

Abstract
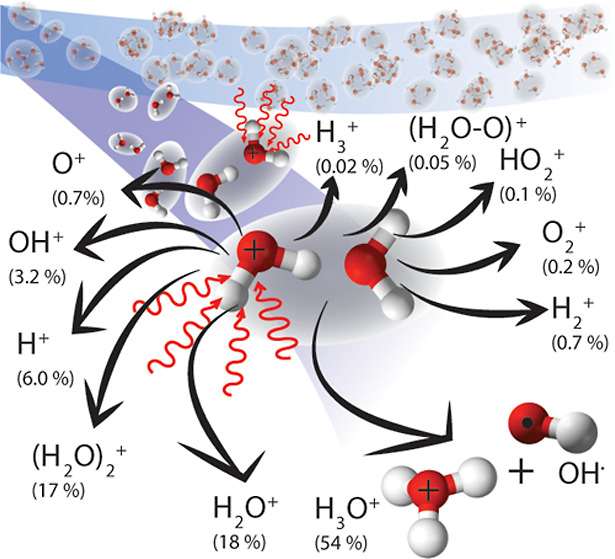
Water dimer (H2O)2—a vital component of the earth’s atmosphere—is an important prototypical hydrogen-bonded system. It provides direct insights into fundamental chemical and biochemical processes, e.g., proton transfer and ionic supramolecular dynamics, occurring in astro- and atmospheric chemistry. Exploiting a purified molecular beam of water dimer and multimass ion imaging, we report the simultaneous detection of all generated ion products of (H2O)+2 fragmentation following single ionization. Detailed information about ion yields and reaction energetics of 13 ion-radical pathways, 6 of which are new, of (H2O)+2 are presented, including strong 18O-isotope effects.
Introduction
Water dimer (H2O)2 is assumed to be a vivid contributor to the radiation budget,1,2 the homogeneous condensation,3 and chemical reactions including degradation of Criegee intermediates4 at low altitudes of the earth’s atmosphere.5−7 Moreover, the decay of the ionic states of water clusters induced by cosmic radiation is considered essential for astrochemistry occurring on ice mantles8−10 and plays a key role as a trigger of chemical evolution in interstellar space.11 Therefore, as (H2O)2 is the smallest water cluster, it is a favorable model system to develop a more thorough understanding of the environmental effect, i.e., hydrogen bonding’s role, in energy, charge, and mass transfer occurring in ion-radical chemistry.12−19
Hydrogen bonding is of major importance for the vast majority of biochemical processes and chemical reactions such as proton transfer in redox reactions of light-harvesting complexes20 or structure and topological stability of biomolecules like the double-helix structure of DNA.21,22 Specifically, understanding how absorbed energy dissipates and charge is redistributed within an ionized aqueous environment is crucial in a biological context, where the interaction of biomolecules with generated low-energy electrons and ion radicals can result in their degradation.
The interest in ionic supramolecular dynamics of the (H2O)2 system led to several theoretical predictions15,16,18,23 of fragmentation pathways of singly and doubly charged water dimer and fragmentation energetics. These were only partly supported by the experimental results of electron-impact ionization24 and of Coulomb explosion12,14,19,25,26 induced by core and valence ionization. This is not surprising, considering the experimental challenge, e.g., the interference of the products of (H2O)+2 fragmentation with those resulting from the fragmentation of higher clusters and isolated H2O molecules. Our experimental approach circumvents these issues by using the electrostatic deflector27 for the purification of (H2O)2 samples and multimass ion imaging28−30 to monitor all the ion-radical channels induced by strong-field ionization at once. The photoionization was set to be predominantly in the multiphoton regime to populate similar states of the (H2O)+2 ion as those populated from VUV/UV- or electron-impact ionization.
Ab initio simulations revealed that the photoionization of (H2O)2 through electron ejection from the four highest occupied molecular orbitals (HOMO to HOMO – 3) preferably leads to proton transfer.15 The first two ionic states of (H2O)+2, i.e., the ground 2A″ and the first excited 2A′ states, are populated by electron ejection from the two nonbonding 1b1 orbitals of oxygen in the proton donor (HOMO) and the proton acceptor (HOMO – 1), respectively.15,18 Both of these states are highly reactive, which is underlined by the estimated time scale for proton migration of less than 100 and 300 fs for (H2O)+2 in its 2A″ and 2A′ states, respectively.15 Recently, the time scale for the proton migration was measured as 55(20) fs in XUV-pump-XUV-probe experiments.26 Subsequently, (H2O)+2 either dissociates into
| 1 |
or survives as an ion-radical pair (<40%)15,16
| 2 |
EA denotes the specific appearance energy, i.e., the minimum energy required to induce the reaction pathway.
The next two excited states of (H2O)+2 are populated through electron ejection from the 3a1 orbital of donor (HOMO–2) and acceptor (HOMO – 3) H2O followed by the prompt decay15 into the 2A″ and 2A′ states with subsequent dissociation via the channel in (1). Additional minor channels (<20%) either lead to an ion-radical pair (H2O)+2 through the channel in (2) or fragmentation without proton transfer as
| 3 |
Furthermore, the electron ejection from the 1b2 orbital of the donor (HOMO–4) and acceptor (HOMO–5) predominantly leads to the three-body fragmentation as
| 4 |
generating four species: three radical fragments and an electron. Those species should eminently increase the possibility of radiation damage in an aqueous environment.15 The two-body fragmentation channels producing H3O+ and H2O+ ions participate only with a minor contribution (<20%) after HOMO–4 and HOMO–5 ionization.15
Additional channels of (H2O)+2 fragmentation appearing at higher ionization energies were described,15,16 i.e.
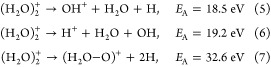 |
5 |
Contrary to valence ionization, stripping of a core electron from the 2a1 or 1a1 orbitals, e.g., by X-ray ionization, induces direct or proton-transfer-mediated13,17 relaxation through local or intermolecular Auger decay12−14,17 and subsequent ejection of a valence electron.
Here, we exploited the combination of electrostatic deflection31 and multimass imaging with a Timepix3 camera28,29 to yield unprecedented details into the ionic reaction pathways of the prototypical ionized (H2O)2 system. We produced a rotationally cold molecular beam of (H2O)2 with ∼92% purity27 and recorded all ionic reaction products, see Methods and the Supporting Information for details. We directly observed all theoretically predicted fragmentation channels of (H2O)+2.15,16 Moreover, we observed multiple new fragmentation channels. The velocity-map-imaging (VMI) detection inherently provided information about the released translational energy and rovibronic excitation of the products, which could affect subsequent reactions.
Methods
All experiments were performed in our recently commissioned transportable endstation for controlled-molecule experiments (eCOMO), which will be described in more detail elsewhere.32Figure 1 provides a sketch of the experimental setup.32,33 A sample of distilled water (0.5 μL, room temperature) was dropped on a glass-filter paper and installed in a sample holder behind the pulsed valve (Amsterdam Piezo Valve).34 The water vapor was mixed with helium buffer gas using a stagnation pressure of 4 bar and the mixture coexpanded into vacuum in 50 μs pulses into the source chamber; pressures with the valve on/off were 2 × 10–6 mbar/2 × 10–8 mbar. The molecular beam of water clusters was extracted by a first skimmer (diameter of 3 mm) from the supersonic jet and further collimated by a second skimmer (diameter of 1.5 mm) before entering the electrostatic deflector.
Figure 1.

Schematic picture of the endstation for controlled-molecule experiments (eCOMO).32,33
To spatially separate the water dimer clusters from the carrier helium gas as well as from isolated water molecules and larger water clusters, we used a b-type deflector,31,35 applying a voltage of 13 kV. The dipole moments of water monomer and dimer are 1.86 and 2.63 D, respectively.27,36,37 Subsequently, the molecular beam was cut in half by a knife edge to increase both, the effective separation of water dimer from the rest of the molecular beam and the column density.33 The source, deflector, knife edge, and skimmers are all movable, which enabled us to optimize their positions for the separation and purification of the water-dimer clusters. The deflected molecules were intersected with short laser pulses in the interaction region of a double-sided VMI spectrometer. The purity of the water-dimer beam was estimated to be 92% by comparing the signals from known fragmentation channels to the background signal, i.e., water monomer, nitrogen, and oxygen, at the deflected-beam position of 2 mm.
To photoionize the molecules, we used an 800 nm Ti/sapphire chirped-pulse-amplifier system (Coherent Astrella) operated at 1 kHz. The pulse duration is estimated to be 40 fs and the pulse energies, 170 μJ. Focusing the laser beam to 53 μm full width at half-maximum intensity yielded a peak intensity up to ∼2 × 1014 W/cm2, corresponding to a Keldysh parameter of ∼0.7 for photoionization of (H2O)2 at Ei = 11.7 eV.38
The generated ions were then accelerated toward the detector in a perpendicular geometry of the double-sided spectrometer39 and projected under VMI conditions. At the end of the spectrometer, a microchannel-plate–phosphor-screen combination (MCP: Photonics, APD 3 PS 75/32/25/8 I 60:1 NR MGO 8″FM; PS: P47) was mounted to produce light flashes for individual ions. The flashes were detected with a Timepix3 camera28,40 (Amsterdam Scientific Instruments) operated and controlled by our open-source-library PymePix,41,42 which was also used to extract the raw physics events from the Timepix3 data stream. The detector’s temporal resolution of ∼1.6 ns28 enabled us to work in multimass-detection mode and to obtain the VMI images of all fragments directly by slicing, i.e., computational event selection, in the time-of-flight coordinate. The synchronization of the whole experiment is provided by a Stanford DG645 delay generator.
The plotted VMI images and beam-plot figures were background-subtracted and also stripped off the contribution from the scattered/nondeflected beam by subtraction of the measured background-subtracted signal with the deflector off. The valve was operated at 200 Hz resulting in 4 successive background measurements due to the 5 times higher repetition rate of the laser with respect to the molecular-beam valve. The data were centroided by calculating the center of mass for each detected ion.28 The 3D velocity (radial) and angular distributions were obtained by standard integration methods for VMI images.43 For momentum calibration,28 we fitted the dependence of the center positions of the VMI images on the time of arrival and converted it to momenta applying the physical size of the ion detector and the magnification factor (M = vi/vmb = 0.843) of the spectrometer estimated by SIMION.44 The resulting velocity of the molecular beam is 2 km/s.
Results and Discussion
Figure 2 depicts an overview of all acquired ions and their fragmentation channels resulting from singly and multiple-charged water dimers after strong-field ionization at 800 nm. The image shows the background-subtracted ion signal for the mass-over-charge ratio and the position of arrival at the detector; see the Supporting Information for details. The mass-over-charge ratio m/q is obtained by a direct transformation from the ion time of flight by m/q ∝ t2. Due to the molecular beam velocity, in this two-dimensional spectrum, all signals from species originating from the molecular beam are shifted upward in the figure with increasing mass-over-charge ratio. Ions with zero velocity in the co-moving frame of the molecular beam appear on the dashed line. Ions in the surrounding structures centered at the dashed line are due to fragmentation of the cluster after ionization. The remaining signals are due to the ionization of the diffuse rest gas in the chamber.
Figure 2.
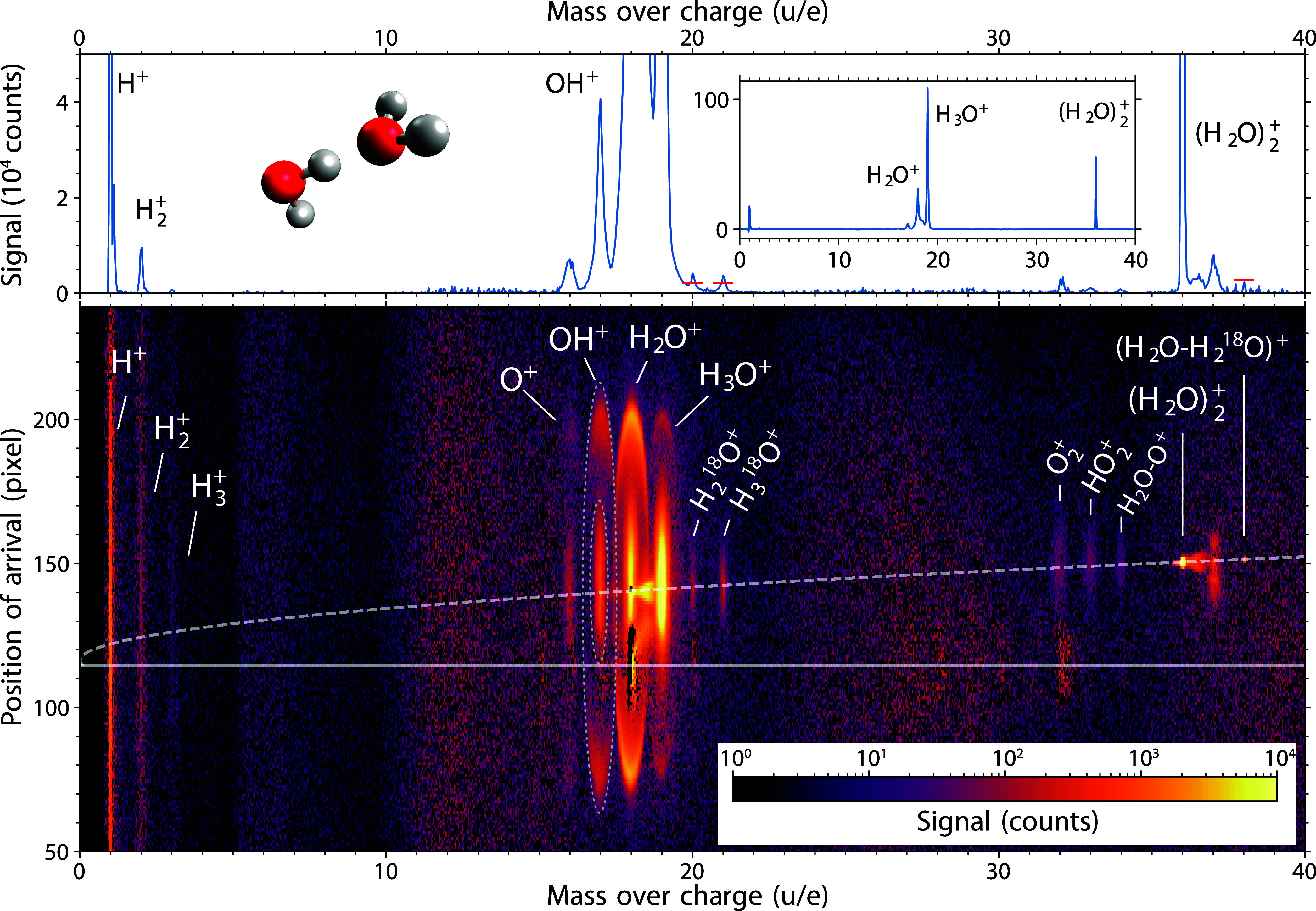
Image mapping the ion signal according to the mass-over-charge ratio and the position of arrival on the detector (lower panel). The upper panel outlines the zoomed-in mass spectrum, with an inset showing the whole vertical range for reference. The observed fragments of (H2O)+2 are H+, H+2, H+3, O+, OH+, H2O+, H3O+, O+2, HO+2, and (H2O–O)+. The observed fragments of (H2O–H218O)+ are H218O+ and H318O+. The expected isotopologue-peak heights of H218O+, H318O+, and (H2O–H218O)+ ions are indicated by the red horizontal bars. Dotted ellipses at m/q = 17 u/e are drawn to mark product regions of (H2O)+2 fragmentation and Coulomb explosion as an example. See text for details.
For a direct comparison between the various fragments in terms of signal strength, the inset in the top graph of Figure 2 depicts the mass spectrum obtained by the summation of the signal in the lower figure along the vertical axis. The top figure itself is a zoom into the inset to highlight weak channels.
The most prominent features in Figure 2 are observed in the region between m/q = 16 and 19 u/e, corresponding to the O+, OH+, H2O+, and H3O+ fragments. These fragments originated from the (H2O)+2 fragmentation and Coulomb-explosion channels of (H2O)2+2. We were able to distinguish between these two sources of the signal using the 3D ion velocity detection with the Timepix3 camera.28 Whereas (H2O)+2 fragmentation is represented by the central broad features around the dashed line, e.g., confined by a inner dotted ellipse at m/q = 17∼u/e, the Coulomb-explosion channels from (H2O)2+2 exhibit sharp ring-like—graphically projected, oval—structures assigned to “fast” ions, e.g., the area between the two dotted ellipses around m/q = 17∼u/e, which will be discussed in a future publication. The strong peaks at m/q = 16 to 19 u/e are also accompanied by weak signals at m/q = 20 and 21 u/e assigned to the isotopologues H218O+ and H318O+, respectively. The signals corresponding to the expected peak heights for isotopes in natural abundance, i.e., 0.2% of the 16O-isotopologue signal, are indicated by the red horizontal lines in the mass spectrum of Figure 2. The red bar assigned to H218O+ was corrected upward to compensate for the contribution from the neighboring H3O+ signal.
The second region with a strong signal, at m/q = 36 u/e, corresponds to the parent ion (H2O)+2. Moreover, it is accompanied by a weak peak of its isotopologue (H2O–H218O)+ at m/q = 38 u/e, again with its expected natural-abundance signal contribution indicated by a red line in the mass spectrum. The signal strength is in very good agreement with the expected abundance. The well-resolved and clear observation of these isotopologues demonstrates the high sensitivity of our experiment. In between the two peaks, there is another structure at m/q = 37 u/e, which we assign to protonated water dimer (H2O)2H+ originating from the fragmentation of larger clusters (H2O)+n, n > 2, i.e., a remaining impurity in the experiment.
For m/q ratios of 32, 33, and 34 u/e, we also observed weak but distinct signals originating in the molecular beam. These signals were assigned to O2+, HO+2, and (H2O–O)+ ions,16 respectively. The origin of these fragments is not straightforwardly linked to any specific ion precursor. Nevertheless, measurements without purification of the reactant by deflection do not exhibit any significantly intense signals between m/q = 32 and 34 u/e, see Figure S1 in the Supporting Information, which excludes their origin from larger water clusters (H2O)n or mixed (H2O)m(O2)n clusters. Furthermore, there are no coincidences of the O+2, HO+2, and (H2O–O)+ ions with H+ and H+2 ions, see Figure S2 in the Supporting Information. Therefore, we assign the signal at 32, 33, and 34 u/e to fragmentation products of (H2O)+2.
The last region with a distinguishable signal is located at m/q = 1–3 u/e. We assign the signal to the hydrogen ions H+, H+2, and H+3, as the isotopic natural-abundance contribution of deuterium D is below 0.02%.
Besides the mass spectrometric information about the created ions and their signal, in VMI, we also obtain insights into the energy redistribution for the specific fragmentation channels through the distributions of kinetic-energy releases, which are displayed for some selected ions in Figure 3; for comparison, see also the released-total-momentum distributions in Figure S3 of the Supporting Information. The kinetic-energy-release spectra were obtained from the measured 3D single-ion momenta, assuming two-body fragmentation of the parent ion.
Figure 3.

Kinetic-energy release of the reaction channels yielding the specified ions, assuming two-body fragmentation.
This is a valid assumption for the H3O+ ions and the larger part of the H2O+ signal according to channels (1) and (3), respectively, with the calculated kinetic-energy release equal to the total kinetic energy Etr. However, the OH+ ions were produced from three-body fragmentation according to channel (5). Here, the most probable scenario is a sequential fragmentation into OH+ with an intermediate H3O, which further dissociates into H2O and H. Thus, the plotted curve of the kinetic-energy release in the OH+ reaction channel illustrates the kinetic energy released in the first step of the three-body fragmentation.
The low-energy peaks up to 2 eV in the Etr spectra are attributed to the decay of various rovibrational states of (H2O)+2. The individual curve shape then results from the convolution of initial rovibronic excitation of (H2O)+2 and subsequent statistical fragmentation. From these distributions, rovibronic excitation energies Erv can be obtained by assuming the conservation of energy given by Etotal = Etr + Erv. In a state-selective experimental approach, such a constraint would allow for the calculation of the exact rovibronic excitation of the generated fragments. However, limitations arise in the ultrashort-pulse ionization regime due to the spectral bandwidth of the ionizing laser, ∼30 nm in our case. Assuming that the deposited energy during the (H2O)2 ionization is given by the lowest number of photons necessary to overcome the energy threshold, i.e., the appearance energy EA, of the selected fragmentation-reaction channel and considering the effective spectral narrowing due to the multiphoton ionization, one could extract low-resolution (>0.04 eV) rovibrational spectra using the dissociation threshold for each detected fragment.
Table 1 provides a summary of previously reported and our new findings: A list of all detected ions from decay of (H2O)+2 is shown in the first column. Seven of the observed ions can be linked to the already reported pathways from simulations.15,16 The additionally detected ions are created through previously unreported fragmentation channels. The fragmentation pathways leading to the ejection of the newly discovered ions and those discussed in the introduction are shown in the last column. These suggested pathways are based solely on energetic arguments and thus should be further investigated, e.g., by computations or through radical detection. From our data, we cannot directly link the detected ions to the exact initial state of the ion. Nevertheless, we can count the number of detected ions and determine their relative ion yields, which are given in the second and third columns, respectively.
Table 1. Summary of All the Observed Ions from (H2O)+2 Dissociation Together With Their Relative Ion Yieldsa.
| fragment | signal (counts) | relative ion yield | EA (eV) | reaction channel: |
|---|---|---|---|---|
| (H2O)+2 → | ||||
| H3O+ | 1,933,751 | 0.541(42) | 11.715 | H3O+ + OH15,16,19 |
| H2O+ | 630,492 | 0.176(11) | 12.815 | H2O+ + H2O15,16,19 |
| 18.215 | H2O+ + OH + H15 | |||
| (H2O)+2 | 613,728 | 0.172(8) | 11.715 | H3O+···OH15,16,19 |
| H+ | 213,905b | 0.06(4) | 19.215 | H+ + H2O + OH15 |
| OH+ | 114,390 | 0.032(8) | 18.515 | OH+ + H2O + H15 |
| O+ | 24,235 | 0.0068(20) | 21.7 | O+ + H2O + 2H |
| H+2 | 13,947 | 0.0068(14) | 21.7 | H+2 + H2O + O |
| H218O+ | 10,225 | 0.0029(1) | ||
| H318O+ | 9268 | 0.0026(3) | ||
| O+2 | 6052 | 0.0017(3) | 27.6 | O+2 + 4H |
| HO+2 | 4206 | 0.0012(2) | 29.2 | HO+2 + 3H |
| (H2O–O)+ | 1792 | 0.0005(1) | 32.616/33.0 | (H2O–O)+ + 2H16 |
| H+3 | 796 | 0.0002(3) | 36.5 | H+3 + HO2 |
Our estimated appearance energies EA are shown in the fourth column in bold, alongside the previously reported appearance energies.15,16 The last column shows previously reported and suggested fragmentation pathways producing the observed ions.
Signal contributions by fast nondetected H+ ions are expected to be below 5% of the integrated H+ signal.
Upon inspecting the ion yields, an intriguing observation is that while the signal intensity of H3O+ is roughly 3 times larger than the signal of H2O+, the situation is changed for their isotopologues H318O+ and H218O+, which exhibit comparable signal intensities, see Figure 4. With the natural abundance of 17O and the sensitivity of our experiment, one would also expect to observe H317O+ and H217O+. Unfortunately, these signals are hidden under the stronger signals of H218O+ and H3O+, respectively. For the H3O+ signal, a background contribution from H317O+ is below 0.04% and thus negligible. For the H218O+ peak at m/q = 20 u/e, a contribution by H317O+ could be as large as 20%. In any case, the difference in the ratios for the H2,316O and H2,318O signals is a surprisingly strong illustration of isotopic substitution modifying chemical-reaction pathways. This could be due to a preferred stereometric position of H218O as a proton donor in (H2O)2, or it could be due to a reduced proton tunneling and transfer probability between the two water moieties in the 18O isotopologue, which could both be a consequence of small reduced-mass changes resulting in zero-point-energy and anharmonic-coupling effects.
Figure 4.

Comparison of the peak-normalized mass spectra including the signals assigned to (blue) H2O+ and H3O+ and (red) H218O+ and H318O+. The red line is background-corrected to exclude the background of the H218O+ peak centered at 20 u/e. This is shown in Figure S6 in the Supporting Information. See also the text for details on possible small 17O backgrounds.
On top of the qualitative analysis, the obtained relative ion yields were also utilized to estimate the appearance energies EA of the newly detected ions and assigned channels. Within our model, the relative ion yields can be expressed as
| 8 |
N is a normalization factor and ySk is the branching ratio of the channel k producing ion I+ after ionization into an initial ionic state S. The probability to ionize into such a state S is described by the distribution D as a function of the ionization energy Ei of the state S and a parameter F corresponding to the strength of the applied external electric field.45 The D was chosen to have an exponential character based on the strong-field ionization approximation.45 The unknown parameters of our model, i.e., F and the branching ratios of H+ and OH+ ions, were calibrated using the theoretical branching ratios and EA of each channel given by Svoboda et al.15 We then calculated the so far unknown EA of the less abundant ions from (8), see the Supporting Information for further details. These estimated EA values are shown in Table 1 including the reported ones.15,16 This includes the EA = 33.0 eV of (H2O–O)+ known from reference (16) to be 32.6 eV. The surprisingly good agreement should be taken with caution and not as an illustration of the high precision of the presented ad hoc model. The calculated and reported appearance energies EA assigned to the observed ions directly reflect the broad range of the initial energies deposited by the strong-field photoionization triggering the (H2O)+2-fragmentation reactions.
Conclusions
Overall, our study provides unique novel experimental observations of the (H2O)+2-fragmentation pathways, which were previously only, and only partly, predicted by molecular dynamics simulations.15,16 These results substantially broaden our perspective on (H2O)+2 fragmentation by showing an additional set of newly observed ion-radical pathways and their relative ion yields. Together with our estimated appearance energies, EA, these indicate the relative significance among all detected channels.
The observed very strong O-isotope effect indicates nonclassical aspects of molecular structure and reactivity in the fragmentation. Together with the experimental information on kinetic energies and angular distributions of all fragments, these data are a very valuable asset for advanced molecular dynamics simulations unraveling the underlying chemistry. Time-resolved investigations of the ionization and subsequent reaction pathways of water dimer could provide further insights into its electronic and nuclear dynamics, including the time scales of proton and hydrogen transfer, the traversal of electronic states, and electronic relaxation processes.
Our findings are relevant for discussions of the role of water ionization and the produced ionic and radical fragments as triggers of the subsequent ion-radical photochemistry, e.g., on ice mantels of dust particles in interstellar space. They also generally help to disentangle a broad inventory of environmentally important radicals.
Acknowledgments
We acknowledge financial support by Deutsches Elektronen-Synchrotron DESY, a member of the Helmholtz Association (HGF), also for the use of the Maxwell computational resources operated at DESY and through the Center for Molecular Water Science (CMWS). This work was further supported by the Matter innovation pool of the Helmholtz Association through the DataX project and through the Helmholtz-Lund International Graduate School (HELIOS, HIRS-0018), and by the federal cluster of excellence “Advanced Imaging of Matter” (AIM, EXC 2056, ID 390715994) of the Deutsche Forschungsgemeinschaft (DFG).
Data Availability Statement
The scripts used to analyze the recorded data and the specified equations are available from the corresponding author upon request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.3c07958.
Experimental ion-imaging map with the deflector-electrodes grounded; photoion–photoion coincidence map; description of a model for calculating the appearance energies; plot of the total-momentum release for specified ions; dependence of the appearance energy on the relative ion yields; and details on the correction of the background in the calculations of the H218O+/H318O+ ratio (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chýlek P.; Geldart D. Water vapor dimers and atmospheric absorption of electromagnetic radiation. Geophys. Res. Lett. 1997, 24, 2015–2018. 10.1029/97GL01949. [DOI] [Google Scholar]
- Vogt E.; Kjaergaard H. G. Vibrational spectroscopy of the water dimer at jet-cooled and atmospheric temperatures. Annu. Rev. Phys. Chem. 2022, 73, 209–231. 10.1146/annurev-physchem-082720-104659. [DOI] [PubMed] [Google Scholar]
- Schenter G. K.; Kathmann S. M.; Garrett B. C. Dynamical nucleation theory: a new molecular approach to vapor-liquid nucleation. Phys. Rev. Lett. 1999, 82, 3484–3487. 10.1103/PhysRevLett.82.3484. [DOI] [Google Scholar]
- Anglada J. M.; Solé A. Impact of the water dimer on the atmospheric reactivity of carbonyl oxides. Phys. Chem. Chem. Phys. 2016, 18, 17698–17712. 10.1039/C6CP02531E. [DOI] [PubMed] [Google Scholar]
- Headrick J.; Vaida V. Significance of water complexes in the atmosphere. Phys. Chem. Earth C 2001, 26, 479–486. 10.1016/S1464-1917(01)00035-6. [DOI] [Google Scholar]
- Tretyakov M. Y.; Koshelev M. A.; Serov E. A.; Parshin V. V.; Odintsova T. A.; Bubnov G. M. Water dimer and the atmospheric continuum. Phys.-Usp. 2014, 57, 1083–1098. 10.3367/UFNe.0184.201411c.1199. [DOI] [Google Scholar]
- Tretyakov M. Y.; Serov E. A.; Koshelev M. A.; Parshin V. V.; Krupnov A. F. Water dimer rotationally resolved millimeter-wave spectrum observation at room temperature. Phys. Rev. Lett. 2013, 110, 093001. 10.1103/PhysRevLett.110.093001. [DOI] [PubMed] [Google Scholar]
- Chaparro Molano G.; Kamp I. The role of OH in the chemical evolution of protoplanetary disks-I. The comet-forming region. Astron. Astrophys. 2012, 537, 138. 10.1051/0004-6361/201015868. [DOI] [Google Scholar]
- Woon D. E. Quantum chemical cluster studies of cation–ice reactions for astrochemical applications: seeking experimental confirmation. Acc. Chem. Res. 2021, 54, 490–497. 10.1021/acs.accounts.0c00717. [DOI] [PubMed] [Google Scholar]
- Ciesla F. J. The phases of water ice in the solar nebula. Astrophys. J. Lett. 2014, 784, L1. 10.1088/2041-8205/784/1/L1. [DOI] [Google Scholar]
- Watanabe N.; Kouchi A. Ice surface reactions: a key to chemical evolution in space. Prog. Surf. Sci. 2008, 83, 439–489. 10.1016/j.progsurf.2008.10.001. [DOI] [Google Scholar]
- Jahnke T.; Sann H.; Havermeier T.; Kreidi K.; Stuck C.; Meckel M.; Schöffler M.; Neumann N.; Wallauer R.; Voss S.; Czasch A.; Jagutzki O.; Malakzadeh A.; Afaneh F.; Weber T.; Schmidt-Böcking H.; Dörner R. Ultrafast energy transfer between water molecules. Nat. Phys. 2010, 6, 139–142. 10.1038/nphys1498. [DOI] [Google Scholar]
- Thürmer S.; Ončák M.; Ottosson N.; Seidel R.; Hergenhahn U.; Bradforth S. E.; Slavíček P.; Winter B. On the nature and origin of dicationic, charge-separated species formed in liquid water on X-ray irradiation. Nat. Chem. 2013, 5, 590–596. 10.1038/nchem.1680. [DOI] [PubMed] [Google Scholar]
- Jahnke T. Interatomic and intermolecular Coulombic decay: the coming of age story. J. Phys. B 2015, 48, 082001. 10.1088/0953-4075/48/8/082001. [DOI] [Google Scholar]
- Svoboda O.; Hollas D.; Ončák M.; Slavíček P. Reaction selectivity in an ionized water dimer: nonadiabatic ab initio dynamics simulations. Phys. Chem. Chem. Phys. 2013, 15, 11531. 10.1039/c3cp51440d. [DOI] [PubMed] [Google Scholar]
- Chalabala J.; Uhlig F.; Slavíček P. Assessment of real-time time-dependent density functional theory (RT-TDDFT) in radiation chemistry: ionized water dimer. J. Phys. Chem. A 2018, 122, 3227–3237. 10.1021/acs.jpca.8b01259. [DOI] [PubMed] [Google Scholar]
- Richter C.; Hollas D.; Saak C. M.; Förstel M.; Miteva T.; Mucke M.; Björneholm O.; Sisourat N.; Slavíček P.; Hergenhahn U. Competition between proton transfer and intermolecular Coulombic decay in water. Nat. Commun. 2018, 9, 4988. 10.1038/s41467-018-07501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada T.; Tachikawa H. Direct ab initio molecular dynamics study on the reactions of multi-valence ionized states of water dimer. J. Phys. B 2021, 54, 145103. 10.1088/1361-6455/ac170b. [DOI] [Google Scholar]
- Zhang C.; Lu J.; Feng T.; Rottke H. Proton transfer dynamics following strong-field ionization of the water dimer. Phys. Rev. A 2019, 99, 053408. 10.1103/PhysRevA.99.053408. [DOI] [Google Scholar]
- Kramer D. M.; Avenson T. J.; Edwards G. E. Dynamic flexibility in the light reactions of photosynthesis governed by both electron and proton transfer reactions. Trends Plant Sci. 2004, 9, 349–357. 10.1016/j.tplants.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Boudaïffa B.; Cloutier P.; Hunting D.; Huels M. A.; Sanche L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- Hanel G.; Gstir B.; Denifl S.; Scheier P.; Probst M.; Farizon B.; Farizon M.; Illenberger E.; Märk T. D. Electron attachment to uracil: effective destruction at subexcitation energies. Phys. Rev. Lett. 2003, 90, 188104. 10.1103/PhysRevLett.90.188104. [DOI] [PubMed] [Google Scholar]
- Svoboda O.; Ončák M.; Slavíček P. Simulations of light induced processes in water based on ab initio path integrals molecular dynamics. II. Photoionization. J. Chem. Phys. 2011, 135, 154302. 10.1063/1.3649943. [DOI] [PubMed] [Google Scholar]
- Buck U.; Winter M. Electron bombardment induced fragmentation of size selected neutral (D2O)n clusters. Z. Phys. D 1994, 31, 291–297. 10.1007/BF01445010. [DOI] [Google Scholar]
- Ch’ng L. C.; Samanta A. K.; Czakó G.; Bowman J. M.; Reisler H. Experimental and theoretical investigations of energy transfer and hydrogen-bond breaking in the water dimer. J. Am. Chem. Soc. 2012, 134, 15430–15435. 10.1021/ja305500x. [DOI] [PubMed] [Google Scholar]
- Schnorr K.; Belina M.; Augustin S.; Lindenblatt H.; Liu Y.; Meister S.; Pfeifer T.; Schmid G.; Treusch R.; Trost F.; Slavíček P.; Moshammer R. Direct tracking of ultrafast proton transfer in water dimers. Sci. Adv. 2023, 9, eadg7864 10.1126/sciadv.adg7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieker H.; Onvlee J.; Johny M.; He L.; Kierspel T.; Trippel S.; Horke D. A.; Küpper J. Pure molecular beam of water dimer. J. Phys. Chem. A 2019, 123, 7486–7490. arXiv:1904.08716 [physics] 10.1021/acs.jpca.9b06460. [DOI] [PubMed] [Google Scholar]
- Bromberger H.; Passow C.; Pennicard D.; Boll R.; Correa J.; He L.; Johny M.; Papadopoulou C.; Tul-Noor A.; Wiese J.; Trippel S.; Erk B.; Küpper J. Shot-by-shot 250 kHz 3D ion and MHz photoelectron imaging using Timepix3. J. Phys. B 2022, 55, 144001.arXiv:2111.14407 [physics] 10.1088/1361-6455/ac6b6b. [DOI] [Google Scholar]
- Cheng C.; Moğol G.; Weinacht T.; Nomerotski A.; Trallero-Herrero C. 3D velocity map imaging of electrons with TPX3CAM. Rev. Sci. Instrum. 2022, 93, 013003. 10.1063/5.0071804. [DOI] [PubMed] [Google Scholar]
- Brouard M.; Halford E.; Lauer A.; Slater C.; Winter B.; Yuen W.; John J.; Hill L.; Nomerotski A.; Clark A.; et al. The application of the fast, multi-hit, pixel imaging mass spectrometry sensor to spatial imaging mass spectrometry. Rev. Sci. Instrum. 2012, 83, 114101. 10.1063/1.4766938. [DOI] [PubMed] [Google Scholar]
- Chang Y.-P.; Horke D. A.; Trippel S.; Küpper J. Spatially-controlled complex molecules and their applications. Int. Rev. Phys. Chem. 2015, 34, 557–590. arXiv:1505.05632 [physics] 10.1080/0144235X.2015.1077838. [DOI] [Google Scholar]
- Jin W.; Trippel S.; Küpper J., et al. , eCOMO, a new endstation for controlled molecule experiments 2023. in preparation. [Google Scholar]
- Trippel S.; Johny M.; Kierspel T.; Onvlee J.; Bieker H.; Ye H.; Mullins T.; Gumprecht L.; Długołęcki K.; Küpper J. Knife edge skimming for improved separation of molecular species by the deflector. Rev. Sci. Instrum. 2018, 89, 096110.arXiv:1802.04053 [physics] 10.1063/1.5026145. [DOI] [PubMed] [Google Scholar]
- Irimia D.; Dobrikov D.; Kortekaas R.; Voet H.; van den Ende D. A.; Groen W. A.; Janssen M. H. M. A short pulse (7 μs FWHM) and high repetition rate (dc–5kHz) cantilever piezovalve for pulsed atomic and molecular beams. Rev. Sci. Instrum. 2009, 80, 113303. 10.1063/1.3263912. [DOI] [PubMed] [Google Scholar]
- Kienitz J. S.; Długołęcki K.; Trippel S.; Küpper J. Improved spatial separation of neutral molecules. J. Chem. Phys. 2017, 147, 024304.arXiv:1704.08912 [physics] 10.1063/1.4991479. [DOI] [PubMed] [Google Scholar]
- Gregory J. K. The dipole moment of the water dimer. Chem. Phys. Lett. 1998, 282, 147–151. 10.1016/S0009-2614(97)01228-1. [DOI] [Google Scholar]
- Gregory J. K.; Clary D. C.; Liu K.; Brown M. G.; Saykally R. J. The water dipole moment in water clusters. Science 1997, 275, 814–817. 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]
- Snow K. B.; Thomas T. F. Mass spectrum, ionization potential, and appearance potentials for fragment ions of sulfuric acid vapor. Int. J. Mass Spectrom. 1990, 96, 49–68. 10.1016/0168-1176(90)80041-Z. [DOI] [Google Scholar]
- Eppink A. T. J. B.; Parker D. H. Velocity map imaging of ions and electrons using electrostatic lenses: application in photoelectron and photofragment ion imaging of molecular oxygen. Rev. Sci. Instrum. 1997, 68, 3477–3484. 10.1063/1.1148310. [DOI] [Google Scholar]
- Zhao A.; van Beuzekom M.; Bouwens B.; Byelov D.; Chakaberia I.; Cheng C.; Maddox E.; Nomerotski A.; Svihra P.; Visser J.; Vrba V.; Weinacht T. Coincidence velocity map imaging using Tpx3Cam, a time stamping optical camera with 1.5 ns timing resolution. Rev. Sci. Instrum. 2017, 88, 113104.arXiv:1707.06253 [physics] 10.1063/1.4996888. [DOI] [PubMed] [Google Scholar]
- Al-Refaie A. F.; Johny M.; Correa J.; Pennicard D.; Svihra P.; Nomerotski A.; Trippel S.; Küpper J. PymePix: a python library for SPIDR readout of Timepix3. J. Instrum. 2019, 14 (10), P10003.arXiv:1905.07999 [physics] 10.1088/1748-0221/14/10/p10003. [DOI] [Google Scholar]
- CFEL Controlled Molecule Imaging PymePix, Code repository, https://github.com/CFEL-CMI/pymepix, 2020.
- Whitaker B.Imaging in Molecular Dynamics: Technology and Applications; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Scientific Instrument Services Inc., USA Simion 8.1, http://simion.com, 2011.
- Ammosov M. V.; Delone N. B.; Krainov V. P. Tunnel ionization of complex atoms and of atomic ions in an alternating electromagnetic field. Sov. Phys. JETP 1986, 64, 1191. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scripts used to analyze the recorded data and the specified equations are available from the corresponding author upon request.