLR2A-3zz) and negative control (BA-dapB-3zz) BaseScope (Advanced Cellular Diagnostics, Inc.) probes. Expression of canonical and cryptic STMN2 transcripts was investigated using custom BaseScope probes (BA-Hs-STMN2–3zz-st and BA-Hs-STMN2-intron1–1zz-st1, respectively) (Advanced Cellular Diagnostics, Inc.) in conjunction with detection of TDP-43 protein using a mouse antibody against TDP-43 (0.25 μg/ml, TDP-43-L95A, Biogen) and Bond Primary Antibody Diluent (Advanced Cellular Diagnostics, Inc., Cat. No. AR9352) and the Bond Polymer Refine Detection Kit (Advanced Cellular Diagnostics, Inc., Cat No. DS9800).
RNA in situ hybridizations were performed on a Leica Bond automated platform using the BaseScope Reagent Kits (Advanced Cell Diagnostics, Inc.) according to manufacturer’s instructions. Briefly, 5um-thick FFPE tissue sections were pretreated with heat (Epitope retrieval (LS ER2) was carried out for 30 min at 95C) and protease (Protease IV, 30 min at 40C) prior to probe hybridization and antibody incubation (15 min, 0.25ug/ml). Pre-preamplifier, preamplifier, amplifier, and HRP/AP-labeled oligos were hybridized sequentially, followed by chromogenic detection. Samples were counterstained with hematoxylin. Brightfield images were collected at 40X using a 3DHistech Pannoramic SCAN II digital slide scanner. Scanned images were processed using a custom image analysis algorithms developed in Visiopharm for threshold-based detection of STMN2 and TDP-43 signal. MNs were annotated by training a Random Forest classifier and the resulting ROIs were manually inspected and adjusted prior to analysis of STMN2 and TDP-43 signal. Resulting data were analyzed using GraphPad Prism and STMN2 RNA expression differences were determined using a Mann Whitney U Test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of differential protein expression between ALS and CTL MNs was performed in R93 as follows: proteins with 3 or more missing values in at least one condition (1,375 out of 2,752 proteins) were excluded from downstream analysis. Data were background corrected and normalized by variance stabilizing transformation (vsn).94 Missing data were imputed following inspection of the pattern of missing data a heatmap and comparison of compared intensity distributions and cumulative fraction of proteins with and without missing values. As data appeared to be MNAR (missing not at random) (i.e., proteins with missing values have, on average, low intensities), missing data were imputed via left-censored imputation methods (MinProb and man). Differential expression analysis was then conducted by application of a linear mixed model to account for the dependence structure among samples from the same donors (using duplicateCorrelation() function from limma). Proteins with an FDR-adjusted p value of <0.05 and with absolute log2 fold changes ≥1.5 were considered significantly differentially abundant. For TDP-43 stratified samples, correction for individual donor contribution was evaluated by considering donor origins as individual variables to correct for within-donor correlations between samples from the same individuals. Raw protein intensity data were log2-normalized and compared across each stratified TDP-43 group (i.e., NON, MLD, MOD, SEV) to the control and to each other, accounting for donor contribution as a variable. Differential protein abundances between groups were determined using limma and p values were corrected for multiple hypothesis testing (Benjamini-Hochberg). Proteins with an FDR-adjusted p value of <0.05 and with absolute log2 fold changes ≥1.5 were considered significantly differentially abundant and were visualized via Volcano plots. Lists of significantly increased- or decreased-in-abundance proteins were made for each TDP-43 stratum relative to the control group. Using these lists, we identified significantly DAPs that were common across multiple strata or unique to an individual stratum, as visualized using upset plots.
Interaction networks for proteins exhibiting significantly differential abundance in ALS vs. CTL MNs were generated in the web-based STRING application (v12)95 with additional filtering requiring a minimum interaction score of 0.7 and with “Experimental” and “Database” active interaction sources enabled. Networks were then imported into Cytoscape (v3.8.2)96 to allow mapping of protein abundance data onto individual nodes. For TDP-43 negative correlators, the physical interaction network was constructed as described above, with the following modifications: minimum interaction score = 0.4; “physical subnetwork” option enabled. Over-representation of gene ontology (GOBP), KEGG, and Reactome pathway terms associated with identified protein subsets were determined using hypergeometric tests (statistical background = whole genome) followed by Benjamini-Hochberg correction for multiple hypothesis testing using the STRING enrichment analysis widget.97 Spearman correlation scores were calculated using the PerseusGUI (v1.6.5.0)98 and GraphPad Prism based on only non-imputed abundance values forindivu(missing data = NaN). Fisher exact testing for TDP-43 protein-protein interaction (PPI) enrichments was performed on contingency tables in GraphPad Prism (2-sided test, 90% CI) using TDP-43 PPI data from BioGRID.
Glial:neuronal abundance ratios were calculated based on published data from a study focused on comprehensive profiling of the mouse brain proteome across cell types and brain regions. Median abundance values were calculated across all ages/DIV stages for each cell population: astrocyte, oligodendroglial, microglial, and neuronal LFQ (label-free quantitation) data published in Sharma et al. 2016, Table S1 (File ID: https://static-content.springer.com/esm/art%3A10.1038%2Fnn.4160/MediaObjects/41593_2015_BFnn4160_MOESM38_ESM.xlsx; PXD001250; 67). Ratios per population were then calculated as the log2-transformed ratios of median abundance in each glial population vs. the median abundance in the neuronal population. A pan-glial ratio for each protein was also calculated as the mean of the individual per-glial-subtype enrichment scores. Resulting data were analyzed using GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse antibody to human TDP-43 protein | Biogen | TDP-43-L95A |
| Rabbit antibody to human ChAT protein, clone EPR16590 | Abcam | ab178850; RRID:AB_2721842 |
|
| ||
| Biological samples | ||
|
| ||
| Frozen human ALS and control spinal cord tissues | University of Miami Brain Bank | B2AE76 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Carboxymethylcellulose | Sigma Aldrich | 419273–100G |
| Superfrost PlusGold slides | Fisher Scientific | 15–188-48 |
| Zeiss PEN membrane slides | Zeiss | 415190–9041-000 |
| 10% Neutral Buffered Formalin | Fisher Scientific | SF100–4 |
| 1X phosphate buffered saline | Abcam | ab128983 |
| Alcohol, 70%, Fisherbrand™, HistoPrep™ | Fisher Scientific | HC-1000–1GL |
| Dimethyl sulfoxide | ThermoFisher Scientific | 022914.M1 |
| Mayer’s Hematoxylin | Sigma Aldrich | MHS16–500mL |
| Eosin Y solution | Sigma Aldrich | 110116–500mL |
| Scott’s bluing reagent | Ricca Chemical Company | 6697–32 |
| Ventana DISCOVERY Red Kit | Roche | 760–228 |
| Ventana DISCOVERY Yellow Kit | Roche | 760–239 |
| Pierce HeLa Protein Digest Standard | ThermoFisher Scientific | 88329 |
| Pierce Formic Acid, LC-MS grade | ThermoFisher Scientific | 28905 |
| Dithiothreitol (DTT) | ThermoFisher Scientific | R0861 |
| Iodoacetamide (IAA) | ThermoFisher Scientific | 122275000 |
| dodecyl-β-d-maltopyranoside (DDM) | ThermoFisher Scientific | 329370010 |
| CHROMASOLV™ LC-MS water | Honeywell | 392531L |
| CHROMASOLV™ LC-MS Acetonitrile | Honeywell | 349674X4L |
| Trypsin, MS-grade | Promega | V5280 |
| Lys-C, MS-grade | Promega | VA1170 |
| Bond Primary Antibody Diluent | Advanced Cellular Diagnostics | AR9352 |
| Bond Polymer Refine Detection Kit | Advanced Cellular Diagnostics | DS9800 |
| BaseScope™ VS Reagent Kit | Advanced Cellular Diagnostics | 323700 |
| BaseScope™ LS Red Reagent Kit | Advanced Cellular Diagnostics | 323600 |
| BaseScope™ Duplex Reagent Kit | Advanced Cellular Diagnostics | 323800 |
|
| ||
| Deposited data | ||
|
| ||
| MassIVE Data Repository | UCSD Center for Computational Mass Spectrometry |
https://massive.ucsd.edu/,Accession: MSV000092119; RRID:SCR_013665 |
| ProteomeXchange/Proteome Central Repository | ProteomeXchange Consortium | http://www.proteomexchange.org/, Accession: PXD042799; RRID:SCR_004055 |
|
| ||
| Oligonucleotides | ||
|
| ||
| BaseScope positive control probe | Advanced Cellular Diagnostics | BA-Hs-POLR2A-3zz |
| BaseScope negative control probe | Advanced Cellular Diagnostics | BA-dapB-3zz |
| BaseScope probe targeting canonical Stathmin2 | Advanced Cellular Diagnostics | BA-Hs-STMN2–3zz-st |
| BaseScope probe targeting cryptic Stathmin2 | Advanced Cellular Diagnostics | BA-Hs-STMN2-intron1–1zz-st1 |
|
| ||
| Software and algorithms | ||
|
| ||
| Proteome Discoverer software, v2.3 | ThermoFisher Scientific | https://thermo.flexnetoperations.com/control/thmo/login; RRID:SCR_014477 |
| Visiopharm Image Analysis software | Visiopharm A/S | https://visiopharm.com/; RRID:SCR_021711 |
| GraphPad Prism software, v9–10 | GraphPad Software | https://www.graphpad.com/features; RRID:SCR_002798 |
| CaseViewer software | 3DHistech | https://www.3dhistech.com/solutions/caseviewer/; RRID:SCR_017654 |
| limma R package | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html; RRID:SCR_010943 |
| Perseus | Juergen Cox Lab, Max Planck Institute of Biochemistry |
https://maxquant.net/perseus/;RRID:SCR_015753 |
| Cytoscape | Cytoscape Tea, Institute for Systems Biology | https://cytoscape.org/; RRID:SCR_003032 |
| String Database | STRING CONSORTIUM | https://string-db.org/; RRID:SCR_005223 |
|
| ||
| Other | ||
|
| ||
| Zeiss PALM MicroBeam Laser Capture Microscope |
Zeiss | PALM MicroBeam; RRID:SCR_020929 |
| Pannoramic SCAN II Whole Slide Imager | 3DHistech | SCAN II |
| Leica CM1520 cryostat | Leica | CM1520; RRID:SCR_017543 |
| UltiMate 3000 RSLCnano HPLC system | ThermoFisher Scientific | UltiMate 3000; RRID:SCR_020563 |
| Thermo Orbitrap Exploris 480 mass spectrometer |
ThermoFisher Scientific | Orbitrap Exploris 480; RRID:SCR_022215 |
| Ventana BenchMark Ultra autostainer system | Roche | Ventana BenchMark Ultra |
| Leica BOND autostainer system | Leica Biosystems | Leica BOND |
Highlights.
Single-cell proteomic analysis of human ALS motor neurons directly captured by LMD
ALS motor neuron deficiencies in splicing, translation, and vesicular transport machinery
Stratification of single-neuron proteomes based on pathologic TDP-43 inclusions
Utility of trace sample proteomics in augmenting the study of human neurological diseases
ACKNOWLEDGMENTS
We express gratitude first and foremost to the individuals and families who generously donated tissues for the advancement of medical research and the University of Miami Brain Endowment Bank (funded by NIMH, NINDS, NICHD) for sample collection and provision. We further thank Biogen and BYU colleagues and group members for their insightful discussions and feedback. This research was supported by NCI/NIH awards R33 CA225248 and R01 GM138931 to R.T.K. and through a sponsored research agreement with Biogen.
Footnotes
DECLARATION OF INTERESTS
A.J.G., J.-H.C., S.X., J.G., and E.D.P. are employees and shareholders of Biogen.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113636.
REFERENCES
- 1.Miller RG, Mitchell JD, and Moore DH (2012). Riluzole for Amyotrophic Lateral Sclerosis (ALS)/motor Neuron Disease (MND) (Cochrane Database Syst Rev). CD001447. [DOI] [PubMed] [Google Scholar]
- 2.Witzel S, Maier A, Steinbach R, Grosskreutz J, Koch JC, Sarikidi A, Petri S, Günther R, Wolf J, Hermann A, et al. (2022). Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 79, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, Karam C, Caress JB, Owegi MA, Quick A, et al. (2020). Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 383, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chiò A, Van Damme P, Ludolph AC, Glass JD, et al. (2022). Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 387, 1099–1110. [DOI] [PubMed] [Google Scholar]
- 5.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, and Oda T (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. [DOI] [PubMed] [Google Scholar]
- 6.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 8.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling JP, Pletnikova O, Troncoso JC, and Wong PC (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito-Armas JM, Kalmar B, Ule A, Yu Y, et al. (2018). Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 37, e98684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, et al. (2022). TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22. 180–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, and Lee EB (2019). Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 27, 1409–1421.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelen-Lee J, Blokhuis AM, Spliet WGM, Pasterkamp RJ, Aronica E, Demmers JAA, Broekhuizen R, Nardo G, Bovenschen N, and Van Den Berg LH (2017). Proteomic profiling of the spinal cord in ALS: decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 210–220. [DOI] [PubMed] [Google Scholar]
- 16.Umoh ME, Dammer EB, Dai J, Duong DM, Lah JJ, Levey AI, Gearing M, Glass JD, and Seyfried NT (2018). A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iridoy MO, Zubiri I, Zelaya MV, Martinez L, Ausín K, Lachen-Montes M, Santamaría E, Fernandez-Irigoyen J, and Jericó I (2018). Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 20, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedl TJ, San Gil R, Cheng F, Rayner SL, Davidson JM, De Luca A, Villalva MD, Ecroyd H, Walker AK, and Lee A (2019). Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 13, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Piehowski PD, Zhao R, Chen J, Shen Y, Moore RJ, Shukla AK, Petyuk VA, Campbell-Thompson M, Mathews CE, et al. (2018). Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho R, Workman MJ, Mathkar P, Wu K, Kim KJ, O’Rourke JG, Kellogg M, Montel V, Banuelos MG, Arogundade OA, et al. (2021). Cross-Comparison of Human iPSC Motor Neuron Models of Familial and Sporadic ALS Reveals Early and Convergent Transcriptomic Disease Signatures. Cell Syst. 12, 159–175.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly RT (2020). Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteomics 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cong Y, Motamedchaboki K, Misal SA, Liang Y, Guise AJ, Truong T, Huguet R, Plowey ED, Zhu Y, Lopez-Ferrer D, and Kelly RT (2020). Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 12, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Clair G, Chrisler WB, Shen Y, Zhao R, Shukla AK, Moore RJ, Misra RS, Pryhuber GS, Smith RD, et al. (2018). Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem. Int. Ed. Engl. 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Dou M, Piehowski PD, Liang Y, Wang F, Chu RK, Chrisler WB, Smith JN, Schwarz KC, Shen Y, et al. (2018). Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell. Proteomics 17, 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehmkuhl EM, and Zarnescu DC (2018). Lost in Translation: Evidence for Protein Synthesis Deficits in ALS/FTD and Related Neurodegenerative Diseases. Adv. Neurobiol. 20, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue YC, Ng CS, Xiang P, Liu H, Zhang K, Mohamud Y, and Luo H (2020). Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 13, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S, Kaufman RJ, Weppler SA, Voncken JW, et al. (2006). Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 25, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheaton WW, and Chandel NS (2011). Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 300, C385–C393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, et al. (2003). VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 34, 383–394. [DOI] [PubMed] [Google Scholar]
- 30.Kim SM, Kim H, Lee JS, Park KS, Jeon GS, Shon J, Ahn SW, Kim SH, Lee KM, Sung JJ, and Lee KW (2013). Intermittent hypoxia can aggravate motor neuronal loss and cognitive dysfunction in ALS mice. PLoS One 8, e81808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner MR, Goldacre R, Talbot K, and Goldacre MJ (2016). Cerebrovascular injury as a risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 87, 244–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagara Y, Tateishi T, Yamasaki R, Hayashi S, Kawamura M, Kikuchi H, Iinuma KM, Tanaka M, Iwaki T, Matsushita T, et al. (2013). Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1alpha in amyotrophic lateral sclerosis. Brain Pathol. 23, 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato K, Morimoto N, Kurata T, Mimoto T, Miyazaki K, Ikeda Y, and Abe K (2012). Impaired response of hypoxic sensor protein HIF-1alpha and its downstream proteins in the spinal motor neurons of ALS model mice. Brain Res. 1473, 55–62. [DOI] [PubMed] [Google Scholar]
- 34.Durrenberger PF, Fernando S, Kashefi SN, Ferrer I, Hauw JJ, Seilhean D, Smith C, Walker R, Al-Sarraj S, Troakes C, et al. (2010). Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J. Neuropathol. Exp. Neurol. 69, 70–81. [DOI] [PubMed] [Google Scholar]
- 35.Lee JW, Ko J, Ju C, and Eltzschig HK (2019). Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 51, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao S, Oyang L, Liang J, Yi P, Han Y, Luo X, Xia L, Lin J, Tan S, Hu J, et al. (2021). Biological Function of HYOU1 in Tumors and Other Diseases. OncoTargets Ther. 14, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jurga AM, Paleczna M, Kadluczka J, and Kuter KZ (2021). Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 11, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodge K, Have ST, Hutton L, and Lamond AI (2013). Cleaning up the masses: exclusion lists to reduce contamination with HPLC-MS/MS. J. Proteomics 88, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mackenzie IRA, and Rademakers R (2008). The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 21, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altman T, Ionescu A, Ibraheem A, Priesmann D, Gradus-Pery T, Farberov L, Alexandra G, Shelestovich N, Dafinca R, Shomron N, et al. (2021). Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat. Commun. 12, 6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boekweg H, Guise AJ, Plowey ED, Kelly RT, and Payne SH (2021). Calculating Sample Size Requirements for Temporal Dynamics in Single-Cell Proteomics. Mol. Cell. Proteomics 20, 100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, Barro C, Kappos L, Comabella M, Fazekas F, et al. (2018). Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 14, 577–589. [DOI] [PubMed] [Google Scholar]
- 44.Pijnenburg YAL, Verwey NA, van der Flier WM, Scheltens P, and Teunissen CE (2015). Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement. 1, 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, Ghidoni R, Pijnenburg YA, Borroni B, Galimberti D, et al. (2016). Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann. Clin. Transl. Neurol. 3, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattsson N, Insel PS, Palmqvist S, Portelius E, Zetterberg H, Weiner M, Blennow K, and Hansson O; Alzheimer’s Disease Neuroimaging Initiative (2016). Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol. Med. 8, 1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weston PSJ, Poole T, Ryan NS, Nair A, Liang Y, Macpherson K, Druyeh R, Malone IB, Ahsan RL, Pemberton H, et al. (2017). Serum neurofilament light in familial Alzheimer disease: A marker of early neurodegeneration. Neurology 89, 2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vagnozzi AN, and Pratico D (2019). Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol. Psychiatry 24, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filippone A, Smith T, and Pratico D (2021). Dysregulation of the Retromer Complex in Brain Endothelial Cells Results in Accumulation of Phosphorylated Tau. J. Inflamm. Res. 14, 7455–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, et al. (2011). VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 195, 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qureshi YH, Berman DE, Marsh SE, Klein RL, Patel VM, Simoes S, Kannan S, Petsko GA, Stevens B, and Small SA (2022). The neuronal retromer can regulate both neuronal and microglial phenotypes of Alzheimer’s disease. Cell Rep. 38, 110262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simoes S, Guo J, Buitrago L, Qureshi YH, Feng X, Kothiya M, Cortes E, Patel V, Kannan S, Kim YH, et al. (2021). Alzheimer’s vulnerable brain region relies on a distinct retromer core dedicated to endosomal recycling. Cell Rep. 37, 110182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimprich A, Benet-Pagè s A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eleuteri S, and Albanese A (2019). VPS35-Based Approach: A Potential Innovative Treatment in Parkinson’s Disease. Front. Neurol. 10, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, and Kim TW (2005). Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 58, 909–919. [DOI] [PubMed] [Google Scholar]
- 57.Tang FL, Zhao L, Zhao Y, Sun D, Zhu XJ, Mei L, and Xiong WC (2020). Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons. Cell Death Differ. 27, 2099–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pérez-Torres EJ, Utkina-Sosunova I, Mishra V, Barbuti P, De Planell-Saguer M, Dermentzaki G, Geiger H, Basile AO, Robine N, Fagegaltier D, et al. (2022). Retromer dysfunction in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 119. e2118755119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muzio L, Sirtori R, Gornati D, Eleuteri S, Fossaghi A, Brancaccio D, Manzoni L, Ottoboni L, Feo LD, Quattrini A, et al. (2020). Retromer stabilization results in neuroprotection in a model of Amyotrophic Lateral Sclerosis. Nat. Commun. 11, 3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao W, Todd TW, Wu Y, Jones CY, Tong J, Jansen-West K, Daughrity LM, Park J, Koike Y, Kurti A, et al. (2022). Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 378, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu G, Coyne AN, Pei F, Vaughan S, Chaung M, Zarnescu DC, and Buchan JR (2017). Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 8, 2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, et al. (2007). TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 171, 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi M, Kitaura H, Kakita A, Kakihana T, Katsuragi Y, Onodera O, Iwakura Y, Nawa H, Komatsu M, and Fujii M (2022). USP10 Inhibits Aberrant Cytoplasmic Aggregation of TDP-43 by Promoting Stress Granule Clearance. Mol. Cell Biol. 42, e0039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watanabe R, Higashi S, Nonaka T, Kawakami I, Oshima K, Niizato K, Akiyama H, Yoshida M, Hasegawa M, and Arai T (2020). Intracellular dynamics of Ataxin-2 in the human brains with normal and frontotemporal lobar degeneration with TDP-43 inclusions. Acta Neuropathol. Commun. 8, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, Tysnes OB, Auburger G, et al. (2011). Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 20, 1697–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, et al. (2015). Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krach F, Batra R, Wheeler EC, Vu AQ, Wang R, Hutt K, Rabin SJ, Baughn MW, Libby RT, Diaz-Garcia S, et al. (2018). Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. 136, 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, and Peles E (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. [DOI] [PubMed] [Google Scholar]
- 70.Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, and Peles E (2001). Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J. Neurosci. 21, 7568–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scott R, Sánchez-Aguilera A, van Elst K, Lim L, Dehorter N, Bae SE, Bartolini G, Peles E, Kas MJH, Bruining H, and Marín O. (2019). Loss of Cntnap2 Causes Axonal Excitability Deficits, Developmental Delay in Cortical Myelination, and Abnormal Stereotyped Motor Behavior. Cereb. Cortex 29, 586–597. [DOI] [PubMed] [Google Scholar]
- 72.Martin-de-Saavedra MD, Dos Santos M, Culotta L, Varea O, Spielman BP, Parnell E, Forrest MP, Gao R, Yoon S, McCoig E, et al. (2022). Shed CNTNAP2 ectodomain is detectable in CSF and regulates Ca(2+) homeostasis and network synchrony via PMCA2/ATP2B2. Neuron 110, 627–643.e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oh S, Y JV, Boxer A, Sockanathan S, and Na C-H (2021). Discovery of Biomarkers for Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration From Human Cerebrospinal Fluid Using Mass Spectrometry-Based Proteomics. Research Square Preprint. (preprint)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ladd AC, Brohawn DG, Thomas RR, Keeney PM, Berr SS, Khan SM, Portell FR, Shakenov MZ, Antkowiak PF, Kundu B, et al. (2017). RNA-seq analyses reveal that cervical spinal cords and anterior motor neurons from amyotrophic lateral sclerosis subjects show reduced expression of mitochondrial DNA-encoded respiratory genes, and rhTFAM may correct this respiratory deficiency. Brain Res. 1667, 74–83. [DOI] [PubMed] [Google Scholar]
- 75.Hartmann H, Hornburg D, Czuppa M, Bader J, Michaelsen M, Farny D, Arzberger T, Mann M, Meissner F, and Edbauer D (2018). Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 1, e201800070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez-Colasante X, Pacut C, et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rabin SJ, Kim JMH, Baughn M, Libby RT, Kim YJ, Fan Y, Libby RT, La Spada A, Stone B, and Ravits J (2010). Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum. Mol. Genet. 19, 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maier T, Güell M, and Serrano L (2009). Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973. [DOI] [PubMed] [Google Scholar]
- 79.Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenäs C, Lundeberg J, Mann M, and Uhlen M (2010). Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Payne SH (2015). The utility of protein and mRNA correlation. Trends Biochem. Sci. 40, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edfors F, Danielsson F, Hallström BM, Käll L, Lundberg E, Pontén F, Forsström B, and Uhlén M (2016). Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 12, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Füllgrabe A, Fuentes AMP, Jupp S, Koskinen S, et al. (2016). Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44, D746–D752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. (2014). Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587. [DOI] [PubMed] [Google Scholar]
- 84.Srzentić K, Zhurov KO, Lobas AA, Nikitin G, Fornelli L, Gorshkov MV, and Tsybin YO (2018). Chemical-Mediated Digestion: An Alternative Realm for Middle-down Proteomics? J. Proteome Res. 17, 2005–2016. [DOI] [PubMed] [Google Scholar]
- 85.Ellwanger DC, Scheibinger M, Dumont RA, Barr-Gillespie PG, and Heller S (2018). Transcriptional Dynamics of Hair-Bundle Morphogenesis Revealed with CellTrails. Cell Rep. 23, 2901–2914.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen H, Albergante L, Hsu JY, Lareau CA, Lo Bosco G, Guan J, Zhou S, Gorban AN, Bauer DE, Aryee MJ, et al. (2019). Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat. Commun. 10, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arrasate M, Mitra S, Schweitzer ES, Segal MR, and Finkbeiner S (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. [DOI] [PubMed] [Google Scholar]
- 88.Subramaniam S, Sixt KM, Barrow R, and Snyder SH (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 324, 1327–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stoica SI, Bleotu C, Ciobanu V, Ionescu AM, Albadi I, Onose G, and Munteanu C (2022). Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery. Biomedicines 10, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hernandez-Gerez E, Fleming IN, and Parson SH (2019). A role for spinal cord hypoxia in neurodegeneration. Cell Death Dis. 10, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cong Y, Liang Y, Motamedchaboki K, Huguet R, Truong T, Zhao R, Shen Y, Lopez-Ferrer D, Zhu Y, and Kelly RT (2020). Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem. 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.R Core Team (2020). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- 94.Välikangas T, Suomi T, and Elo LL (2018). A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, and Jensen LJ (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper are available upon request form the lead contact. Mass spectrometry data generated in this study have been deposited to ProteomeXchange (PXD042799) and MassIVE (MSV000092119).
Custom code is available on GitHub (https://doi.org/10.5281/zenodo.10268532).
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.