

Abstract
Background
Hereditary transthyretin amyloid cardiomyopathy (hATTR‐CM) is a progressive and fatal disease. Recent evidence indicates that bone scintigraphy may serve as a tool to monitor the effectiveness of hATTR‐CM treatment. The objective of this study was to examine how eplontersen therapy influences the semiquantitative uptake of technetium‐99m‐pyrophosphate in individuals diagnosed with hATTR‐CM.
Methods and Results
We retrospectively analyzed a prospective cohort from the NEURO‐TTRansform trial, including patients with hATTR‐CM receiving eplontersen (45 mg/4 weeks). A control group comprised patients with hATTR‐CM who had not received eplontersen, inotersen, tafamidis, or patisiran. Technetium‐99m‐pyrophosphate single‐photon emission computed tomography/computed tomography was conducted at baseline and during follow‐up. Thirteen patients with hATTR‐CM were enrolled, with 6 receiving eplontersen and 7 serving as the control group. The median follow‐up time was 544 days. The eplontersen group exhibited a significant decrease in volumetric heart and lung ratio (3.774 to 2.979, P=0.028), whereas the control group showed no significant change (4.079 to 3.915, P=0.237). Patients receiving eplontersen demonstrated a significantly greater reduction in volumetric heart and lung ratio compared with the control group (−20.7% versus −3.4%, P=0.007).
Conclusions
The volumetric heart and lung ratio used to quantify technetium‐99m‐pyrophosphate uptake showed a significant reduction subsequent to eplontersen treatment in individuals diagnosed with hATTR‐CM. These findings suggest the potential efficacy of eplontersen in treating hATTR‐CM and highlight the value of technetium‐99m‐pyrophosphate single‐photon emission computed tomography/computed tomography as a tool for monitoring therapeutic effectiveness.
Keywords: 99mTc‐pyrophosphate, eplontersen, hereditary transthyretin amyloidosis, single‐photon emission computed tomography/computed tomography
Subject Categories: Cardiomyopathy, Heart Failure, Diagnostic Testing
Nonstandard Abbreviations and Acronyms
- 99mTc‐PYP
99mTc‐pyrophosphate
Clinical Perspective.
What Is New?
Technetium‐99m‐pyrophosphate single‐photon emission computed tomography/computed tomography has emerged as a potential modality to track the efficacy of transthyretin amyloid cardiomyopathy therapy, and volumetric heart to lung ratio measured by single‐photon emission computed tomography/computed tomography may provide a better metric of change in quantifying technetium‐99m‐pyrophosphate single uptake.
Eplontersen, a ligand‐conjugated investigational antisense oligonucleotide that preferentially targets hepatic transthyretin mRNA, significantly decreased cardiac technetium‐99m‐pyrophosphate single uptake in patients with hereditary transthyretin amyloid cardiomyopathy.
What Are the Clinical Implications?
Eplontersen treatment may be effective in patients with hereditary transthyretin amyloid cardiomyopathy, and technetium‐99m‐pyrophosphate single‐photon emission computed tomography/computed tomography can be used to monitor the efficacy of eplontersen therapy.
Hereditary transthyretin amyloidosis (hATTR) is a rare systemic disorder resulting from autosomal dominant mutations in the gene encoding transthyretin. 1 , 2 , 3 , 4 , 5 The continuous deposition and accumulation of amyloid in multiple organ systems result in the progressive development of peripheral polyneuropathy, cardiomyopathy, nephropathy, and gastrointestinal dysfunction. 6 , 7 The presence of cardiomyopathy is associated with a worse prognosis, 8 , 9 , 10 underscoring the significance of early diagnosis, timely treatment initiation, and diligent monitoring of treatment response of hATTR cardiomyopathy (ATTR‐CM).
Technetium‐99m‐pyrophosphate (99mTc‐PYP) scintigraphy stands out as 1 of the most frequently used imaging modalities, offering accurate diagnosis of ATTR‐CM and thereby eliminating the necessity for an invasive cardiac biopsy. 10 , 11 , 12 , 13 Emerging evidence suggests that bone scintigraphy can also be used to monitor the efficacy of ATTR‐CM treatment. 14 , 15 , 16 , 17 , 18 , 19 A 2‐dimensional imaging approach to interpret pyrophosphate images using visual score, and planar heart and contralateral lung (H/CL) ratio can be influenced by blood pooling and rib activity. 11 , 12 Therefore, volumetric calculation of heart to lung (H/L) ratio with single‐photon emission computed tomography/computed tomography (SPECT/CT) was introduced to overcome these shortcomings. In our prior study, we explored the impact of 99mTc‐PYP SPECT/CT in patients with A97S hATTR‐CM undergoing tafamidis treatment. Our findings revealed a noteworthy reduction in 99mTc‐PYP cardiac uptake among patients treated with tafamidis. In addition, visual score, planar H/CL ratio, and volumetric H/L ratio all exhibited significant decreases after an average 19 months of tafamidis treatment. 20
Eplontersen (formerly ION‐682884; Ionis Pharmaceuticals, Carlsbad, CA) is a ligand‐conjugated investigational antisense oligonucleotide that preferentially targets hepatic transthyretin mRNA, thereby inhibiting transthyretin protein synthesis. 21 Positive results were reported in a planned 35‐week interim analysis of the phase 3 NEURO‐TTRansform study (NCT04136184) of eplontersen in patients with hereditary TTR‐mediated amyloid polyneuropathy. 22 Eplontersen resulted in a significant mean reduction of 81.2% (P<0.0001) in serum transthyretin concentration compared with baseline, signifying a substantial decrease in transthyretin protein production. Furthermore, eplontersen exhibited a notable therapeutic impact on neuropathic disease progression, measured by modified Neuropathy Impairment Score+7. 23 This benefit was confirmed by a statistically significant difference in mean change from baseline, as compared with the external placebo group (P<0.0001).
Due to the promising results of the NEURO‐TTRansform study, we hypothesized that eplontersen may also be effective in patients with ATTR‐CM. Therefore, the aim of this study was to longitudinally assess 99mTc‐PYP SPECT/CT imaging findings to determine whether eplontersen treatment results in changes in semiquantitative measures of tracer uptake in patients with hereditary A97S (Ala97Ser) ATTR‐CM.
METHODS
The study obtained approval from the institutional review board of National Taiwan University Hospital (number 201912120MSA, 202208006RIND), and the requirement for informed consent was waived. Data supporting the findings of this study are accessible from the corresponding author upon reasonable request.
Patient Population
Patients with confirmed hATTR‐CM who received pretherapeutic and follow‐up 99mTc‐PYP SPECT/CT at National Taiwan University Hospital over a 3‐year period between July 2019 and October 2022 were retrospectively identified from a prospectively collected cohort.
The inclusion criteria were as follows: (1) patients with heart failure symptoms or evidence of amyloid cardiomyopathy on the basis of noninvasive diagnostic criteria (end‐diastolic interventricular septal wall thickness >12 mm without another cause of hypertrophy) 24 , 25 and (2) a pretherapeutic 99mTc‐PYP scan demonstrating myocardial enhancement with a visual score ≥2, a planar H/CL ratio ≥1.3, and the exclusion of light chain disease. 26 , 27 The exclusion criteria were (1) patients who died or were lost to follow‐up within 1 year and (2) patients without complete baseline and follow‐up 99mTc‐PYP planar and SPECT/CT imaging.
Participants in NEURO‐TTRansform (NCT04136184; EudraCT:2019‐001698‐10), an international, open‐label, phase 3 study, who met the inclusion criteria were identified as the eplontersen group. Patients with hATTR‐CM who met the above inclusion criteria but were not enrolled in the NEURO‐TTRansform study and did not receive tafamidis, patisiran, inotersen, or eplontersen were retrospectively identified as the control group. All patients were enrolled before 99mTc‐PYP SPECT/CT imaging analysis.
Correlations with and the progression of NT‐proBNP (N‐terminal pro‐B‐type natriuretic peptide) (Pfizer, New York, NY) concentration and estimated glomerular filtration rate, estimated with the 4‐variable Modification of Diet in Renal Disease study equation, 28 were investigated for comparison. Both at the time of diagnosis and during serial measurements, these 2 independent biomarkers show a correlation with survival outcomes in patients with cardiac ATTR amyloidosis. 29 , 30
Echocardiography
Chamber quantification, valvular pathology, and myocardial function were defined according to the standards outlined by the American Society of Echocardiography. 31 Interventricular septal diameter, left ventricular posterior wall diameter, left ventricular end‐diastolic diameter, and left ventricular ejection fraction were obtained. The calculation of left ventricular mass used the formula as reported by Devereux and Reichek, 32 and the results was normalized by body surface area to derive the left ventricular mass index. Two‐dimensional speckle‐tracking echocardiography analysis of the left ventricle was analyzed offline using the commercially available postprocessing software QLAB (Philips Healthcare Taiwan, Taipei, Taiwan).
99mTc‐PYP Planar and SPECT/CT Scintigraphy
Planar and SPECT/CT scans of the chest were conducted 3 hours after the intravenous administration of 20 mCi of 99mTc‐PYP. The image acquisition parameters and protocol adhere to the joint guidelines established by the Taiwan Society of Cardiology and the Society of Nuclear Medicine of the Republic of China, 33 as well as consensus recommendations from multiple expert societies. 34 , 35
The images were analyzed visually using visual score and semiquantitatively using planar H/CL ratio and volumetric H/L ratio. A visual score of tracer uptake in the left ventricular (LV) myocardium grade ≥2 was interpreted as being positive for ATTR‐CM according to previous consensus and guidelines. 33 In separate sessions, 2 expert nuclear medicine physicians, without access to the patient's clinical information, independently analyzed the visual score and the planar H/CL ratio. A clinical manager documented any differences in their evaluations, which were then resolved by reaching a consensus during a joint review session. The reconstructed SPECT/CT images were processed using PMOD version 4.2 software (PMOD Technologies, Zurich, Switzerland) for volumetric H/L ratio analysis after patient enrollment. An automated algorithm, using a Hounsfield unit threshold of −400, was used for lung segmentation on the CT images. Volumes of interest within the LV myocardium were delineated using an automatic contouring process set to capture 40% to 50% of the peak heart count. The volumetric H/L ratio was derived by dividing the mean count of the heart by the count of the lungs (Figure 1).
Figure 1. Workflow of 99mTc‐PYP SPECT/CT with volumetric H/L ratio.
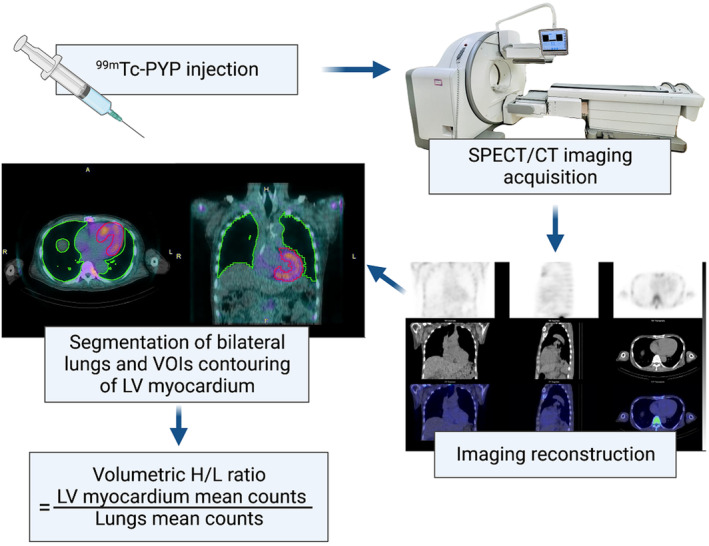
Segmentation of the lungs took place on CT images using an automated algorithm based on an HU threshold value of −400. The volumes of interest in the LV myocardium were generated by auto contouring 40% to 50% of maximum heart count. The volumetric H/L ratio was calculated as mean count of the heart divided by that of the lungs. 99mTc‐PYP indicates technetium‐99m‐pyrophosphate; CT, computed tomography; H/L, heart to lung; HU, Hounsfield unit; LV, left ventricular; SPECT, single‐photon emission computed tomography; and VOIs, volume of involvement.
Statistical Analysis
The data were presented as mean±SD for continuous variables and as relative percentages for categorical variables. The normality of continuous data was assessed through the Kolmogorov‐Smirnov goodness‐of‐fit test. Paired tests, including the Wilcoxon signed rank test for nonnormally distributed data and parametric tests for normally distributed data, were used to compare changes between baseline and follow‐up. All statistical analyses were conducted using Stata version 17 (StataCorp, College Station, TX).
RESULTS
Patient Characteristics
Thirteen patients with ATTR‐CM were analyzed, of whom 6 were treated with eplontersen and the other 7 were enrolled as the control group. Baseline demographic and clinical variables of the 2 groups are shown in Table 1. All patients in the eplontersen group (100%) carried the Ala97Ser (A97S) mutation. One of the 7 patients in the control group (14%) carried the Glu89Lys (E89K) mutation, whereas the other 6 carried the A97S mutation. The 6 A97S patients in the control group were included in our prior study. 20 The baseline clinical characteristics were comparable between the eplontersen and control groups, including age (59.33±5.5 versus 62.43±6.2 years, P=0.517), sex (83% versus 71% men, P=0.626), mean platelet count (214.50±28.877 versus 195.00±38.792 k/μL, P=0.394), median baseline NT‐proBNP (374.90 [69–4271] versus 470.60 [176–1286] pg/mL, P=0.886), mean left ventricular posterior wall diameter (13.50±1.517 versus 12.68±3.007 mm, P=0.807), mean left ventricular mass index (141.30±43.645 versus 148.80±39.222 g/m2, P=0.831), and mean LV global longitudinal strain (−15.40±4.489 versus −15.78±3.056, P=0.866). All patients in the eplontersen group were treated with eplontersen 45 mg SC every 4 weeks. There were no discontinuations of eplontersen treatment due to adverse effects among patients, and adherence to the treatment was ascertained at ≥1 visit within the period from baseline to 1 year. The average time spans from baseline to follow‐up 99mTc‐PYP scans were 607.51 days (range, 526–850) in the eplontersen group and 491.00 days (range, 195–644) in the control group (P=0.445). There were no instances of mortality, hospitalization, or significant adverse events during the follow‐up period in both groups.
Table 1.
Baseline Characteristics Between the Eplontersen Group and Control Group*
| Characteristic | Eplontersen group (n=6) | Control group (n=7) | P value |
|---|---|---|---|
| Age, y | 59.33±5.538 | 62.43±6.214 | 0.517 |
| Male sex | 5 (83.3) | 5 (71.4) | 0.626 |
| BMI, kg/m2 | 23.26±1.832 | 22.53±3.398 | 0.937 |
| NYHA Fc I‐II | 6 (100) | 7 (100) | 1.000 |
| Follow‐up duration, d | 607.51 [526–850] | 491.00 [195–644] | 0.445 |
| Genetics | |||
| Ala97Ser (A97S) | 6 (100) | 6 (86) | |
| Glu89Lys (E89K) | 0 (0) | 1 (14) | |
| Biochemistry | |||
| Platelets, k/μL | 214.50±28.877 | 195.00±38.792 | 0.394 |
| NT‐proBNP, pg/mL | 374.90 [69–4271] | 470.60 [176–1286] | 0.886 |
| Creatinine, mg/dL | 0.83±0.197 | 0.73±0.206 | 0.306 |
| eGFR, mL/min per 1.73 m2 | 100.22±20.857 | 116.27±38.532 | 0.475 |
| Echocardiography | |||
| LV ejection fraction, % | 63.33±11.572 | 63.03±11.330 | 0.873 |
| IVS thickness, mm | 14.67±2.422 | 13.77±4.809 | 0.744 |
| LV posterior wall thickness, mm | 13.50±1.517 | 12.68±3.007 | 0.807 |
| LV end‐diastolic diameter, mm | 43.00±4.243 | 46.17±5.492 | 0.296 |
| LV mass index, g/m2 | 141.30±43.645 | 148.80±39.222 | 0.831 |
| LVGLS, % | −15.40±4.489 | −15.78±3.056 | 0.866 |
BMI indicates body mass index; eGFR, estimated glomerular filtration rate; IVS, interventricular septal; LV, left ventricular; LVGLS, left ventricular global longitudinal strain; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; and NYHA Fc, New York Heart Association functional classification.
Data are given as mean±SD, number (percentage), or median [first quartile–third quartile].
Biomarkers and Echocardiography
The treatment response to eplontersen in the eplontersen group with regard to biochemistry, LV geometry, and function is shown in Table 2. After eplontersen treatment, NT‐proBNP (from 374.90 [69–4271] to 198.25 [67–3045] pg/mL, P=0.463), creatinine (from 0.83±0.197 to 0.80±0.200 mg/dL, P=0.589), and estimated glomerular filtration rate (from 100.22±20.857 to 105.75±27.058 mL/min per 1.73 m2, P=0.600) numerically improved, but without statistical significance. However, left ventrticular ejection fraction (from 63.33±11.572% to 64.50±11.540%, P=0.463), left ventricular posterior wall diameter (from 13.50±1.517 to 13.50±2.540 mm, P=1.000), left ventricular end‐diastolic diameter (from 43.00±4.243 to 44.17±2.483 mm, P=0.196), left ventricular mass index (from 141.30±43.645 to 140.53±40.306 g/m2, P=0.917), and LV global longitudinal strain (−15.40±4.489 versus −16.37±3.655, P=0.499) did not significantly change during the study period.
Table 2.
Biochemical, Echocardiographic, and 99mTc‐PYP Data Before and After Treatment in the Eplontersen Group and the Control Group*
| Clinical parameters | Eplontersen group (n=6) | Control group (n=7) | ||||
|---|---|---|---|---|---|---|
| Baseline | Follow‐up | P value | Baseline | Follow‐up | P value | |
| Biochemistry | ||||||
| Platelet, k/μL | 214.5±28.877 | 204.6±33.620 | 0.786 | 195.0±38.792 | 187.0±38.972 | 0.116 |
| NT‐proBNP, pg/mL | 374 [69–4271] | 198 [67–3045] | 0.463 | 471 [176–1286] | 462 [184–898] | 0.612 |
| Creatinine, mg/dL | 0.83±0.197 | 0.80±0.200 | 0.589 | 0.73±0.206 | 0.76±0.151 | 0.739 |
| eGFR, mL/min per 1.73 m2 | 100.2±20.857 | 105.8±27.058 | 0.600 | 116.3±38.532 | 107.8±36.037 | 0.612 |
| Echocardiography | ||||||
| LVEF, % | 63.33±11.572 | 64.50±11.540 | 0.463 | 63.03±11.330 | 65.87±15.913 | 0.500 |
| IVS thickness, mm | 14.67±2.422 | 13.83±2.041 | 0.197 | 13.77±4.809 | 14.33±2.422 | 0.680 |
| LVPW thickness, mm | 13.50±1.517 | 13.50±2.540 | 1.000 | 12.68±3.007 | 14.00±2.550 | 0.216 |
| LVEDd, mm | 43.00±4.243 | 44.17±2.483 | 0.196 | 46.17±5.492 | 47.17±3.371 | 1.000 |
| LVMI, g/m2 | 141.3±43.645 | 140.5±40.306 | 0.917 | 148.80±39.222 | 153.41±33.430 | 0.465 |
| LVGLS, % | −15.40±4.489 | −16.37±3.655 | 0.499 | −15.78±3.056 | −14.58±3.897 | 0.121 |
| 99mTc‐PYP scintigraphy | ||||||
| Planar H/CL ratio | 1.561±0.105 | 1.473±0.116 | 0.173 | 1.701±0.171 | 1.657±0.130 | 0.735 |
| Volumetric H/L ratio | 3.774±0.474 | 2.979±0.354 | 0.028 | 4.079±0.534 | 3.915±0.386 | 0.237 |
99mTc‐PYP indicates technetium‐99m‐pyrophosphate; eGFR, estimated glomerular filtration rate; H/CL ratio, heart and contralateral lung ratio; H/L ratio, heart and lung ratio; IVS, interventricular septum; LVEDd, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVGLS, left ventricular global longitudinal strain; LVMI, left ventricular mass index; LVPW, left ventricular posterior wall; and NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide.
Data are given as mean±SD or median [first quartile–third quartile].
Serial 99mTc‐PYP Cardiac Imaging
The results of serial 99mTc‐PYP cardiac imaging analysis are shown in Table 3. The baseline 99mTc‐PYP parameters were comparable between the eplontersen and control groups, including planar H/CL ratio (1.561±0.105 versus 1.701±0.171, P=0.234) and volumetric H/L ratio (3.774±0.474 versus 4.079±0.534, P=0.317). Semiquantitative visual scores remained stationary between the 2 groups. In the eplontersen group, 33.3% of the patients had an improvement in visual score, whereas 66.7% remained stationary, and none of the patients had a worse visual score. In comparison, none (0%) of the patients improved, 71.4% remained stationary, and 28.6% had a worse visual score in the control group.
Table 3.
99mTc‐PYP Cardiac Data Changes Between the Eplontersen Group and the Control Group*
| Semi quantitative analysis | Eplontersen group (n=6) | Control group (n=7) | P value |
|---|---|---|---|
| Baseline planar H/CL ratio | 1.561±0.105 | 1.701±0.171 | 0.234 |
| Baseline volumetric H/L ratio | 3.774±0.474 | 4.079±0.534 | 0.317 |
| ΔPlanar H/CL ratio | −0.086±0.138 | −0.044±0.181 | 0.721 |
| ΔVolumetric H/L ratio | −0.797±0.351 | −0.164±0.349 | 0.010† |
| Change in planar H/CL ratio, % | −5.299±8.879 | −1.930±10.397 | 0.475 |
| Change in volumetric H/L ratio, % | −20.703±7.821 | −3.402±8.128 | 0.007‡ |
| Follow‐up time, d | 607.51 [526–850] | 491.00 [195–644] | 0.445 |
99mTc‐PYP indicates technetium‐99m‐pyrophosphate; H/CL ratio, heart and contralateral lung ratio; and H/L ratio, heart and lung ratio.
Data are given as mean±SD or median [first quartile–third quartile].
P<0.05.
P<0.01.
In the eplontersen group, a significant reduction in volumetric H/L ratio (3.774±0.474 to 2.979±0.354, P=0.028) was noted after treatment, but planar H/CL ratio (1.561±0.105 to 1.473±0.116, P=0.173) remained stationary (Figure 2). In the control cohort, there were no significant changes between both scans in volumetric H/L ratio (4.079±0.534 to 3.915±0.3864, P=0.237) and planar H/CL ratio (1.701±0.171 to 1.657±0.130, P=0.735). In the comparison between the 2 groups, the eplontersen group exhibited a significantly greater reduction in volumetric H/L ratio compared with the control group (−20.7±7.8% versus −3.4±8.1%, P=0.007) (Figure 3). The interobserver and intraobserver correlation of the volumetric H/L ratio were 0.9663 and 0.9817, respectively.
Figure 2. Representative 99mTc‐PYP planar and SPECT/CT images at baseline and after eplontersen treatment in a 51‐year‐old man with A97S hereditary ATTR‐CM.
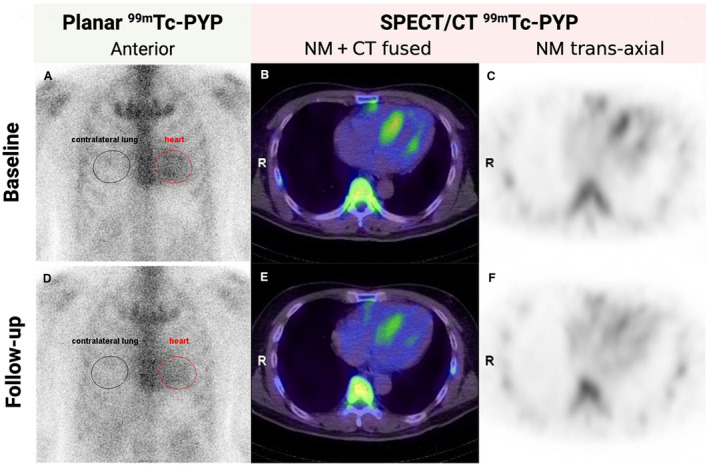
Both planar H/CL ratio (from 1.59 to 1.44, A and D) and volumetric H/L ratio (from 4.50 to 3.59, B and C, E and F) decreased after a 1.5‐year course of eplontersen treatment. 99mTc‐PYP indicates technetium‐99m‐pyrophosphate; ATTR‐CM, transthyretin amyloid cardiomyopathy; CT, computed tomography; H/CL, heart to contralateral lung; H/L, heart to lung; NM, nuclear medicine; and SPECT, single‐photon emission computed tomography.
Figure 3. Changes in volumetric H/L ratio on 99mTc‐PYP between the eplontersen and control groups.
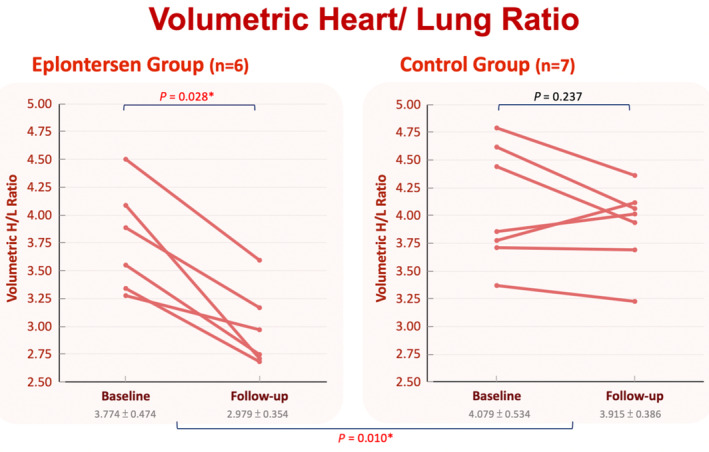
*P<0.05. 99mTc‐PYP indicates technetium‐99m‐pyrophosphate; and H/L, heart to lung.
DISCUSSION
This study represents the first to illustrate the potential effectiveness of eplontersen in treating patients with hereditary A97S ATTR‐CM and underscores the prospective usefulness of 99mTc‐PYP SPECT/CT in monitoring ATTR‐CM following treatment. Our approach incorporated volumetric H/L ratio, alongside planar H/CL ratio and visual score, providing a comprehensive and precise assessment of cardiac tracer uptake. Our results showed that 99mTc‐PYP uptake significantly decreased in quantitative volumetric H/L ratio in the patients with hereditary A97S ATTR‐CM after a mean 608 days of eplontersen treatment.
ATTR‐CM is characterized by deposition of amyloid fibrils, consisting of misfolded transthyretin protein, in the heart. This accumulation of misfolded transthyretin in the myocardium can result in conduction disturbance, heart failure, and sudden cardiac death. 36 Previous studies using 99mTc‐PYP to diagnose ATTR‐CM have shown that the method was reproducible at multiple sites with high accuracy. 12 , 13 Consequently, 99mTc‐PYP has been incorporated into several consensus algorithms for the noninvasive diagnosis of ATTR‐CM.10,13,34 Although cardiac scintigraphy has been shown to be a significantly sensitive tool for diagnosing ATTR‐CM, serial 99mTc‐PYP has failed to show a remarkable clinical decline during the natural course of ATTR‐CM using semiquantitative visual score and planar H/CL ratio. 37 Segmental evaluation and attenuation correction with SPECT/CT offer the potential advantages of being more quantitative and a better metric of change. 38 By quantifying natural disease progression with quantitative 99mTc‐3,3‐diphosphonol‐1,2‐propanodicarboxylic acid scintigraphy, Ross et al demonstrated cohort‐wide progression. 39 This suggests the possibility of monitoring changes using quantitative scintigraphy.
In our study, we used the volumetric H/L ratio in conjunction with the planar H/CL ratio and visual score to provide an objective and precise quantification of cardiac tracer uptake. A reduction in 99mTc‐PYP cardiac uptake was observed in the patients treated with eplontersen, as assessed through the semiquantitative volumetric H/L ratio. Our research group previously demonstrated decreases in planar H/CL ratio and volumetric H/L ratio on 99mTc‐PYP scintigraphy in 21 patients with hereditary A97S ATTR‐CM receiving at least 1 year of tafamidis treatment. 20 Another study reported similar findings, with a decrease in hydroxydimethylene diphosphonate cardiac uptake in a patient with ATTR‐CM treated with tafamidis. 16
Eplontersen, a ligand‐conjugated antisense medication, is specifically engineered for targeted delivery to hepatocytes, the predominant cells responsible for the systemic circulation of transthyretin. 22 This innovative antisense design incorporates a triantennary GalNAc (N‐acetylgalactosamine), facilitating efficient receptor‐mediated uptake by hepatocytes through the high‐capacity asialoglycoprotein receptors. In a comprehensive analysis of 8 GalNAc‐conjugated antisense oligonucleotides, conducted through phase 1 randomized placebo‐controlled trials with healthy human volunteers, a potency increase of 20 to 30 times was observed in comparison with their parent unconjugated antisense oligonucleotides. 21 They were well tolerated, with no discontinuations due to adverse events. The 35‐week interim results of the NEURO‐TTRansform phase III trial showed that eplontersen achieved statistically significant and clinically meaningful changes from baseline for the coprimary end points of percent change in serum transthyretin concentration and change from baseline in Neuropathy Impairment Score+7 versus an external placebo group. 23 Another global, double‐blind, randomized, placebo‐controlled phase 3 cardiovascular outcome study of eplontersen, CARDIO‐TTRansform (NCT04136171), recently achieved its original enrollment goal. 40 Data from this study are expected in the first half of 2025.
This study offers important insights into the use of 99mTc‐PYP scintigraphy to track eplontersen treatment effects in hereditary A97S ATTR‐CM. A97S is the predominant mutation in patients with hATTR‐CM in Taiwan and has also been identified in China, Malaysia, Thailand, and among those of Chinese descent. 41 , 42 , 43 , 44 Over one‐third (34.3%) of the non‐V30M NEURO‐TTRansform cohort had this sequence variant. 45 Refractory heart failure is the major cause of death in these patients. Although the median NT‐proBNP values were not high in both groups, indicating mild cardiac involvement in some of the patients, significant improvements were demonstrated by volumetric H/L ratio on 99mTc‐PYP scintigraphy in these patients after eplontersen treatment. Considering the consistent and unchanging radiotracer uptake in previous studies among patients experiencing progression in the natural course of ATTR‐CM 37 , 39 and within our control cohort, our findings may imply potential benefits associated with eplontersen in these patients. This study is a pilot study, and further larger studies with longer follow‐up are needed to confirm our findings.
Limitations
This study has several limitations. First, the sample size was small, and this was a prospective cohort with retrospective analysis. Matching the control group with the treatment group in terms of baseline characteristics is currently not possible due to the small sample size. However, considering the fact that ATTR‐CM is a rare disease, we still provide valuable data on the application of 99mTc‐PYP SPECT/CT to monitor the effect of eplontersen in patients with hereditary A97S ATTR‐CM. Second, although our cohort predominantly consists of patients with the A97S variant, this may limit generalizability. However, A97S is the most common mutation among patients with hATTR‐CM in Taiwan. Given its significance as an important variant observed in patients of Chinese descent and in the Southeast Asian region, we believe it is our responsibility to report on the potential treatment response in this crucial variant. Third, the mean follow‐up period was only 608 days, and thus the long‐term effect of eplontersen on A97S ATTR‐CM using 99mTc‐PYP SPECT/CT cannot be demonstrated in the present study. Finally, in a recent case report, Smiley et al observed significant cardiac uptake using I124‐evuzamitide positron emission tomography‐computed tomography imaging, despite negative 99mTc‐PYP results and unchanged echocardiographic parameters. This discrepancy highlights the need for further comparative studies of these imaging techniques. 46 The relationships among cardiac fibril deposition, clinical symptoms, and cardiac radiotracer uptake should be clarified, and the underlying mechanisms should be investigated.
CONCLUSIONS
In this study, a significant reduction in 99mTc‐PYP uptake, as assessed by the semiquantitative volumetric H/L ratio, was observed after eplontersen treatment in patients with hATTR‐CM. These results indicate the potential efficacy of eplontersen in management of hATTR‐CM and highlight the potential usefulness of 99mTc‐PYP SPECT/CT for monitoring the effectiveness of eplontersen therapy and assessing cardiac amyloid burden.
Sources of Funding
None.
Disclosures
None.
Acknowledgments
The authors thank all colleagues, especially Y.H. Huang and T.Y. Lee, in the Nuclear Medicine Department, for image acquisition and processing.
This article was sent to Barry London, MD, PhD, Senior Guest Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 9.
Contributor Information
Mei‐Fang Cheng, Email: meifang@ntuh.gov.tw.
Chi‐Chao Chao, Email: ccchao@ntu.edu.tw.
References
- 1. Coelho T, Maurer MS, Suhr OB. THAOS—the transthyretin amyloidosis outcomes survey: initial report on clinical manifestations in patients with hereditary and wild‐type transthyretin amyloidosis. Curr Med Res Opin. 2013;29:63–76. doi: 10.1185/03007995.2012.754348 [DOI] [PubMed] [Google Scholar]
- 2. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15:387–404. doi: 10.1038/s41582-019-0210-4 [DOI] [PubMed] [Google Scholar]
- 3. Cuddy SAM, Falk RH. Amyloidosis as a systemic disease in context. Can J Cardiol. 2020;36:396–407. doi: 10.1016/j.cjca.2019.12.033 [DOI] [PubMed] [Google Scholar]
- 4. Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease‐modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86:1036–1043. doi: 10.1136/jnnp-2014-308724 [DOI] [PubMed] [Google Scholar]
- 5. Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante‐Bordeneuve V, Schmidt HH, Merlini G. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66:2451–2466. doi: 10.1016/j.jacc.2015.09.075 [DOI] [PubMed] [Google Scholar]
- 6. Conceicao I, Gonzalez‐Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML, Amass L. "Red‐flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21:5–9. doi: 10.1111/jns.12153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beirao I, Lobato L, Costa PM, Fonseca I, Mendes P, Silva M, Bravo F, Cabrita A, Porto C. Kidney and anemia in familial amyloidosis type I. Kidney Int. 2004;66:2004–2009. doi: 10.1111/j.1523-1755.2004.00971.x [DOI] [PubMed] [Google Scholar]
- 8. Lane T, Fontana M, Martinez‐Naharro A, Quarta CC, Whelan CJ, Petrie A, Rowczenio DM, Gilbertson JA, Hutt DF, Rezk T, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140:16–26. doi: 10.1161/CIRCULATIONAHA.118.038169 [DOI] [PubMed] [Google Scholar]
- 9. Rintell D, Heath D, Braga Mendendez F, Cross E, Cross T, Knobel V, Gagnon B, Turtle C, Cohen A, Kalmykov E, et al. Patient and family experience with transthyretin amyloid cardiomyopathy (ATTR‐CM) and polyneuropathy (ATTR‐PN) amyloidosis: results of two focus groups. Orphanet J Rare Dis. 2021;16:70. doi: 10.1186/s13023-021-01706-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2019;73:2872–2891. doi: 10.1016/j.jacc.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc‐pyrophosphate scintigraphy for differentiating light‐chain cardiac amyloidosis from the transthyretin‐related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6:195–201. doi: 10.1161/CIRCIMAGING.112.000132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, Pozniakoff T, Buberg FL, Miller EJ, Berk JL, et al. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol. 2016;1:880–889. doi: 10.1001/jamacardio.2016.2839 [DOI] [PubMed] [Google Scholar]
- 13. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612 [DOI] [PubMed] [Google Scholar]
- 14. Odouard S, Abulizi M, Kharoubi M, Oghina S, Guendouz S, Zaroui A, Teiger E, Itti E, Damy T, Galat A. Tafamidis decreases cardiac uptake of (99m)Tc‐HMDP in transthyretin cardiac amyloidosis. JACC Cardiovasc Imaging. 2022;15:2149–2151. doi: 10.1016/j.jcmg.2022.06.013 [DOI] [PubMed] [Google Scholar]
- 15. Fontana M, Martinez‐Naharro A, Chacko L, Rowczenio D, Gilvertson JA, Whelan CJ, Strehina S, Thirusha L, Moon J, Hutt DF, et al. Reduction in CMR derived extracellular volume with patisiran indicates cardiac amyloid regression. JACC Cardiovasc Imaging. 2021;14:189–199. doi: 10.1016/j.jcmg.2020.07.043 [DOI] [PubMed] [Google Scholar]
- 16. Bellevre D, Bailliez A, Marechaux S, Manrique A, Mouquet F. First follow‐up of cardiac amyloidosis treated by tafamidis, evaluated by absolute quantification in bone scintigraphy. JACC Case Rep. 2021;3:133–135. doi: 10.1016/j.jaccas.2020.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elsadany M, Rivas CG, Arora S, Jaiswal A, Weissler‐Snir A, Duvall WL. The use of SPECT/CT quantification of 99mTc‐PYP uptake to assess tafamidis treatment response in ATTR cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2021;22:iii72. doi: 10.1093/ehjci/jeab111.058 [DOI] [Google Scholar]
- 18. Doumas A, Zegkos T, Parcharidou D, Gossios T, Ntelios D, Sofia Chatzileontiadou MD, Emmanouil Papanastasiou MD, Evdoxia Hatjiharissi MD, Ioannis Iakovou MD, Efthimiadis GK. A novel quantitative method for assessing the therapeutic response to tafamidis therapy in patients with cardiac TTR amyloidosis. A preliminary report. Hell J Nucl Med. 2022;25:216–219. [DOI] [PubMed] [Google Scholar]
- 19. Wu YA, Tsai CH, Su MY, Chao CC, Cheng MF, Shun CT, Hsieh ST, Lin YH. Reverse cardiac remodelling and dysfunction in A97S transthyretin cardiac amyloidosis after tafamidis treatment. ESC Heart Fail. 2022;9:4335–4339. doi: 10.1002/ehf2.14165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu AL, Chen YC, Tsai CH, Chao CC, Su MY, Jaung JM, Lee MJ, Hsieh ST, Cheng MF, Lin YH. Tafamidis treatment decreases 99mTc‐pyrophosphate uptake in patients with hereditary Ala97Ser transthyretin amyloid cardiomyopathy. JACC Cardiovasc Imaging. 2023;16:866–867. doi: 10.1016/j.jcmg.2022.12.016 [DOI] [PubMed] [Google Scholar]
- 21. Viney NJ, Guo S, Tai LJ, Baker BF, Aghajan M, Jung SW, Yu RZ, Booten S, Murray H, Machemer T, et al. Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: preclinical and phase 1 data. ESC Heart Fail. 2021;8:652–661. doi: 10.1002/ehf2.13154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coelho T, Ando Y, Benson MD, Berk JL, Waddington‐Cruz M, Dyck PJ, Gillmore JD, Khella SL, Litchy WJ, Obici L, et al. Design and rationale of the global phase 3 NEURO‐TTRansform study of antisense oligonucleotide AKCEA‐TTR‐L(Rx) (ION‐682884‐CS3) in hereditary transthyretin‐mediated amyloid polyneuropathy. Neurol Ther. 2021;10:375–389. doi: 10.1007/s40120-021-00235-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dyck PJB, Gonzalez‐Duarte A, Obici L, Polydefkis M, Wiesman JF, Antonio I, Litchy WJ, Dyck PJ. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS+7. J Neurol Sci. 2019;405:116424. doi: 10.1016/j.jns.2019.116424 [DOI] [PubMed] [Google Scholar]
- 24. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Krisen AV, Grogan M, Witteles R, Damy T, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016. doi: 10.1056/NEJMoa1805689 [DOI] [PubMed] [Google Scholar]
- 25. Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, Merlini G, Moreau P, Ronco P, Sanchorawala V, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th international symposium on amyloid and amyloidosis, Tours, France, 18‐22 April 2004. Am J Hematol. 2005;79:319–328. doi: 10.1002/ajh.20381 [DOI] [PubMed] [Google Scholar]
- 26. Kitaoka H, Izumi C, Izumiya Y, Inomata T, Ueda M, Kubo T, Koyama J, Sano M, Sekijima Y, Tahara N, et al. JCS 2020 guideline on diagnosis and treatment of cardiac amyloidosis. Circ J. 2020;84:1610–1671. doi: 10.1253/circj.CJ-20-0110 [DOI] [PubMed] [Google Scholar]
- 27. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135:1357–1377. doi: 10.1161/CIRCULATIONAHA.116.024438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, Kusek JW, Lente FV, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145:247–254. doi: 10.7326/0003-4819-145-4-200608150-00004 [DOI] [PubMed] [Google Scholar]
- 29. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinex‐Naharro A, Quata CC, Rezk T, Whelan CJ, Gonzalez‐Lopez E, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39:2799–2806. doi: 10.1093/eurheartj/ehx589 [DOI] [PubMed] [Google Scholar]
- 30. Law S, Petrie A, Chacko L, Cohen OC, Ravichandran S, Gilbertson JA, Rowczenio D, Wechalekar A, Martinez‐Naharro A, Lachmann HJ, et al. Disease progression in cardiac transthyretin amyloidosis is indicated by serial calculation of National Amyloidosis Centre transthyretin amyloidosis stage. ESC Heart Fail. 2020;7:3942–3949. doi: 10.1002/ehf2.12989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lang RM, Badano LP, Mor‐Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–270. doi: 10.1093/ehjci/jev014 [DOI] [PubMed] [Google Scholar]
- 32. Devereux RB, Reichek N. Echocardiographic determination of left ventricular mass in man. Anatomic validation of the method. Circulation. 1977;55:613–618. doi: 10.1161/01.CIR.55.4.613 [DOI] [PubMed] [Google Scholar]
- 33. Huang CC, Chen YH, Lin MS, Lin CH, Li HY, Chiu MJ, Chao CC, Wu YW, Chen YA, Lee JK, et al. Association of the recovery of objective abnormal cerebral perfusion with neurocognitive improvement after carotid revascularization. J Am Coll Cardiol. 2013;61:2503–2509. doi: 10.1016/j.jacc.2013.02.059 [DOI] [PubMed] [Google Scholar]
- 34. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2‐evidence base and standardized methods of imaging. Circ Cardiovasc Imaging. 2021;14:e000029. doi: 10.1161/HCI.0000000000000029 [DOI] [PubMed] [Google Scholar]
- 35. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2‐diagnostic criteria and appropriate utilization. Circ Cardiovasc Imaging. 2021;14:e000030. doi: 10.1161/HCI.0000000000000030 [DOI] [PubMed] [Google Scholar]
- 36. Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A. Transthyretin‐related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398–408. doi: 10.1038/nrcardio.2010.67 [DOI] [PubMed] [Google Scholar]
- 37. Castano A, DeLuca A, Weinberg R, Pozniakoff T, Blaner WS, Pirmohamed A, Bettencourt B, Gollob J, Karsten V, Vest JA, et al. Serial scanning with technetium pyrophosphate ((99m)Tc‐PYP) in advanced ATTR cardiac amyloidosis. J Nucl Cardiol. 2016;23:1355–1363. doi: 10.1007/s12350-015-0261-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh V, Falk R, Di Carli MF, Kijewski M, Rapezzi C, Dorbala S. State‐of‐the‐art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol. 2019;26:158–173. doi: 10.1007/s12350-018-01552-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ross JC, Hutt DF, Burniston M, Grigore SF, Fontana M, Page J, Hawkins PN, Gilvertson JA, Rowczenio D, Gillmore JD. The role of serial (99m)Tc‐DPD scintigraphy in monitoring cardiac transthyretin amyloidosis. Amyloid. 2022;29:38–49. doi: 10.1080/13506129.2021.1991302 [DOI] [PubMed] [Google Scholar]
- 40. Ionis Pharmaceuticals, Inc . CARDIO‐TTRansform: A Study to Evaluate the Efficacy and Safety of Eplontersen (Formerly Known as ION‐682884, IONIS‐TTR‐LRx and AKCEA‐TTR‐LRx) in Participants With Transthyretin‐Mediated Amyloid Cardiomyopathy (ATTR CM). ClinicalTrials.gov identifier: NCT04136171. Updated August 3, 2023. Accessed November 10, 2023. https://classic.clinicaltrials.gov/ct2/show/NCT04136171.
- 41. Chao HC, Liao YC, Liu YT, Guo YC, Chang FP, Lee YC, Lin KP. Clinical and genetic profiles of hereditary transthyretin amyloidosis in Taiwan. Ann Clin Transl Neurol. 2019;6:913–922. doi: 10.1002/acn3.778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du K, Li F, Wang H, Miao Y, Lv H, Zhang W, Wang Z, Yuan Y, Meng L. Hereditary transthyretin amyloidosis in mainland China: a unicentric retrospective study. Ann Clin Transl Neurol. 2021;8:831–841. doi: 10.1002/acn3.51328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Low SC, Tan CY, Md Sari NA, Ahmad‐Annuar A, Wong KT, Lin KP, Shahrizaila N, Tan CT, Goh KJ. Ala97Ser mutation is common among ethnic Chinese Malaysians with transthyretin familial amyloid polyneuropathy. Amyloid. 2019;26:7–8. doi: 10.1080/13506129.2019.1582479 [DOI] [PubMed] [Google Scholar]
- 44. Pasutharnchat N, Taychargumpoo C, Vorasettakarnkij Y, Amornvit J. Ala97Ser transthyretin amyloidosis‐associated polyneuropathy, clinical and neurophysiological profiles in a Thai cohort. BMC Neurol. 2021;21:206. doi: 10.1186/s12883-021-02243-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coelho T, Waddington Cruz M, Chao CC, Parman Y, Wixner J, Weiler M, Barroso FA, Dasgupta NR, Jung SW, Schneider E, et al. Characteristics of patients with hereditary transthyretin amyloidosis‐polyneuropathy (ATTRv‐PN) in NEURO‐TTRansform, an open‐label phase 3 study of eplontersen. Neurol Ther. 2023;12:267–287. doi: 10.1007/s40120-022-00414-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smiley DA, Einstein AJ, Mintz A, Shetty M, Chan N, Helmke ST, Bhatia K, Goldner K, Brannagan TH, Santana DD, et al. Gene silencing therapy in hereditary (variant) transthyretin cardiac amyloidosis: a puzzling case of decreasing pyrophosphate uptake on scintigraphy. Circ Cardiovasc Imaging. 2023;16:e015243. doi: 10.1161/CIRCIMAGING.123.015243 [DOI] [PMC free article] [PubMed] [Google Scholar]