

Abstract
Background
Pediatric dilated cardiomyopathy often leads to death or cardiac transplantation. We sought to determine whether changes in left ventricular (LV) end‐diastolic dimension (LVEDD), LV end‐diastolic posterior wall thickness, and LV fractional shortening (LVFS) over time may help predict adverse outcomes.
Methods and Results
We studied children up to 18 years old with dilated cardiomyopathy, enrolled between 1990 and 2009 in the Pediatric Cardiomyopathy Registry. Changes in LVFS, LVEDD, LV end‐diastolic posterior wall thickness, and the LV end‐diastolic posterior wall thickness:LVEDD ratio between baseline and follow‐up echocardiograms acquired ≈1 year after diagnosis were determined for children who, at the 1‐year follow‐up had died, received a heart transplant, or were alive and transplant‐free. Within 1 year after diagnosis, 40 (5.0%) of the 794 eligible children had died, 117 (14.7%) had undergone cardiac transplantation, and 585 (73.7%) had survived without transplantation. At diagnosis, survivors had higher median LVFS and lower median LVEDD Z scores. Median LVFS and LVEDD Z scores improved among survivors (Z score changes of +2.6 and −1.1, respectively) but remained stable or worsened in the other 2 groups. The LV end‐diastolic posterior wall thickness:LVEDD ratio increased in survivors only, suggesting beneficial reverse LV remodeling. The risk for death or cardiac transplantation up to 7 years later was lower when LVFS was improved at 1 year (hazard ratio [HR], 0.83; P=0.004) but was higher in those with progressive LV dilation (HR, 1.45; P<0.001).
Conclusions
Progressive deterioration in LV contractile function and increasing LV dilation are associated with both early and continuing mortality in children with dilated cardiomyopathy. Serial echocardiographic monitoring of these children is therefore indicated.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: NCT00005391.
Keywords: cardiac transplantation, dilated cardiomyopathy, heart failure, pediatrics, remodeling
Subject Categories: Pediatrics, Heart Failure, Remodeling, Cardiomyopathy
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- LVFS
left ventricular fractional shortening
- LVEDD
left ventricular end‐diastolic dimension
- LVPWT
left ventricular end‐diastolic posterior wall thickness
- PCMR
Pediatric Cardiomyopathy Registry
Clinical Perspective.
What Is New?
This study demonstrates the value of serial echocardiographic changes in the prediction of risk in pediatric dilated cardiomyopathy.
What Are the Clinical Implications?
Serial echocardiographic assessment of left ventricular remodeling in children with dilated cardiomyopathy is able to predict death or cardiac transplantation both in the first year after diagnosis, and later in their course.
LV dilation and contractile performance are important and persistent markers of disease prognosis during and beyond the first year following pediatric dilated cardiomyopathy diagnosis.
Serial echocardiography is a valuable means of tracking individual patient progress in pediatric dilated cardiomyopathy and should be routinely performed and endorsed by published appropriate use criteria.
Dilated cardiomyopathy (DCM) in children is a heterogeneous group of disorders identified by the echocardiographic characteristics of left ventricular (LV) dilation and LV systolic dysfunction. 1 Although DCM phenotypes are determined by these characteristics, it is now clear that the same echocardiographic appearance may have many underlying causes and therefore differences in prognoses. 2 , 3 , 4 , 5 Identifying patients at highest risk for clinical deterioration and death is challenging as the cause of DCM is undefined or idiopathic in at least 40% of children. 6
Echocardiographic LV remodeling in children with DCM at presentation has prognostic value, but only the presenting characteristics of remodeling have been studied. 4 , 7 Larger LV dimension at presentation of myocarditis, a well‐recognized cause of DCM in children, is associated with poor recovery; however, some children with DCM without evident myocarditis may also eventually recover and regain normal echocardiographic values. 5 , 8 Therefore, we sought to determine whether the degree of LV remodeling, as measured on serial echocardiograms in the first 12‐months following DCM diagnosis, in the absence of myocarditis, was associated with mortality in these children.
Methods
Data were abstracted from the PCMR (Pediatric Cardiomyopathy Registry), a National Heart, Lung, and Blood Institute‐funded research registry that enrolled more than 3500 children with various phenotypic forms of cardiomyopathy from 98 North American centers between 1990 and 2009. Entry criteria for the PCMR include a diagnosis of familial and certain metabolic, genetic (including dystrophinopathy), and idiopathic causes of DCM. [1] For this investigation, we included all children evaluated between 1990 and 2009 who met PCMR phenotypic criteria for DCM, except for those with a coexisting non‐DCM phenotype or with myocarditis as an underlying cause of DCM. We have relied on clinical diagnosis and supportive investigations as determined by the center providing care to make the distinction between myocarditis and nonmyocarditis causes. The diagnosis of myocarditis was therefore based on clinical presentation, endomyocardial biopsy, or core myocardial sample at ventricular assist device placement; viral polymerase chain reaction studies; and cardiac magnetic resonance imaging assessment when available. We have shown previously that acute myocarditis, whether biopsy confirmed or clinically suspected, has a more fulminant course in childhood. Although the outcome of death or cardiac transplant is uncommon, when it occurs, it is attained more rapidly than in cases of pediatric DCM. 4 , 5 Familial DCM was defined to be present if 1 or more additional family members were confirmed to have a diagnosis of cardiomyopathy. Children with cardiomyopathies from systemic diseases or in association with malformation syndromes were also excluded. Children with mixed phenotypes, such as LV noncompaction cardiomyopathy with dilation, were excluded, because remodeling in LV noncompaction cardiomyopathy can pose challenges in assessment by serial echocardiography. We have previously described the outcomes of LV noncompaction cardiomyopathy, which is modified by phenotypic overlap with LV dilation or hypertrophy, and others have alluded to the propensity for both progressive and reverse remodeling to occur in some patients with LV noncompaction cardiomyopathy. 9 , 10 The authors declare that all supporting data are available within the article (and its online supplementary files).
The designation of DCM in the PCMR requires both an LV end‐diastolic dimension (LVEDD) Z score >2 (referenced to means of a healthy population for body surface area) and an LV fractional shortening (LVFS) Z score <−2.0 on baseline echocardiogram. We applied both of these parameters as well as LV posterior wall thickness (LVPWT) and its relationship to LV diastolic dimension (LVPWT:LVEDD) as indicative of remodeling, because there is conflicting evidence as to how closely related these may be over time and whether LVFS change is related to baseline LVEDD or not. 11 , 12 The baseline echocardiogram may have been acquired on, or within 3 months before, the date of diagnosis. Our analysis was then restricted to children in whom at least both baseline and 1‐year follow‐up echocardiograms were available.
The baseline echocardiogram was that performed at or most recently before registration in PCMR and is equivalent to the date of diagnosis assigned. Follow‐up echocardiographic measures were derived from annual repeat echocardiograms indexed annually to the date of diagnosis and must have been obtained within 6 months of that anniversary date. If death or cardiac transplantation occurred before the anniversary date, the most proximate echocardiogram obtained within 3 months before the outcome event was assessed. Therefore, all outcomes were indexed to the original baseline echocardiogram. We restricted this analysis to children who remained in follow‐up, died, or who underwent cardiac transplantation and echocardiography at or beyond 6 months after their baseline echocardiogram.
Demographic, family history, and clinical data relevant to cardiomyopathy, including vital status and cardiac transplant status and echocardiographic values, were all evaluated. We determined whether symptomatic heart failure (HF) was evident at original presentation. Medical therapies for HF, including angiotensin‐converting enzyme inhibitors, calcium channel blockers, beta blockers, digoxin, antiarrhythmics, and diuretics were recorded. Children were grouped by the outcomes of all‐cause mortality, cardiac transplantation, or cardiac transplant‐free survival. Death and cardiac transplantation were considered censoring events for further analysis. All participating centers obtained institutional review board approval to enroll children in the PCMR. In all cases, a waiver of the requirement for informed consent was granted.
Statistical Analysis
Echocardiographic measurements were converted to Z scores based on normative pediatric reference ranges, to adjust for differences related to age or body surface area. Categorical baseline patient characteristics were compared among the 3 groups using chi‐square tests. The Z scores for baseline LVFS, LVEDD, and LVPWT at end‐diastole, and the LVPWT:LVEDD were compared among groups with the Kruskal–Wallis test. Changes in serial echocardiographic measures within these groups were also assessed with these tests. Increasing LV dilation or decreasing LV thickness‐to‐dimension ratio was interpreted as indicating pathologic LV remodeling.
Secondary analyses used outcome data following the baseline echocardiogram. The Kaplan–Meier method was used to determine the probability of death, cardiac transplantation (whichever occurred first), or transplantation‐free survival from baseline as mutually exclusive outcomes. In event‐free children followed beyond the first year after diagnosis, Cox proportional hazard modeling was used to determine whether any baseline echocardiographic value or change in any echocardiographic value during the first year of follow‐up predicted death or cardiac transplantation after the first year and up to 17 years thereafter. Echocardiographic values significant in the unadjusted analysis were combined into a final model based on a bivariate P value of <0.05. Interactions between patient age and changes in the echocardiographic values were evaluated by treating age as a continuous variable and as dichotomous variables of <1 year and 1 year or more. The variable LVPWT:LVEDD ratio was removed from the model because it was colinear with other independent echocardiographic values.
In addition to the risk assessment, logistic regression was used to fit models for probability of death or cardiac transplantation versus survival, while receiver‐operator characteristic curves were used to determine the sensitivity and specificity of baseline LVEDD and LVFS Z scores for the outcome of death or cardiac transplantation versus survival. Outcome was assessed at both 1 and 7 years after baseline. Alpha was set at 0.05, and all tests were 2 tailed. Data were analyzed using statistical analysis software SAS 9.4 (SAS Institute Inc., Cary, NC).
Results
Of the eligible 1450 children, 619 (43%) did not have follow‐up echocardiograms available in the PCMR database (Figure 1). This number includes 230 children (37%) who died or required cardiac transplantation within 6 months following baseline assessment. The analysis was based on the remaining 794 children with baseline and follow‐up echocardiograms.
Figure 1. Cohort study flow chart.
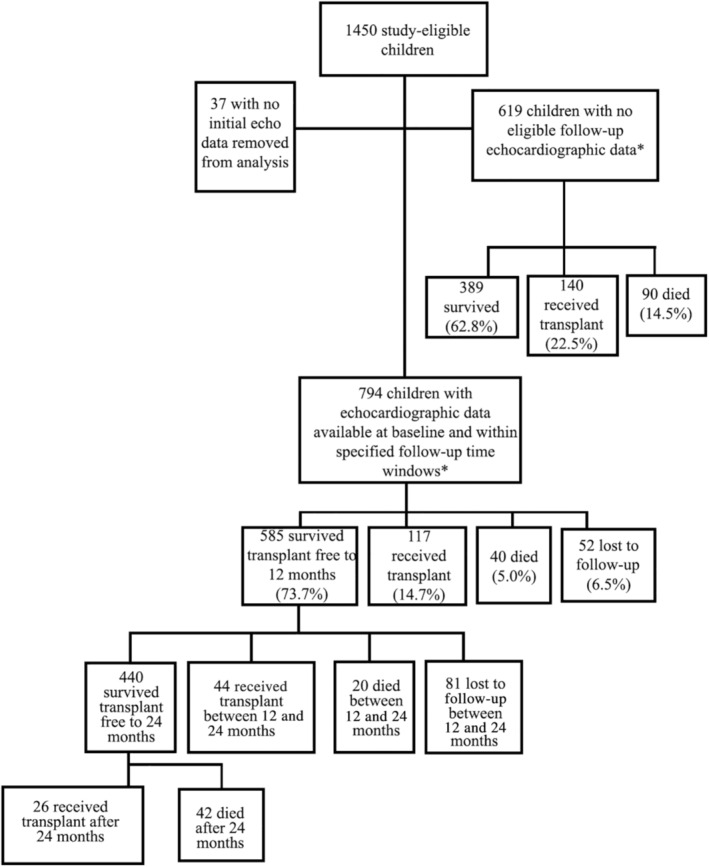
Cohort selection of children with dilated cardiomyopathy from the Pediatric Cardiomyopathy Registry in a study to determine the prognostic value of serial changes in echocardiographic measurements. *Children not having an echocardiogram within 90 days before death or cardiac transplant in the first year, or within 6 months of their 1‐year anniversary of the first echocardiogram, if event free during the first year.
Baseline Characteristics
The clinical features of the cohort subjected to analysis are shown in Table 1. Baseline echocardiograms were acquired on the actual day of diagnosis for 67% of children. Median age at diagnosis ranged from 1 to 2 years (Table 1). Race or ethnicity and cause of cardiomyopathy we marginally associated with outcomes in the adjusted analysis, with the highest rate of death or transplant occurring in 27.9% of non‐Hispanic Black children. Idiopathic cardiomyopathy was the most common diagnosis and was marginally associated with cardiac transplantation. Clinical HF at diagnosis was far more likely in children destined for cardiac transplantation or death within the first year (P<0.001). Similarly, a lower LVFS Z score (P<0.001), a higher LVEDD Z score (P<0.001), and a thinner LVPWT Z score (P<0.007) at baseline were all associated with death or cardiac transplantation (Table 2). Median (interquartile range) follow‐up duration of the cohort was 2.55 (0.96–5.79) years.
Table 1.
Clinical Characteristics of 794 Children With Dilated Cardiomyopathy at Diagnosis by Outcome During the First Year After Diagnosis
| Characteristic at diagnosis | Died (N=40) | Cardiac transplant (N=117) | Event‐free survival (N=585) | Lost to follow‐up (N=52) | P value* |
|---|---|---|---|---|---|
| Age at diagnosis, y | |||||
| Median (interquartile range) | 1.1 (0.4, 11.9) | 2.1 (0.4, 11.3) | 1.2 (0.3, 11.6) | 1.0 (0.3, 5.7) | |
| Age <1 y at diagnosis, n (%) | 18 (45.0) | 52 (44.4) | 274 (46.8) | 28 (53.8) | 0.88 |
| Male sex, n (%) | 20 (50.0) | 65 (55.6) | 310 (53.0) | 27 (51.9) | 0.80 |
| Race or ethnicity, n (% by column) | 0.02 | ||||
| White, non‐Hispanic | 17 (42.5) | 60 (51.3) | 334 (57.1) | 28 (53.8) | |
| Black, non‐Hispanic | 10 (25.0) | 31 (26.5) | 102 (17.4) | 4 (7.7) | |
| Hispanic | 6 (15.0) | 17 (14.5) | 107 (18.3) | 14 (26.9) | |
| Other | 7 (17.5) | 6 (5.1) | 35 (6.0) | 5 (9.6) | |
| Type of DCM, n (% by column) | 0.02 | ||||
| Familial | 2 (5.0) | 4 (3.4) | 39 (6.7) | 2 (3.8) | |
| Idiopathic | 33 (82.5) | 108 (92.3) | 463 (79.1) | 48 (92.3) | |
| Other | 5 (12.5) | 5 (4.3) | 83 (14.2) | 2 (3.8) | |
| Congestive heart failure at diagnosis, n (% by column) | 33 (82.5) | 99 (84.6) | 355 (60.7) | 36 (69.2) | <0.001 |
| Medication use, n/N (%)† | |||||
| Antiarrhythmic | 7/23 (30.4) | 16/59 (27.1) | 69/317 (21.8) | 2/14 (14.3) | 0.46 |
| Angiotensin‐converting enzyme inhibitor | 15/23 (65.2) | 46/59 (78.0) | 226/318 (71.1) | 12/14 (85.7) | 0.43 |
| Beta blocker | 3/23 (13.0) | 11/58 (19.0) | 31/315 (9.8) | 3/14 (21.4) | 0.13 |
| Digoxin and diuretics | 36/38 (94.7) | 106/115 (92.2) | 459/542 (84.7) | 44/48 (91.7) | 0.03 |
DCM indicates dilated cardiomyopathy.
Compares death, cardiac transplant, and survival to 1 year after diagnosis.
n, number of children on the medication; N, group total. Type of DCM “Other” refers to defined diagnoses including dystrophinopathy (Duchenne, Becker, or Emery–Dreifuss muscular dystrophy).
Table 2.
Echocardiographic Characteristics of 794 Children With Dilated Cardiomyopathy at Diagnosis and by Outcome at 12 Months
| Characteristic at diagnosis | Died (N=40) | Cardiac transplanted (N=117) | Event‐free survival (N=585) | Lost to follow‐up (N=52) | P value* |
|---|---|---|---|---|---|
| Left ventricular fractional shortening Z score | |||||
| Mean (SD) | −9.39 (2.99) | −9.73 (2.62) | −7.84 (3.86) | −8.50 (4.94) | <0.001 |
| Median (IQR) | −10.02 (−11.62 to −7.10) | −9.87 (−11.77 to −8.02) | −8.65 (−10.58 to −5.15) | −9.41 (−11.70 to −6.96) | |
| LVEDD Z score | <0.001 | ||||
| Mean (SD) | 5.68 (2.01) | 5.82 (2.00) | 4.09 (2.70) | 4.88 (2.32) | |
| Median (IQR) | 5.61 (4.47 to 6.83) | 5.74 (4.23 to 7.33) | 3.97 (2.15 to 5.94) | 5.00 (3.14 to 6.59) | |
| LVPWT Z score | 0.07 | ||||
| Mean (SD) | 0.28 (1.47) | −0.98 (1.93) | −0.70 (2.33) | −0.12 (1.82) | |
| Median (IQR) | 0.02 (−0.82 to 1.16) | −1.11 (−1.79 to 0.22) | −0.78 (−2.11 to 0.63) | −0.41 (−1.51 to 0.71) | |
| LVPWT:LVEDD ratio Z score | <0.001 | ||||
| Mean (SD) | −1.83 (1.84) | −2.67 (1.27) | −1.41 (2.17) | −1.47 (1.83) | |
| Median (IQR) | −1.99 (−3.37 to −0.84) | −2.80 (−3.38 to −2.17) | −1.71 (−2.71 to −0.41) | −1.74 (−2.86 to −0.45) | |
IQR indicates interquartile range; LVEDD, left ventricular end‐diastolic dimension; and LVPWT, left ventricular end‐diastolic posterior wall thickness.
The type and frequency of medications used at the time of diagnosis were similar among the 3 outcome groups, although anticongestive therapy and angiotensin‐converting enzyme inhibitors were more common. Beta blockers were used in a minority of cases at diagnosis.
Left Ventricular Remodeling
Of the 794 children, 585 (74%) survived without transplant, 52 (7%) were lost to follow‐up, 40 (5%) had died, and 117 (15%) had received a cardiac transplant by 1 year of follow‐up. The 1‐year follow‐up echocardiograms were acquired at a median 8.6 (interquartile range, 6.0–10.4) months following the baseline study. Changes in the first year after diagnosis differed between groups (Figure 2). Baseline LVEDD Z scores were significantly lower in children who survived for 1 year than for those who died or underwent cardiac transplant (median Z scores, 4.0, 5.6, and 5.7 respectively; P<0.001). This difference was further evident at 1 year when the median LVEDD Z score was reduced by 1.1 (improved and closer to normal) in survivors (P<0.001), remained relatively constant in children undergoing cardiac transplant, and increased by 0.6 (P<0.001) in children who died.
Figure 2. Progression of LV dimension Z score.
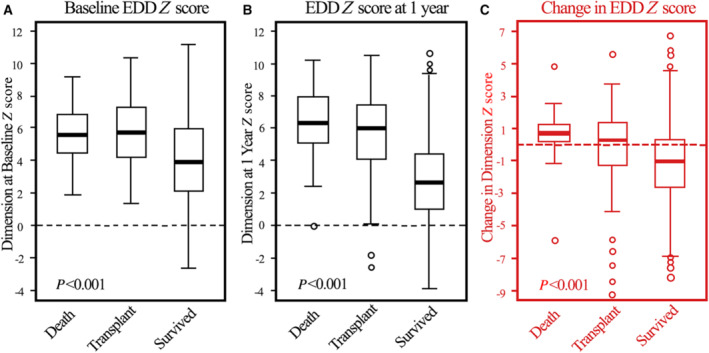
Left ventricular end‐diastolic dimension Z scores at (A) diagnosis, (B) 1 year later, and (C) changes between baseline and 1 year for children with dilated cardiomyopathy who died, underwent heart transplant, or survived during the first year after diagnosis. Data in the box‐and‐whisker plots are medians, interquartile ranges, minimum and maximum within median±1.5 interquartile range and outliers. Survivors differed significantly from both other groups at time points A and B. The changes in left ventricular end‐diastolic dimension Z scores between survivors, those transplanted, and nonsurvivors (C) were also significant. EDD indicates end‐diastolic dimension; and LV, left ventricular.
In contrast to the progression of LV dilation, changes in LVPWT (Figure 3) differed only slightly among the groups. For the most part, LVPWT was normal for all groups, both at baseline and at 1 year (median Z scores ranged between 0 and − 2). However, baseline LVPWT was higher in the children who later died. The LVPWT:LVEDD ratio Z score differed significantly among groups, both at baseline and at follow‐up, with the Z score marginally higher in survivors at both times than in children in the other groups (Figure 4). Changes in the LVPWT:LVEDD ratio from baseline over the first year did not differ significantly among the 3 groups.
Figure 3. Progression of LV posterior wall thickness Z score.
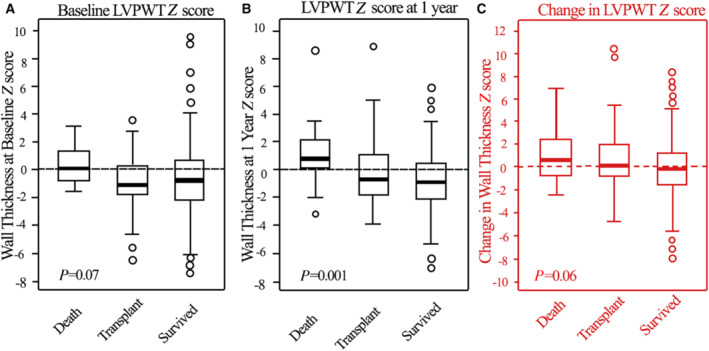
LVPWT Z scores at (A) diagnosis, (B) 1 year later, and (C) changes between diagnosis and 1 year for children with dilated cardiomyopathy who died, underwent heart transplant, or survived during the first year after diagnosis. Data in the box‐and‐whisker plots are medians, interquartile ranges, minimum and maximum within median±1.5 interquartile range and outliers. Differences were only marginally significant at baseline but were more evident at 1 year. Changes in left ventricular wall thickness (C) were similar in all 3 groups, with no clear between‐group differences from baseline. LVPWT indicates left ventricular end‐diastolic posterior wall thickness.
Figure 4. Progression of LV wall thickness to dimension ratio Z score.
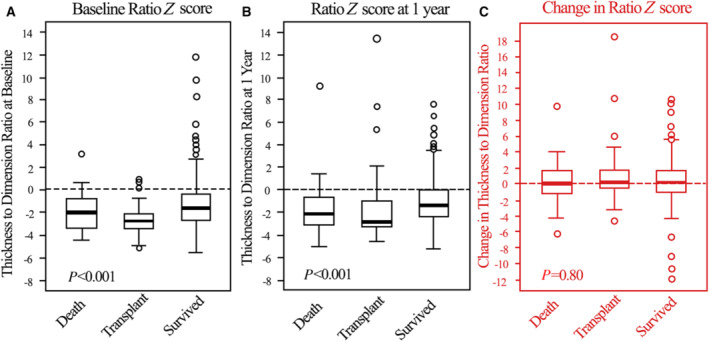
The ratio of left ventricular posterior wall thickness:left ventricular end diastolic dimension Z scores at (A) diagnosis, (B) 1 year later, and (C) changes between baseline and 1 year for children with dilated cardiomyopathy who died, underwent heart transplant, or survived during the first year after diagnosis. Data in the box‐and‐whisker plots are medians, interquartile ranges, minimum and maximum within median±1.5 interquartile range and outliers. Differences between the 3 groups were significant at both time points but not with regard to the magnitude of change during the first year shown in panel (C). LV indicates left ventricular.
Baseline LV systolic function, as measured by LVFS (Figure 5), was depressed in all groups but significantly less so in survivors, in whom LVFS improved substantially (change in LVFS Z score of +2.6, P<0.001). LVFS remained unchanged in the other 2 groups.
Figure 5. Progression of LV fractional shortening Z score.
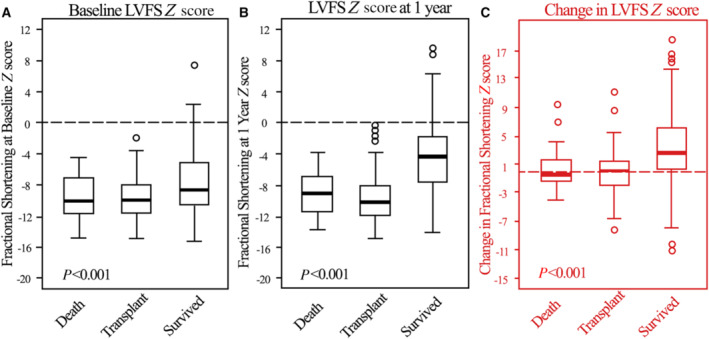
Left ventricular fractional shortening Z scores at (A) diagnosis/(baseline), (B) 1 year later, and (C) changes between baseline and 1 year for children with dilated cardiomyopathy who died, underwent heart transplant, or survived during the first year after diagnosis. Data in the box‐and‐whisker plots are medians, interquartile ranges, minimum and maximum within median±1.5 interquartile range and outliers. The groups differed significantly at both time points, as well as in respect of the improvement in LVFS from 0 to 1 year (C). LVFS indicates left ventricular fractional shortening.
Survival Beyond 12 Months After DCM Diagnosis
We determined whether the progressive changes in LVEDD, LVPWT, LVPWT:LVEDD ratio, and LVFS were associated with survival beyond 1 year for the 585 children who did not undergo early cardiac transplantation. Of these children, 440 (75.2%) survived beyond 24 months, after which 42 (9.5%) died and 26 (5.9%) received a cardiac transplant (Figure 6B). We determined the hazard function for death or cardiac transplantation after survival to 1 year, both at baseline and at follow‐up assessment (Table 3). Changes in LVEDD (P<0.001) and LVFS (P=0.004) measured in the first year were important determinants of the time to death or cardiac transplant beyond 1 year after diagnosis. In this multivariable model, baseline LVEDD and progressive dilation during the first year after diagnosis were associated with an increased hazard for death or cardiac transplantation. Conversely, a higher baseline LVFS and an increase in interval LVFS were associated with better cardiac transplant‐free survival. We assessed for any interaction with age at presentation (<1 year of age versus base case >1 year of age) in the multivariable model and found no significant interaction with age at presentation.
Figure 6. Transplant‐free survival probability distribution in children with dilated cardiomyopathy.
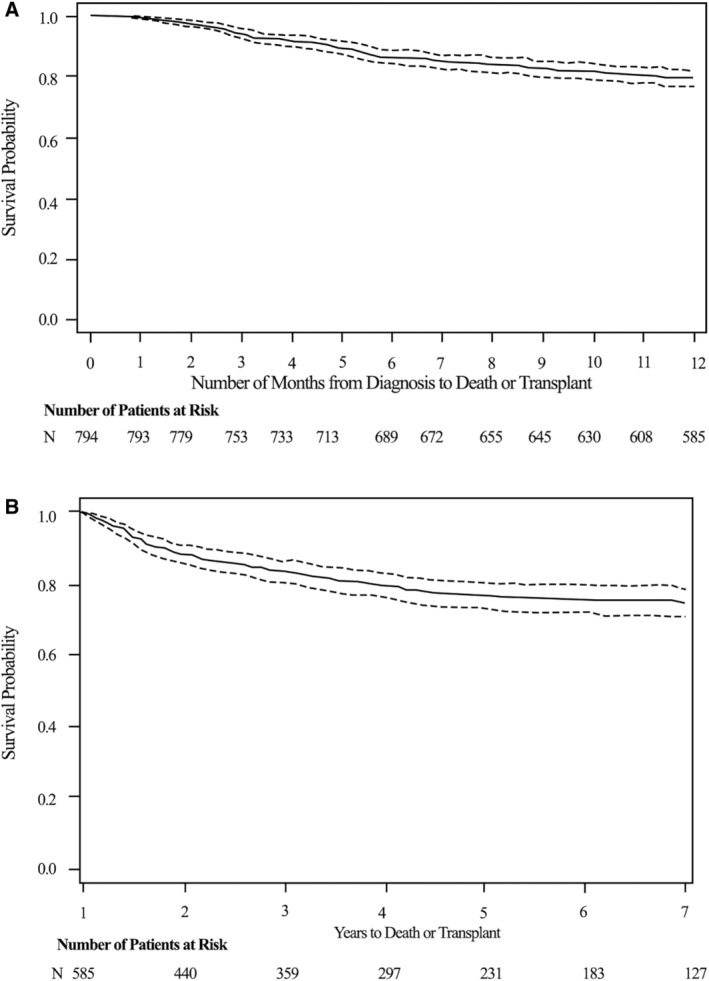
A, From diagnosis to 12 months after baseline echocardiogram. B, For those surviving beyond 1 year after diagnosis (x axis is in years). Dotted lines are 95% confidence limits.
Table 3.
Hazard Ratios From Multivariable Modeling of Incremental Risk of Death or Cardiac Transplant Per 1‐SD Change in Z Score for 585 Children With Dilated Cardiomyopathy Surviving 1 Year After Diagnosis
| Separate bivariate models (baseline and change) | Multivariable model | |||
|---|---|---|---|---|
| Characteristic | Hazard ratio (95% CI) | P value | Hazard ratio (95% CI) | P value |
| Baseline LVEDD Z score | 1.57 (1.40–1.75) | <0.001 | 1.22 (1.01–1.49) | 0.04 |
| Change in LVEDD Z score | 1.73 (1.54–1.94) | <0.001 | 1.45 (1.21–1.75) | <0.001 |
| Baseline LVPWT Z score | 0.96 (0.82–1.12) | 0.61 | ||
| Change in LVPWT Z score | 1.15 (1.01–1.32) | 0.03 | 1.10 (1.00–1.21) | 0.06 |
| Baseline LVFS Z score | 0.77 (0.71–0.83) | <0.001 | 0.86 (0.75–0.99) | 0.04 |
| Change in LVFS Z score | 0.79 (0.74–0.84) | <0.001 | 0.83 (0.73–0.94) | 0.004 |
| Baseline LVPWT:LVEDD ratio Z score | 0.72 (0.60–0.87) | <0.001 | ||
| Change in LVPWT:LVEDD ratio Z score | 0.78 (0.66–0.92) | 0.004 | ||
All analyses are adjusted for age and sex. LVEDD indicates left ventricular end‐diastolic dimension; LVFS, left ventricular fractional shortening; and LVPWT, left ventricular end‐diastolic posterior wall thickness.
Prognostic Value of Echocardiographic Changes
The area under the receiver‐operator characteristic curve (AUC) for a particular baseline LVEDD z‐score was relatively low for 1‐year survival (0.67), and even lower for 7‐year survival (0.60; data not shown). However, specificity for a 1‐year outcome of death or cardiac transplantation was 77.8% at a baseline LVEDD Z score of 6.4 or greater (Table 4). The prognostic value of LVEDD was improved somewhat by measuring the change in LVEDD Z score during the first year, both for death or cardiac transplantation at 1 year (area under the curve, 0.69) and at 7 years (area under the curve, 0.74). Progressive dilation, signified by an increase in the LVEDD Z score of only 0.49 (the 75th percentile of the changes we observed), was ≈79% specific for the outcome of death or cardiac transplantation at 1 year and > 83% specific for death or transplantation at 7 years (Table 4).
Table 4.
Baseline LVEDD Z Scores and Changes in Z Score in Children With Dilated Cardiomyopathy by Percentile and by Outcome
| EDD percentile | EDD‐Z | Death/Transplant | Survived | Sensitivity % | Specificity % | ||
|---|---|---|---|---|---|---|---|
| Baseline ≥EDD‐Z (true +) | Baseline <EDD‐Z (false ‐) | Baseline <EDD‐Z z (true ‐) | Baseline ≥EDD‐Z (false +) | ||||
| Outcome at 1 y | |||||||
| (n=135) | (n=496) | ||||||
| 25th | 2.51 | 128 | 7 | 153 | 343 | 94.8 | 30.8 |
| 50th | 4.69 | 92 | 43 | 275 | 221 | 68.1 | 55.4 |
| 75th | 6.35 | 46 | 89 | 386 | 110 | 34.1 | 77.8 |
| Outcome at 7 y | |||||||
| (n=135) | (n=103) | ||||||
| 25th | 2.51 | 206 | 26 | 30 | 73 | 88.8 | 29.1 |
| 50th | 4.69 | 142 | 90 | 49 | 54 | 61.2 | 47.6 |
| 75th | 6.35 | 71 | 161 | 79 | 24 | 30.6 | 76.7 |
| Change in EDD percentile | Change in EDD‐Z score | Change ≥EDD‐Z (true +) | Change <EDD‐Z (false ‐) | Change <EDD‐Z (true ‐) | Change ≥EDD‐Z (false +) | Sensitivity % | Specificity % |
|---|---|---|---|---|---|---|---|
| Outcome at 1 y | |||||||
| (n=118) | (n=440) | ||||||
| 25th | −2.52 | 104 | 14 | 114 | 326 | 88.1 | 25.9 |
| 50th | −0.81 | 90 | 28 | 236 | 204 | 76.3 | 53.6 |
| 75th | 0.49 | 53 | 65 | 347 | 93 | 44.9 | 78.9 |
| Outcome at 7 y | |||||||
| (n=200) | (n=95) | ||||||
| 25th | −2.52 | 181 | 19 | 27 | 68 | 90.5 | 28.4 |
| 50th | −0.81 | 156 | 44 | 61 | 34 | 78.0 | 64.2 |
| 75th | 0.49 | 88 | 112 | 79 | 16 | 44.0 | 83.2 |
Sensitivity and specificity of baseline echocardiographic LVEDD Z score and change in baseline LVEDD Z at follow‐up assessment for the prediction of death or cardiac transplant vs survival at 1 year and at 7 years in children with dilated cardiomyopathy for whom echocardiographic values were available. Values at the 25th, 50th, and 75th percentile of the range are shown. EDD indicates end‐diastolic dimension; and LVEDD, left ventricular end‐diastolic dimension.
Baseline LVFS Z scores (a lower Z score indicates worse LV function) did not discriminate well (32% sensitivity and 78% specificity) for values at or below the 25th percentile: z≤−10.9 (Table 5). However, again, a decline in LVFS Z score of only −0.18 during the first year (the 25th percentile observed) was more specific for death or cardiac transplantation (79% at 1 year and 81% at 7 years) (Table 5).
Table 5.
Baseline Fractional Shortening Z Scores and Changes in Z Score in Children With Dilated Cardiomyopathy by Percentile and by Outcome
| FS Percentile | FS‐z | Death/Transplant | Survived | Sensitivity % | Specificity % | ||
|---|---|---|---|---|---|---|---|
| Baseline <FS‐Z (true +) | Baseline ≥FS‐Z (false −) | Baseline ≥FS‐Z (true −) | Baseline <FS‐Z (false +) | ||||
| Outcome at 1 y | |||||||
| (n=137) | (n=518) | ||||||
| 25th | −10.94 | 44 | 93 | 402 | 116 | 32.1 | 77.6 |
| 50th | −9.04 | 83 | 54 | 277 | 241 | 60.6 | 53.5 |
| 75th | −5.94 | 119 | 18 | 153 | 365 | 86.9 | 29.5 |
| Outcome at 7 y | |||||||
| (n=241) | (n=112) | ||||||
| 25th | −10.94 | 67 | 174 | 82 | 30 | 27.8 | 73.2 |
| 50th | −9.04 | 132 | 109 | 53 | 59 | 54.8 | 47.3 |
| 75th | −5.94 | 200 | 41 | 29 | 83 | 83.0 | 25.9 |
| Change in FS percentile | Change in FS Z score | Change<FS‐Z (true +) | Change ≥ FS‐Z (false −) | Change ≥ FS‐Z (true −) | Change<FS‐Z (false +) | Sensitivity % | Specificity % |
|---|---|---|---|---|---|---|---|
| Outcome at 1 y | |||||||
| (n=117) | (n=479) | ||||||
| 25th | −0.18 | 50 | 67 | 380 | 99 | 42.7 | 79.3 |
| 50th | 1.95 | 95 | 22 | 267 | 212 | 81.2 | 55.7 |
| 75th | 5.50 | 111 | 6 | 134 | 345 | 94.9 | 28.0 |
| Outcome at 7 y | |||||||
| (n=210) | (n=105) | ||||||
| 25th | −0.18 | 85 | 125 | 85 | 20 | 40.5 | 81.0 |
| 50th | 1.95 | 160 | 50 | 64 | 41 | 76.2 | 61.0 |
| 75th | 5.50 | 197 | 13 | 33 | 72 | 93.8 | 31.4 |
Sensitivity and specificity of baseline echocardiographic LV fractional shortening Z score and change in baseline LV fractional shortening Zscore at follow‐up assessment for the prediction of death or cardiac transplant vs survival at 1 year and at 7 years in children with dilated cardiomyopathy for whom echocardiographic values were available. Values at the 25th, 50th, and 75th percentile of the range are shown. FS indicates fractional shortening; and LV, left ventricular.
Discussion
We found important echocardiographic milestones of disease progression or recovery for children with DCM in the first year after diagnosis, which should help clinicians define the evolution of risk in specific patients. In describing the relationship of serial changes in LVFS, LVEDD, and LVPWT to the outcomes of survival, death, or cardiac transplantation in a large cohort of children with DCM, the risk for death or cardiac transplantation was associated with severity of LV dilation and LV systolic dysfunction at diagnosis. Furthermore, the progression of these abnormalities was associated with reduced survival at 1 year and beyond. Serial changes in LV dimension and function predicted later outcomes of survival versus death or cardiac transplant better than a single assessment at diagnosis. To date, the field of risk prediction in adult nonischemic HF has relied on multiple clinical attributes to create a single point risk estimate, exemplified by the Meta‐Analysis Global Group in Chronic Heart Failure (MAGGIC) calculator for HF risk score and others. Although these have fair discriminant value, it is worth noting that no risk score has yet been deployed that leverages the degree of progressive or reverse remodeling that is accruing over time as a risk modifier. 13 , 14
Dilation of the systemic ventricle is considered to be a signal event in LV remodeling. 15 In nonischemic DCM, the mechanisms leading to LV remodeling are multifactorial and have been linked to changes in the expression or function of structural sarcomeric proteins or the interstitial matrix, ultimately leading to myocardial cell death with associated interstitial fibrosis. 16 , 17 , 18 , 19 Although children with HF had different patterns of LV remodeling, the children we studied by definition had LV dilation and diminished LV systolic function. 19
In such children, LV mass, sphericity index, and LV stroke volume are all closely associated, and a reduction in the LV ejection fraction <20% is associated with an even more pronounced reduction in LV stroke volume. 20 In children, reduced LV ejection fraction or LVFS at presentation are important risk factors for death. 4 , 21 , 22 The degree of LV dilation and reduced LV function are additive in increasing the hazards of death in these children. 22 , 23 We found here that serial changes in LVFS, LVEDD, and in the LVPWT:LVEDD ratio were associated with survival at 1 year after diagnosis and relevant to longer‐term survival.
A higher baseline LVFS Z score and improvement of LVFS Z scores during the first year predicted improved survival beyond the first year. In this respect, our findings are similar to those of the Australian Childhood Cardiomyopathy study, in that a low baseline LVFS and a failure of improvement in LVFS during follow‐up were associated with higher risk for death or transplantation. 11 Similarly, in the first year, a progressive Z score reduction (improvement) of LV dilation was associated with survival, whereas a progressive increase in LV dilation was associated with death or cardiac transplantation. Simultaneously, an increase in the LVPWT:LVEDD ratio, consistent with LV reverse remodeling, in the first year, was associated with improved survival. Moreover, a lower LVEDD at baseline and at 1 year, and a reduction in LVEDD between diagnosis and 1 year had similar favorable prognostic values.
Overall, in our population with pediatric DCM, marked LV remodeling seems to have occurred by the time of diagnosis, with LVPWT:LVEDD ratio Z scores ≤−2. A failure to compensate with LV hypertrophy in the presence of chronic LV volume loading is associated with progressive contractile dysfunction and HF in valvular heart disease and is also a hallmark of pediatric DCM. 15 , 24 Poorer survival in the first year after diagnosis was associated with a lower LVPWT:LVEDD ratio, which is consistent with pathologic LV remodeling in other populations. However, this effect was not evident in the multivariable hazard analysis after 1 year, suggesting that the effect of this LV remodeling may be exerted early in the disease course. In addition, children with greater LV dilation and a lower LVPWT:LVEDD ratio Z score early in the clinical course (Table 1) may have been listed for cardiac transplantation more often, as we have noted. 23
The clinical utility of changes in LVEDD and LVFS over time relative to absolute values is indicated by the receiver‐operator characteristic analysis that, without setting specific cutoff values, indicates the improved predictive value of changes in LV dimension and contractility over baseline values. The specificity of death or transplant for a hypothetical patient whose LVEDD is greater than the 75th percentile at diagnosis is 77.8% by 1 year and 76.7% by 7 years. Serial assessment increases that specificity to 78.9% in year 1 and to more than 83% specificity by year 7. For LVFS, the specificity for death or transplant was 77.6% at 1 year and 73.2% at 7 years, if baseline LVFS is below the 25th percentile (Table 5). By accounting for changes in the first year, a slight reduction in Z score (further reduced LV function) increases that specificity to 79.3% and 81% for such an outcome at years 1 and 7, respectively. These changing values may be important in treatment planning and reinforce the evidence that adverse ventricular remodeling in children portends a poor outcome.
In response to the perceived misuse or overuse and increasing expenses of echocardiography, appropriate use criteria were developed in 2011. 25 , 26 , 27 , 28 For adults with systolic or diastolic HF, these criteria state that serial echocardiography is inappropriate in patients at risk for HF, or even in those with established HF, at <1 year intervals unless clinical cardiac status changes. 29 , 30 , 31 , 32 Yet, several studies of adults with HF have suggested that early improvement, indicated by serial echocardiograms, predicts better long‐term survival and that changes in New York Heart Association functional class do not always correlate with ventricular function. 32 , 33 , 34 Although the goal of optimizing clinical testing is laudable, our data do not support eliminating serial diagnostic echocardiography in children with HF. In the developing child, ventricular anatomy and function may substantially change soon after diagnosis of cardiomyopathy. 8 Indeed, the careful mapping of echocardiographic remodeling in children with DCM is likely to prove both clinically valuable and cost effective if it guides early and appropriate intervention. Conversely, if appropriate use criteria are rigidly applied without clinical judgment, disease progression may be overlooked and outcomes may be affected. Our data support the benefit of serial assessment at relevant intervals for children regardless of any change in symptoms.
Finally, our results suggest that the clinical course and risk factors in children with DCM, most of whom have systolic HF, may differ from that of adults with the same phenotype: in fact, progression in several forms of systolic HF in children appears to be closely associated with echocardiographic evidence of remodeling. 35 , 36 This observation is consistent with data showing differential gene expression and remodeling pathways in adults and children with HF. 37 , 38 , 39 These findings indicate that disease causation, outcomes, and management of HF differ substantially between children and adults, differences that should be acknowledged in future pediatric HF guidelines. 40 , 41
Study Limitations
The results of the study cannot be generalized to patients presenting with DCM who expired or received a transplant within 6 months of presentation and to those with a clinical diagnosis of myocarditis. Ascertainment bias for the degree and extent of remodeling is also possible, because the time of onset is usually unknown in DCM and is assigned as the date of first echocardiographic diagnosis in the PCMR. The observed rate and extent of LV remodeling may be influenced by the duration of unrecognized disease and may differ by the cause of disease. Nonetheless, the value of echocardiographic assessment described here was independent of any specific diagnosis. Similarly, our assessment assumes a constant trajectory of remodeling, but whether the actual changes in LV dimensions over time are more complex (with early improvement followed by deterioration) is unknown.
The low interobserver and intertest reliability of echocardiographic measurements is a limitation to any serial assessment of changes in LV dimensions. However, our echocardiographic measurements were acquired in real‐world settings, which indicates their pragmatic value. Overall, the most important changes were in LVEDD values whose variability (2%–4%) is the lowest among echocardiographic measures. 42 It is important to note that small changes in LVPWT can result in a large change in the LVEDD:LVPWT ratio, which is an inherent limitation in the echocardiographic measurement of LVPWT. Therefore, the impact of changes we observed in LVEDD:LVPWT ratio may well have been driven mainly by LVEDD.
Outcomes of cardiac transplantation are likely to be somewhat skewed by the subjective nature of cardiac transplantation listing decisions. Nevertheless, this subjectivity represents an important realistic end point and should be considered. A study from the Netherlands illustrates that, in more recent cohorts, cardiac transplantation rate may be lower than in our experience. 43 The Netherlands series, however, is from a later era and included patients with myocarditis and other causes not represented in our DCM cohort, which may have reduced the likelihood for cardiac transplantation as an outcome in that series.
Finally, we did not formally address the effect of medical therapy on the trajectory of LV remodeling. Although no clear association of medical therapy with any outcome‐defined group was shown, we were unable to examine this in detail, given our registry design.
Conclusions
Although children with DCM are collectively at high risk for death in the first 2 years after diagnosis, the risk profile can be refined according to whether LV dilation is progressive or not. Serial changes in both LVEDD and LVFS are more sensitive and specific for predicting an adverse event by 1 year or later than are the initial echocardiographic values. Thus, serial monitoring of echocardiographic remodeling in pediatric DCM in the first year after diagnosis is important in determining prognosis. Furthermore, because changes in LVEDD and LVFS in the first year following diagnosis affect long‐term survival, therapy that limits this progressive LV remodeling is likely to improve later cardiac transplant‐free survival.
Sources of Funding
The Pediatric Cardiomyopathy Registry is supported by grants to S.E.L. from the National Heart, Lung, and Blood Institute (HL53392, HL111459, and HL109090) and the Children's Cardiomyopathy Foundation. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the Children's Cardiomyopathy Foundation. There were no industry relationships involved in the presented work.
Disclosures
Dr Lipshultz is on the medical advisory board of Secretome Therapeutics, which is a paid position. He is also chairman of the medical advisory board of the Children's Cardiomyopathy Foundation, which is an unpaid position.
Supporting information
Data S1
Acknowledgments
We thank the participating centers for subject recruitment and follow‐up data collection. We also thank the Children's Cardiomyopathy Foundation and the Kyle John Rymiszewski Foundation for their ongoing support of the PCMR's research efforts. The authors would also like to acknowledge Danielle Dauphin Megie and Stacy DiCenso for playing a central role in the coordination and development of this article.
A complete list of the Pediatric Cardiomyopathy Registry Investigators can be found in the Supplemental Material.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022557
For Sources of Funding and Disclosures, see page 13.
References
- 1. Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, Lurie PR, Sleeper LA, Orav EJ, Lipshultz SE. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–S95. doi: 10.1067/mhj.2000.103933 [DOI] [PubMed] [Google Scholar]
- 2. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–1655. doi: 10.1056/NEJMoa021715 [DOI] [PubMed] [Google Scholar]
- 3. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752–762. doi: 10.1016/S0140-6736(09)62023-7 [DOI] [PubMed] [Google Scholar]
- 4. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867 [DOI] [PubMed] [Google Scholar]
- 5. Foerster SR, Canter CE, Cinar A, Sleeper LA, Webber SA, Pahl E, Kantor PF, Alvarez JA, Colan SD, Jefferies JL, et al. Ventricular remodeling and survival are more favorable for myocarditis than for idiopathic dilated cardiomyopathy in childhood: an outcomes study from the Pediatric Cardiomyopathy Registry. Circ Heart Fail. 2010;3:689–697. doi: 10.1161/circheartfailure.109.902833 [DOI] [PubMed] [Google Scholar]
- 6. Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. doi: 10.1016/j.cardfail.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arola A, Tuominen J, Ruuskanen O, Jokinen E. Idiopathic dilated cardiomyopathy in children: prognostic indicators and outcome. Pediatrics. 1998;101:369–376. doi: 10.1542/peds.101.3.369 [DOI] [PubMed] [Google Scholar]
- 8. Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, Addonizio LJ, Towbin JA, Rossano J, Singh RK, et al. Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol. 2014;63:1405–1413. doi: 10.1016/j.jacc.2013.11.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, Kantor PF, Everitt MD, Webber SA, Kaufman BD, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the Pediatric Cardiomyopathy Registry. J Card Fail. 2015;21:877–884. doi: 10.1016/j.cardfail.2015.06.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minamisawa M, Koyama J, Kozuka A, Miura T, Ebisawa S, Motoki H, Okada A, Izawa A, Ikeda U. Regression of left ventricular hypertrabeculation is associated with improvement in systolic function and favorable prognosis in adult patients with non‐ischemic cardiomyopathy. J Cardiol. 2016;68:431–438. doi: 10.1016/j.jjcc.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 11. Alexander PM, Daubeney PE, Nugent AW, Lee KJ, Turner C, Colan SD, Robertson T, Davis AM, Ramsay J, Justo R, et al. Long‐term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population‐based study of childhood cardiomyopathy. Circulation. 2013;128:2039–2046. doi: 10.1161/circulationaha.113.002767 [DOI] [PubMed] [Google Scholar]
- 12. Kotlyar E, Hayward CS, Keogh AM, Feneley M, Macdonald PS. The impact of baseline left ventricular size and mitral regurgitation on reverse left ventricular remodelling in response to carvedilol: size doesn't matter. Heart. 2004;90:800–801. doi: 10.1136/hrt.2002.009696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sartipy U, Dahlström U, Edner M, Lund LH. Predicting survival in heart failure: validation of the MAGGIC heart failure risk score in 51,043 patients from the Swedish heart failure registry. Eur J Heart Fail. 2014;16:173–179. doi: 10.1111/ejhf.32 [DOI] [PubMed] [Google Scholar]
- 14. Dong Y, Wang D, Lv J, Pan Z, Xu R, Ding J, Cui X, Xie X, Guo X. MAGGIC risk model predicts adverse events and left ventricular remodeling in non‐ischemic dilated cardiomyopathy. Int J Gen Med. 2020;13:1477–1486. doi: 10.2147/ijgm.s288732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Colan S. Normal echocardiographic values for cardiovascular structures. In: Lai WWCM, Geva T, Mertens L, eds Echocardiography in Pediatric and Congenital Heart Disease Edition 2. Wiley‐Blackwell; 2015. doi: 10.1002/9781118742440.app1 [DOI] [Google Scholar]
- 16. Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, et al. Familial dilated cardiomyopathy caused by an alpha‐tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:320–329. doi: 10.1016/j.jacc.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rasi K, Piuhola J, Czabanka M, Sormunen R, Ilves M, Leskinen H, Rysa J, Kerkela R, Janmey P, Heljasvaara R, et al. Collagen XV is necessary for modeling of the extracellular matrix and its deficiency predisposes to cardiomyopathy. Circ Res. 2010;107:1241–1252. doi: 10.1161/CIRCRESAHA.110.222133 [DOI] [PubMed] [Google Scholar]
- 18. Vigliano CA, Cabeza Meckert PM, Diez M, Favaloro LE, Cortes C, Fazzi L, Favaloro RR, Laguens RP. Cardiomyocyte hypertrophy, oncosis, and autophagic vacuolization predict mortality in idiopathic dilated cardiomyopathy with advanced heart failure. J Am Coll Cardiol. 2011;57:1523–1531. doi: 10.1016/j.jacc.2010.09.080 [DOI] [PubMed] [Google Scholar]
- 19. Gaasch WH, Delorey DE, St John Sutton MG, Zile MR. Patterns of structural and functional remodeling of the left ventricle in chronic heart failure. Am J Cardiol. 2008;102:459–462. doi: 10.1016/j.amjcard.2008.03.081 [DOI] [PubMed] [Google Scholar]
- 20. Ky B, Plappert T, Kirkpatrick J, Silvestry FE, Ferrari VA, Keane MG, Wiegers SE, Chirinos JA, St John Sutton M. Left ventricular remodeling in human heart failure: quantitative echocardiographic assessment of 1,794 patients. Echocardiography. 2012;29:758–765. doi: 10.1111/j.1540-8175.2012.01701.x [DOI] [PubMed] [Google Scholar]
- 21. Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN. The impact of changing medical therapy on transplantation‐free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:1377–1384. doi: 10.1016/j.jacc.2009.11.059 [DOI] [PubMed] [Google Scholar]
- 22. Molina KM, Shrader P, Colan SD, Mital S, Margossian R, Sleeper LA, Shirali G, Barker P, Canter CE, Altmann K, et al. Predictors of disease progression in pediatric dilated cardiomyopathy. Circ Heart Fail. 2013;6:1214–1222. doi: 10.1161/circheartfailure.113.000125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alvarez JA, Orav EJ, Wilkinson JD, Fleming LE, Lee DJ, Sleeper LA, Rusconi PG, Colan SD, Hsu DT, Canter CE, et al. Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. Circulation. 2011;124:814–823. doi: 10.1161/circulationaha.110.973826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brower GL, Janicki JS. Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol. 2001;280:H674–H683. doi: 10.1152/ajpheart.2001.280.2.H674 [DOI] [PubMed] [Google Scholar]
- 25. Douglas PS, Garcia MJ, Haines DE, Lai WW, Manning WJ, Patel AR, Picard MH, Polk DM, Ragosta M, Ward RP, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. A report of the American College of Cardiology Foundation appropriate use criteria task force, American Society of Echocardiography, American Heart Association, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Critical Care Medicine, Society of Cardiovascular Computed Tomography, and Society for Cardiovascular Magnetic Resonance endorsed by the American College of Chest Physicians. J Am Coll Cardiol. 2011;57:1126–1166. doi: 10.1016/j.jacc.2010.11.002 [DOI] [PubMed] [Google Scholar]
- 26. Mansour IN, Razi RR, Bhave NM, Ward RP. Comparison of the updated 2011 appropriate use criteria for echocardiography to the original criteria for transthoracic, transesophageal, and stress echocardiography. J Am Soc Echocardiogr. 2012;25:1153–1161. doi: 10.1016/j.echo.2012.08.008 [DOI] [PubMed] [Google Scholar]
- 27. Bhatia RS, Carne DM, Picard MH, Weiner RB. Comparison of the 2007 and 2011 appropriate use criteria for transthoracic echocardiography in various clinical settings. J Am Soc Echocardiogr. 2012;25:1162–1169. doi: 10.1016/j.echo.2012.07.018 [DOI] [PubMed] [Google Scholar]
- 28. Wiener DH. Achieving high‐value cardiac imaging: challenges and opportunities. J Am Soc Echocardiogr. 2014;27:1–7. doi: 10.1016/j.echo.2013.08.027 [DOI] [PubMed] [Google Scholar]
- 29. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Failure Society of America. Circulation. 2017;136:e137–e161. doi: 10.1161/cir.0000000000000509 [DOI] [PubMed] [Google Scholar]
- 30. Doherty JU, Kort S, Mehran R, Schoenhagen P, Soman P. ACC/AATS/AHA/ASE/ASNC/HRS/SCAI/SCCT/SCMR/STS 2019 appropriate use criteria for multimodality imaging in the assessment of cardiac structure and function in Nonvalvular heart disease: a report of the American College of Cardiology Appropriate use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and the Society of Thoracic Surgeons. J Nucl Cardiol. 2019;26:1392–1413. doi: 10.1007/s12350-019-01751-7 [DOI] [PubMed] [Google Scholar]
- 31. Porter TR, Shillcutt SK, Adams MS, Desjardins G, Glas KE, Olson JJ, Troughton RW. Guidelines for the use of echocardiography as a monitor for therapeutic intervention in adults: a report from the American Society of Echocardiography. J Am Soc Echocardiogr. 2015;28:40–56. doi: 10.1016/j.echo.2014.09.009 [DOI] [PubMed] [Google Scholar]
- 32. Hebert K, Macedo FY, Trahan P, Tamariz L, Dias A, Palacio A, Arcement LM. Routine serial echocardiography in systolic heart failure: is it time for the heart failure guidelines to change? Congest Heart Fail. 2011;17:85–89. doi: 10.1111/j.1751-7133.2011.00213.x [DOI] [PubMed] [Google Scholar]
- 33. Agha SA, Kalogeropoulos AP, Shih J, Georgiopoulou VV, Giamouzis G, Anarado P, Mangalat D, Hussain I, Book W, Laskar S, et al. Echocardiography and risk prediction in advanced heart failure: incremental value over clinical markers. J Card Fail. 2009;15:586–592. doi: 10.1016/j.cardfail.2009.03.002 [DOI] [PubMed] [Google Scholar]
- 34. Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM. Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail. 2012;5:720–726. doi: 10.1161/circheartfailure.111.966366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lipshultz SE, Easley KA, Orav EJ, Kaplan S, Starc TJ, Bricker JT, Lai WW, Moodie DS, Sopko G, Colan SD. Cardiac dysfunction and mortality in HIV‐infected children: the prospective P2C2 HIV multicenter study. Pediatric Pulmonary and Cardiac Complications of Vertically Transmitted HIV Infection (P2C2 HIV) Study Group. Circulation. 2000;102:1542–1548. doi: 10.1161/01.cir.102.13.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fisher SD, Easley KA, Orav EJ, Colan SD, Kaplan S, Starc TJ, Bricker JT, Lai WW, Moodie DS, Sopko G, et al. Mild dilated cardiomyopathy and increased left ventricular mass predict mortality: the prospective P2C2 HIV multicenter study. Am Heart J. 2005;150:439–447. doi: 10.1016/j.ahj.2005.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Patel MD, Mohan J, Schneider C, Bajpai G, Purevjav E, Canter CE, Towbin J, Bredemeyer A, Lavine KJ. Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight. 2017;2:2. doi: 10.1172/jci.insight.94382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tatman PD, Woulfe KC, Karimpour‐Fard A, Jeffrey DA, Jaggers J, Cleveland JC, Nunley K, Taylor MR, Miyamoto SD, Stauffer BL, et al. Pediatric dilated cardiomyopathy hearts display a unique gene expression profile. JCI Insight. 2017;2:2. doi: 10.1172/jci.insight.94249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jana S, Zhang H, Lopaschuk GD, Freed DH, Sergi C, Kantor PF, Oudit GY, Kassiri Z. Disparate remodeling of the extracellular matrix and proteoglycans in failing pediatric versus adult hearts. J Am Heart Assoc. 2018;7:e010427. doi: 10.1161/jaha.118.010427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pahl E, Sleeper LA, Canter CE, Hsu DT, Lu M, Webber SA, Colan SD, Kantor PF, Everitt MD, Towbin JA, et al. Incidence of and risk factors for sudden cardiac death in children with dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol. 2012;59:607–615. doi: 10.1016/j.jacc.2011.10.878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kantor PF, Lougheed J, Dancea A, McGillion M, Barbosa N, Chan C, Dillenburg R, Atallah J, Buchholz H, Chant‐Gambacort C, et al. Presentation, diagnosis, and medical management of heart failure in children: Canadian Cardiovascular Society guidelines. Can J Cardiol. 2013;29:1535–1552. doi: 10.1016/j.cjca.2013.08.008 [DOI] [PubMed] [Google Scholar]
- 42. Lee CK, Margossian R, Sleeper LA, Canter CE, Chen S, Tani LY, Shirali G, Szwast A, Tierney ES, Campbell MJ, et al. Variability of M‐mode versus two‐dimensional echocardiography measurements in children with dilated cardiomyopathy. Pediatr Cardiol. 2014;35:658–667. doi: 10.1007/s00246-013-0835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. den Boer SL, Lennie van Osch‐Gevers M, van Ingen G, du Marchie Sarvaas GJ, van Iperen GG, Tanke RB, Backx AP, Ten Harkel AD, Helbing WA, Delhaas T, et al. Management of children with dilated cardiomyopathy in The Netherlands: implications of a low early transplantation rate. J Heart Lung Transplant. 2015;34:963–969. doi: 10.1016/j.healun.2015.01.980 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1