

Summary
Neonatal mouse hearts have transient renewal capacity, which is lost in juvenile and adult stages. In neonatal mouse hearts, myocardial infarction (MI) causes an initial loss of cardiomyocytes. However, it is unclear which type of regulated cell death (RCD) occurs in stressed cardiomyocytes. In the current studies, we induced MI in neonatal and juvenile mouse hearts and showed that ischemic cardiomyocytes primarily undergo ferroptosis, a non-apoptotic and iron-dependent form of RCD. We demonstrated that cardiac fibroblasts (CFs) protect cardiomyocytes from ferroptosis through paracrine effects and direct cell-cell interaction. CFs show strong resistance to ferroptosis due to high ferritin expression. The fibrogenic activity of CFs, typically considered detrimental to heart function, is negatively regulated by paired-like homeodomain 2 (Pitx2) signaling from cardiomyocytes. In addition, Pitx2 prevents ferroptosis in cardiomyocytes by regulating ferroptotic genes. Understanding the regulatory mechanisms of cardiomyocyte survival and death can identify potentially translatable therapeutic strategies for MI.
Subject areas: cardiovascular medicine, Physiology, cell biology
Graphical abstract
Highlights
-
•
Neonatal and juvenile mouse cardiomyocytes mainly undergo ferroptosis after MI
-
•
Cardiac fibroblasts protect cardiomyocytes through paracrine effect
-
•
Cardiac fibroblasts interact with cardiomyocytes to share iron burden
-
•
Pitx2 pathway protects cardiomyocytes from ferroptosis and controls fibrosis
Cardiovascular medicine; Physiology; Cell biology
Introduction
Heart failure caused by MI is one of the main causes of death worldwide.1 In the infarcted myocardium, ischemia and increased reactive oxygen species (ROS) promote cardiomyocyte death and maladaptive fibrosis.2,3 Tightly regulated cell death (RCD) and cellular injury response are crucial for tissue repair and maintenance of cardiac function. It is unclear to what extent each type of RCDs contributes to the loss of cardiomyocytes in the infarcted myocardium. Widely studied RCDs in heart disease includes apoptosis, necroptosis, and autophagy.4,5,6 Ferroptosis, a recently added RCD member, is a non-apoptotic form of RCD caused by excessive production of lipid hydroperoxides in the presence of iron.7 Ferroptotic cells do not demonstrate apoptotic hallmarks such as caspase activation or cell blebbing.7 Morphologically, ferroptosis is characterized by shrinkage of mitochondria and mitochondrial membrane rupture.8,9 Cellular iron, normally stored in a protein complex composed of ferritins, is required for the onset of ferroptosis.10 Strong rationale for studying ferroptosis in the heart includes the known cardiotoxicity of iron and the presence of iron overload in the border zone of infarcted myocardium.11 Two enzymatic proteins have been identified as the major inhibitors of ferroptosis. The glutathione peroxidase 4 (Gpx4) inhibits ferroptosis by reducing lipid peroxides to lipid alcohols using its main cofactor glutathione (GSH).12 Meanwhile, the ferroptosis suppressor protein 1 (Fsp1) functions as an oxidoreductase on the cell membrane to reduce coenzyme Q10 (CoQ) and generates antioxidants to halt the propagation of lipid peroxides.13,14 Recently, increased ferroptosis was observed in adult mouse models of cardiomyopathy.15,16 Little is known about the transcriptional regulation of ferroptosis in cardiomyocytes and how ferroptotic cells affect surrounding cells after myocardial injury.
Recent studies in cultured tumor cells showed that high cell density and confluency protected the cells from ferroptosis.17 In infarcted myocardium, cell density decreases with the loss of cardiomyocytes and is restored when resident fibroblasts are activated and migrate to the infarct zone.18 CFs are fast responders to cardiac injury. They lay down extracellular matrix (ECM) to form fibrotic scarring, which is vital for the maintenance of myocardial integrity after heart attack.19 Cardiomyocyte-fibroblast interaction plays a role in maintaining cardiac function,20,21 although how this cell-cell interaction impacts cardiomyocyte death remains unanswered.
Neonatal mammalian hearts are regenerative due to residual cell-cycle activity in cardiomyocytes that is lost in juvenile and adult hearts.22,23 Utilizing mouse heart surgery models and in vitro human cardiac cell cultures, we aimed to address whether and how ferroptosis was involved in the regeneration of young hearts after injury. We found that ferroptosis contributed significantly to the loss of cardiomyocytes after MI in postnatal day (P) 1 and P7 hearts. CFs protect cardiomyocytes from ferroptosis through cell-cell interaction and paracrine signaling. CFs are resistant to ferroptosis and can tolerate high iron load. Mechanistically, the Pitx2 signaling in cardiomyocyte prevents ferroptosis and negatively regulates the fibrotic activity of CFs.
Results
Mouse cardiomyocytes mainly undergo ferroptosis after myocardial infarction in regenerative and non-regenerative stages
To analyze the prevalence of common types of RCDs in cardiomyocytes during MI, we performed left anterior descending coronary artery occlusion (LAD-O) in the regenerative P1 mouse hearts and the non-regenerative P7 hearts.24 Immunofluorescence staining of cleaved Caspase 3 (cCasp3) in tissue sections from the infarcted left ventricular free wall showed a low rate of apoptosis in cardiomyocytes at 1, 3, and 6 days post-MI (DPMI) after P1 or P7 LAD-O (Figures 1A–1G). The ratio of necroptotic cardiomyocytes, marked by phosphorylated mixed lineage kinase domain-like pseudokinase (pMLKL),25 was below 1% at all time points (Figures 1H–1N). However, more necroptotic cardiomyocytes were observed at 6 h after MI, likely caused by acute ischemic injury (Figures S1A–S1D). Next, we examined the level of prostaglandin-endoperoxide synthase 2 (Ptgs2), a commonly used ferroptotic marker (Figures 1O–1U).12 Ptgs2 was detected in 7% of cardiomyocytes at 1 DPMI after P1 LAD-O, and the ratio dropped at 3 DPMI and decreased further at 6 DPMI (Figures 1O–1Q and 1U). In comparison, after P7 LAD-O, around 10% of ferroptotic cardiomyocytes were detected in all three time points, indicating a consistent loss of cardiomyocytes through ferroptosis (Figures 1R–1U). Ptgs2 was barely detectable in cardiomyocytes after sham-operated procedure (Figures S1E–S1I), making it a suitable protein marker for studying ferroptosis in cardiomyocytes. These data suggest that ferroptosis is the major form of RCD in cardiomyocytes after either P1 or P7 injury, matching the previous report in adult mouse models of cardiomyopathy.15
Figure 1.

Ferroptosis is more prevalent in cardiomyocytes than apoptosis and necroptosis in infarcted postnatal hearts
LAD-O was performed at either P1 or P7, heart tissue section prepared at 1, 3, or 6 DPMI.
(A–G) Infarct zone stained for cCasp3 (magenta), MF20 (green), and DAPI (blue). Arrow: cardiomyocytes positive for cCasp3, ratio quantified in (G). Arrowhead: non-cardiomyocyte positive for cCasp3.
(H–N) Infarct zone stained for pMLKL (magenta), cTnT (green), and DAPI (blue). Arrow: cardiomyocytes positive for pMLKL, ratio quantified in (N). Arrowhead: non-cardiomyocyte positive for pMLKL.
(O–U) Infarct zone stained for Ptgs2 (magenta), cTnT (green), and DAPI (blue). Arrow: cardiomyocytes positive for Ptgs2, ratio quantified in (U). All bar graphs represent mean ± SD. ∗p < 0.05; NS, not significant by t test. NS in (G), no significance among all groups. #, infarct zone. Scale bar, 25 μm (A–F, H–M, O–T). See also Figures S1 and S2.
Ferroptosis is comparatively new to the family of RCDs, and the identification of reliable protein markers has not reached a consensus. Therefore, we confirmed our findings by immunofluorescence staining of 4-hydroxynonenal (4-HNE), a byproduct of lipid peroxidation (Figures S1J–S1L).15 The level of 4-HNE was detectable in cardiomyocytes after P1 LAD-O at a significantly lower rate compared with P7 LAD-O (Figures S1J–S1L). In mouse hearts, MI led to increased level of lipid peroxidation as confirmed by malondialdehyde (MDA) assay (Figure S2A). Noticeably, although LAD-O at P7 caused higher 4-HNE level at 1 and 3 DPMI compared with the P1 group, it dropped to comparable level at 6 DPMI (Figure S1L). Interestingly, 4-HNE is still present in cardiomyocytes at 21 days after P7 LAD-O, suggesting a persistent oxidative stress in the non-regenerative myocardium after injury (Figures S2B and S2C). Ptgs2 is not detectable at this point (Figure S2D), indicating that the infarct is stabilized after remodeling and the death rate of cardiomyocytes is low at this stage.
To examine if the loss of a ferroptosis inhibitor impacts neonatal heart regeneration, we generated a mouse model with cardiomyocyte-specific knockout of Gpx4 (Gpx4-CKO). Adeno-associated virus 9 (AAV9) expressing a pair of guide RNAs (gdRNAs) targeting the Gpx4 locus was injected into a mouse strain harboring a transgenic Cas9 driven by myosin heavy chain 6 (Myh6) promoter or wild-type littermates as controls (Figure S2E).26 AAV9 injection efficiently decreased the protein level of Gpx4 (Figures S2F and S2G).12 With this master inhibitor of ferroptosis knocked out in cardiomyocytes, the Gpx4-CKO mice had larger scarring area in the infarct zone at 15 days after P1 LAD-O, compared with controls (Figures S2H–S2J). Echocardiography showed compromised ventricular contractility in the Gpx4-CKO mice (Figures S2K and S2L).
So far, our data indicate that ferroptosis is a major form of RCD in both P1 and P7 hearts after LAD-O. Cardiomyocytes in P7 hearts seemed to be more vulnerable to ferroptosis compared with P1 hearts (Figure 1U). To investigate the underlying mechanism of the age-dependent differences in MI-induced ferroptosis, we examined the expression of ferritin components in myocardial tissue of P1 and P7 hearts. The onset of ferroptosis depends on available free iron. As the key components of the anti-ferroptotic, iron-storing ferritins, ferritin light chain 1 (Ftl1) and ferritin heavy chain 1 (Fth1) form protein cages while the Fth1 also oxidizes ferrous iron (Fe2+) to ferric iron (Fe3+), which is then stably stored in the mineral core of the ferritin cage.10,27,28 Immunoblotting showed higher level of Fth1 and Ftl1 protein in P1 myocardium compared with P7 myocardium (Figures S2M–S2O), suggesting a stronger ability to contain free iron and resistance to ferroptosis in neonatal cardiomyocytes.
Cardiac fibroblasts protect cardiomyocytes from ferroptosis through paracrine signaling and cell-cell interaction
Although ferroptosis has not been widely studied in models of cardiovascular disease, the ferroptotic pathway has become a popular target for anti-tumor research. It has been reported that high density/confluency of tumor cells protects them from ferroptosis through signaling initiated through cell-cell contact (Figure 2A).17 On the other hand, cardiomyocytes are packed in the myocardium at a high density, which decreases after MI (Figure 2B). We hypothesized that the influx and expansion of CFs can restore cardiac cell density and therefore protect the remaining cardiomyocytes from ferroptosis (Figure 2B). To test this possibility, human-induced pluripotent stem cells (hiPSCs) expressing GFP-tagged TITIN were differentiated into iPSC-derived cardiomyocytes (iCMs) and cultured at low, mid, or high density (Figure S3A).29 Cells were treated with erastin, a ferroptosis-inducing system Xc-inhibitor.30 Erastin caused comparable death rate in iCMs cultured at different cell densities (Figure 2C). On the contrary, cultured primary human cardiac fibroblasts (HCFs) had significantly higher resistance to erastin at mid and high density, compared with low density (Figure 2D). Next, we tested if iCMs can be protected from erastin treatment when co-cultured with HCFs to increase total cell density. iCMs were either cultured alone or co-cultured to confluency with HCFs (Figures 2E–2H). Co-culture with HCFs significantly decreased the level of PTGS2 in iCMs after erastin treatment (Figures 2E–2I), suggesting a protective effect caused by HCFs. It is noteworthy that although the iCMs are sensitive to erastin even at high culture density, a human cardiomyocyte cell line called AC16 showed density-dependent resistance to erastin (Figures S3B and S3C).31 Compared with the iCMs that beat spontaneously and do not proliferate efficiently, the immortalized AC16 cells do not beat and are highly proliferative.31 The AC16 cells do not have organized sarcomere structure and resemble the morphology of embryonic cardiomyocytes. The immature and immortalized features of AC16 cells mimic the tumor cell lines that may explain their resistance to ferroptosis. Therefore, primary culture of cardiomyocytes and iCMs is more suitable for studying ferroptosis in the heart.
Figure 2.

Cardiac fibroblasts protect cardiomyocytes from ferroptosis
(A) Schematic of high cell density protecting tumor cells from ferroptosis.
(B) Schematic showing the change of cardiac cell density during MI.
(C) Survival (negative for trypan blue) rates of iCMs cultured at low, mid, or high density after erastin treatment (15 μM, 5 h).
(D) Survival rates of HCFs cultured at low, mid, or high density after erastin treatment (15 μM, 5 h).
(E and F) PTGS2 (red) and DAPI (blue) were stained and imaged with endogenous TITIN-GFP (green) in iCM after erastin (30 μM) treatment.
(G and H) PTGS2 (red), αSMA (gray), and DAPI (blue) were stained and imaged with endogenous TITIN-GFP (green) in co-cultured iCMs and HCFs after erastin (30 μM) treatment.
(I) Fold change of PTGS2 fluorescent intensity in iCMs and HCFs.
(J–Q) Heart tissue of controls (PostnMCM/+, J, K, N, O) and PostnMCM/+;ROSA-DTA (L, M, P, Q) mice were stained for 4-HNE (magenta, J-M) or Ptgs2 (magenta, N-Q) at 4 DPMI after P1 LAD-O; tamoxifen was administered daily at 1–3 DPMI. Green, MF20; blue, DAPI. #, infarct zone. (R) Ratio of cardiomyocytes positive for Ptgs2 or 4-HNE.
(S–V) Heart sections of 2-month-old control (ROSA-DTA, S, T) and Pdgfrα-CreERT2;ROSA-DTA (U, V) mice stained for 4-HNE (red), cTnT (green), Pdgfrα (gray), and DAPI (blue) after tamoxifen administration (see Figure S3).
(W) Density of Pdgfrα-positive cells in control and mutant groups.
(X) Average 4-HNE intensity in cardiomyocytes from both groups. #, infarct zone. All bar graphs represent mean ± SD. ∗p < 0.05; ∗∗p < 0.01; NS, not significant by t test. NS in (A), no significance among three groups. Scale bar, 50 μm (E–H), 25 μm (J–Q, S–V). See also Figure S3.
Next, we determined if CFs protect cardiomyocytes from ferroptosis in vivo. We crossbred PostnMCM/+ mice, a tamoxifen-inducible myofibroblast-specific Cre strain, to Rosa-DTA strain.19 Upon tamoxifen injection, Cre-induced diphtheria toxin A subunit (DTA) expression eliminates the myofibroblasts with Periostin (Postn) gene activity (Figures S3D–S3G). Myofibroblasts, known as activated fibroblasts, are essential for myocardial remodeling after MI and can be identified by the expression of alpha smooth muscle actin (αSMA), Vimentin (Vim), and Postn.18 At four days after P1 LAD-O, PostnMCM/+;Rosa-DTA mice had significantly higher level of 4-HNE (Figures 2J–2M) and Ptgs2 (Figures 2N–2Q) in the infarct and border zone compared with the control group (Figure 2R). These results support the hypothesis that the influx of CFs can restore cardiac cell density and protect remaining cardiomyocytes from further ferroptosis (Figure 2B). The Postn lineage of CFs was reported to regulate myocardial growth and cardiac neuronal remodeling during postnatal cardiac maturation.32 At four days after P1 injury, eliminating Postn lineage cells did not affect cell-cycle activity in cardiomyocytes, as shown by immunofluorescence staining of phosphorylated histone H3 (pHH3) (Figures S3H–S3J) nor did it impact the population of myocardial nerve, marked by tyrosine hydroxylase (TH) (Figures S3K and S3L). However, loss of myofibroblasts impeded neonatal heart regeneration after MI (Figures S3M–S3Q). Notably, ablation of Postn lineage cells did not result in increased scar area, consistent with the role of myofibroblasts as the main depositor of fibrotic collagen (Figures S3M and S3N). However, it did lead to the thinning of left ventricular free wall, likely due to increased cardiomyocyte loss during initial injury (Figures S3M–S3O). In addition, ejection fraction (EF) and fractional shortening (FS) of hearts with Postn lineage ablation decreased at 15 DPMI, compared with controls (Figures S3P and S3Q).
We investigated if CFs in the adult mouse heart protect cardiomyocytes. In adult hearts, loss of myofibroblasts after MI compromises cardiac muscle integrity and affects myocardial remodeling,19,33 which may cause a secondary defect in cardiomyocytes that obstruct our investigation. To rule out this possibility, we bred Pdgfrα-CreERT2;Rosa-DTA adult mice and administered tamoxifen to eliminate cardiac interstitial fibroblasts without LAD-O procedure (Figure S3R).34 To avoid potential secondary effects, our strategy was optimized so that the myocardial interstitial fibrosis and contractile functions were not affected (Figures S3S–S3U). Previous report by the Tallquist group using the same mouse model showed that ablation of 63% of the Pdgfrα-lineage cells did not affect cardiac function but mildly altered the interstitial collagen network.35 In our studies, with effective removal of over 95% of the myocardial mesenchymal cell population, the Pdgfrα-CreERT2;Rosa-DTA hearts had significantly higher 4-HNE level in cardiomyocytes compared with controls (Figures 2S–2X), suggesting that CFs directly regulate cardiomyocyte homeostasis in adult heart. Interestingly, the Pdgfrα-CreERT2;Rosa-DTA hearts did not have increased Ptgs2 expression compared with controls (Figures S3V–S3W). It is likely that the increased lipid peroxidation caused by loss of mesenchymal cell population is not sufficient to induce ferroptosis in the absence of severe myocardial injury.
Compared with cardiomyocytes, CFs showed higher resistance to ferroptosis (Figure 2D). We characterized and authenticated the primary HCFs used in our studies. HCFs demonstrated high level of αSMA, TGFβR2, and nucleus-localized pSMAD2 and pSMAD3 (Figures 3A–3C), suggesting that HCFs have phenotype of myofibroblasts. Cardiomyocytes and CFs are exposed to increased ROS after myocardial injury.36 In vitro, H2O2 treatment reduced survival rate of both HCFs and iCMs on a dosage-dependent pattern, indicating their sensitivity to oxidative stress (Figures 3D and 3E).36 When treated with erastin at low to high dosages, HCFs survival rate decreased with dosage dependency from 2 μM to 10 μM but recovered when erastin concentration reached 20 μM (Figures 3F and 3G). It is possible that HCFs activate their stress-responding mechanisms when the severity of the challenge reaches certain threshold. When treated with 30 μM of erastin, HCF survival rate further increased compared with the group treated with 20 μM of erastin (Figures 3F and 3G). The same resistance to high erastin dosage was not observed in iCMs (Figure 3H). To validate the observed resistance of HCFs to ferroptosis in mouse models, we prepared primary cultures of CFs from P1 and P7 mouse heart ventricles.37 A similar pattern of resistance was observed in P1 CFs, with the survival rate decreased and subsequently recovered as the erastin dosage increased (Figure 3I). The P7 group also showed an increased but milder resistance compared with the P1 group (Figure 3J). In comparison, HEK-293, a human embryonic kidney epithelial cell line, had decreased survival in a dosage-dependent manner when treated with either H2O2 or erastin (Figures 3K and 3L). High confluency did not protect the HEK-293 cells from erastin-induced ferroptosis (Figure 3M). It is likely that the ferroptosis resistance is not ubiquitous but restricted to certain cell types under specific types of injury or stress.
Figure 3.

Cardiac fibroblasts are resistant to ferroptosis
(A–C) HCFs were stained for pSMAD2 [green in (A)], pSMAD3 [green in (B)], and TGFβR2 [green in (C)], with αSMA (magenta) and DAPI (blue).
(D) Survival rate of HCFs after treatment with 50, 100, or 200 μM of H2O2, compared with vehicle (H2O).
(E) Survival rate of iCM after treatment with 50, 100, or 200 μM of H2O2, compared with vehicle (H2O).
(F and G) Brightfield of HCFs after erastin or DMSO treatment. Survival rate quantified in (G).
(H) Survival rate of iCMs after erastin treatment at 2, 10, 15, 20, or 30 μM, compared with DMSO control.
(I and J) Survival rate of primary mouse cardiac fibroblasts (CFs), prepared from P1 (I) and P7 (J) hearts, after erastin treatment.
(K) Survival rate of HEK293 cells after H2O2 or vehicle treatment.
(L) Survival rate of HEK293 cells treated with erastin at gradient concentration.
(M) Survival rate of HEK293 cells cultured at low, mid, and high density after erastin treatment at 15 or 30 μM, compared with DMSO groups. All bar graphs represent mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant by t test. Scale bar, 100 μm (A–C, E). See also Figure S3.
We determined the cellular and molecular mechanisms underlying the protective roles of CFs. Considering the proximity between cardiomyocytes and CFs in myocardium, we hypothesized a paracrine-mediated and a contact-directed mechanism. HCFs were pretreated with H2O2 to mimic the oxidative stress in infarcted myocardium. H2O2 was then removed, and HCF-conditioned medium was prepared (Figure 4A). Conditioned media from H2O2-pretreated HCFs, but not vehicle-pretreated HCFs, partially rescued iCMs from erastin-induced ferroptosis, compared with non-conditioned (fresh) medium (Figure 4B). We performed a protein array assay to examine a pool of common paracrine cytokines and chemokines in the HCF-conditioned medium (Table S1). H2O2 treatment significantly increased the secretion of interleukin-8 (IL-8) and showed a trending increment of epidermal growth factor (EGF) level in HCFs (Figures 4C and 4D). IL-8, also known as CXCL8, is typically released by macrophages and epithelium to attract neutrophils during inflammation.38 Meanwhile, the EGF family ligands such as neuregulins are well known for their key roles in cell proliferation, including in cardiomyocyte.39 Treating iCM with either IL-8 or EGF recombinant protein promoted iCM survival after erastin treatment (Figures 4E and 4F). These results suggest a stoichiometric effect of a series of paracrine factors from CFs that contribute to cardiomyocyte survival after MI. One remaining question is how cardiomyocytes respond to IL-8 efficiently. Cxcr1 and Cxcr2 are known receptors of IL-8.40 To examine whether Cxcr1 and Cxcr2 are expressed in regenerating neonatal cardiomyocytes, we prepared RNA from purified cardiomyocyte nuclei at four DPMI after P1 LAD-O. qPCR showed similar level of Cxcr1 in cardiomyocytes compared with total blood cell samples (Figure 4G). RNA level of Cxcr2 is lower but detectable in cardiomyocyte (Figure 4G). In iCM culture, the protein level of CXCR1 and CXCR2 was easily detected and increased after erastin treatment (Figures 4H and 4I). These findings support the hypothesis that fibroblasts protect cardiomyocytes from ferroptosis through paracrine signaling.
Figure 4.

Fibroblast-derived cytokine and chemokine promote iCM survival after erastin treatment
(A) Flowchart of conditioned media and cytokine array experiment.
(B) Survival rate of iCMs, cultured in control medium (DMEM), HCF conditioned medium, or H2O2-treated HCF conditioned medium, after erastin treatment, normalized to respective DMSO control groups.
(C) Cytokine-chemokine protein array blotting image of conditioned media from vehicle- or H2O2-treated HCFs.
(D) Blotting signal intensity of IL-8 and EGF.
(E and F) Survival rate of iCMs after erastin treatment in the presence of IL-8 (E) or EGF (F), normalized to DMSO control groups.
(G) qPCR of CXCR1 and CXCR2 in total blood cells and purified cardiomyocyte nuclei after P1 LAD-O.
(H) Western blot of CXCR1, CXCR2, and α-tubulin in human iCMs after DMSO or erastin treatment.
(I) Normalized band (immuno-blotting) intensity of CXCR1 and CXCR2 in (H). All bar graphs represent mean ± SD. ∗p < 0.05; ∗∗p < 0.01; NS, not significant by t test.
In terms of the contact-directed mechanism for CFs to rescue ferroptotic cardiomyocytes, we observed an interesting expression pattern of Fth1 and Ftl1 in infarcted mouse hearts. At one day after P7 LAD-O, a group of Fth1-high cardiac cells lined the border of ischemic (cTnT-low) and healthy (cTnT-high) myocardium (Figure 5A). A similar pattern was observed when staining for Ftl1 (Figure 5B). Further immunofluorescent studies showed that these cells were a subgroup of Pdgfrα-expressing fibroblasts (Figures 5C and 5D). The presence of this subpopulation of fibroblasts persisted into the later period of myocardial regeneration (Figures 5E and 5F) and was also observed after P1 LAD-O (Figures S4A and S4B). We hypothesized that these ferritin-high fibroblasts establish cell-cell contact and share the iron burden with cardiomyocytes at the border zone (Figure 5G).11 Cardiac connexins (Cx), such as Cx45 and Cx43, can form gap junctions between cardiomyocytes and fibroblasts.41 After LAD-O in P1 and P7 hearts, both Cx45 and Cx43 were observed in the infarct zone between cardiomyocytes and CFs (Figures 5H, 5I, S4C, and S4D), indicating interactions between these cardiac cell types through gap junctions.
Figure 5.

Cardiac fibroblasts interact with cardiomyocytes through gap junctions to share free iron
(A and B) Wild-type mouse heart tissue stained for Fth1 [magenta, (A)] or Ftl [magenta, (B)], with cTnT (green) and DAPI (blue) at 1DPMI after P7 LAD-O. Arrows: non-cardiomyocytes positive for Fth1 (A) or Ftl (B).
(C and D) Mouse heart tissue stained for Fth1 [red, (C)] or Ftl [red, (D)], with Pdgfrα (gray), cTnT (green), and DAPI (blue) at 1 DPMI after P7 LAD-O. Arrows: cells positive for Pdgfrα and Fth1 (C) or Ftl (D).
(E and F) Mouse heart tissue stained for Fth1 [green, (E)] or Ftl [green, (F)], with Pdgfrα (red), MF20 (gray), and DAPI (blue) at 6 DPMI after P7 LAD-O. Arrows: cells positive for Pdgfrα and Fth1 (E) or Ftl (F).
(G) Diagram of cardiomyocyte-fibroblast interaction after MI.
(H and I) Mouse heart section stained for Cx45 [green, (H)] or Cx43 [green, (I)], with Pdgfrα (red), MF20 (gray), and DAPI (blue) after P7 LAD-O. Arrows: potential locations of gap junctions between cardiomyocytes and fibroblasts.
(J–L) Co-cultured iCM and HCF stained for VIMENTIN (VIM, gray), free Fe2+ (red), DAPI (blue), and imaged with TITIN-GFP (green) after DMSO (J) or erastin (15 μM) (K, L) treatment. Asterisks: HCFs with accumulation of Fe2+.
(M) siRNA knockdown of CX43 and CX45 simultaneously in iCM-HCF co-culture; Fe2+ fluorescent intensity ratio of iCMs over HCFs was quantified after erastin or DMSO treatment. LV, left ventricle. All bar graphs represent mean ± SD. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001 by t test. Scale bar, 75 μm (A, B, E, F, H), 25 μm (C, D, I, J–L). See also Figures S4 and S5.
Gap junctions have an internal diameter large enough for shuttling free iron.42 In vivo, ATAC-Seq of CFs showed increased accessibility at Gja1 (encoding Cx43) and Gjc1 loci (encoding Cx45) after MI (Figure S4E),43 suggesting higher gene activity in response to heart injury. In HCF culture, CX43 and CX45 were expressed and conjugated at the perinuclear area when the cell density was low but distributed more to the cytoplasm and plasma membrane when cell density was high (Figures S4F–S4I′). Co-culture of iCMs and HCFs showed the localization of CX43 and CX45 between the two types of cells (Figures S4J and S4K).
Iron is required for the onset of ferroptosis and is present in the culture media.7 When cultured separately, iCMs had a 55% increase of free Fe2+ after erastin treatment, whereas the HCFs had a 17% increase (Figures S4L–S4P). When co-cultured, Fe2+ accumulated in HCFs after erastin treatment, compared with adjacent iCMs (asterisks in Figures 5J–5L). This shift of Fe2+ burden was likely mediated by gap junctions, as simultaneous knockdown of CX43 and CX45 led to increased free Fe2+ in iCMs (Figure 5M). To better assess the roles of CX43 and CX45 in iron transferring activity, we overloaded the iCM-HCF co-culture with Fe2+ and performed live imaging. HCFs demonstrated a dynamic iron distribution patten with initial increment and later decline, likely due to the ferroxidase activity of FTH1 (arrow in Video S1). With CX43 and CX45 being knocked down, no obvious iron transferring activity was observed (arrows in Video S2). These data support the hypothetical model of fibroblasts sharing iron burden with cardiomyocytes after MI. Moreover, the fact that HCFs can tolerate high iron load suggests a potential mechanism underlying their high resilience under ferroptotic stress (Figure 3G). It is known that CF density increases in the infarct zone after MI. In HCF culture, we found out that increased cell density promotes FTH1 and FTL protein level (Figures S5A–S5C). Efficient knockdown of either FTH1 or FTL led to lower survival rate of HCFs when treated with 30 μM of erastin (Figures S5D–S5F), the dosage at which HCFs showed resistance (Figure 3G). These findings suggest a unique mechanism for CFs to promote cardiomyocyte survival in a pro-ferroptotic micro-environment.
Pitx2 signaling negatively regulates fibrosis and cardiomyocyte ferroptosis
We described a beneficial role of CFs during myocardial regeneration. However, the detrimental effect caused by a large population of activated fibroblasts during a heart attack cannot be neglected. Compared with P7 LAD-O, the activation of CFs into myofibroblasts occurred slightly earlier after P1 LAD-O (Figures S6A–S6D). However, P7 LAD-O resulted in a bigger population of myofibroblasts at 6 DPMI, shown by fate-mapping using PostnMCM;ROSAmT/mG mice (Figures 6A–6C). We examined key regulatory factors of fibroblast-to-myofibroblast transition using fluorescence staining and western blot. Expression of thrombospondin 1 (Tsp1), encoded by Thbs1, was significantly higher at 6 DPMI after P7 LAD-O, compared with the P1 group (Figures 6D–6F and S6E). Tsp1 activates latent TGFβ1, which is essential for myofibroblast transition, collagen deposition, and tissue scarring.44 We attempted to identify the upstream regulator of these fibrotic genes. Previously, we reported Pitx2 as a redox regulator that promotes neonatal mouse heart regeneration.36 Loss of Pitx2 in cardiomyocytes impeded neonatal heart regeneration; meanwhile, overexpression of Pitx2 in mature cardiomyocytes promoted heart repair after LAD-O.36 RNA-seq analysis showed over 30 fibrotic genes regulated by Pitx2 in regenerating neonatal heart and unveiled Pitx2 as a regulator of fibrotic response (Figures S6F–S6H).36 Cardiomyocyte-specific knockout of Pitx2 promoted the expression of a series of collagens and fibrotic regulators including Thbs1 and Smad3 in injured myocardium (Figures S6F and S6G). ChIP-Seq data showed Thbs1 as a direct target of Pitx2 in regenerating neonatal hearts (Figure S6I). Tamoxifen-induced, cardiomyocyte-specific knockdown of Pitx2 (Mhccre-ERT2;Pitx2f/f, Pitx2-iCKO)36 after P1 LAD-O led to increased Tsp1 level in the infarct zone, compared with controls (Mhccre-ERT2) (Figures 6G–6I and S6J). On the contrary, overexpression of Pitx2 specifically in cardiomyocytes (Mhccre-ERT2;Pitx2GOF, Pitx2-OE)36 reduced myocardial Tsp1 after P7 LAD-O (Figures 6J–6L and S6K). Lower Tsp1 level in the infarcted Pitx2-OE myocardium led to lower level of pSmad3, a major fibrotic regulator of the TGFβ signaling (Figures 6M–6O).45 Together, these findings support a regulatory role of Pitx2 in fibrosis after MI.
Figure 6.

Pitx2 negatively regulates fibrosis by downregulating Tsp1 expression after MI
(A and B) PostnMCM/+;ROSA-mTmG heart sections imaged for endogenous GFP at six days after P1 (A) or P7 (B) LAD-O or sham procedure. Tamoxifen administrated daily at 1, 2, 3, and 5 days after surgery.
(C) GFP-positive cell ratio over total nuclei number in sham and LAD-O groups.
(D and E) PostnMCM/+;ROSA-mTmG heart sections stained for Tsp1 (red) and DAPI (blue) at 6 DPMI after P1 (D) or P7 (E) LAD-O.
(F) Normalized band (immuno-blotting) intensity of myocardial Tsp1 at 4 DPMI after P1 or P7 LAD-O.
(G and H) Control (Mhccre-Ert) (G) and Pitx2-iCKO (Mhccre-Ert;Pitx2f/f). (H) Heart tissue stained for Tsp1 (green) and DAPI (blue) at 3 DPMI after P1 LAD-O. Tamoxifen was given at P0–P2.
(I) Normalized band intensity of Tsp1 in control and Pitx2-iCKO left ventricle at 3 DPMI after P1 LAD-O.
(J and K) Control (Mhccre-Ert) (J) and Pitx2-OE (Mhccre-Ert;Pitx2gof). (K) Hearts were stained for Tsp1 (green) and DAPI (blue) at 3 DPMI after P7 LAD-O. Tamoxifen was given at P6–P8.
(L) Normalized band intensity of Tsp1 in control and Pitx2-OE left ventricle at 3 DPMI after P7 LAD-O.
(M and O) Control (Mhccre-Ert) (M) and Pitx2-OE (Mhccre-Ert;Pitx2gof). (N) Hearts were stained for pSmad3 (green) and DAPI (blue) at 3 DPMI after P7 LAD-O. Tamoxifen was given at P6–P8. Number of pSmad3-positive cells was quantified in (O). All bar graphs represent mean ± SD. ∗p < 0.05; ∗∗p < 0.01; NS, not significant by t test. LV, left ventricle. Scale bar, 100 μm (A, B, D, E, G, H, J, K, M, N). See also Figure S6.
Further analysis of RNA-Seq data generated in regenerating neonatal mouse hearts and iCMs indicated that Pitx2 is directly involved in a cell autonomous, anti-ferroptotic mechanism.36 Pitx2 positively regulated a series of anti-ferroptotic genes including Fth1, Ftl1 (mouse)/Ftl (human), glutathione synthetase (Gss), CDGSH iron sulfur domain 1 (Cisd1) (Figures 7A and 7B),10,12,46 as well as Gpx4, as reported previously.36 Pitx2 also repressed lysophosphatidylcholine acyltransferase 3 (Lpcat3), required for the onset of ferroptosis (Figures 7A and 7B).47 The expression of these Pitx2 target genes demonstrated an anti-ferroptotic pattern in control hearts after surgery at P1, compared with the control sham hearts (Figure 7A). However, this anti-ferroptotic pattern was not noted in hearts with Pitx2 knocked out specifically in the cardiomyocytes (Figure 7A). The regulation of ferroptotic genes by PITX2 was validated using qPCR in iCMs with erastin treatment and lentivirus overexpressing PITX2 (Figure 7C). Furthermore, our earlier ChIP-Seq data showed the direct regulation of Ftl, Fth1, Gpx4, and Cisd1 by Pitx2 in regenerating neonatal mouse hearts (Figure 7D).36 In vitro, PITX2 transcript level increased in response to erastin treatment (Figure 7E), further supporting the direct involvement of PITX2 in the regulation of cardiomyocyte ferroptosis. In mouse models, immunofluorescence staining showed decreased 4-HNE in Pitx2-OE cardiomyocytes after LAD-O, compared with controls (Figures 7F–7H). Immunofluorescence staining and western blot showed lower level of Ptgs2 in Pitx2-OE hearts compared with controls, indicating lower rate of ferroptosis (Figures 7I and 7L). These data support an anti-ferroptotic role of Pitx2 during heart regeneration.
Figure 7.

Pitx2 prevents ferroptosis in cardiomyocytes by regulating ferroptotic genes
(A) Heatmap shows transcript level of ferroptotic genes in Pitx2-CKO (MCKcre;Pitx2f/f) ventricles and controls (Pitx2f/f) at five days after apex resection (DPR) or sham.
(B) Heatmap shows transcript level of ferroptotic genes in human iCMs after siRNA knockdown of PITX2, with scramble controls.
(C) qPCR of ferroptotic targets in iCMs after vehicle or erastin treatment, with PITX2-expressing or control lentivirus transduction.
(D) ChIP-Seq showing Pitx2 binding region near ferroptotic gene loci in regenerating neonatal ventricles.
(E) qPCR of PITX2 in AC16 cells treated with vehicle or erastin.
(F–H) Heart tissue of control (F) and Pitx2-OE (G) were stained for 4HNE (magenta), MF20 (green), and DAPI (blue) at three days after P7 LAD-O. Tamoxifen was administrated daily from P6–P8. Number of 4-HNE-positive cardiomyocytes was quantified in (H).
(I and J) Heart tissue of control (I) and Pitx2-OE (J) were stained for Ptgs2 (magenta), MF20 (green), and DAPI (blue) at three days after P7 LAD-O.
(K and L) Western blot of Ptgs2 in control and Pitx2-OE left ventricles at three days after sham or LAD-O at P7. Ptgs2 band intensity quantified in (L).
(M) Working model of cardiomyocytes interacting with fibroblasts to regulate ferroptosis after MI. All bar graphs represent mean ± SD. ∗p < 0.05; ∗∗p < 0.01; NS, not significant by t test. Scale bar, 75 μm (F, G), 25 μm (I, J).
Discussion
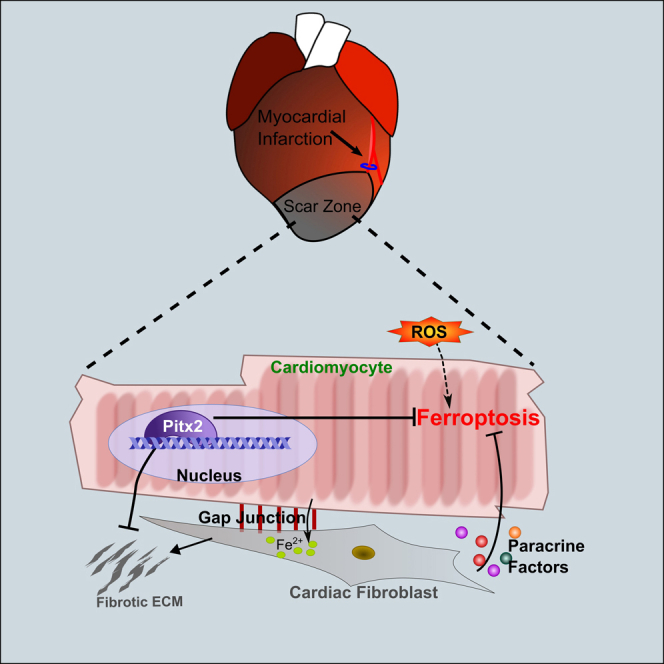
The discovery of a transient regenerative capacity in neonatal mammalian hearts was an important milestone in the field of cardiovascular regenerative medicine.22,48,49 Neonatal heart regeneration models provide an effective platform for identifying “healing-factors,” which could promote adult cardiomyocyte repair.50 Here, we showed that cardiomyocyte survival and the interaction between different cardiac cell types are also important aspects of heart regeneration. After P1 and P7 LAD-O, cardiomyocytes preferably underwent ferroptosis as compared with apoptosis and necroptosis. CFs showed resistance to ferroptosis and can protect cardiomyocytes from ferroptosis through paracrine signaling and cell-cell contact. The fibrogenic activity of CFs was kept under control by cardiomyocyte-derived Pitx2 signaling, which also regulated an intrinsic anti-ferroptotic program in cardiomyocytes.
Although cCasp3- or TUNEL-positive cardiomyocytes have been observed in animal models of heart attack, the number of apoptotic cells seemed to be minimal.51 In a pig model of Ischemia-Reperfusion injury, TUNEL reagent marks roughly 17 cardiomyocytes in every 1 cm2 of area.52 This low number of apoptotic cardiomyocytes could not explain the large number of cardiomyocytes lost during MI. Therefore, it was imperative to assess alternative RCD in mammalian MI models. Ferroptosis is comparatively new to the family of RCDs.7 As such, the field has not reached consensus on reliable molecular markers.46 The expression of Ptgs2, for example, is associated with ferroptosis but may not be a part of the driving mechanism.12 In fact, the molecular mechanism of ferroptosis may vary based on cell types and diseases, suggesting that cardiomyocytes may have their own specified ferroptotic machinery. Interestingly, our data showed a large quantity of lipid peroxidation accumulated in the nuclear and peri-nuclear area of cardiomyocytes instead of plasma membrane.53,54 This could be a protective mechanism for cardiomyocytes to avoid cell membrane damage and rupture. However, whether cardiomyocytes can efficiently fix the damaged nuclear envelope and other associated membranous structures will require further studies.
CFs, normally considered detrimental in cardiac fibrosis after injury, are required for maintaining the integrity of infarcted myocardium after heart attack.19 They promote cardiomyocyte proliferation during development.55 The fact that CFs can endure the oxidative microenvironment in an infarcted myocardium makes them candidates as the “guardians” of stressed cardiomyocytes. Here, we described two mechanisms through which the CFs can protect cardiomyocytes from ferroptosis: the production of protective paracrine factors (including IL-8 and EGF) and the direct interaction that facilitates iron transfer (Figure 7M).
Interestingly, CFs in both P1 and P7 mouse heart demonstrated resilience to ferroptosis-inducing reagent. They also showed high ferritin levels and interacted with cardiomyocytes at the border zone of infarcted myocardium. Despite the mouse heart being considered non-regenerative at P7, the protective role of CFs seemed to persist. Neonatal mouse hearts can re-establish blood supply to the infarct zone by forming collateral coronary arteries at 4 DPMI.56 Therefore, ischemic cardiomyocytes, if survive through the initial days after MI, could be rescued and recover. On the other hand, collateral coronary arteries are not formed after P7 LAD-O, and more cardiomyocytes will die eventually albeit being protected by CFs.
Importantly, we showed that the protective effect of CFs was not restricted to myofibroblasts. Adult cardiomyocytes showed increased level of 4-HNE when resident CFs were removed by genetic approaches. This observation supports the role of fibroblasts in the maintenance of myocardial homeostasis. In addition, fibroblast-to-myofibroblast transition was not required for this protective mechanism, and the insight could be taken into consideration when designing anti-fibrotic treatment.57,58 Ideally, the anti-ferroptotic function of CFs should not conflict with the anti-fibrotic treatment, which could mainly target the myofibroblasts. The timing of these therapeutic treatment could also be optimized for more favorable outcome.
We previously reported that Pitx2 efficiently promoted myocardial regeneration but had only a mild effect on cardiomyocyte proliferation.36 Our current studies provided alternative explanations as to how Pitx2 promotes heart repair. In infarcted myocardium, high ROS level and the presence of free iron likely push cardiomyocytes toward a ferroptotic fate. As a key regulator of redox balance, Pitx2 drives an intrinsic anti-ferroptotic mechanism in cardiomyocytes as shown in our studies. We have reported Gpx4 as a part of the antioxidant mechanisms regulated by Pitx2 during heart repair. Here, we showed that Gpx4 is required for neonatal heart regeneration. Its specific role in cardiomyocyte death, proliferation, and/or repair will need further investigation.
One important hallmark of ferroptotic cells is mitochondrial abnormalities.7,8 We previously reported 505 direct target genes of Pitx2 in regenerating neonatal mouse hearts. Gene Ontology analysis of these target genes revealed enrichment in mitochondria and respiratory chain activity.36 Pitx2 is deeply involved in the maintenance of the function and homeostasis of mitochondria, adding more rationale to further dissect the roles of Pitx2 signaling in ferroptosis. Although the CFs protect cardiomyocytes from ferroptosis, their fibrotic activity is kept under control by the Pitx2 signaling (Figure 7M). The mechanistic insight into the role of Pitx2 signaling could inform the invention of future therapeutic approaches for treating MI.
Limitations of the study
In the current studies, we used the common markers to examine ferroptotic cardiomyocytes. However, it is necessary to understand that the field of ferroptotic studies has not reached a consensus on reliable ferroptotic markers. The process of cardiomyocyte death is highly dynamic. Therefore, trying to depict cardiomyocyte death with fixed molecular pathways may conceal the biochemical mechanisms and features that drive the progress of cardiomyocyte death. For example, lipid peroxidation, the major cause of ferroptosis, occurs at a basal rate in cardiomyocytes as shown in our current studies. The threshold of lipid peroxidation required for initiating ferroptosis is unknown but deserves our attention in the future studies. In addition, it will be necessary to identify a protein marker of ferroptosis that can define the “point-of-no-turning-back” in stressed cardiomyocytes and other cell types.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myosin, sarcomere (MHC) antibody | Developmental Studies Hybridoma Bank | Cat# MF 20; RRID: AB_2147781 |
| Cleaved Caspase 3 | Cell Signaling Technology | Cat# 9664; RRID: AB_2070042 |
| 4-Hydroxynonenal | Bioss | Cat# bs-6313R; RRID: AB_2827741 |
| Phospho-MLKL (Ser345) | Cell Signaling Technology | Cat# 37333; RRID: AB_2799112 |
| Cox2/Cyclooxygenase 2/PTGS2 | Proteintech | Cat# 12375-1-AP; RRID: AB_2085127 |
| Anti-Troponin T, Cardiac Isoform Ab-1 Monoclonal Antibody, Unconjugated | Lab Vision | Cat# MS-295-P1ABX; RRID:AB_61809 |
| Monoclonal Anti-Actin, alpha-Smooth Muscle – Cy3™ | Sigma-Aldrich | Cat# C6198; RRID: AB_476856 |
| Recombinant Anti-pSmad3 (phosphor S423 + S425) | Abcam | Cat# ab52903; RRID: AB_882596 |
| Anti-pSmad2 (phospho S255) | Abcam | Cat#ab188334; RRID: AB_2732791 |
| Anti-TGF beta Receptor II | Abcam | Cat# ab186838; RRID: AB_2728775 |
| Ferritin Light Chain Antibody | Abcam | Cat# ab109373; RRID: AB_10862715 |
| Anti-Connexin 45/GJA7/Cx45 Antibody | Abcam | Cat# ab135474; RRID: AB_2753195 |
| Anti-Human/Rat Connexin-43 Antibody | Sigma-Aldrich | Cat# C6219; RRID: AB_476857 |
| Mouse PDGF R alpha Antibody | R and D Systems | Cat# AF1062; RRID: AB_2236897 |
| Vimentin antibody | Abcam | Cat# ab92547; RRID: AB_10562134 |
| Thrombospondin antibody | Abcam | Cat# ab85762; RRID: AB_10674322 |
| Ferritin Heavy Chain (B-12) | Santa Cruz Biotechnology | Cat# sc-376594; RRID: AB_11150859 |
| Monoclonal Anti-alpha-Tubulin antibody produced in mouse | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| CD181 (CXCR1) Monoclonal Antibody | Thermo Fisher Scientific | Cat# 12-1819-42; RRID: AB_2848258 |
| FITC anti-mouse CD182 (CXCR2) | BioLegend | Cat# 149310; RRID:AB_2566148 |
| CXCR2 Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5-83213; RRID: AB_2790369 |
| Cardiac Troponin T Monoclonal Antibody | Thermo Fisher Scientific | Cat# MA5-12960; RRID: AB_11000742 |
| GPX4 antibody | Proteintech | Cat# 67763-1-Ig; RRID: AB_2909469 |
| ACSL4 Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5-27137; RRID: AB_2544613 |
| AMID Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5-103183; RRID: AB_2852551 |
| Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody Alexa FluorTM Plus 488 | Thermo Fisher Scientific | Cat# A32723; RRID: AB_2633275 |
| Chicken anti-Mouse IgG (H + L) Cross-adsorbed Secondary Antibody, Alexa FluorTM 488 | Thermo Fisher Scientific | Cat# A-21200; RRID: AB_2535786 |
| Donkey anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa FluorTM 647 | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Donkey anti-Goat IgG (H + l) Cross-Adsorbed Secondary Antibody, Alexa FluorTM 568 | Thermo Fisher Scientific | Cat#: A-11057; RRID: AB_2534104 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa FluorTM 488 | Thermo Fisher Scientific | Cat# A-11008; RRID: AB_143165 |
| Donkey anti-Goat IgG (H + L) Antibody, Alexa Fluor® 633 | Thermo Fisher Scientific | Cat#A21082; RRID: AB_10562400 |
| Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa FluorTM 633 | Thermo Fisher Scientific | Cat# A-21050; RRID: AB_2535718 |
| Anti-Pitx2ABC | Capra Science | Cat# PA-1020; RRID: AB_2819162 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 32430; RRID: AB_1185566 |
| Mouse anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2357; RRID: AB_628497 |
| Recombinant Anti-Ferritin Heavy Chain antibody | Abcam | Cat# AB183781; RRID: AB_2940987 |
| Bacterial and virus strains | ||
| NEB® Stable Competent E. coli | New England Biolabs Inc. | C3040H |
| NEB® 5-alpha Competent E. coli | New England Biolabs Inc. | C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | SigmaAldrich | Cat# T5648 CAS: 10540-29-1 |
| IWR-1-endo | SelleckChem | Cat# S7086 CAS: 1127442-82-3 |
| Ammonium iron (II) sulfate hexahydrate 98.5–101.5% ACS | Thermo Fisher Scientific | Cat# AA13448-36 CAS:7783-85-9 |
| Erastin | MilliporeSigma | Cat# E7781 CAS: 571203-78-6 |
| Picric Acid Solution | SigmaAldrich | Cat# P6744 CAS: 88-89-1 |
| Acetic acid | Sigma-Aldrich | Cat# A6283 CAS: 64-19-7 |
| Direct Red | Sigma-Aldrich | Cat# 365548 CAS: 2610-10-8 |
| DAPI | Sigma-Aldrich | Cat# D9542 CAS: 28718-90-3 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A3059 CAS: 9048-46-8 |
| TritonTM X-100 | Sigma-Aldrich | Cat# T8787 CAS: 9036-19-5 |
| Ponceau S | Sigma-Aldrich | Cat# P3504 CAS: 6226-79-5 |
| Ferrous Ammonium Sulfate | Thermo Fisher Scientific | Cat# A15178.36 CAS: 7782-63-0 |
| Critical commercial assays | ||
| Proteome Profiler Human XL Cytokine Array Kit | R and D Systems | ARY022B |
| MilliporeSigma PROTEIN-CONCENTRATE Kit | Thermo Fisher Scientific | 50-525-36 |
| PierceTM BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Gibson Assembly Kit | New England BioLabs | E2611 |
| AAVpro® Purification Kit Midi (All Serotypes) | Takara Bio USA, Inc. | 6675 |
| Deposited data | ||
| Raw and Analyzed Data | This paper | GEO: GSE211568 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | Cat# CRL-1573, RRID: CVCL_0045 |
| AC16 (Human hybrid cardiomyocyte) | MilliporeSigma | RRID: CVCL_4U18 |
| WTC-mEGFP-TTN-cl39 | Coriell Institute for Medical Research | RRID: CVCL_UD16 |
| Primary Human Cardiac Fibroblasts (HCF) | Cell Applications | Cat# 306V-05A N/A |
| AAVpro® 293T cell Line | Takara Bio USA, Inc. | Cat# 632273 |
| Lenti-X 293T | Clontech Laboratories, Inc. | Cat# 632180 |
| Experimental models: Organisms/strains | ||
| Mouse:FVB/NJ | The Jackson Laboratory | RRID: IMSR_JAX:001800 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: B6.FVB(129)-A1cfTg(Myh6−cre/Esr1∗)JmK/J | The Jackson Laboratory | RRID: IMSR_JAX:005657 |
| Mouse: B6.129S-Pdgfratm1.1(cre/ERT2)Blh/J | The Jackson Laboratory | RRID: IMSR_JAX:032770 |
| Mouse: B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J | The Jackson Laboratory | RRID: IMSR_JAX:009669 |
| Mouse: B6.129S-Postntm2.1(cre/Esr1∗)Jmol/J | The Jackson Laboratory | RRID: IMSR_JAX:029645 |
| Mouse: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,−EGFP)Luo/J | The Jackson Laboratory | RRID: IMSR_JAX:007676 |
| Mouse: Mhccre-Er | The Jackson Laboratory | IMSR_JAX:005657 |
| Mouse: Pitx2GoF | Tao et al., Nature 201636 | N/A |
| Mouse: Pitx2f/f | Gage PJ et al., Development 199959 | N/A |
| Mouse: Myh6Cas9 | Carroll et al., PNAS 201626 | N/A |
| Oligonucleotides | ||
| siRNA targeting sequence FTH1 (hs.Ri.FTH1.13.2) rGrUrUrUrArCrCrUrGrUrCrCrArUrGrUrCrUrUrArCrUrAC U rArGrUrArGrUrArArGrArCrArUrGrGrArCrArGrGrUrArArArCrGrU |
Integrated DNA Technologies | Cat# hs.Ri.FTH1.13.2 |
| siRNA targeting sequence FTL (hs.Ri.FTL.13.1) rGrArGrGrArArGrUrGrArArGrCrUrUrArUrCrArArGrArAGA rUrCrUrUrCrUrUrGrArUrArArGrCrUrUrCrArCrUrUrCrCrUrCrArU |
Integrated DNA Technologies | Cat# hs.Ri.FTL.13.1 |
| siRNA targeting sequence CX43 (hs.Ri.GJC1.13.1) rArGrArArUrGrGrArCrUrUrArCrArUrGrUrUrArUrCrArGUU rArArCrUrGrArUrArArCrArUrGrUrArArGrUrCrCrArUrUrCrUrUrC |
Integrated DNA Technologies | Cat# hs.Ri.GJC.13.1 |
| siRNA targeting sequence CX45 (hs.Ri.GJA1.13.2) rGrUrGrGrUrArCrArUrCrUrArUrGrGrArUrUrCrArGrCrUUG rCrArArGrCrUrGrArArUrCrCrArUrArGrArUrGrUrArCrCrArCrUrG |
Integrated DNA Technologies | Cat# hs.Ri.GJA1.13.2 |
| Scrambled Negative Control rCrUrUrCrCrUrCrUrCrUrUrUrCrUrCrUrCrCrCrUrUrGrUGA rUrCrArCrArArGrGrGrArGrArGrArArArGrArGrArGrGrArArGrGrA |
Integrated DNA Technologies | Cat# 51-01-19-08 Cat# 51-01-19-09 |
| Recombinant DNA | ||
| AAV-U6-GPX4gRNA1-U6gGPX4RNA2-TnT-Cre | This paper | N/A |
| AAV-U6gRNA1-U6gRNA2-TnT-Cre | Addgene | 87682 |
| pAAV2/9n | Addgene | 112865 |
| pAdDeltaF6 | Addgene | 112867 |
| pWPI | Addgene | 12254 |
| pMD2.G | Addgene | 12259 |
| psPAX2 | Addgene | 12260 |
| Software and algorithms | ||
| ImageJ | NIH | RRID: SCR_00370 https://imagej.net/software/ |
| STAR (Spliced Transcripts Alignment to a Reference) | Dobin et al.60 | RRID:SCR_004463 |
| DESeq2 | Love et al.61 | RRID:SCR_015687 |
| Graphpad Prism | GraphPad Software | RRID:SCR_002798 https://www.graphpad.com/ |
| R | R core team | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ge Tao (taog@musc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-Seq data have been deposited to GEO (GSE211568) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.
Experimental model and study participant details
Mouse lines
All animal protocols and procedures complied with the NIH guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical University of South Carolina (Charleston, South Carolina 29425, USA). The FVB (JAX 001800), C57BL/6J (JAX 000664), Mhccre-Ert (JAX 005657), Pdgfrα-CreERT2 (JAX 032770), ROSA-DTA (JAX 009669), PostnMCM (JAX 029645), ROSA-mTmG (JAX 007676) were purchased from the Jackson Laboratory. The Pitx2GOF transgene for overexpressing Pitx2 and the floxed allele for Pitx2 (Pitx2f/f) were described previously.36,59 The Myh6Cas9 strain was previously described.26 All mice were housed in a temperature- and humidity-controlled room and maintained on a 12:12 light:dark cycle, with standard chow and water. DNA was extracted from tail or ear biopsies for genotyping using primers provided by JAX or described previously.36 For all mouse studies, both male and female mice were used and randomly distributed among groups. 8–16 weeks old adult male and female mice were placed in same cage for mating. The neonatal mice (1–7 days after birth) were used in surgical models and cell isolation. 10–12 weeks old Pdgfrα-CreERT2;ROSA-DTA were used as adult mouse model in Figure 2. Genotyping primer sequence are as follows. Pitx2f/f: Forward, 5′-TCGTGTCTTAAAAGGATGTGTTTCTTC-3’; Reverse, 5′-CGCCCACATCCTCATTCTTTCC-3’. Mhccre-Ert: Common forward, 5′-TCTATTGCACACAGCAATCCA-3’; wild type reverse, 5′-CCAACTCTTGTGAGAGGAGCA-3’; mutant reverse, 5′-CCAGCATTGTGAGAACAAGG-3’. ROSA-mTmG: Wild type forward, 5′-AGGGAGCTGCAGTGGAGTAG-3’; Mutant forward, 5′-TAGAGCTTGCGGAACCCTTC-3’; Common reverse, 5′-CTTTAAGCCTGCCCAGAAGA-3’. Pitx2GOF: Forward, 5′-CAAACATAGACTGGAGGTGCAT-3’; Reverse, 5′-TCCCTATAAACGTACGGAGGAG-3’. ROSA-DTA: Wild type forward, 5′-CCAAAGTCGCTCTGAGTTGTTATC-3’; Wild type reverse, 5′-GAGCGGGAGAAATGGATATG-3’; Mutant forward, 5′-CGACCTGCAGGTCCTCG-3’; Mutant reverse, 5′-CTCGAGTTTGTCCAATTATGTCAC-3’. Pdgfrα-CreERT2: Wide type forward, 5′-GCCTTAAGCTGGGACATGCT-3’; Mutant forward, 5′-ATCGCATTCCTTGCAAAAGT-3’; Common reverse, 5′-AGGCCCACAGAACATGGAC-3’. PostnMCM: Wild type forward, 5′-GGTGCTTCTGTAAGGCCATC-3’; Mutant forward, 5′-GGTGGGACATTTGAGTTGCT-3’; Common reverse, 5′-CCTTGCAATAAGTAAAACAGC TC-3’. sMyh6Cas9: Forward, 5′-ACATGGCCGTCATCAAAGA-3’; Reverse, 5′-CTTGTACAGCTCGTCCATGC-3’.
Cell culture
HEK293 cells (ATCC CRL-1573) were cultured in Eagle’s Minimum Essential Medium (ATCC, 30–2003), supplemented with 10% FBS (Corning, 35-011-CV) and 1% penicillin/streptomycin (Corning, 30-002-CI). Lenti-X 293T and AAVpro 293T cell Lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose (4.5 g/L), 4 mM L-glutamine, and 3.7 g/L sodium bicarbonate (Sigma-Aldrich Co., No. D5796); supplemented with 10% fetal bovine serum and 1 mM sodium pyruvate. AC16 human cardiomyocyte cell line (MilliporeSigma, SCC109) was cultured in DMEM/Hams F-12 50/50 Mix (Corning, 10-092-CV), supplemented with 12.5%FBS and 1% penicillin/streptomycin. Human Cardiac Fibroblasts (HCF) (Cell Applications Inc, 306V-05a) were cultured in HCF Growth Medium (Cell Applications Inc, 316–500). Cell lines have been authenticated either by the vendor, or by our group using qPCR of cell-type specific gene markers. All cell lines were tested for mycoplasma contamination at the beginning of the projects and no contamination was observed.
Differentiation of human iPSCs to cardiomyocytes (iCMs)
iPSCs (Coriell Institute for Medical Research, AICS-0048-039) were cultured in mTeSR1media (STEMCELL Technologies, 85850) on Matrigel (Gibco, A1413302)-coated plates. At 80% of confluency, iPSCs were differentiated into cardiomyocytes as previously described.29 Briefly, the iPSCs were treated with 8μM CHIR-99021 (SelleckChem, S2924) in RPMI (Gibco, 11875093)-B27(no insulin) (Gibco, A1895601) from day 0–1. Media was changed on day 2 and the cells were treated with 5 μM IWR1 (SelleckChem, S7086) in RPMI-B27(no insulin) from day 3–4. Starting from day 7, RPMI-B27(with insulin) (Gibco, 17504044) media were given to iPSC-derived cardiomyocytes (iCMs). Two rounds of glucose starvation from day 12–15 and from day 20–23 were performed to eliminate non-cardiomyocyte cells. At day 30, iCMs were transferred to Matrigel-coated 24-well plates and maintained for further use. For co-culture of iCM and HCF, iCM cells were seeded at 2.5 × 104 cells per well in a Matrigel-coated 24-well plate on day 1, and HCF cells were seeded at 1.5 × 104 cells per well on day 2. Cells were cultured in RPMI-B27 (with insulin). Co-culture was used on day 3 for experiments and analysis.
Primary culture of mouse cardiac fibroblasts
Isolated wild type hearts (P1 or P7) were minced into 1 mm3 pieces and placed in 0.1% gelatin (Sigma, G1890)-coated 6-well plates with 300 μL explant media (DMEM, 20%FBS). Three hours later, when tissue pieces settled, another 2mL media was slowly added and replaced every 3 days. Migrated cells were harvested at day 7 and filtered through 40 μM cell strainers for further experiments.37
Method details
Left anterior descending coronary artery occlusion (LAD-O)
For all mouse survival surgeries, littermate controls were used whenever possible. Both male and female mice were randomly distributed among groups. All surgeries were done blinded from mouse genotype, sex, or treatment. LAD-O was performed on P1 and P7 mice as previously described.24 Briefly, mice were placed under ice to anesthetize. Nylon sutures (AD Surgical, S-G618R13-U) were used to occlude the left anterior descending artery (LAD). Occlusion was confirmed by blanching of the myocardium. VetBond tissue adhesive (Santa Cruz Biotechnology, NC0846393) was used to close the thoracic cavity. The entire procedure lasted approximately 10 min from hypothermia induction until recovery. Sham procedures were done identically without the occlusion of coronary artery. PostnMCM/+;ROSA-mTmG mice underwent LAD-O at P1 or P7; tamoxifen (SigmaAldrich, T5648) was administered subcutaneously at 1, 2, 3 and 5 days-post-surgery before heart tissue collection at 6 days post-surgery. PostnMCM/+;ROSA-DTA mice were subjected to LAD-O surgery at P1 with tamoxifen administered at P2-4 before heart collection for analysis at P5. Pitx2-iCKO (Mhccre-ERT;Pitx2f/f) and control (Mhccre-ERT) mice were subjected to LAD-O at P1 and tamoxifen injection at P0-2; hearts were collected 3 days post-surgery for analysis. Pitx2-OE (Mhccre-ERT;Pitx2GOF) and control (Mhccre-ERT) mice were subjected to LAD-O at P7 with tamoxifen injection at P6-8, hearts collected at 3 days post-surgery for analysis.
Echocardiography
8-week-old Pdgfrα-CreERT2;ROSA-DTA mice had intraperitoneal tamoxifen injection as described in Figure S3H. Echocardiography was performed using a Vevo 3100 ultrasound system (Fujifilm VisualSonics), equipped with a MX550D transducer. B-mode and M-mode data were acquired following the manufacturer’s guidelines.
HCF-conditioned medium
HCF cells were cultured in 10 cM dishes to confluency, washed with PBS, and then treated with 200 μM H2O2 in DMEM basal media. After 2 h, H2O2 was removed, cells were washed with PBS 3 times and cultured in DMEM basal media overnight. Conditioned media were collected and filtered through a 0.2 μm Supor membrane (Pall Laboratory, 4612). Control conditioned medium was prepared in the same way but treated with H2O.
Cytokine array assay
HCF-conditioned media were applied to the Proteome Profiler Human XL Cytokine Array Kit (R&D Systems, ARY022B). 5mL of conditioned medium from H2O2 or H2O treated HCFs was concentrated into 500μL using MilliporeSigma Protein-Concentrate Kit (Fisher Scientific, 50-525- 36) according to the manufacturer’s instructions. Equal amount of total protein from all groups was loaded on the cytokine array. Blotting signal was visualized using chemiluminescence detection and a ChemiDoc Touch Imaging System (Bio-Rad, 12003154).
Transfection of siRNA in HCF and iCM cells
Lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific, 13778030) was used to deliver siRNA into HCFs and iCMs following the manufacturer’s guidelines. Scrambled siRNA was used as control. The siRNA oligonucleotides were pre-designed DsiRNA Duplex from Integrated DNA Technologies (IDT) with oligonucleotide sequences as follow:
FTH1 (hs.Ri.FTH1.13.2)
rGrUrUrUrArCrCrUrGrUrCrCrArUrGrUrCrUrUrArCrUrACU rArGrUrArGrUrArArGrArCrArUrGrGrArCrArGrGrUrArArArCrGrU
FTL (hs.Ri.FTL.13.1)
rGrArGrGrArArGrUrGrArArGrCrUrUrArUrCrArArGrArAGA rUrCrUrUrCrUrUrGrArUrArArGrCrUrUrCrArCrUrUrCrCrUrCrArU
CX43 (hs.Ri.GJC1.13.1)
rArGrArArUrGrGrArCrUrUrArCrArUrGrUrUrArUrCrArGUU rArArCrUrGrArUrArArCrArUrGrUrArArGrUrCrCrArUrUrCrUrUrC
CX45 (hs.Ri.GJA1.13.2)
rGrUrGrGrUrArCrArUrCrUrArUrGrGrArUrUrCrArGrCrUUG rCrArArGrCrUrGrArArUrCrCrArUrArGrArUrGrUrArCrCrArCrUrG
Scrambled negative control:
rCrUrUrCrCrUrCrUrCrUrUrUrCrUrCrUrCrCrCrUrUrGrUGA rUrCrArCrArArGrGrGrArGrArGrArArArGrArGrArGrGrArArGrGrA
Lentivirus transduction
Coding sequence of human PITX2C (NM_000325.6) was cloned into lentivirus transfer plasmid (pWPI, Addgene#12254) using Gibson Assembly kit (NEB, E2611).
Primers used for amplifying PITX2C insert:
Forward: 5′- CTAGCCTCGAGGTTTAT GAACTGCATGAAAGGCCC-3’
Reverse: 5′- TGCAGCCCGTAGTTTCCGGCAAGG TCCTAGGATCC-3’.
Empty pWPI was used for preparing control lentivirus. For lentivirus packaging, pWPI, pMD2.G (Addgene#12259) and psPAX2 (Addgene#12260) plasmids were co-transfected into Lenti-XTM 293T cells (Clontech Laboratories, 632180) using Lipofectamine 3000 (Fisher, L3000008). Virus particles in the supernatant was collected, filtered and stored in −80°C freezer until be used.
AAV9 viral particle preparation
A pair of guide RNAs targeting mouse Gpx4 locus were designed using CRISPick (Broad Institute Genetic Perturbation Platform): gdRNA#1:GTCGGCGGCGCCTTGGCTAC; gdRNA#2: GGTGACTACCTACGGTGAGT. Both gdRNAs were cloned into an AAV-U6gRNA1-U6gRNA2-TnT-Cre plasmid (Addgene#87682), and co-transfected with pAAV2/9n (Addgene#112865) and pAdDeltaF6 (Addgene#112867) into AAVpro 293T cell Line (Takara, Cat#632273) following protocol described previously.62 AAV9 particles were harvested from the cell culture using AAVpro Purification Kit (Takara 6675) following manufacturer’s guidelines.
Live imaging of HCF and iCM co-culture with iron overload
iCMs were seeded and allowed to acclimate for 3 days until spontaneously beating was observed. siRNA was used to knockdown CX43 and CX45 for 24 h as described above. iCMs were then washed with PBS. Similar number of HCFs were added to iCM culture to reach confluency. The co-culture was allowed to acclimate overnight. The following morning cells were washed with PBS and treated with FluoroBrite DMEM media containing 25 μM of ferrous ammonium sulfate (Thermo, A15178.36) for 1 h. Following treatment cells were washed with PBS three times and BioTracker FerroOrange Live Cell Dye (MilliporeSigma, SCT210) (1000×) diluted in FluoroBrite DMEM was added to the cells. After 1 h of incubation, cells were washed twice with FluoroBrite DMEM and imaged on a Leica SP8 confocal microscope with live imaging chamber.
Cell viability measurement
For low density, HCF, HEK293, and iCM cells were seeded at 3×103 cells per well in a 24-well plate. For middle density, HCF and HEK293 cells were seeded at 3×104 cells per well while iCM cells were seeded at 2×104 per well in a 24-well plate. For high density, HCF and HEK293 cells were seeded at 105 cells per well and iCMs were seeded at 6×104 cells per well in 24-well plate (Figure S3A). Cells were cultured overnight and then treated with erastin (Sigma, E7781) at varying concentrations in DMEM or RPMI basal medium for 5 or 16 h. DMSO-treated wells were used as controls. HCF and HEK293 cells were treated with varying concentrations of H2O2 for 6 h in DMEM basal medium and iCMs were treated similarly but in RPMI basal medium. Primary culture of P1 and P7 mouse cardiac fibroblasts were treated with erastin or DMSO for 16 h in DMEM basal medium. Cell viability was assessed using Trypan Blue (Invitrogen, T10282) according to the manufacturer’s instructions. Cell viability was reported as a ratio of Trypan-Blue negative cell number in experimental group over the vehicle control group. AC16 cells were seeded at low (3×104) or high (8×104) density in a 24-well plate and cultured overnight before being treated with erastin at varying concentrations in F12/DMEM basal medium, with DMSO as controls for 9 h. Cells were then washed with HEPES then stained with DAPI and Sytox Green (Thermo Fisher, S7020) at 37°C for 20 min. Following staining, cells were washed with HEPES and immediately imaged using a Lionheart FX Automated Live Cell Imager (Agilent).
Fe2+ staining
iCMs were stained with FerroOrange Live Cell Dye according to the manufacturer’s guideline. Briefly, cells were rinsed with PBS. The dye was pre-diluted to 1X in Fluorobrite DMEM (Thermo Fisher, A1896701) and added to cells. After 30 min of incubation at 37°C, cells were rinsed with PBS twice and fixed in 10% Neutral Buffered Formalin (Leica, 3800598) for 15 min at room temperature before imaging using a Leica SP8 Confocal Microscope (Leica Microsystems).
Tissue processing, histology, and immunohistochemistry
Hearts were fixed in 10% formalin (VWR, 10015-192) at room temperature overnight with continuous rocking. The tissue was then processed for paraffin embedding. Briefly, tissue was dehydrated through ethanol and xylene before embedding in paraffin wax. Tissue was sectioned at 7 μm thickness on VistaVision microscope slides (VWR, 16004-406), deparaffinized in xylene, and rehydrated through ethanol. For immunofluorescent staining, tissue slides were heated in 6% citrate buffer for antigen retrieval and permeabilized using 0.2% Triton X-100 (Sigma, T8787). Primary antibody was diluted in 1% BSA in PBS and incubated on slides at 4°C in a humid chamber. After overnight incubation, slides were washed in PBS and incubated for 1 h at room temperature in secondary antibody diluted in 1% BSA in PBS. Slides were washed with PBS and counterstained with DAPI (Sigma, D9542) then mounted in VECTASHIELD hardset mounting medium (Vector Laboratories, H1400). HCFs were fixed in formalin for 15 min followed by PBS wash and permeabilization in 0.2% Triton X-100 (Sigma, T8787) in PBS. Cells were blocked for 1 h in 5% BSA (Sigma, A3059) in PBS at room temperature before the incubation with primary antibody for 2 h and fluorophore-labeled secondary antibody for 1 h. Cells were counterstained with DAPI (Sigma, D9542) and placed in cold PBS for imaging. Immunofluorescent images were acquired using a Leica SP8 confocal microscope (Leica Microsystems). Primary antibodies used include: cleaved Caspase 3 (1:200, Cell Signaling, 9664), pMLKL (1:100, Cell Signaling, D6E3G), PTGS2 (1:100, Proteintech, 12375-1-AP), 4-HNE (1:100, Bioss, bs-6313R), αSMA (1:400, Sigma, C6198), pSMAD2 (1:200, Abcam, ab188334), pSMAD3 (1:200, Abcam, ab52903), TGFβRII (1:200 Abcam, ab186838), FTH1 (1:200, Abcam, ab183781), FTL (1:100, Abcam, ab109373) CX45 (1:200, Abcam, ab135474), CX43 (1:200, Sigma, C6219), PDGFRα (1:100, R&D Systems, AF1062-SP), MF20 (1:400, Developmental Studies Hybridoma Bank, AB_2147781), Vimentin (1:400, Abcam, ab92547), TSP1 (1:100, Abcam, ab85762), and cTNT (1:400, Fisher, ms-295p1abx). Secondary antibodies used include: Goat Anti-Mouse IgG 488 (1:400, Santa Cruz Biotechnology, A32723), Chicken Anti-Mouse IgG 488 (1:400, Thermo Invitrogen, A21200), Donkey Anti-Rabbit IgG 647 (1:400, Thermo Invitrogen, A31573), Alexa 488 Goat Anti-Rabbit (1:400, Thermo Invitrogen, A11008), Donkey Anti-Goat IgG 633 (1:400, Thermo Invitrogen, A21082), Alexa 633 Goat Anti-Mouse (1:400, Thermo Invitrogen, A21050), Donkey Anti-Goat 568 (1:200, Thermo Invitrogen, A11057). For Picro-Sirius Red staining, tissue slides were deparaffinized, washed with water, before immersion in 0.1% Picro-Sirius Red made with picric acid (Sigma, P6744-1GA) and Direct Red (Sigma, 365548). Slides were then washed in 0.5% glacial acetic acid (Sigma, A6283), dehydrated in ethanol, cleared in xylene, and mounted using Shurmount (General Data, LC-A).
Western blot
HCFs or mouse heart tissue were lysed in RIPA buffer. Protein concentration was determined using Pierce BCA protein assay kit (Thermo Fisher, PI23227) according to manufacturer’s instructions. Western blot was performed as previously described.63 Total protein was examined using Pierce Reversible Protein Stain Kit for PVDF membranes (Thermo Fisher, PI24585) and imaged using Bio-Rad ChemiDoc Imaging System (Bio-Rad, 12003154). Target protein was detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher, 34577). Target band intensities were quantified using ImageJ software (National Institutes of Health). Primary Antibodies used include: FTH1 (1:1000, Santa Cruz Biotechnology, sc-376594), FTL (1:1000, Abcam, ab109373), PTGS2 (1:1000, Proteintech, 12375-1-AP), TSP1 (1:1000, Abcam, ab85762), and α-TUBULIN (1:1000, Sigma, T5168-100UL). Secondary antibodies include goat-anti-mouse (1:2500, Thermo Invitrogen, 32430) or goat-anti-rabbit (1:2500, Santa Cruz Biotechnology, sc2357) horseradish peroxidase (HRP)-conjugated antibodies.
qPCR
RNA was prepared from cells or heart tissue using TRIzol (Fisher, 15596018) according to manufacturer’s guideline. cDNA was prepared from 1 μg of RNA with iScript Reverse Transcription Supermix (Bio-Rad, 1708841). qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 172–5271) and a CFX96 Touch Real-Time PCR machine (BioRad). Primers used for human genes were as follows: PITX2, forward: 5′-AGCGGACTCACTTTACCAGC-3′, reverse: 5′-CCGTAAGGTTGGTCCACACA-3’; FTL, forward: 5′-TGGGCTTCTATTTCGACCGC-3′, reverse: 5′-TTTCATGGCGTCTGGGGTTT-3’; FTH1, forward: 5′- GCTCTACGCCTCCTACGTTT-3′, reverse: 5′-AAGGAAGATTCGGCCACCTC-3’; CISD1, forward: 5′- GCTGTGTACTGCCGTTGTTG-3′, reverse: 5′-TGATCAGAGGGCCCACATTG-3’; GPX4, forward: 5′-GAGGCAAGACCGAAGTAAACTAC-3′, reverse: 5′- CCGAACTGGTTACACGGGAA-3’; ACTB, forward: 5′- CAATGAGCTGCGTGTGGCT-3′, reverse: 5′-GGATAGCACAGCCTGGATAGCAA-3’; HSP90, forward: 5′- CGAAGTTGGACAGTGGTAAAGAG-3′, reverse: 5′- TGCCCAATCATGGAGATGTCT-3’;
Primers used for mouse genes were as follows: Cxcr1, forward: 5′-TGTCCCTTCTGAGCTTGCTG-3′, reverse: 5′- CCAAGAAGGGCAGGGTCAAT-3’; Cxcr2, forward: 5′-CGCTGCTCATCATGCTGTTC-3′, reverse: 5′- GCAGGAAGACAAGGACGACA-3’. Col6a2, forward: 5′- GGAGATAAAGGGGAGCCAGC -3′, reverse: 5′- ATCCACAGGTCTCCCTCACA -3’. Col1a1, forward: 5′- GTGGAAACCCGAGGTATGCT -3′, reverse: 5′- GGGTCCCTCGACTCCTACAT -3’. Col5a1, forward: 5′- GGGCTCCAACGATGAGGAAA -3′, reverse: 5′- ATTTCTGTGAGGCTTCGCCA -3’. Col15a1, forward: 5′- CATGGTGTCCGTCTTGTGGA -3′, reverse: 5′- CGATGCACAGCACGATTAGC -3’. Col4a2, forward: 5′- CGGCGTAATCTCAAAAGGCG -3′, reverse: 5′- GGCCTCTGCTTCCTTTCTGT -3’. Col4a1, forward: 5′- ACAACATCCGGCCCTTCATT -3′, reverse: 5′- TTAGGGCACTGCGGAATCTG -3’. Col6a6, forward: 5′- CTGCTCACGGCGATTAAAGC -3′, reverse: 5′- ACTGCTAAAGAACGTGGCGA -3’. Col6a3, forward: 5′- GAGGTGGGCAAGATAGCCTC -3′, reverse: 5′- CAGGCTTTGGAGACATCGGT -3’. Col5a3, forward: 5′- TCGATTCCACAACCACAGGG -3′, reverse: 5′- ATGTGGAGGGAGGTCTGGTT -3’. Col1a2, forward: 5′- TAGCCAACCGTGCTTCTCAG -3′, reverse: 5′- TGCCCTCAGCAACAAGTTCA -3’. MMP15, forward: 5′- ACCTGCATGGAATCAGCCTC -3′, reverse: 5′- ACCATCTGGGGAGCCATACA -3’. MMP23, forward: 5′- CCCACACGCTACAGTTGGAA -3′, reverse: 5′- AGTTCATCCTGGGACAGTGC -3’. MMP9, forward: 5′- GCGTCATTCGCGTGGATAAG -3′, reverse: 5′- CCTGGTTCACCTCATGGTCC -3’. Timp4, forward: 5′- TGCCAATGCCATGTACCATCT -3′, reverse: 5′- TCATGCAGACATAGTGCTGGG -3’. Fbn1, forward: 5′- GCCACCGCCAGAATATCCAT -3′, reverse: 5′- AGCGCCCATTTTGACAGAGA -3’. Hsp90ab1, forward: 5′- TGGAGGAGAGCAAGGCAAAG -3′, reverse: 5′- GTGCTTGTCACAATGCAGCA -3’. Actb, forward: 5′- TACTGCTCTGGCTCCTAGCA -3′, reverse: 5′- CGGACTCATCGTACTCCTGC -3’.
RNA-Seq
Total RNA was extracted from iCMs treated with siRNA targeting PITX2 or scramble control using TRIzol (Fisher, 15596018). RNA-Seq was performed using Novogene’s Illumina NovaSeq PE150 for nextgeneration sequencing according to the manufacturer’s direction. Sequenced reads in Fastq format were aligned to mm10 reference genome using STAR (Spliced Transcripts Alignment to a Reference) alignment software.60 R package DESeq2 was used to normalize and quantify the aligned RNA-seq reads with threshold p ≤ 0.05 and log2foldchange ≤ −0.58 to compare each treatment groups to another.61 Heatmaps were created using R package Pheatmap. The RNA-Seq data from this study have been deposited in GEO under accession number GEO: GSE211568.
Quantification and statistical analysis
All quantitative experiments (qPCR, western blot, cell count) included at least 3 biological replicates. Animal studies included at least 3 mice per group. Statistical significance was determined by t-test. Equal variance was assumed. Outliers were determined by Grubb’s test. All statistical work was done on GraphPad Prism (GraphPad Software). All bar graphs included scattered dots. All bar graphs represent mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 were considered statistically significant.
Acknowledgments
This project was supported by grants from the National Institutes of Health (NIH) (1R01HL148728 to G.T.; HL144938 to H.M.S.). G.T. was supported in part by American Heart Association (AHA) (17SDG33400141), Saving Tiny Hearts Society, NSF EPSCoR RII Track-1: Materials Assembly and Design Excellence in South Carolina (MADE in SC) OIA-1655740, and NIH (1R21AI162775-01A1/412712-19270). M.E.M. was supported in part by training grant from NIH (R25GM072643). A.M.T. was supported in part by training grant from NIH (T32GM132055). R.A.S. was supported in part by training grant from NIH (HL007260). C.L. was supported by AHA (19CDA34760019). M.Z. was supported by National Natural Science Foundation of China (81901489). The Molecular Analytics Core at the Medical University of South Carolina (MUSC) is supported by NIGMS GM103499 and MUSC’s Office of the Vice President for Research. We thank all members of the Tao and Sucov laboratories for their constructive feedback during the preparation of manuscript. We thank Dr. Eric Olson (UT Southwestern Medical Center) and Dr. Takako Makita (MUSC) for sharing reagents and mouse models. We thank the husbandry and veterinary staff at MUSC Division of Laboratory Animal Resources. We also thank the Department of Regenerative Medicine and Cell Biology at the MUSC for providing common equipment.
Author contributions
G.T. conceived and supervised the project. G.T., M.E.M., and S.L. designed the experiments. M.E.M, S.L., G.T., A.M.T., R.A.S., P.L.R., and C.L. performed experiments and analyzed data. H.M.S. provided reagents and scientific advice. C.L. provided reagents and advice on hiPSC platform. A.M.T., G.T., and M.Z. performed bioinformatics and statistical analyses. G.T., M.E.M., S.L., and A.M.T. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109219.
Supplemental information
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Lopaschuk G.D., Collins-Nakai R.L., Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc. Res. 1992;26:1172–1180. doi: 10.1093/cvr/26.12.1172. [DOI] [PubMed] [Google Scholar]
- 3.Puente B.N., Kimura W., Muralidhar S.A., Moon J., Amatruda J.F., Phelps K.L., Grinsfelder D., Rothermel B.A., Chen R., Garcia J.A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Re D.P., Amgalan D., Linkermann A., Liu Q., Kitsis R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019;99:1765–1817. doi: 10.1152/physrev.00022.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu K., Lan D., Li C., Liu S., Dai X., Song T., Wang X., Kong Q., He Z., Tan J., Zhang J. A double-edged sword: role of apoptosis repressor with caspase recruitment domain (ARC) in tumorigenesis and ischaemia/reperfusion (I/R) injury. Apoptosis. 2023;28:313–325. doi: 10.1007/s10495-022-01802-4. [DOI] [PubMed] [Google Scholar]
- 6.Maslov L.N., Popov S.V., Naryzhnaya N.V., Mukhomedzyanov A.V., Kurbatov B.K., Derkachev I.A., Boshchenko A.A., Khaliulin I., Prasad N.R., Singh N., et al. The regulation of necroptosis and perspectives for the development of new drugs preventing ischemic/reperfusion of cardiac injury. Apoptosis. 2022;27:697–719. doi: 10.1007/s10495-022-01760-x. [DOI] [PubMed] [Google Scholar]
- 7.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedmann Angeli J.P., Schneider M., Proneth B., Tyurina Y.Y., Tyurin V.A., Hammond V.J., Herbach N., Aichler M., Walch A., Eggenhofer E., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang X., Cai Z., Wang H., Han D., Cheng Q., Zhang P., Gao F., Yu Y., Song Z., Wu Q., et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020;127:486–501. doi: 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- 11.Bulluck H., Rosmini S., Abdel-Gadir A., White S.K., Bhuva A.N., Treibel T.A., Fontana M., Ramlall M., Hamarneh A., Sirker A., et al. Residual Myocardial Iron Following Intramyocardial Hemorrhage During the Convalescent Phase of Reperfused ST-Segment-Elevation Myocardial Infarction and Adverse Left Ventricular Remodeling. Circ. Cardiovasc. Imaging. 2016;9 doi: 10.1161/CIRCIMAGING.116.004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S., Cheah J.H., Clemons P.A., Shamji A.F., Clish C.B., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bersuker K., Hendricks J.M., Li Z., Magtanong L., Ford B., Tang P.H., Roberts M.A., Tong B., Maimone T.J., Zoncu R., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doll S., Freitas F.P., Shah R., Aldrovandi M., da Silva M.C., Ingold I., Goya Grocin A., Xavier da Silva T.N., Panzilius E., Scheel C.H., et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 15.Fang X., Wang H., Han D., Xie E., Yang X., Wei J., Gu S., Gao F., Zhu N., Yin X., et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baba Y., Higa J.K., Shimada B.K., Horiuchi K.M., Suhara T., Kobayashi M., Woo J.D., Aoyagi H., Marh K.S., Kitaoka H., Matsui T. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2018;314:H659–H668. doi: 10.1152/ajpheart.00452.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J., Minikes A.M., Gao M., Bian H., Li Y., Stockwell B.R., Chen Z.N., Jiang X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–406. doi: 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu X., Khalil H., Kanisicak O., Boyer J.G., Vagnozzi R.J., Maliken B.D., Sargent M.A., Prasad V., Valiente-Alandi I., Blaxall B.C., Molkentin J.D. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 2018;128:2127–2143. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanisicak O., Khalil H., Ivey M.J., Karch J., Maliken B.D., Correll R.N., Brody M.J., J Lin S.C., Aronow B.J., Tallquist M.D., Molkentin J.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016;7 doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasquez C., Mohandas P., Louie K.L., Benamer N., Bapat A.C., Morley G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010;107:1011–1020. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miragoli M., Gaudesius G., Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 22.Porrello E.R., Mahmoud A.I., Simpson E., Hill J.A., Richardson J.A., Olson E.N., Sadek H.A. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergmann O., Zdunek S., Felker A., Salehpour M., Alkass K., Bernard S., Sjostrom S.L., Szewczykowska M., Jackowska T., Dos Remedios C., et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015;161:1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Heallen T., Morikawa Y., Leach J., Tao G., Willerson J.T., Johnson R.L., Martin J.F. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–4690. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Carroll K.J., Makarewich C.A., McAnally J., Anderson D.M., Zentilin L., Liu N., Giacca M., Bassel-Duby R., Olson E.N. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA. 2016;113:338–343. doi: 10.1073/pnas.1523918113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suarez-Ortegon M.F., Ensaldo-Carrasco E., Shi T., McLachlan S., Fernandez-Real J.M., Wild S.H. Ferritin, metabolic syndrome and its components: A systematic review and meta-analysis. Atherosclerosis. 2018;275:97–106. doi: 10.1016/j.atherosclerosis.2018.05.043. [DOI] [PubMed] [Google Scholar]
- 28.Chen X., Yu C., Kang R., Tang D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lian X., Zhang J., Azarin S.M., Zhu K., Hazeltine L.B., Bao X., Hsiao C., Kamp T.J., Palecek S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013;1:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolma S., Lessnick S.L., Hahn W.C., Stockwell B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 31.Davidson M.M., Nesti C., Palenzuela L., Walker W.F., Hernandez E., Protas L., Hirano M., Isaac N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Hortells L., Valiente-Alandi I., Thomas Z.M., Agnew E.J., Schnell D.J., York A.J., Vagnozzi R.J., Meyer E.C., Molkentin J.D., Yutzey K.E. A specialized population of Periostin-expressing cardiac fibroblasts contributes to postnatal cardiomyocyte maturation and innervation. Proc. Natl. Acad. Sci. USA. 2020;117:21469–21479. doi: 10.1073/pnas.2009119117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis J., Burr A.R., Davis G.F., Birnbaumer L., Molkentin J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell. 2012;23:705–715. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung M.I., Bujnis M., Barkauskas C.E., Kobayashi Y., Hogan B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 2018;145 doi: 10.1242/dev.163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuwabara J.T., Hara A., Bhutada S., Gojanovich G.S., Chen J., Hokutan K., Shettigar V., Lee A.Y., DeAngelo L.P., Heckl J.R., et al. Consequences of PDGFRalpha(+) fibroblast reduction in adult murine hearts. Elife. 2022;11 doi: 10.7554/eLife.69854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao G., Kahr P.C., Morikawa Y., Zhang M., Rahmani M., Heallen T.R., Li L., Sun Z., Olson E.N., Amendt B.A., Martin J.F. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L., Liu Z., Yin C., Zhou Y., Liu J., Qian L. Improved Generation of Induced Cardiomyocytes Using a Polycistronic Construct Expressing Optimal Ratio of Gata4, Mef2c and Tbx5. J. Vis. Exp. 2015;105 doi: 10.3791/53426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harada A., Sekido N., Akahoshi T., Wada T., Mukaida N., Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J. Leukoc. Biol. 1994;56:559–564. [PubMed] [Google Scholar]
- 39.Bersell K., Arab S., Haring B., Kühn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 40.Holmes W.E., Lee J., Kuang W.J., Rice G.C., Wood W.I. Structure and functional expression of a human interleukin-8 receptor. Science. 1991;253:1278–1280. doi: 10.1126/science.1840701. [DOI] [PubMed] [Google Scholar]
- 41.Brown T.R., Krogh-Madsen T., Christini D.J. Illuminating Myocyte-Fibroblast Homotypic and Heterotypic Gap Junction Dynamics Using Dynamic Clamp. Biophys. J. 2016;111:785–797. doi: 10.1016/j.bpj.2016.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007;94:120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao Y., Hill M.C., Li L., Deshmukh V., Martin T.J., Wang J., Martin J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33:1491–1505. doi: 10.1101/gad.329763.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crawford S.E., Stellmach V., Murphy-Ullrich J.E., Ribeiro S.M., Lawler J., Hynes R.O., Boivin G.P., Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 45.Dobaczewski M., Bujak M., Li N., Gonzalez-Quesada C., Mendoza L.H., Wang X.F., Frangogiannis N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang D., Chen X., Kang R., Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon S.J., Winter G.E., Musavi L.S., Lee E.D., Snijder B., Rebsamen M., Superti-Furga G., Stockwell B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye L., D'Agostino G., Loo S.J., Wang C.X., Su L.P., Tan S.H., Tee G.Z., Pua C.J., Pena E.M., Cheng R.B., et al. Early Regenerative Capacity in the Porcine Heart. Circulation. 2018;24:2798–2808. doi: 10.1161/CIRCULATIONAHA.117.031542. [DOI] [PubMed] [Google Scholar]
- 49.Zhu W., Zhang E., Zhao M., Chong Z., Fan C., Tang Y., Hunter J.D., Borovjagin A.V., Walcott G.P., Chen J.Y., et al. Regenerative Potential of Neonatal Porcine Hearts. Circulation. 2018;138:2809–2816. doi: 10.1161/CIRCULATIONAHA.118.034886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deshmukh V., Wang J., Martin J.F. Leading progress in heart regeneration and repair. Curr. Opin. Cell Biol. 2019;61:79–85. doi: 10.1016/j.ceb.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Empel V.P.M., Bertrand A.T.A., Hofstra L., Crijns H.J., Doevendans P.A., De Windt L.J. Myocyte apoptosis in heart failure. Cardiovasc. Res. 2005;67:21–29. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Weil B.R., Young R.F., Shen X., Suzuki G., Qu J., Malhotra S., Canty J.M., Jr. Brief Myocardial Ischemia Produces Cardiac Troponin I Release and Focal Myocyte Apoptosis in the Absence of Pathological Infarction in Swine. JACC. Basic Transl. Sci. 2017;2:105–114. doi: 10.1016/j.jacbts.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doll S., Proneth B., Tyurina Y.Y., Panzilius E., Kobayashi S., Ingold I., Irmler M., Beckers J., Aichler M., Walch A., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riegman M., Sagie L., Galed C., Levin T., Steinberg N., Dixon S.J., Wiesner U., Bradbury M.S., Niethammer P., Zaritsky A., Overholtzer M. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020;22:1042–1048. doi: 10.1038/s41556-020-0565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ieda M., Tsuchihashi T., Ivey K.N., Ross R.S., Hong T.T., Shaw R.M., Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das S., Goldstone A.B., Wang H., Farry J., D'Amato G., Paulsen M.J., Eskandari A., Hironaka C.E., Phansalkar R., Sharma B., et al. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019;176:1128–1142.e18. doi: 10.1016/j.cell.2018.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qian L., Huang Y., Spencer C.I., Foley A., Vedantham V., Liu L., Conway S.J., Fu J.D., Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aghajanian H., Kimura T., Rurik J.G., Hancock A.S., Leibowitz M.S., Li L., Scholler J., Monslow J., Lo A., Han W., et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gage P.J., Suh H., Camper S.A. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S., Guo Y., Pu W.T. AAV Gene Transfer to the Heart. Methods Mol. Biol. 2021;2158:269–280. doi: 10.1007/978-1-0716-0668-1_20. [DOI] [PubMed] [Google Scholar]
- 63.Li L., Tao G., Hill M.C., Zhang M., Morikawa Y., Martin J.F. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development. 2018;145 doi: 10.1242/dev.168609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data have been deposited to GEO (GSE211568) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.