

Abstract

The ability of antimicrobial peptides to efficiently kill their bacterial targets depends on the efficiency of their binding to the microbial membrane. In the case of enterocins, there is a three-part interaction: initial binding, unpacking of helices on the membrane surface, and permeation of the lipid bilayer. Helical unpacking is driven by disruption of the peptide hydrophobic core when in contact with membranes. Enterocin 7B is a leaderless enterocin antimicrobial peptide produced from Enterococcus faecalis that functions alone, or with its cognate partner enterocin 7A, to efficiently kill a wide variety of Gram-stain positive bacteria. To better characterize the role that tertiary structural plasticity plays in the ability of enterocin 7B to interact with the membranes, a series of arginine single-site mutants were constructed that destabilize the hydrophobic core to varying degrees. A series of experimental measures of structure, stability, and function, including CD spectra, far UV CD melting profiles, minimal inhibitory concentrations analysis, and release kinetics of calcein, show that decreased stabilization of the hydrophobic core is correlated with increased efficiency of a peptide to permeate membranes and in killing bacteria. Finally, using the computational technique of adaptive steered molecular dynamics, we found that the atomistic/energetic landscape of peptide mechanical unfolding leads to free energy differences between the wild type and its mutants, whose trends correlate well with our experiment.
Introduction
Bacteriocins comprise a large and diverse family of low molecular weight antibacterial peptides. They function through a mechanism of bacterial cell membrane disruption1 via a receptor-dependent or receptor-independent initial binding event.2−4 The peptides are produced by both Gram-stain positive and Gram-stain negative bacteria; they exhibit activity against bacterial species that are related to the species that produces bacteriocin.5 Hence, in nature, they act as a defense mechanism against competing bacteria.6 Current interest in the application of bacteriocins involves food preservation,7,8 biofilm reduction,9 treating infection,10 and antibiotic-resistant infections.11 The latter include, for example, tuberculosis,12 wound healing,13 and cancer therapeutics.14,15 Enterocins are members of the broader bacteriocin family of antimicrobial and cell penetrating peptides.5 Antimicrobial peptides differ widely in their three-dimensional structure, degree of posttranslational modification, whether they function alone or as a two-peptide system, and their exact mechanism of action.16−18 The simplest peptide form is a single α helix as seen in the peptide magainin-2.19 Larger bacteriocins like Lacticin Q20 are composed of multiple α helices. The antimicrobial peptide laterosporulin is composed of a series of β strands held in place via disulfide bonds.21 A mixed α/β motif is seen in leucocin A.22 Bacteriocins can also be circularized at their N- and C-termini via a peptide bond (e.g., enterocin AS-4823). Finally, bacteriocins can be translated with or without an N-ter leader sequence that directs the secretion process. For a complete review of bacteriocin structure, see ref (24).
The mechanism of bacteriocin-mediated bacterial cell death is pleotrophic19 and complex.25 Bacteriocins can produce huge toroidal pores (HTPs) (e.g., lacticin Q), smaller pores or slits (e.g., AS-48), or generalized membrane permeation without pore formation (e.g., aureocin A53). The generalized mechanism is reviewed by Bechinger and Gorr26 and Marquette and Bechinger.19 Disruption of Gram-stain positive membranes is a more difficult process due to the existence of a peptidoglycan cell wall27,28 and may involve other features that are normally associated with bacteriocin activity being critical: the degree of cationic character, surface hydrophobicity, and the position of amphiphilic helices. In fact, tryptophan may play an important role in interactions with the bacterial lipid bilayers and other membrane components.29 Pore formation may be in fact primarily driven by surface tension exerted on the membrane by the peptide30 or be proton motive force driven.31
Enterocins 7A and 7B are a Class II (subclass IIb) enterocin pair that are produced by the Gram-stain positive bacterium Enterococcus faecalis 710C.32 The peptides are active against several species of the genera Enterococcus, Lactobacillus, and Bacillus.5 The peptides are leaderless and linear (versus circularized) and do not contain disulfide bonds. Their amino acid sequence revealed that enterocins 7A/7B were completely identical to enterocins MR10A and MR10B33 and highly identical to the enterocin L50A/L50B pair that is produced by the related bacterium, Enterococcus faecium L50 (98 and 95% amino acid identity respectively34). The selected peptides are 74% identical with each other. They are less identical to but still related to lacticin Q and aureocin A53. Enterocins 7A and 7B function together (although they do not form heterodimers in solution35), but also have significant activity against bacterial targets when acting alone. This is an unusual feature for a two-peptide bacteriocin system36 and as we show in this paper antimicrobial activity and membrane disruption occurs with enterocin 7B alone. Although they are produced with a formylated N-terminus in vivo, N-formylation is not required for activity. An interesting observation is that both enterocin 7 peptides are fully structured in water35 unlike other bacteriocins. The nuclear magnetic resonance solution structures of enterocin 7A and 7B were determined.35 For simplicity, in this work, the focus is solely on enterocin 7B.
The major characteristics of enterocin 7B are that it is a 43 amino acid peptide of 5182 Da with a pI of 10.2, it contains 8 positively charged amino acid residues and 3 tryptophan residues, and has an 86.3 aliphatic index, where all the aliphatic residues are in the hydrophobic core. The structure (PDB code 2M60(35)) indicates that the peptide is nearly completely α-helical; comprising an N-terminal helix (residues 4–10), a second helix (residues 14–29), and a third helix (residues 33–43). The second helix is kinked at residue 19. Helices one and two are nearly coplanar, while helix three rises above the two as can also be seen in Figure 1a. The helices are amphiphilic and bury a hydrophobic core of approximately 940 Å3. The distribution of cationic amino acid residues results in a positively charged surface. The structure of enterocin 7B suggests a possible interaction scheme with the membranes. The first step in the interaction is the initial contact between the membrane and the peptide. This could be driven by surface electrostatics and hydrophobicity. The second step could be unpacking the helices on the membrane, which exposes the hydrophobic core and drives peptide insertion into the membrane. The final step could be either pore formation or membrane permeabilization in the absence of pore formation. Of those steps, the exposure of the hydrophobic core and the degree to which three-dimensional structure plasticity drives the efficiency of that exposure are the subjects of this work. A series of arginine single-site mutants were designed to disrupt the hydrophobic core and the activity of those mutants to kill Bacillus subtilis and to interact with synthetic membranes was determined. In addition, the change of free energy along the unfolding paths was obtained using adaptive steered molecular dynamics (ASMD)37 to observe the hydrophobic core energetics and dynamics at the atomistic level.
Figure 1.

Structure of enterocin 7B (PDB 2M60, WT) in panel a shown as a ribbon diagram with the inner and outer faces shown in cyan and red, highlighting the residue locations (in licorice) that are muted to construct the seven mutants shown in panels b–h, as labeled. In the latter seven panels, the (WT) is also included in blue to highlight the resulting differences in the minimum energy structures. The orientation of the WT in panels b–h is the same, but differs from that in panel a in order to highlight the differences in structure to the mutants.
ASMD was developed as an enhanced sampling method based on SMD to efficiently investigate the protein unfolding process in stages.38 In SMD, several nonequilibrium trajectories can be sampled to reveal the underlying mechanisms and obtain the equilibrium free energy over the reaction coordinate with the use of the Jarzynski average (JE). However, the determination of the change in free energy through the averaging of Boltzmann-weighted work values in the JE can impede the use of SMD because the work values spread with the extent of steering, leading to decreasing accuracy and increasing error bars. Thus, the ASMD introduces stages to limit the spread of the work values along the steered direction. In between stages, a contraction of the space of configurations represented by the end of the trajectories from the previous stages is performed39,40 to retain a representative sample space for the equilibrium distribution. The choice of contraction leads to several methodological variants.37−42 In this work, naïve ASMD37−39 was selected as it is the most efficient method, and the unfolding of the protein did not appear to require sampling across a landscape with multiple barrier regions.
We hypothesize that the mutations shown in Figure 1 destabilize the enterocin 7B hydrophobic core. This might be expected because molten globule proteins possess dramatically increased ability to permeabilize and disrupt membranes,43−45 and because we eliminated several alternative hypotheses as detailed in the root-mean-square error (Supporting Information). The effect from the selected mutations can, in general, help to further clarify the mechanism of helical unpacking in that the membrane surface can itself serve as a moderately denaturing agent46 by lowering the energy barrier for the transition to a partially unfolded peptide form—viz, an unpacked helix.47 This effect would be magnified with a system consisting of an already destabilized hydrophobic core. In conjunction with intrinsic membrane effects, the efficiency of peptide-membrane interactions may be predominantly a function of the native flexibility of the peptide.48
A variety of measures were utilized to test the hypothesis that a more plastic hydrophobic core results in an increase in the level of enterocin 7B activity. These techniques included near and far UV circular dichroism (CD), determination of the melting temperature, measurement of the minimal inhibitory concentration (MIC) against a bacterial target, calcein release kinetics from large unilamellar liposomes, and ASMD-based dynamics calculations. Taken together, a clearer picture emerges for the role of the hydrophobic core in enterocin 7B in determining its interactions with membranes.
Materials and Methods
Biochemical Experimental Methods
Chemicals and Reagents
All general chemicals, bacterial growth media, chromatography reagents, calcein, and laboratory supplies were obtained from Millipore-Sigma, Inc. (St. Louis, MO). Enterocin 7B peptides were synthesized using standard solid phase chemistry and were HPLC purified to approximately 98% homogeneity by New England Peptide (Framingham, MA; now owned by Biosynth, Inc., Louisville, KY). Phosphatidylglycerol (PG), phosphoethanolamine (PE), and liposome making equipment were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Bacillus subtilis (strain 168, subspecies subtilis, ATCC 23857) was purchased from the American Type Culture Collection (Manassas, VA).
Bacterial Growth and Determination of MIC
Cultures of B. subtilis were grown aerobically at 30 °C in Potato Extract Media (ATCC medium 415). Typically, growth experiments utilized a fresh bacterial culture taken from a freezer stock and passaged twice in liquid media. Lyophilized peptides were diluted to a stock concentration of 1.0 mM in 5.0 mM HEPES (pH 7.5), aliquoted, and frozen at −80 °C until needed. Thawed peptides were not refrozen or used if they were unfrozen longer than 2 days. Growth assays were performed in 96-well microtiter plates in a well volume of 300 μL. Optical density (at 650 nm) was recorded as a function of time in a Molecular Devices, Inc. SpectraMax M5 spectrophotometer. To determine MIC values, peptides were diluted to concentrations between 1.0 nM and 0.1 mM (final concentration) in distilled/deionized water and added to wells containing either a newly inoculated B. subtilis culture (1:20 dilution from an overnight culture) or to wells containing bacteria in mid log phase (approximately 0.4 OD at 650 nm). Growth was monitored over the course of the experiment (typically 10 h). For initial experiments, MIC determinations were monitored for 24 h and were conducted according to Kowalska-Krochmai and Dudek-Wicher49 as part of the European Society of Clinical Microbiology and Infectious Diseases MIC recommendations. Subsequent determinations were shortened to 10 h as that was the optimal time frame for B. subtilis growth. Multiple MIC determinations were conducted (and reported as the mean ± standard deviation) once the optimal enterocin dilutions were determined. This is because biological antimicrobial effects are more pleiotropic in MIC than are traditional small molecule antimicrobials. The low standard deviations reported give confidence to the repeatability/robustness of the MIC values.
Circular Dichroism
Peptides were dissolved to a final concentration of between 5 and 20 μM (far UV CD, 200–250 nm) or between 80 and 125 μM (near UV CD, 250–320 nm) in 10 mM NaPO4 (pH 7.2). Peptide concentration was determined using an extinction coefficient of 17,990 M–1 cm–1 at 280 nm. All CD spectra reported here are presented as the average of three independent determinations, and a buffer-only spectrum was subtracted from the raw scans. No baseline correction was performed. Spectra were obtained using an Applied Photophysics Chirascan spectrophotometer in a 1 mm (far UV) or 2 mm (near UV) path length quartz cuvette. Raw CD signal was converted to mean molar ellipticity according to
| 1 |
where m is the raw CD reading in millidegrees, l is the cell path length in millimeters, n is the number of amino acids in the peptide, and C is the micromolar concentration. For experiments to determine the melting temperature of the peptides, far UV CD spectra were obtained at 1 °C intervals after a 5 min equilibration at the new temperature, with an averaging time of 5 s. Data were best fit to a simple 2-state model (F ↔ U) by global analysis using the fitting software from Applied Photophysics. Thermal denaturation was fully reversible as evidenced by the recovery of 99% of the CD signal upon cooling and the confirmation that the forward and reverse melting curves were fully superimposable. Data represented in this paper are the average of three independent (replicate) experiments, which are all within 0.3 °C of Tm.
Liposome Construction and Calcein Release Assay
Calcein-encapsulated unilamellar liposomes were formed from an equimolar mixture of PG/PE. The lipid mixture was dissolved in chloroform and was dried under a nitrogen stream followed by two h of vacuum drying at room temperature. The resulting film was rehydrated in 5.0 mM HEPES (pH 7.5) and 100 mM calcein. The suspension was freeze–thawed five times followed by extrusion through two stacked polycarbonate filters (100 nm pore size) for a total of 10 times. Free calcein was removed from the liposome preparation via gel filtration chromatography using a Sephadex G-50 column (10 × 1.5 cm). Column buffer consisted of 10 mM HEPES (pH 7.5), 100 mM NaCl, and 1.0 mM sodium EDTA. Calcein-free unilamellar liposomes were made in a similar fashion and were utilized to adjust the final liposome concentration. The calcein release assay measures the increase in fluorescence emission intensity at 520 nm (excitation at 500 nm) that results from the movement of calcein from inside the unilamellar liposome (where it is quenched) to the bulk medium due to peptide-induced membrane damage. The maximum fluorescence associated with 100% leakage is determined by the addition of 10% Triton X-100. The percent calcein released (meaning the percent leakiness) is calculated as
| 2 |
where Fp, F0, and FT are the fluorescence emission intensities in the presence of the peptide, in the absence of the peptide, and in the presence of Triton X-100; respectively. The assays were conducted in a total volume of 400 μL in black 96-well microtiter plates in Molecular Devices, Inc. SpectraMax M5 spectrophotometer. Reactions contained 10 mM HEPES (pH 7.5), 100 mM NaCl, 1.0 mM sodium EDTA, 200 μM unilamellar liposomes, and peptide between 10 nM and 10 μM. Reactions were conducted at room temperature for 10 min.
Computational Methods
SMD is a nonequilibrium sampling method that can be used to characterize protein unfolding or ligand-unbinding processes. Within SMD simulations, an external force is applied on a pseudoatom attached to the system with a harmonic potential to stretch it along the pulling direction. This lends itself to the use of the JE50 which was combined with SMD to calculate free-energy differences along the path,51,52 and implemented successfully in other enhanced sampling methods.53−55 Specifically, the free energy along the path provided by the JE is
| 3 |
where 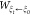 is the work performed by the pseudoatom,
β is 1/kBT, kB is the Boltzmann constant, and T as the absolute temperature, SMD formally provides the equilibrium
free-energy difference between two states from a series of nonequilibrium
work values. However, the convergence of the average of the exponential-Boltzmann-weighted
calculation in the JE is slow and becomes worse with increasing steering.
Both issues are further exacerbated with the increasing complexity
of the protein and its energy landscape.
is the work performed by the pseudoatom,
β is 1/kBT, kB is the Boltzmann constant, and T as the absolute temperature, SMD formally provides the equilibrium
free-energy difference between two states from a series of nonequilibrium
work values. However, the convergence of the average of the exponential-Boltzmann-weighted
calculation in the JE is slow and becomes worse with increasing steering.
Both issues are further exacerbated with the increasing complexity
of the protein and its energy landscape.
ASMD offers an improvement in the efficiency of this type of nonequilibrium sampling. In ASMD, the steering path is completed in several stages to limit the deviations sampled by the nonequilibrium trajectories as they grow with the extent of steering. Within each stage, SMD simulations are performed with the JA value determined via the JE. The chosen contraction criterion—discussed below—leads to the selection of one or several representative structures at the end of each stage to initiate the subsequent stage. The JA values across the stages are combined according to
| 4 |
where Wj is the nonequilibrium work from tj–1 to t evaluated for each
of the N trajectories 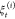 in the jth stage.37
in the jth stage.37
There exist several variants of ASMD37−41,56 according to the choice of contraction criteria as appropriate for the system of interest. A detailed discussion of the contraction criteria, and relevant advantages can be found in ref (37). For the naïve ASMD37−39 that was applied in this work, the trajectory that has the work value closest to the JA at the end of each stage is selected to represent the overall ensemble space, and used as the starting configuration for the next stage. This simple contraction criterion keeps the relevant information that contributes most to the JA calculations and discards the trajectories that fluctuate far away from the potential of mean force (PMF) results, thus achieving convergence in a computationally efficient manner. In addition, ASMD can also be combined with the telescoping boxes42 to adjust the necessary size of the solvent box between stages to accommodate for the expanding protein along the steering direction and limit the number of solvent molecules.
ASMD has been previously benchmarked on neuropeptide Y38 and its mutants,57 deca-alanine in vacuum, implicit and explicit solvents,39,41,56 and β-hairpin.58,59 It has also been utilized successfully by several groups to investigate a large number of biophysical phenomena, such as mutagenesis,60,61 host–guest interactions,62 protein–ligand interactions,63 and dissociation mechanisms.64
Observables
In addition to the energetic profile of the steering process, observables—e.g., hydrogen bonds—along the steered path of the ASMD simulation can also be determined using exponential-Boltzmann-weighting. The number of hydrogen bonds along each trajectory can be obtained using MDAnalysis (2.0)65−68 with the distance and angle cutoff as 3.5 Å between two electronegative atoms and 120° with H atom as the vertex. The expectation average of the hydrogen bonds are
| 5 |
where 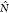 records the number of hydrogen bonds formed
between two specified groups of residues, S1 and S2, and the Boltzmann weighting
is specified by the work values along a given nonequilibrium trajectory
records the number of hydrogen bonds formed
between two specified groups of residues, S1 and S2, and the Boltzmann weighting
is specified by the work values along a given nonequilibrium trajectory 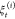 as in eq 4.
as in eq 4.
Simulation Protocols
Both the wild type (WT) Enterocin 7B and its mutants are modeled using the CHARMM36 force field69 because its CMAP correction terms are known to be necessary for accurate representation of α-helices.
Equilibration Protocol
A given peptide is equilibrated in an explicit water (TIP3P model) solvent environment before the ASMD simulations. Each peptide is initially neutralized with capped ends, rotated to set the pulling direction along the z-axis, and solvated in a rectangular solvent box (70 × 70 × 170 Å) to accommodate the extended structures after it is steered–viz., stretched. The solvated systems were then neutralized with Na and Cl ions. Subsequently, a three-step equilibration procedure is performed using Scalable MD (NAMD)70 and our ASMD71 scripts to obtain the equilibrated system. First, the whole system is equilibrated for 5 ns under NPT conditions at 300 K. The Nosé–Hoover Langevin piston method is applied to maintain the pressure at 1 atm with a decay period of 100 fs, a damping time constant of 50 fs and a damping coefficient as 5 ps–1.72 During the NPT equilibration, the Cα ends of each peptide are constrained to allow the water solvent to reach the appropriate density. The system is then equilibrated under NVT conditions at 300 K and constrained by a harmonic potential with a force constant (equal to 10.0 kcal/mol Å2) applied initially on the backbone of each peptide. A series of 200 ps relaxations are then performed with the harmonic potential decreasing to 5.0 and 1.0 kcal/mol Å2 sequentially. The peptide is then allowed to freely equilibrate under NVT conditions for an additional 1 ns. At the end of this equilibration substep, the peptide ends are no longer on the z-axis. Therefore, the whole system is once again rotated to set the ends along the z-axis in parallel with the stretch direction. Finally, the system is again equilibrated under NPT conditions for 5 ns at 300 K with the ends constrained. Each equilibrated structure is confirmed based on the root mean-square deviation (rmsd) values.
Production Phase
ASMD simulations for the WT enterocin 7B and the selected mutants are carried out in explicit solvent under NPT conditions at 300 K and 1 atm with NAMD.70 The solvent box was slightly larger than the size of the unfolded peptide and accommodates the peptide throughout the stretch without requiring a telescoping box. Within a simulation, the Cα atom of the first residue is fixed, and the Cα atom of the other end of the peptide is stretched through its attachment to a pseudoatom by a harmonic spring with a force constant of 7.2 kcal/mol Å2. Within each stage, 100 SMD trajectories are performed by pulling the pseudoatom at 10 Å/ns pulling speed for 100 Å pulling distance along the z-axis direction. A total of 50 stages are required to break most of the intramolecular interactions within reasonable computational time, and to reach the unfolded protein, but not necessarily a linear amino acid sequence. Trajectories are visualized and analyzed using the visual MD (VMD),73 ChimeraX74 and MDAnalysis65,66 packages, and in-house python scripts.
Results
Accommodation of the Arginine Single-Site Mutations
The arginine single-site mutations were originally designed to disrupt the enterocin 7B hydrophobic core. The locations for the substitutions were made based on a visual inspection of the core environment of WT enterocin 7B peptide via the 2M60 PDB structure. Arginine was selected to provide both steric and electrostatic destabilization. All of the single-site mutations resulted in peptides that could be successfully synthesized and purified with the exception of the I23R mutant. The reason for this failure was not further investigated. All proteins were highly soluble in aqueous buffers. The structure and the location of the mutants is shown in Figure 1a. The native hydrophobic core is naturally surprisingly plastic in that arginine single-site mutation replacements were tolerated both in the modeling and in the actual peptides. Using the VMD MUTATE tool, arginine was substituted, and the most common rotomer was selected. After the equilibration steps for the solvated systems constrained at 300 K and 1 atm, as described in the Computational Methods section, several metrics of the final peptide models revealed the a priori effects of the substitutions. The seven single-site mutants are shown in Figure 1b–h, illustrating the position of the replacement. ChimeraX was used to calculate peptide volume and solvent accessible surface area (SASA), and VMD was utilized to calculate the radius of gyration (Rg) and the main-chain rmsd compared to the WT peptide. Results of these calculations are available in Table S1 in the Supporting Information. Peptide volume is similar for all seven mutants compared to that of WT, all within 100 Å3 of that of WT. There are more observed differences in SASA values, with all peptides having greater SASA than WT: L40R, I23R, and L7R have the largest increase in SASA (547, 424, and 426 Å3, respectively) and V8R and F15R have SASA values closest to WT. As might be expected, the pattern is the same for Rg. L7R and I23R have the largest rmsds versus WT (3.27 and 3.46 Å, respectively), with V8R and F15R having the lowest rmsds (1.53 and 1.73 Å, respectively). The mainchain rmsds between all the peptides are available in Table S2 of the SM. Values range from a low of 1.48 Å (V8R vs F15R) to a high of 4.12 Å (I23R vs L7R), and the trend is listed in column a of Table 3.
Table 3. Comparison of Measured Observables among the WT 2M60 and 7 Mutantse.
| rmsda | CDb | Tmc | MICd | PEPt | PEP0f | [ΔCAL]g | k[CAL]h | Wi |
|---|---|---|---|---|---|---|---|---|
| 1.I23R | 1.WT | 1.I4R | 1.I4R | 1.I23R | 1.I4R | 1.F15R | 1.I4R | 1.I23R |
| 1.L7R | 2.I4R | 1.I23R | 1.I23R | 2.I4R | 1.I23R | 1.I23R | 1.I23R | 2.I4R |
| 1.I4R | 2.L7R | 1.L40R | 1.L40R | 2.L40R | 1.L40R | 1.L40R | 1.L40R | 2.L40R |
| 4.L40R | 2.V8R | 4.WT | 4.WT | 4.WT | 4.WT | 4.WT | 4.WT | 4.WT |
| 5.F26R | 2.F26R | 4.V8R | 4.V8R | 4.V8R | 4.V8R | 4.I4R | 4.V8R | 4.V8R |
| 5.F15R | 6.F15R | 4.F15R | 4.F15R | 4.F15R | 4.F15R | 4.V8R | 4.F15R | 4.F15R |
| 7.V8R | 6.I23R | 7.F26R | 4.F26R | 4.F26R | 4.F26R | 4.F26R | 4.F26R | 4.F26R |
| 6.L40R | 7.L7R | 8.L7R | 8.L7R | 8.L7R | 8.L7R | 8.L7R | 8.L7R |
rmsd (Å), from highest to lowest.
Near UV CD signal magnitude, from more to less signal.
Melting temperature, from lowest to highest.
MIC value, from better to worse.
Peptide added to growing bacterial culture, from more to less effective.
Peptide added at the start of a bacterial culture, from more to less effective.
Calcein release, from more to less release.
Calcein release rate, from fast to slow release.
Work at ree = 60 Å, from low to high work value.
Computational modeling and MD minimization resulted in mutant peptides with an intact secondary structure. This was experimentally verified by UV CD analysis. As is shown in Figure 2A, the spectra between 200 and 250 nm are virtually identical, with an α helical minimum at 222 nm and a coil minima at 209 nm. Near UV CD between 250 and 320 nm is a powerful method to probe tertiary interactions involving aromatic amino acids. The near UV CD spectra are predominated by a tryptophan single peak centered around 280 nm, phenylalanine signals at 261 and 268 nm, and a smaller contribution by the single enterocin 7B tyrosine at 279 and 286 nm. The spectra are shown in Figure 2B. Variations in the minimum observed at 280 nm leads to the conclusion that there may be differences in the tertiary microenvironment (i.e., the geometry of the hydrophobic core).75,76 WT peptide has the largest near UV CD molar ellipticity signal at 280 nm (−9.6). Three mutants, F15R, I23R, and L40R, have significantly less overall near UV CD signal than WT (−7.2, −6.1, and −7.6; respectively). The remaining mutants have an intermediate value for the CD signal at 280 nm (I4R, −8.9; L7R, −8.7; V8R, −8.9; F26R, −8.8). If one equates the overall near UV CD signal to the degree of hydrophobic core relaxation, then all of the mutants exhibit increased hydrophobic core plasticity relative to WT.
Figure 2.

Secondary and tertiary structural analysis of the enterocin peptides. All presented CD spectra are presented as the average of three independent determinations. (A) Far UV CD spectra plotted as the mean molar ellipticity versus wavelength at 10 μM peptide. (B) Near UV spectra plotted as the mean molar ellipticity versus wavelength at 90 μM peptide.
Thermal Stability of Selected Enterocin 7B Mutants
The stability effect of the arginine single-site mutations was directly measured via thermal unfolding. Global fitting of the temperature vs far UV CD showed that the data were best fit to a simple two-state unfolding reaction (F ↔ U). All peptides evidenced a gradual unfolding that began at approximately 35 °C and was complete between 48 and 56 °C depending on the peptide (see Figure 3). Three mutants exhibited thermal unfolding curves that were distinct from those of the WT and the other mutants. I4R, I23R, and L40R all begin thermal unfolding transitions at lower temperatures and have unfolding curves with slopes higher than those of the other five peptides. Two peptides have nearly superimposable unfolding curves compared to WT (V8R and F15R) and two mutants are actually more stable than WT (L7R and F26R) with similar unfolding curve slopes but with midpoints at slightly higher temperatures. The melting temperature (defined as the midpoint of the unfolding curve) for each of the peptides is listed in Table 1.
Figure 3.

Fraction of unfolded peptide as a function of temperature determined by a global fit of far UV CD melting profiles. The inset shows the entire span from 4 to 90 °C.
Table 1. Melting Temperature of Enterocin Peptidesa.
| peptide | Tm (°C) |
|---|---|
| WT | 48.2 ± 0.3 |
| I4R | 44.6 ± 0.3 |
| L7R | 49.1 ± 0.2 |
| V8R | 48.3 ± 0.2 |
| F15R | 47.6 ± 0.3 |
| I23R | 43.4 ± 0.3 |
| F26R | 49.1 ± 0.4 |
| L40R | 44.4 ± 0.3 |
Each listed value is the mean and standard deviation of three independent replicates.
Functional Characterization of the Enterocin 7B Mutants
MIC values were determined for the eight peptides versus the Gram stain (+) bacterium Bacillus subtilis. All of the peptides displayed the ability to kill B. subtilis, but with varying degrees of effectiveness. The MIC values are shown in Table 2 and the lower the value, the more lethal the peptide is in killing B. subtilis. The effect of the peptides on the growth of B. subtilis is shown in Figure S7. In panel S7a, the μM peptide is added to an early exponential culture of B. subtilis. An effect on increased optical density (at 650 nm) is seen after approximately 25 min post addition. Compared to the no peptide control bacterial growth curve, all of the peptides inhibit bacterial growth. L7R is the least efficient in preventing further bacterial growth, as evidenced by the observation that the L7R curve is closest to the control curve with regard to slope and final bacterial density. I23R is the most effective peptide. Its addition at 2 μM (closest concentration to its MIC of 3.1 μM) results in full arrest of bacterial growth and significant lysis of the culture. The remaining peptides cluster into two distinct groups between L7R and I23R. More effective than WT are peptides I4R and L40R. Equally effective as WT are the peptides V8R, F15R, and F26R. The functional analysis clarifies the interaction between the peptides and the bacterium by illustrating the direct effects of enterocin mutants on B. subtilis at concentrations below the MIC. The effect of 2 μM peptide on B. subtilis growth kinetics when the peptide is added to a newly inoculated culture is shown in Figure S7b. Three peptides show greater killing efficiency compared to WT, that is, I4R, I23R, and L40R. The peptide L7R is again least effective in killing B. subtilis, and the remaining four peptides are roughly as effective as WT in preventing bacterial growth.
Table 2. Functional Properties of Enterocin Peptides.
| peptide | MICa (μM) | initial calcein release rateb (dFo/dt)c | percent calcein released at MIC (%) |
|---|---|---|---|
| WT | 10.6 ± 0.1 | 9.2 ± 1.1 | 30.5 ± 1.3 |
| I4R | 6.1 ± 0.2 | 18.9 ± 1.4 | 24.0 ± 1.5 |
| L7R | 22.2 ± 0.1 | 6.4 ± 0.8 | 27.7 ± 1.2 |
| V8R | 11.5 ± 0.1 | 8.2 ± 0.5 | 30.1 ± 1.1 |
| F15R | 10.9 ± 0.1 | 9.1 ± 0.9 | 33.4 ± 1.3 |
| I23R | 3.1 ± 0.1 | 19.8 ± 0.7 | 24.5 ± 1.0 |
| F26R | 10.5 ± 0.1 | 9.9 ± 1.0 | 26.1 ± 1.5 |
| L40R | 6.4 ± 0.2 | 18.1 ± 1.2 | 25.6 ± 1.4 |
MIC.
Calcein release rate from 1:1 PG/PE liposomes at 25 °C.
dFo/dt is the time derivative of the emission intensity at 520 nm and t = 0.
%calcein release is relative to Trion X-100 plateau (which measures 100% release).
The ability of the enterocin peptides to disrupt synthetic PG/PE unilamellar liposomes was measured using a calcein release assay, as shown in Figure 4. In this assay, the dequenching of calcein is monitored fluorometrically (calcein is self-quenched due to its relatively high concentration within the liposome). The maximum fluorescence emission intensity, which corresponds to 100% release, was determined by fully destroying the liposomes with the surfactant Triton X-100. The initial kinetic release rates using 200 μM liposomes and 10 μM peptide (Figure 4a) for the linear portion of the curve (typically 0 to 100 s) are shown in Table 2. Three peptides (F15R, I23R, and L40R) are more effective in PG/PE membrane disruption than the WT enterocin, as evidenced by higher initial release rates and a higher percent of total calcein released by the end of the assay. The percentage of calcein released is linearly proportional to the peptide: lipid ratio for 5–20 μM peptide (or peptide/lipid ratios from 1:40 to 1:10). The percentage of calcein release at the MIC for each peptide is given in Table 2. Percentages range from 24% (I4R) to 33.4% (F15R) with an average value of 27.7%. At the MIC value for each peptide, the percent calcein release should be similar if the mechanism of unilamellar liposome interaction is the same. In this case, it is.
Figure 4.

(a) Release kinetics of calcein from 1:1 PG/PE liposomes (200 μM) as a function of time. At t = 50 s, either peptide (at a final concentration of 10 μM) or Triton-X100 (final concentration of 10% w/v) was added to each well and calcein release was measured with an excitation wavelength of 500 nm and an emission wavelength of 520 nm. (b) Percent release of calcein from 1:1 PG/PE liposomes (200 μM) as a function of peptide concentration. The 100% release level was determined using Triton-X detergent. Curves are the mean of three independent experiments with standard deviation error bars for WT (closed squares), I4R (open circles), L7R (closed triangles), V8R (open triangles), V8R (open triangles), F15R (closed inverted triangles), I23R (open squares), F26R (pluses), and L40R (crosses).
Structural Analysis
The equilibrated structure of the Enterocin 7B WT structure is compared to that of each mutant in Figure 1. The hydrophobic core of WT is stabilized by several aromatic rings (e.g., Phe, Tyr, and Trp) within the core. The overall stability of the hydrophobic core of the WT is affected by the introduction of steric and electrostatic effect through Arg mutations. Specifically they disrupt charge–charge interactions and π–π stacking, though to varying degrees depending on the position of the substitution. The L7R mutation also has the potential to form a salt-bridge with Glu37 on the other end of the peptide, leading to stabilization of the overall protein.77 The F26R brings guanidinium interactions (Arg) with Trp31 and Trp39 and could potentially form favorable π–π interactions,78 leading to stabilization as observed in high Tm value. On the other hand, the V8R mutant retains almost the same configuration as the WT and the F15R mutant is the second closest in structure. This should be apparent in the comparisons of Figure 1, and is also supported by the rmsd values in Table S1 of the Supporting Information. In comparison, the other mutants displayed higher rmsd values and larger structural differences. L40R had a prominent structural change at the C-terminus caused by the mutation, while I4R and I23R had the most significant structural variation at the N-terminus and the “turn” structure formed between the middle helical region and the C-terminus. Thus, the introduction of Arg at these positions disrupts the van der Waals interactions between the residues of the core and makes the mutants more flexible.
Furthermore, the averaged SASA profiles were also generated for all peptides and are compared in Figure S8. After equilibration, the I23R and L40R mutants have an obvious increase in SASA for residues in both end regions, indicating that these mutants adopted a more extended conformation and might be attributed to the more flexible helices on the end regions or the hydrophobic core after the mutations. Since I23R and L40R peptides have mutants on the middle and C-terminal helical regions, those residue positions may play an important role in involving the N-terminal helical region when forming the hydrophobic core. Both I4R and L7R mutations result in a local area that is more hydrophilic and with an increased SASA value around mutated residues. The F26R mutation also moderately increases the level of exposure of the “kink” to water. Finally, the SASA values did not change significantly for F15R and V8R.
Energetics
The PMFs displayed in Figure 5 provide a comparison of the mechanical unfolding energetics and structural rigidity differences between WT Enterocin 7B and the mutants, revealing, in part, the effects of the hydrophobic core. The pairwise comparison between the WT and each mutant, including an error analysis, is available in Figure S9 in the Supporting Information. Within the unfolding simulations, all peptides were initially compressed to an end-to-end distance equal to 15 Å so that they could explore configurations near the local minima of the PMFs. We found that the selected mutations had different end-to-end distance minima from that of the WT enterocin 7B. The F15R, F26R and V8R had similar end-to-end distance minima as the WT at around 20 Å, while I4R had a larger value at 23 Å and L7R had a smaller value at 19 Å. However, the I23R and L40R mutants had broad local minimum regions, which indicated that they adopted quite flexible structures.
Figure 5.

Comparison of the energetics of the WT enterocin 7B and each mutant. The PMF have been obtained using 100 tps at 10 Å/ns.
Peptides were stretched beyond their minima to break some of the intrapeptide hydrogen bonds as reflected by the initial increases in the PMF. The plateau observed at around 60 Å in the PMF for the WT is coincident with the disruption event of the hydrophobic core and was also seen in other mutants. In terms of that disruption event, I4R, I23R, and L40R mutants displayed a pronounced destabilizing influence reflected in the PMF profiles near 60 Å, which is presumably an indication of their weaker hydrophobic core. The structural origin of this outcome appears to be rooted in the position of residue 18 as its ϕ and ψ angles as seen to be out of the “allowed” regions in the Ramachandran plots in Figure S10b,f,h. This, in tandem with the trends in the SASA profiles and the end-to-end distance minima, suggests that the residues are in extended conformations. Residue 18 in F26R also exhibits less favorable orientations near the edges of the “allowed” regions. However, the introduction of the guanidinium interactions discussed above may offset these effects. The L7R mutant, on the other hand, displayed a stronger hydrophobic core in comparison to other peptides. Although its SASA values are increased around residue 7, stabilization of the hydrophobic core can be explained by the introduction of the salt-bridge after mutation and the favorable orientation of most of the residues. Both factors retained the overall structure of L7R with the smallest end-to-end distance at 19 Å over other mutants and WT. The remaining peptides, F15R and V8R, exhibited similar trends in the PMF. In combination with their similar SASA profiles and rmsd values, these two mutations thus had small effects on the overall stability of the WT protein.
As the peptides are stretched further, the hydrophobic core is fully disrupted and the peptides adopt nearly linear shapes. The increasing PMF beyond 60 Å was related to the ongoing hydrogen bond breaking and the covalent bonds stretching to different extents. Thus, the work values of the partially unfolded proteins at the end-to-end distance near 60 Å where the hydrophobic core was disrupted are a meaningful measure of stability.
Secondary Structure
The secondary structure evolution for each peptide was also revealed using ASMD simulations, as shown in Figure 6. The trajectory that has the nearest work value to the JA is presumably most representative of the unfolding pathway and is therefore the one analyzed for each peptide. The extended configuration and isolated bridge conformations are not observed and thus omitted in the plot. The initial structure of each peptide is dominated by α-helical conformations. As they are unfolded, the structures generally transit into 310 conformations, and finally to coils, as also observed in smaller helical proteins.79 The WT also formed some π-helical conformations during the unfolding. In addition, F26R and L40R formed a few π-helical contacts through the end of unfolding. Most of the mutants displayed a pattern similar to that of the WT. However, it is worth noting that L7R, in comparison to the other peptides, had π-helix formation at the middle-stage of stretch, where the hydrophobic core was disrupted. Moreover, I23R, unlike the rest of the peptides, did not display obvious changes in α- and 310-helical contacts before the hydrophobic core was broken.
Figure 6.

Evolution of the secondary structure of the overall unfolding process for WT 2M60 (a) and the seven mutants considered here: I4R (b), L7R (c), V8R (d), F15R (e), I23R (f), F26R (g) and L40R (h).
As the peptides were mainly composed of helices, the total intrapeptide hydrogen bonds shown in Figure S11 are strongly correlated with the helical structure and the extent of unfolding. In addition, the change in hydrogen bonds of each helical region and the fraction of native contacts over the unfolding process are also available in Figures S12 and S13 of the Supporting Information. From the hydrogen bond profiles, the WT Enterocin 7B first lost hydrogen bonds at the stretched end—viz, the terminus. The hydrophobic core was subsequently disrupted, as seen by the plateau areas in both the PMF and hydrogen bond profiles at around a 60 Å end-to-end distance. After the hydrophobic core structure is lost, the remaining hydrogen bonds within the helices are next to break. The F15R and F26R mutants had more hydrogen bonds than the WT after the disruption of the hydrophobic core, which may be due to the change in the size of the side chain after mutation. The mutation for F15R and F26R in the middle helical region necessarily converts the side chain from bulky residues to the more linear arginine, and this increases the tendency for the formation and reformation of the hydrogen bonds with other residues with a more extended configuration. During the competing processes between the disruption event of the hydrophobic core and the breaking event of hydrogen bonds, I23R, I4R, and L40R initially unwind at the hydrophobic core while keeping most of the intrapeptide hydrogen bonds. Moreover, I4R had the mutation of a shorter side chain (I) to a longer one (R), slightly disrupting the helix while the other helical regions remained in place and adopting a more extended structure (23 Å). The largest differences in the native contacts between the WT and mutants were seen for L7R and I23R as reported in detail in Figure S3. All of the peptides were seen to begin to lose native contact due to the stretch and experienced a plateau upon reaching 60 Å as they transitioned into linear-like conformations. However, L7R reached an almost linear structure only until ≈70 Å in stretch, which suggests that its hydrophobic core is more stable. On the other hand, I23R did not display a noticeable plateau behavior in the native contact profile, suggesting that the overall hydrophobic core was disrupted along with the stretch and that therefore I23R possesses a weaker hydrophobic core.
In summary, the structural, functional and computational values reported above lead to the relative rankings of WT and its mutants listed across Table 3. With a few exceptions, the trends in the rankings of the proteins correlate well across the entire set of measures. For instance, L7R requires the greatest amount of work to disrupt the hydrophobic core in the ASMD simulations, and it has one of the highest melting temperatures (tied with F26R) and a higher degree of near UV CD signal.
Discussion
This paper sought to harness a variety of measures: structural, computational, and experimental, to better characterize the role that tertiary structural plasticity plays in the role of enterocin 7B interactions with Gram-stained (+) bacterial membranes. To that end, a series of designed arginine single-site replacement mutations were made to potentially destabilize the hydrophobic core. All the measures reported in Table 3 indicate that L7R is dynamically stable. However, in all of the functional assays, it is uniformly least able to disrupt natural or artificial bacterial membranes. In contrast, I23R requires a minimum amount of work to disrupt the hydrophobic core and has less near UV CD signal and a lower melting temperature than most of the other peptides. This indicates that I23R is less dynamically stable. It is, however, the best, or nearly the best, at disrupting bacterial or artificial membranes. In between these two extremes is the peptide V8R. Although it has the lowest rmsd compared to that of WT, it is always in the middle of the pack for all the other measures. It is moderately functional and requires a moderate amount of work to disrupt the hydrophobic core.
Through ASMD simulations, we found that L7R introduces a salt-bridge interaction with a more compact conformation at the minimum energy, and V8R (along with F15R and F26R) has a similar ree to that of WT, while I23R (along with L40R) adopts a more flexible structure with broad local minimum regions in their PMF. The V8R mutation also has a small effect on the overall stability of the protein as seen in comparison across the PMFs, and this finding is also supported by the SASA profile and rmsd values. In comparison, the L7R mutation impacts the SASA values around residue 7. However, the introduction of a salt-bridge interaction maintained the overall structure near the end-to-end distance of 19 Å, stabilizing the hydrophobic core and thus exhibiting the highest PMF values at 60 Å. The I23R mutation destabilizes the protein the most, and this is likely rooted in the fact that residue 18 is located around the “kink” that connects the helical regions. According to the secondary structure evolution results and intramolecular interaction analysis, L7R prefers to lose more hydrogen bonds than WT in retaining its hydrophobic core configuration and reaches linear-like conformation much later than the rest of peptides. Meanwhile during the stretch, I23R prefers to lose the hydrophobic core first with little hydrogen bond change, and V8R exhibits a similar pattern as the WT with slightly more intrapeptide hydrogen bonds retained.
Taken together, these results, also shown in Figure 7, indicate that the membrane disruption efficiency of leaderless, noncovalently restrained (e.g., internal disulfide bonds or circularization) antimicrobial peptides is increased with the ability to unpack tertiary structure more easily. The higher the plasticity of the hydrophobic core, the higher the efficiency of membrane disruption. The results may also shed light on the driver of the overall mechanism of enterocin–membrane interactions. The enterocin 7B structure is characterized by amphipathic helices, distributed cationic amino acid residues, and three-dimensionally clustered hydrophobic residues. Hence, as is common with all antimicrobial peptides, initial membrane binding may be driven by a combination of electrostatic and hydrophobic interactions. Once initial contact with the membrane is made, the helices unpack to expose the hydrophobic core and the linear peptide (with intact secondary structure) inserts into the membrane (in general, see Sani and Separovic80). Our data suggest that insertion efficiency in enterocin 7B is mediated primarily by the ability to unpack the helices and that a less stable hydrophobic core results in an increased rate (and magnitude) of cell membrane disruption and bacterial cell death.
Figure 7.
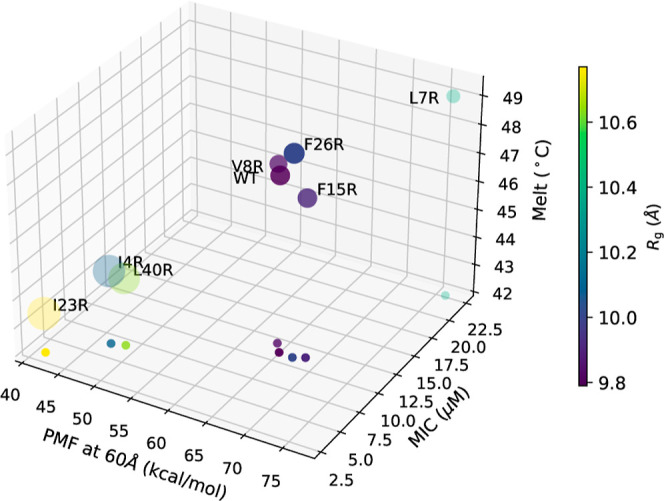
Comparisons of WT and each mutant in terms of MIC, Tm, Rg, calcein release and PMF at 60 Å. The values of the PMF, MIC, and Tm are plotted along the x, y, and z axis, respectively, in a 3D rendering that is fully specified by the projection of the points onto the xy plane. In addition, the color of each data point represents the value of Rg as indicated on the color scale bar, and the size of the circle represents the relative magnitude of calcein release.
There are many mechanisms by which antimicrobial peptides can disrupt lipid membranes.81 It is currently unknown whether enterocins 7A/B form HTPs, smaller pore-like structures, or cause generalized cell permeabilization (e.g., membrane carpets as opposed to discrete pores of any diameter or shape). Structurally related peptides, like lacticin Q82 that form HTPs as well as peptides like aureocin A5383,84 that cause generalized membrane permeability have been described. Enterocin AS-48 depermeabilizes bacterial membranes by forming smaller nonselective pores,85 although it is a circular enterocin. So, although there is a degree of primary sequence and tertiary structure identity in this family of antimicrobial peptides, the final membrane-peptide structure is heterogeneous. Our calcein release experiments most likely rule out the formation of HTPs with enterocin 7B alone as the release of calcein occurred at much lower lipid/peptide ratios than was seen for lacticin Q82 even though the time course of calcein release was similar. Although not formally tested here, our results support the formation of smaller pores or slits in the membrane. This is the first report that we are aware of that shows that enterocin 7B, like lacticin Q,86 can disrupt lipid membranes in the absence of a specific receptor.
Enterocins 7A and 7B function as a pair, as do other enterocins such as L50A and L50B. Unlike enterocin 7A/B,32 L50A/B action is extremely synergistic.34 The observation that enterocin 7B functions so well in the absence of enterocin 7A makes our results easier to analyze and the computational system possible. It also shows how ASMD can be used to lead and augment functional bench experimentation, but more importantly, ASMD provides atomistic-level detail of the behavior of these biological macromolecules. Specifically, in ASMD, the components of the protein contributing the least to the structural rigidity of the protein unravel first. Comparison between enterocin 7B and its mutants further reveals the underlying cause of destabilization (or lack thereof) from the mutations and provides insight into its function. We found the implication that the WT’s ability to unpack the hydrophobic core during unfolding plays an important role in enterocin 7B function. For the most effective mutants, the hydrophobic core appears to be the least stable as it is disrupted initially upon unfolding, while the helical regions are more stable because the hydrogen bonds within them are retained longer. In other mutants, these latter hydrogen bonds are broken first, allowing the hydrophobic core to stay intact longer and thus presumably limiting its function.
Conclusions
In this work, several arginine single-site mutants were selected on the WT enterocin 7B peptide to investigate how the stability of the hydrophobic core of enterocin 7B affects the efficiency of its activity and function through both experimental and computational methods. While arginine is known to have high interaction potential to lipids,87 we found that its impact on the stability of the core is not so clear as it was highly dependent on the position and environment of the mutation. In particular, we found that the tertiary plasticity of the protein impacts the efficiency of enterocin 7B. The less stable the hydrophobic core of the enterocin 7B mutant, the faster the disruption of the cell membrane and bacterial cell death.
In the PMF profiles, near UV (CD) signal, and melting temperatures, it turns out that L7R has the most relatively stable hydrophobic core and exhibits limited disruptive effects on bacterial membranes. However, I23R has the least stable hydrophobic core and displays almost the best performance on disruption of the bacterial membranes. Furthermore, through hydrogen bond profiles and secondary structure evolution analysis, the unfolding mechanism was also characterized. During the steered unfolding of WT enterocin 7B, the C-ter helical region starts losing hydrogen bonds before the hydrophobic core is disrupted. However, in I23R, the hydrophobic core is disrupted from the beginning, reflecting its weak tertiary structure plasticity. The ASMD simulations also suggest that this disruption is due to residue 18 which is located around the “kink.” In comparison, L7R lost more hydrogen bonds than did WT in retaining its hydrophobic core configuration, indicating its stronger tertiary plasticity.
Combined with the structural information on enterocin 7B, our analysis, such as PMF and calcein release experiments, revealed that the likely critical step in the mechanism of bacterial cell membrane disruption for enterocin 7B is the unpacking of its helices to expose the hydrophobic core to drive the insertion without a specific receptor, and supports a possible mechanism involving formation of smaller pores or slits in the membrane. The energetic analysis focusing on the relative disruption of the core and the helices suggests that the corresponding residues affecting those regions would be possible targets for future mutations, including alanine substitutions, aimed at improving the activity.
Acknowledgments
This work has been partially supported by the National Science Foundation (NSF) through grant no. CHE 2102455. The computing resources necessary for this work were provided, in part, through allocation CTS090079 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation (NSF) grants #2138259, #2138286, #2138307, #2137603, and #2138296.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.3c08199.
The evidence that led us to conclude that tertiary plasticity is critical in providing the effects we found in 2M60, optical density as a function of the concentration of the enterocin mutant, the effect of the peptides on the growth of B. subtilis, pairwise comparisons of the PMF of unfolding at pulling speeds under 10 Å/ns, and analysis on the change in hydrogen bonds (PDF)
Author Contributions
Y.Z. performed research, developed methods, analyzed data, and wrote the manuscript. S.Q. designed research, conducted experiments, developed methods, analyzed results, and wrote the manuscript. E.R.S. conducted numerical simulations. H.R.B. and C.R.A. conducted preliminary numerical simulations. R.H. designed research, developed methods, analyzed results, and wrote the manuscript. All authors participated in editing the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Avci F. G.; Akbulut B. S.; Ozkirimli E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. 10.3390/biom8030077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane S. A.; Li X.; He S.; Yu M.; Wu M.; Vederas J. C. Synthesis of Tridecaptin–Antibiotic Conjugates with in Vivo Activity against Gram-Negative Bacteria. J. Med. Chem. 2015, 58, 9779–9785. 10.1021/acs.jmedchem.5b01578. [DOI] [PubMed] [Google Scholar]
- Atanaskovic I.; Mosbahi K.; Sharp C.; Housden N. G.; Kaminska R.; Walker D.; Kleanthous C. Targeted Killing of Pseudomonas aeruginosa by Pyocin G Occurs via the Hemin Transporter Hur. J. Mol. Biol. 2020, 432, 3869–3880. 10.1016/j.jmb.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Zeng J.; Wang C.; Wang J. Structural Basis of Pore Formation in the Mannose Phosphotransferase System by Pediocin PA-1. Appl. Environ. Microbiol. 2022, 88, e0199221 10.1128/aem.01992-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpiński T. M.; Szkaradkiewicz A. K. Characteristic of bacteriocines and their application. Pol. J. Microbiol. 2013, 62, 223–235. 10.33073/pjm-2013-030. [DOI] [PubMed] [Google Scholar]
- Niehus R.; Oliveira N. M.; Li A.; Fletcher A. G.; Foster K. R. The evolution of strategy in bacterial warfare via the regulation of bacteriocins and antibiotics. Elife 2021, 10, e69756 10.7554/elife.69756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter P. D.; Hill C.; Ross R. P. Bacteriocins: developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Xiong T.; Huang T.; Xu X.; Fan P.; Qiao B.; Xie M. Factors affecting production and effectiveness, performance improvement and mechanisms of action of bacteriocins as food preservative. Crit. Rev. Food Sci. Nutr. 2022, 63, 12294–12307. 10.1080/10408398.2022.2100874. [DOI] [PubMed] [Google Scholar]
- Kranjec C.; Kristensen S. S.; Bartkiewicz K. T.; Brønner M.; Cavanagh J. P.; Srikantam A.; Mathiesen G.; Diep D. B. A bacteriocin-based treatment option for Staphylococcus haemolyticus biofilms. Sci. Rep. 2021, 11, 13909. 10.1038/s41598-021-93158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinseth I.; Tønnesen H. H.; Carlsen H.; Diep D. B. Exploring the Therapeutic Potenital of the Leaderless Enterocins K1 and EJ97 in the Treatment of Vancomycin-Resistant Enterococcal Infection. Front. Microbiol. 2021, 12, 649339. 10.3389/fmicb.2021.649339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telhig S.; Ben Said L.; Zirah S.; Fliss I.; Rebuffat S. Bacteriocins to Thwart Bacterial Resistance in Gram Negative Bacteria. Front. Microbiol. 2020, 11, 586433. 10.3389/fmicb.2020.586433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y.; Wang L.; Wang M.; Meng X.; Qiao J. Antimicrobial Peptides: A Promising Strategy for Anti-tuberculosis Therapeutics. Protein Pept. Lett. 2023, 30, 280–294. 10.2174/0929866530666230315113624. [DOI] [PubMed] [Google Scholar]
- Saravanan P.; Pooja R.; Balachander N.; Kesav Ram Singh K.; Silpa S.; Rupachandra S. Anti-inflammatory and wound healing properties of lactic acid bacteria and its peptides. Folia Microbiol. 2023, 68, 337–353. 10.1007/s12223-022-01030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader J. S.; Hoskin D. W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Invest. Drugs 2006, 15, 933–946. 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- Deslouches B.; Di Y. P. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. 10.18632/oncotarget.16743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouhir A.; Hammami R.; Fliss I.; Hamida J. B. A new structure-based classification of gram-positive bacteriocins. Protein J. 2010, 29, 432–439. 10.1007/s10930-010-9270-4. [DOI] [PubMed] [Google Scholar]
- Hammami R.; Zouhir A.; Le Lay C.; Ben Hamida J.; Fliss I. BACTIBASE second release: a database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. 10.1186/1471-2180-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etayash H.; Azmi S.; Dangeti R.; Kaur K. Peptide Bacteriocins–Structure Activity Relationships. Curr. Top. Med. Chem. 2015, 16, 220–241. 10.2174/1568026615666150812121103. [DOI] [PubMed] [Google Scholar]
- Marquette A.; Bechinger B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. 10.3390/biom8020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K.; Ichimasa S.; Zendo T.; Koga S.; Yoneyama F.; Nakayama J.; Sonomoto K. Structural Analysis and Characterization of Lacticin Q, a Novel Bacteriocin Belonging to a New Family of Unmodified Bacteriocins of Gram-Positive Bacteria. Appl. Environ. Microbiol. 2007, 73, 2871–2877. 10.1128/AEM.02286-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P. K.; Solanki V.; Sharma S.; Thakur K. G.; Krishnan B.; Korpole S. The intramolecular disulfide-stapled structure of laterosporulin, a class IId bacteriocin, conceals a human defensin-like structural module. FEBS J. 2015, 282, 203–214. 10.1111/febs.13129. [DOI] [PubMed] [Google Scholar]
- Bodapati K. C.; Soudy R.; Etayash H.; Stiles M.; Kaur K. Design, synthesis and evaluation of antimicrobial activity of N-terminal modified Leucocin A analogues. Bioorg. Med. Chem. 2013, 21, 3715–3722. 10.1016/j.bmc.2013.04.045. [DOI] [PubMed] [Google Scholar]
- González C.; Langdon G. M.; Bruix M.; Gálvez A.; Valdivia E.; Maqueda M.; Rico M. Bacteriocin AS-48, a microbial cyclic polypeptide structurally and functionally related to mammalian NK-lysin. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 11221–11226. 10.1073/pnas.210301097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towle K. M.; Vederas J. C. Structural features of many circular and leaderless bacteriocins are similar to those in saposins and saposin-like peptides. MedChemComm 2017, 8, 276–285. 10.1039/c6md00607h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobone S.; Stella L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. Adv. Exp. Med. Biol. 2019, 1117, 175–214. 10.1007/978-981-13-3588-4_11. [DOI] [PubMed] [Google Scholar]
- Bechinger B.; Gorr S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. 10.1177/0022034516679973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanovic N.; Lohner K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Expert Opin. Invest. Drugs 2016, 9, 59. 10.3390/ph9030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanovic N.; Lohner K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta 2016, 1858, 936–946. 10.1016/j.bbamem.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Khemaissa S.; Walrant A.; Sagan S. Tryptophan, more than just an interfacial amino acid in the membrane activity of cationic cell-penetrating and antimicrobial peptides. Q. Rev. Biophys. 2022, 55, e10 10.1017/s0033583522000105. [DOI] [PubMed] [Google Scholar]
- Cruz V. L.; Ramos J.; Martinez-Salazar J.; Montalban-Lopez M.; Maqueda M. The Role of Key Amino Acids in the Antimicrobial Mechanism of a Bacteriocin Model Revealed by Molecular Simulations. J. Chem. Inf. Model. 2021, 61, 6066–6078. 10.1021/acs.jcim.1c00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll G. N.; Konings W. N.; Driessen A. J. Bacteriocins: mechanism of membrane insertion and pore formation. Antonie Leeuwenhoek 1999, 76, 185–198. 10.1023/A:1002002718501. [DOI] [PubMed] [Google Scholar]
- Liu X.; Vederas J. C.; Whittal R. M.; Zheng J.; Stiles M. E.; Carlson D.; Franz C. M. A. P.; McMullen L. M.; van Belkum M. J. Identification of an N-Terminal Formylated, Two-Peptide Bacteriocin from Enterococcus faecalis 710C. J. Agric. Food Chem. 2011, 59, 5602–5608. 10.1021/jf104751v. [DOI] [PubMed] [Google Scholar]
- Martín-Platero A. M.; Valdivia E.; Ruíz-Rodríguez M.; Soler J. J.; Martín-Vivaldi M.; Maqueda M.; Martínez-Bueno M. Characterization of Antimicrobial Substances Produced by Enterococcus faecalis MRR 10–3, Isolated from the Uropygial Gland of the Hoopoe (Upupa epops). Appl. Environ. Microbiol. 2006, 72, 4245–4249. 10.1128/AEM.02940-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintas L. M.; Casaus P.; Holo H.; Hernandez P. E.; Nes I. F.; Håvarstein L. S. Enterocins L50A and L50B, two novel bacteriocins from Enterococcus faecium L50, are related to staphylococcal hemolysins. J. Bacteriol. 1998, 180, 1988–1994. 10.1128/JB.180.8.1988-1994.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohans C. T.; Towle K. M.; Miskolzie M.; McKay R. T.; van Belkum M. J.; McMullen L. M.; Vederas J. C. Solution Structures of the Linear Leaderless Bacteriocins Enterocin 7A and 7B Resemble Carnocyclin A, a Circular Antimicrobial Peptide. Biochemistry 2013, 52, 3987–3994. 10.1021/bi400359z. [DOI] [PubMed] [Google Scholar]
- Nissen-Meyer J.; Rogne P.; Oppegard C.; Haugen H. S.; Kristiansen P. E. Structure-function relationships of the non-lanthionine-containing peptide (class II) bacteriocins produced by gram-positive bacteria. Curr. Pharm. Biotechnol. 2009, 10, 19–37. 10.2174/138920109787048661. [DOI] [PubMed] [Google Scholar]
- Zhuang Y.; Bureau H.; Quirk S.; Hernandez R. Adaptive Steered Molecular Dynamics of Biomolecules. Mol. Simulat. 2021, 47, 408–419. 10.1080/08927022.2020.1807542. [DOI] [Google Scholar]
- Ozer G.; Valeev E.; Quirk S.; Hernandez R. Adaptive Steered Molecular Dynamics of the Long-Distance Unfolding of Neuropeptide Y. J. Chem. Theory Comput. 2010, 6, 3026–3038. 10.1021/ct100320g. [DOI] [PubMed] [Google Scholar]
- Ozer G.; Quirk S.; Hernandez R. Adaptive steered molecular dynamics: Validation of the selection criterion and benchmarking energetics in vacuum. J. Chem. Phys. 2012, 136, 215104. 10.1063/1.4725183. [DOI] [PubMed] [Google Scholar]
- Ozer G.; Keyes T.; Quirk S.; Hernandez R. Multiple Branched Adaptive Steered Molecular Dynamics. J. Chem. Phys. 2014, 141, 064101. 10.1063/1.4891807. [DOI] [PubMed] [Google Scholar]
- Bureau H. R.; Merz D. Jr.; Hershkovits E.; Quirk S.; Hernandez R. Constrained Unfolding of a Helical Peptide: Implicit Versus Explicit Solvents. PLoS One 2015, 10, e0127034 10.1371/journal.pone.0127034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y.; Thota N. K.; Quirk S.; Hernandez R. Implementation of Telescoping Boxes in Adaptive Steered Molecular Dynamics. J. Chem. Theory Comput. 2022, 18, 4649–4659. 10.1021/acs.jctc.2c00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K.; Bose H. S.; Harris F. M.; Miller W. L.; Bell J. D. Binding of Steroidogenic Acute Regulatory Protein to Synthetic Membranes Suggests an Active Molten Globule. J. Biol. Chem. 2001, 276, 17044–17051. 10.1074/jbc.M100903200. [DOI] [PubMed] [Google Scholar]
- Berne S.; Sepčić K.; Anderluh G.; Turk T.; Maček P.; Ulrih N. P. Effect of pH on the Pore Forming Activity and Conformational Stability of Ostreolysin, a Lipid Raft-Binding Protein from the Edible Mushroom Pleurotus ostreatus. Biochemistry 2005, 44, 11137–11147. 10.1021/bi051013y. [DOI] [PubMed] [Google Scholar]
- Shi Y.-J.; Chiou J.-T.; Huang C.-H.; Lee Y.-C.; Wang L.-J.; Chang L.-S. Modification of carboxyl groups converts α-lactalbumin into an active molten globule state with membrane-perturbing activity and cytotoxicity. Int. J. Biol. Macromol. 2020, 163, 1697–1706. 10.1016/j.ijbiomac.2020.09.095. [DOI] [PubMed] [Google Scholar]
- Bychkova V. E.; Basova L. V.; Balobanov V. A. How membrane surface affects protein structure. Biochemistry 2014, 79, 1483–1514. 10.1134/S0006297914130045. [DOI] [PubMed] [Google Scholar]
- Shin I.; Kreimer D.; Silman I.; Weiner L. Membrane-promoted unfolding of acetylcholinesterase: A possible mechanism for insertion into the lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 2848–2852. 10.1073/pnas.94.7.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halskau O.; Underhaug J.; Frøystein N. Å.; Martínez A. Conformational Flexibility of α-Lactalbumin Related to its Membrane Binding Capacity. J. Mol. Biol. 2005, 349, 1072–1086. 10.1016/j.jmb.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Kowalska-Krochmal B.; Dudek-Wicher R. The Minimum Inhibitory Concentration of Antibiotics: Methods, Interpretation, Clinical Relevance. Pathogens 2021, 10, 165. 10.3390/pathogens10020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzynski C. Equalities and Inequalities: Irreversibility and the Second Law of Thermodynamics at the Nanoscale. Annu. Rev. Condens. Matter Phys. 2011, 2, 329–351. 10.1146/annurev-conmatphys-062910-140506. [DOI] [Google Scholar]
- Park S.; Khalili-Araghi F.; Tajkhorshid E.; Schulten K. Free Energy Calculation from Steered Molecular Dynamics Simulations Using Jarzynski’s Equality. J. Chem. Phys. 2003, 119, 3559–3566. 10.1063/1.1590311. [DOI] [Google Scholar]
- Park S.; Schulten K. Calculating Potentials of Mean Force from Steered Molecular Dynamics Simulations. J. Chem. Phys. 2004, 120, 5946–5961. 10.1063/1.1651473. [DOI] [PubMed] [Google Scholar]
- Ramírez C.; Martí M.; Roitberg A.. Computational Approaches for Studying Enzyme Mechanism Part B; Methods in Enzymology; Academic Press, 2016; Vol. 578, pp 123–143. [DOI] [PubMed] [Google Scholar]
- Dinner A. R.; Mattingly J. C.; Tempkin J. O. B.; Koten B. V.; Weare J. Trajectory Stratification of Stochastic Dynamics. SIAM Rev. 2018, 60, 909–938. 10.1137/16M1104329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; He Q. Seeding the multi-dimensional nonequilibrium pulling for Hamiltonian variation: indirect nonequilibrium free energy simulations at QM levels. Phys. Chem. Chem. Phys. 2022, 24, 8800–8819. 10.1039/D2CP00355D. [DOI] [PubMed] [Google Scholar]
- Ozer G.; Quirk S.; Hernandez R. Thermodynamics of Decaalanine Stretching in Water Obtained by Adaptive Steered Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 4837–4844. 10.1021/ct300709u. [DOI] [PubMed] [Google Scholar]
- Quirk S.; Hopkins M.; Bureau H.; Lusk R.; Allen C.; Hernandez R.; Bain D. L. Mutational Analysis of Neuropeptide Y Reveals Unusual Thermal Stability Linked to Higher-Order Self-Association. ACS Omega 2018, 3, 2141–2154. 10.1021/acsomega.7b01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau H. R.; Hershkovits E.; Quirk S.; Hernandez R. Determining the Energetics of Small β-Sheet Peptides using Adaptive Steered Molecular Dynamics. J. Chem. Theory Comput. 2016, 12, 2028–2037. 10.1021/acs.jctc.5b01110. [DOI] [PubMed] [Google Scholar]
- Bureau H.; Quirk S.; Hernandez R. The Relative Stability of Trpzip1 and Its Mutants Determined by Computation and Experiment. RSC Adv. 2020, 10, 6520–6535. 10.1039/D0RA00920B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantz E. R.; Lindert S. Adaptative Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J. Chem. Inf. Model. 2021, 61, 3052–3057. 10.1021/acs.jcim.1c00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thota N. K.; Quirk S.; Zhuang Y.; Stover E. R.; Lieberman R. L.; Hernandez R. Correlation Between Chemical Denaturation and the Unfolding Energetics of Acanthamoeba Actophorin. Biophys. J. 2023, 122, 2921–2937. 10.1016/j.bpj.2022.11.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Liyana Gunawardana V. W.; Finnegan T. J.; Xie W.; Badjić J. D. Picking on Carbonate: Kinetic Selectivity in the Encapsulation of Anions. Angew. Chem. 2022, 134, e202116518 10.1002/ange.202116518. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Bie L.; Gao J.; Wang X. Insights into Selectin Inhibitor Design from Endogenous Isomeric Ligands of SLea and SLex. J. Chem. Inf. Model. 2021, 61, 6085–6093. 10.1021/acs.jcim.1c01356. [DOI] [PubMed] [Google Scholar]
- de Freitas Fernandes A.; Leonardo D. A.; Cavini I. A.; Rosa H. V. D.; Vargas J. A.; D’Muniz Pereira H.; Nascimento A. S.; Garratt R. C. Conservation and divergence of the G-interfaces of Drosophila melanogaster septins. Cytoskeleton 2023, 80, 153–168. 10.1002/cm.21740. [DOI] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers R.; Linke M.; Barnoud J.; Reddy T.; Melo M.; Seyler S.; Domański J.; Dotson D.; Buchoux S.; Kenney I.; etal. In In Proceedings of the Python in Science Conference; Benthall S., Rostrup S., Eds.; SciPy, 2016; pp 98–105.
- Gowers R. J.; Carbone P. A Multiscale Approach to Model Hydrogen Bonding: The Case of Polyamide. J. Chem. Phys. 2015, 142, 224907. 10.1063/1.4922445. [DOI] [PubMed] [Google Scholar]
- Smith P.; Ziolek R. M.; Gazzarrini E.; Owen D. M.; Lorenz C. D. On the Interaction of Hyaluronic Acid with Synovial Fluid Lipid Membranes. Phys. Chem. Chem. Phys. 2019, 21, 9845–9857. 10.1039/C9CP01532A. [DOI] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; Mackerell A. D. Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalé L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 1999, 151, 283–312. 10.1006/jcph.1999.6201. [DOI] [Google Scholar]
- Allen C.; Bureau H. R.; McGee T. D.; Quirk S.; Hernandez R. Benchmarking Adaptive Steered Molecular Dynamics (ASMD) on CHARMM Force Fields. ChemPhysChem 2022, 23, e202200175 10.1002/cphc.202200175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterlini M. G.; Ferguson D. M. Constant temperature simulations using the Langevin equation with velocity Verlet integration. Chem. Phys. 1998, 236, 243–252. 10.1016/S0301-0104(98)00214-6. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD - Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Meng E. C.; Couch G. S.; Croll T. I.; Morris J. H.; Ferrin T. E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S. M.; Jess T. J.; Price N. C. How to Study Proteins by Circular Dichroism. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1751, 119–139. 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Ranjbar B.; Gill P. Circular Dichroism Techniques: Biomolecular and Nanostructural Analyses- A Review. Chem. Biol. Drug Des. 2009, 74, 101–120. 10.1111/j.1747-0285.2009.00847.x. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Tsai C.-J.; Ma B.; Nussinov R. Contribution of Salt Bridges Toward Protein Thermostability. J. Biomol. Struct. Dyn. 2000, 17, 79–85. 10.1080/07391102.2000.10506606. [DOI] [PubMed] [Google Scholar]
- Vernon R. M.; Chong P. A.; Tsang B.; Kim T. H.; Bah A.; Farber P.; Lin H.; Forman-Kay J. D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, e31486 10.7554/elife.31486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y.; Bureau H. R.; Lopez C.; Bucher R.; Quirk S.; Hernandez R. Energetics and structure of alanine-rich α-helices via adaptive steered molecular dynamics. Biophys. J. 2021, 120, 2009–2018. 10.1016/j.bpj.2021.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sani M.-A.; Separovic F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. 10.1021/acs.accounts.6b00074. [DOI] [PubMed] [Google Scholar]
- Sun D.; Forsman J.; Woodward C. E. Current Understanding of the Mechanisms by which Membrane-Active Peptides Permeate and Disrupt Model Lipid Membranes. Curr. Top. Med. Chem. 2015, 16, 170–186. 10.2174/1568026615666150812121241. [DOI] [PubMed] [Google Scholar]
- Yoneyama F.; Shioya K.; Zendo T.; Nakayama J.; Sonomoto K. Effect of a Negatively Charged Lipid on Membrane-Lacticin Q Interaction and Resulting Pore Formation. Biosci. Biotechnol. Biochem. 2010, 74, 218–221. 10.1271/bbb.90666. [DOI] [PubMed] [Google Scholar]
- Netz D. J. A.; Bastos M. d. C. d. F.; Sahl H.-G. Mode of Action of the Antimicrobial Peptide Aureocin A53 from Staphylococcus aureus. Appl. Environ. Microbiol. 2002, 68, 5274–5280. 10.1128/aem.68.11.5274-5280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento J. D. S.; Coelho M. L. V.; Ceotto H.; Potter A.; Fleming L. R.; Salehian Z.; Nes I. F.; do Carmo de Freire Bastos M. Genes involved in immunity to and secretion of aureocin A53, an atypical class II bacteriocin produced by Staphylococcus aureus A53. J. Bacteriol. 2012, 173, 886–892. 10.1128/jb.06203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez A.; Maqueda M.; Martínez-Bueno M.; Valdivia E. Permeation of bacterial cells, permeation of cytoplasmic and artificial membrane vesicles, and channel formation on lipid bilayers by peptide antibiotic AS-48. J. Bacteriol. 1991, 173, 886–892. 10.1128/jb.173.2.886-892.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama F.; Imura Y.; Ichimasa S.; Fujita K.; Zendo T.; Nakayama J.; Matsuzaki K.; Sonomoto K. Lacticin Q, a Lactococcal Bacteriocin, Causes High-Level Membrane Permeability in the Absence of Specific Receptors. Appl. Environ. Microbiol. 2009, 75, 538–541. 10.1128/aem.01827-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Vorobyov I.; Allen T. W. The Different Interactions of Lysine and Arginine Side Chains with Lipid Membranes. J. Phys. Chem. B 2013, 117, 11906–11920. 10.1021/jp405418y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.