

Abstract
Background
Increasing survival among patients with congenital heart disease (CHD) has recently been reported. However, the impact of Down syndrome (DS) in patients with CHD is still debated. We aimed to estimate survival in patients with CHD with versus without DS compared with matched controls from the general population without CHD or DS.
Methods and Results
We linked data from Swedish health registries to identify patients with CHD born between 1970 and 2017. Data from the Total Population Register were used to match each patient with CHD by sex and birth year with 8 controls without CHD or DS. A Cox proportional regression model was used to estimate mortality risk, and Kaplan–Meier curves were analyzed for the survival analysis. We identified 3285 patients with CHD‐DS, 64 529 patients with CHD without DS, and 26 128 matched controls. The mortality risk was 25.1 times higher (95% CI, 21.3–29.5) in patients with CHD‐DS versus controls. The mortality rate was 2 times higher (95% CI, 1.94–2.31) for patients with CHD with versus without DS. Lower mortality was found during the second versus first birth periods in patients with CHD‐DS compared with controls; hazard ratio: 46.8 (95% CI, 29.5–74.0) and 17.7 (95% CI, 12.8–24.42) in those born between 1970 and 1989 versus 1990 and 2017, respectively.
Conclusions
In this retrospective cohort study, the mortality risk among patients with CHD‐DS was 25 times higher compared with matched controls and 2 times higher compared with patients with CHD without DS. Survival was higher in patients with CHD‐DS born after versus before 1990, coinciding with the modern era of congenital heart care.
Keywords: congenital heart disease, Down syndrome, nationwide study, survival
Subject Categories: Epidemiology, Congenital Heart Disease, Mortality/Survival
Nonstandard Abbreviations and Acronyms
- DS
Down syndrome
- IR
incidence rate
Clinical Perspective.
What Is New?
The overall mortality risk in patients with congenital heart disease (CHD)–Down syndrome (DS) was 25 times higher compared with matched controls and 2 times higher compared with patients with CHD without DS.
Patients with CHD‐DS born in the later birth cohort (1990–2017) still had a higher risk of mortality compared with matched controls and patients with CHD without DS.
What Are the Clinical Implications?
The results of this study confirm increased survival among patients with CHD‐DS born after 1990 versus patients born before 1990, coinciding with the modern era of congenital heart care.
However, patients with CHD‐DS still have an increased risk of mortality compared with the general population and patients with CHD without DS.
Future studies are needed to determine the underlying causes for this excess risk and evaluate whether there is a need for better follow‐up for patients with CHD‐DS.
The birth prevalence of congenital heart disease (CHD) is 1% to 2% globally and is associated with several syndromes. 1 , 2 Previous reports have shown that half of the children with Down syndrome (DS) have associated CHD, and the most common types are atrioventricular septal defect, ventricular septal defect, and tetralogy of Fallot. 3 , 4 Trisomy 21, Down syndrome, comprises a complex of malformations, with a higher incidence of many other conditions that affect survival, such as respiratory diseases, leukemia, and early‐onset Alzheimer disease. 5
Generally, patients with complex CHD undergo at least 1 congenital heart surgery during early childhood to survive to adulthood. However, heart and lung machines for newborns were not fully developed until the mid‐1980s in developed countries, including Sweden. Before corrective heart surgery was available, or in a few cases, depending on the severity of CHD, a palliative procedure was performed. 6 Patients with DS were not highly prioritized for surgery previously, partly because the prognosis was considered poor; therefore, most patients with CHD‐DS did not undergo congenital heart surgery until the late 1980s. 7 This lack of surgery led to the development of pulmonary hypertension and Eisenmenger syndrome, which is 1 of the most complex conditions in CHD and which is associated with high morbidity and mortality during early adulthood. 8 Since the early 1990s, surgical heart treatment standards have changed, and surgical correction of heart defects, even for the most complex CHD, has become feasible. Therefore, long‐term outcomes in patients with CHD‐DS are of great interest. 9 The present study aimed to determine the survival trends in patients with CHD‐DS born between 1970 and 2017 in Sweden and estimate their mortality risk compared with age‐ and sex‐matched controls from the general population and with patients with CHD without DS.
Methods
All raw data from the Swedish National Board of Health and Welfare are available provided that ethical permission can be obtained from the Swedish Ethical Review Authority. For more information, please contact the corresponding author.
Data Source
This study used data from the Swedish National Patient Register and Cause of Death Register, which are held by the National Board of Health and Welfare in Sweden. The National Patient Register was created in 1964 and has had nationwide coverage since 1987. The register contains primary and secondary discharge diagnoses and surgical procedures for all hospital admissions, and since 2001, it also includes data on diagnosis from outpatient clinics. The Cause of Death Register contains information on deaths from 1961 onwards, and is complete since 1968 and updated yearly. The registers are regulated by law, and it is mandatory for hospitals and clinics to participate. All diagnoses are coded in accordance with the International Classification of Diseases, Eighth Revision (ICD‐8), International Classification of Diseases, Ninth Revision (ICD‐9), and International Classification of Diseases, Tenth Revision (ICD‐10).
Study Population
The Swedish National Patient Register and Cause of Death Register were used to identify all patients with CHD diagnoses who were born between January 1970 and December 2017. All patients with CHD with DS were then identified, and each patient was matched for sex and birth year with 8 controls without a diagnosis of CHD or DS from the Total Population Register in Sweden. The follow‐up data for the study population were obtained through the Swedish National Patient Register and Cause of Death Register from January 1970 until December 2017, or death.
Definitions
Patients with CHD were defined as individuals with at least 1 registered ICD code diagnosis of CHD between 1970 and 2017 from an outpatient visit, hospital discharge, or death certificate. The complexity of CHD was classified as complex and noncomplex, which were defined using the hierarchical classification first suggested by Botto et al 10 (Table S1).
The complex group comprised lesion groups 1 and 2, which were defined as patients with conotruncal (ie, tetralogy of Fallot, common arterial trunk, transposition of the great vessels, and aortopulmonary septal defect) and nonconotruncal (ie, endocardial cushion defects, hypoplastic left heart syndrome, and a common ventricle) defects. The noncomplex group comprised lesion groups 3, 4, 5, and 6, which were defined as patients with coarctation of the aorta, ventricular septal defect, atrial septal defect, and all other remaining CHD diagnoses, respectively.
Cardiovascular death was identified from the Cause of Death Register. Comorbidities registered before death or at the end of the study were identified from the National Patient Register as hypertension, diabetes, hyperlipidemia, atrial fibrillation, ischemic stroke, and myocardial infarction (Table S2).
Statistical Analysis
The main outcome was death. The baseline characteristics that were recorded were sex, complexity of CHD, and birth period. Descriptive statistics were used to analyze the characteristics of the study population. Numbers and percentages were used to present categorical data, and numerical data were presented as mean and SD or median and interquartile ranges.
The absolute risk of mortality was estimated as the incidence rate (IR), which was calculated by dividing the number of deaths by the total follow‐up time and reported as the number of events per 1000 person‐years with 95% CIs based on Poisson CI. Kaplan–Meier survival analysis was used to calculate survival probability with 95% CI and was performed for patients with CHD with DS, matched controls, and patients with CHD without DS. To compare the mortality risk, a Cox proportional hazard regression model was used to calculate the hazard ratio (HR) with 95% CI, with the control population as the reference group. Analyses were performed for the group as a whole and on the basis of the complexity of the lesion, sex, and the 2 birth periods (1970–1989 and 1990–2017). In addition, all patients with CHD and controls were divided into age groups (0–0.9, 1–9, 10–17, and ≥18 years), with separate calculations for each age interval. This was performed because of nonproportionality caused by the long follow‐up duration. All final models met the requirement of proportionality. P<0.05 was considered statistically significant, and all statistical analyses were performed with R software, version 4.2.0 (www.r‐project.org).
Ethical Approval
This study was approved by the Regional Ethics Review Board (Gbg 912‐16, T619‐18). The requirement to obtain informed consent for patients to participate was waived because this study used administrative data. All national personal identity numbers are replaced by a code by the National Board of Health and Welfare in Sweden; therefore, all data used in this study were anonymized.
Results
Baseline Characteristics of the Study Population
A total of 67 814 patients with CHD were identified; 3285 patients with CHD‐DS (4.8%), 64 529 patients with CHD without DS, and 26 128 matched controls were included in the present study (Figure S1).
The characteristics of the study population are presented in Table 1. Among the patients with CHD‐DS, 48.4% were female patients and 43.8% had a complex CHD. During a mean follow‐up of 16.5 (±12.5) years, 558 patients with CHD‐DS (17%), 5131 patients with CHD without DS (7.95%), and 198 controls (0.76%) died. The most common comorbidity in patients with CHD‐DS was diabetes, whereas hypertension, atrial fibrillation, and ischemic stroke were common in patients with CHD without DS. The baseline characteristics of the patients with CHD‐DS by birth period are presented in Table S3, and by birth year in Table S4. During the earlier birth period, 54.1% of the patients with CHD‐DS had a complex CHD compared with only 39.5% in the later birth period. We found that 155 patients with CHD‐DS (4.7%) within the complex lesion group had a univentricular heart defect diagnosis, and 47.1% of those died during the study period; however, most patients with CHD‐DS with a univentricular heart (55/155) who died were born in early birth period (1970–1989).
Table 1.
Characteristics of the Study Population
| Characteristic | Patients with CHD‐DS (n=3285) | Patients with CHD without DS (n=64 529) | Controls without DS or CHD (n=26 128) |
|---|---|---|---|
| Female sex | 1591 (48.4) | 32 050 (49.7) | 12 507 (47.9) |
| Year of birth, mean±SD | 1996.9±12.9 | 1999.6±12.8 | 1998.3±12.7 |
| Year of birth, median (IQR)* | 1999 (1987–2008) | 2003 (1991–2010) | 2001 (1990–2009) |
| Follow‐up, mean±SD, y | 16.5±12.5 | 16.0±12.6 | 192±12.6 |
| Follow‐up, median (IQR)*, y | 14.5 (5.8–25.5) | 12.8 (5.6–24.5) | 16.7 (8.7–27.7) |
| Birth period | |||
| 1970–1989 | 956 (29.1) | 14 737 (22.8) | 6460 (24.7) |
| 1990–2017 | 2329 (70.9) | 49 792 (77.2) | 19 668 (75.3) |
| Lesion complexity | |||
| Complex CHD | 1438 (43.8) | 7202 (11.2) | N/A |
| Noncomplex CHD | 1847 (56.2) | 57 327 (88.8) | N/A |
| Down syndrome | 3285 (100.0) | 0 (0.0) | 0 (0.0) |
| Comorbidities† | |||
| Hypertension | 29 (0.9) | 1237 (1.9) | 116 (0.4) |
| Diabetes | 62 (1.9) | 589 (0.9) | 190 (0.7) |
| Hyperlipidemia | 8 (0.2) | 209 (0.3) | 44 (0.2) |
| Atrial fibrillation | 29 (0.9) | 856 (1.3) | 26 (0.1) |
| Ischemic stroke | 29 (0.9) | 777 (1.2) | 22 (0.1) |
| Myocardial infarction | 11 (0.3) | 180 (0.3) | 4 (<0.1) |
| Deaths | |||
| Total | 558 (17.0) | 5131 (8.0) | 198 (0.8) |
| Cardiovascular death | 4 (0.1) | 73 (0.1) | 1 (<0.1) |
Data are given as number (percentage) unless otherwise indicated. CHD indicates congenital heart disease; DS, Down syndrome; IQR, interquartile range; and N/A, not applicable.
Continuous variable.
Comorbidities defined at the last registration before death or at the end of the study.
Mortality
The IR of mortality for the entire observed period was 10.29 per 1000 person‐years in patients with CHD‐DS compared with 4.98 per 1000 person‐years for patients with CHD without DS and 0.39 per 1000 person‐years for controls without DS or CHD (Table 2). Patients with CHD‐DS in the complex lesion group had a higher IR of mortality (15.97 per 1000 person‐years) compared with the noncomplex lesion group (5.81 per 1000 person‐years). The IR of mortality differed between the 2 birth periods. For 1970 to 1989, the IR was 15.16 per 1000 person‐years; and for 1990 to 2017, the IR decreased to 6.25 per 1000 person‐years for patients with CHD‐DS. Furthermore, the IR of mortality was highest during the first year of life for patients with CHD‐DS, then patients with CHD without DS, and controls; and it was highest for patients with CHD‐DS (79.09 per 1000 person‐years) compared with patients with CHD without DS (58.49 per 1000 person‐years) and with controls (2.93 per 1000 person‐years).
Table 2.
Incidence of Mortality for Patients With CHD and DS, Patients With CHD Without DS, and Matched Controls
| Variable | Patients with CHD‐DS | Patients with CHD without DS | Controls without DS or CHD | |||
|---|---|---|---|---|---|---|
| Deaths events, n | IR per 1000 person‐years (95% CI) | Deaths events, n | IR per 1000 person‐years (95% CI) | Deaths events, n | IR per 1000 person‐years (95% CI) | |
| All | 558 | 10.29 (9.46–11.18) | 5131 | 4.98 (4.84–5.12) | 198 | 0.39 (0.34–0.45) |
| Male sex | 270 | 9.98 (8.83–11.25) | 2776 | 5.31 (5.11–5.51) | 86 | 0.45 (0.37–0.54) |
| Female sex | 288 | 10.60 (9.41–11.90) | 2355 | 4.64 (4.45–4.83) | 112 | 0.34 (0.27–0.42) |
| Birth period | ||||||
| 1970–1989 | 373 | 15.16 (13.66–16.78) | 2875 | 6.33 (6.10–6.57) | 116 | 0.49 (0.40–0.58) |
| 1990–2017 | 185 | 6.25 (5.38–7.21) | 2256 | 3.91 (3.75–4.08) | 82 | 0.31 (0.25–0.39) |
| Lesion complexity | ||||||
| Complex CHD | 382 | 15.97 (14.41–17.66) | 2189 | 19.79 (18.97–20.63) | 112 | 0.46 (0.38–0.55) |
| Noncomplex CHD | 176 | 5.81 (4.98–6.73) | 2942 | 3.20 (3.08–3.32) | 86 | 0.34 (0.27–0.41) |
| Age group, y | ||||||
| 0–0.9 | 249 | 79.09 (69.57–89.55) | 3608 | 58.49 (56.60–60.43) | 76 | 2.93 (2.31–3.67) |
| 1–9 | 190 | 8.44 (7.28–9.73) | 798 | 1.82 (1.70–1.95) | 30 | 0.15 (0.10–0.21) |
| 10–17 | 34 | 2.53 (1.75–3.54) | 228 | 0.93 (0.82–1.06) | 7 | 0.06 (0.02–0.12) |
| ≥18 | 85 | 5.62 (4.49–6.94) | 497 | 1.73 (1.59–1.89) | 85 | 0.55 (0.44–0.68) |
CHD indicates congenital heart disease; DS, Down syndrome; and IR, incidence rate.
Survival Probability
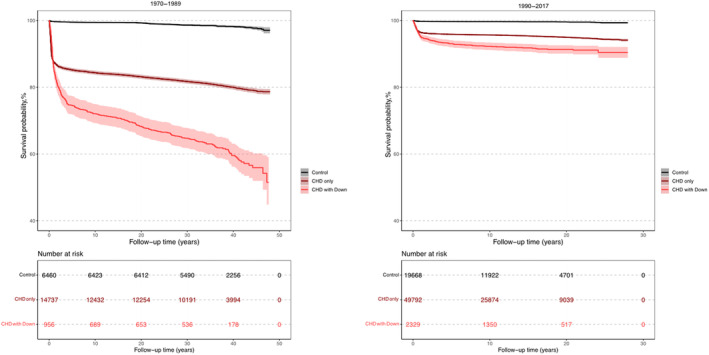
The survival probability of patients with CHD‐DS was markedly lower compared with patients with CHD without DS and matched controls (Figure 1). Patients born in the birth period 1970 to 1989 had a longer follow‐up time compared with patients born in the birth period 1990 to 2017 (Figure 2). The survival probability after the first years of life was much lower for both patients with CHD‐DS and patients with CHD without DS in the birth period 1970 to 1989 compared with the birth period 1990 to 2017. Survival probability by complexity for patients with CHD‐DS was higher compared with patients with CHD without DS in the complex group during the first 30 years of follow‐up (Figure S2). However, in the noncomplex group, patients with CHD‐DS had a lower survival probability compared with both controls and patients with CHD without DS. In patients with CHD‐DS, female patients had a slightly lower survival probability compared with male patients (Figure S3).
Figure 1. Kaplan–Meier survival curves of patients with congenital heart disease (CHD) and Down syndrome (DS), matched controls, and patients with CHD without DS.
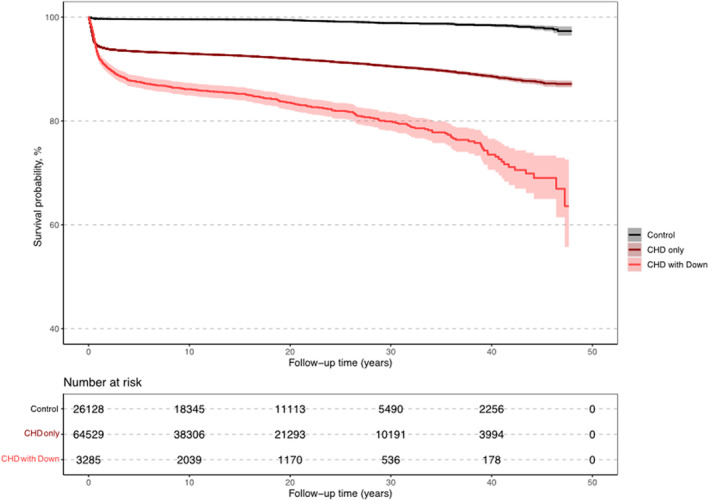
Figure 2. Kaplan–Meier survival curves of patients with congenital heart disease (CHD) and Down syndrome (DS), matched controls, and patients with CHD without DS by birth period.
Risk of Mortality
Patients with CHD‐DS had 25.11 times higher risk of mortality (95% CI, 21.3–29.5) compared with controls and 2.12 times higher risk of mortality (95% CI, 1.94–2.31) compared with patients with CHD without DS (Table 3). The risk of mortality was higher in female versus male patients with CHD‐DS. Patients with CHD‐DS with complex CHD had a lower mortality risk (HR, 0.80 [95% CI, 0.72–0.89]) compared with patients with CHD without DS. An overall decrease in mortality for patients with CHD‐DS was found between the 2 birth periods in all age groups (Tables S5 and S6). The risk of mortality during the first year of life was 46.8 (95% CI, 29.5–74.0) from 1970 to 1989 and 17.7 (95% CI, 12.8–24.42) from 1990 to 2017, compared with controls. The mortality risk was highest before adulthood in patients with CHD‐DS compared with controls. However, adult patients with CHD‐DS still had a 10.43 times higher risk of mortality (HR, 10.43 [95% CI, 7.72–14.10]) compared with controls. Compared with patients with CHD without DS, patients with CHD‐DS had the highest mortality risk in the 1‐ to 9‐year age group (HR, 4.70 [95% CI, 4.01–5.50]).
Table 3.
Risk of Mortality in Patients With CHD and DS Compared With Patients With CHD Without DS and Compared With Matched Controls, Overall and According to Sex, Birth Period, Complexity of Lesion, and Age Intervals
| HR for mortality in patients with CHD‐DS | ||||
|---|---|---|---|---|
| Variable | Patients with CHD without DS as a reference | 95% CI | Controls without CHD or DS as a reference | 95% CI |
| All | 2.12 | 1.94–2.31 | 25.11 | 21.35–29.54 |
| Male sex | 1.87 | 1.65–2.12 | 21.56 | 17.29–26.88 |
| Female sex | 2.42 | 2.14–2.74 | 29.81 | 23.42–37.94 |
| Birth period | ||||
| 1970–1989 | 2.09 | 1.88–2.33 | 28.29 | 22.95–34.86 |
| 1990–2017 | 1.72 | 1.48–2.00 | 19.68 | 15.17–25.52 |
| Lesion complexity | ||||
| Complex | 0.80 | 0.72–0.89 | 32.28 | 26.14–39.87 |
| Noncomplex | 1.85 | 1.59–2.15 | 17.27 | 13.34–22.35 |
| Age group, y | ||||
| 0–0.9 | 1.35 | 1.19–1.53 | 26.91 | 20.81–34.79 |
| 1–9 | 4.70 | 4.01–5.50 | 55.64 | 37.86–81.77 |
| 10–17 | 2.71 | 1.89–3.88 | 44.19 | 19.59–99.69 |
| ≥18 | 3.28 | 2.61–4.13 | 10.43 | 7.72–14.10 |
CHD indicates congenital heart disease; DS, Down syndrome; and HR, hazard ratio.
Discussion
In this retrospective register‐based cohort study, 1 of the main findings was that patients with CHD‐DS had a 25‐fold higher risk of mortality compared with matched controls. In addition, there was an improvement in survival in the later birth period (1990–2017) compared with the earlier cohort (1970–1989), particularly during the first years of life. This improvement in survival was not surprising and can be explained by better surgical care and treatment being prioritized to all patients with CHD, including patients with DS.
Surgical procedures for patients with DS have changed over time and have resulted in better care and prognosis for patients with DS and CHD. 7 A previous study based in the northeast of England showed improved 1‐year survival in patients with CHD‐DS when comparing the birth cohorts 1985 to 1995 and 1996 to 2006, similar to the results seen in our study. 11 However, in our study, the mortality risk was still considerably higher in patients with CHD‐DS compared with matched controls, which leads one to consider whether the elevated risk is related to CHD or DS diagnosis. To investigate this question, we performed a primary analysis comparing patients with CHD‐DS with unmatched patients with only CHD. The results showed that patients with CHD‐DS had a 2 times higher mortality risk compared with patients with CHD only. Although the analysis was limited because matching was not performed because of an insufficient number of patients with CHD without DS, the results suggest that patients with CHD‐DS have a worse prognosis compared with patients with CHD only. Further studies are needed to determine why there is a difference in mortality between these 2 groups. As patients with DS often have other related conditions, it is not unlikely that a DS diagnosis could be a negative prognostic factor for patients with CHD. Thus, it is not the presence of CHD or DS, but the combination of the 2, that has the most impact on the prognosis.
A Danish population‐based study on survival among people with DS reported declining mortality over time, likely because of improved treatment of CHD. 12 The study also reported that deaths among people with DS before the age of 20 years were most often CHD related, and deaths after the age of 20 years were most often attributable to respiratory diseases. Another study evaluating causes of death in patients with DS after CHD surgery did not find that DS was a predictor of CHD‐related mortality but did find that DS was a risk factor for shortened long‐term survival after CHD surgery. 13 This could suggest that coexistent conditions in patients with DS are the main factor affecting long‐term survival when the heart defect has been surgically corrected.
The second birth period in this study (1990–2017) comprised 27 years and was longer than the first birth period (1970–1989), which comprised 19 years. This may partly explain why there were more than twice as many patients with CHD‐DS in the second birth period (n=2329) compared with the first (n=956). Another possible factor causing this increase of patients with CHD‐DS over time could be that more patients with less severe CHD were diagnosed as diagnostic technology improved. This possibility is supported by the increased percentage of noncomplex CHDs over time, when comparing the 2 birth periods. From 1970 to 1989, 45.9% of the patients had noncomplex CHD compared with 60.5% from 1990 to 2017. Prenatal screening for CHD has become more frequent over time and makes it possible to detect heart defects before birth. This has led to higher numbers of terminated pregnancies for fetal anomaly, especially in cases with severe CHD. 14 Therefore, this is also a possible explanation for the decrease in complex CHDs over time. A recent Swedish study supports this theory as the study reported a changed spectrum of diagnoses and a lower risk of complex cardiac lesions among infants with DS and CHD over time. 15 Increasing maternal age and the increased use of antenatal diagnostics over time may also have affected the number of children born with DS. In Sweden's neighboring countries, Norway and Denmark, the incidence of DS has increased (in association with increasing maternal age) 16 or decreased (in association with antenatal diagnostics and increased termination rates) over time. 17 In Sweden, the number of children born with DS/1000 live births remained stable, despite increasing maternal age until 2015 to 2016, when it declined markedly. Thus, for most of the present study period, the birth prevalence of DS remained stable.
There were slightly more male than female patients with CHD‐DS included in this study, and female patients had a higher mortality rate compared with male patients. Patients with complex lesions had an almost 2 times higher mortality risk compared with patients with noncomplex lesions. A Danish study investigating the population with DS born between 1994 and 2009 using a similar classification of CHD as that in the present study showed similar results for mortality according to severity. 18 In addition, the study found a similar 5‐year mortality rate for patients with DS and nonsevere CHD and patients with DS only. This is an interesting observation as it was not possible to include a group of patients with DS only, in the present study. Ideally, future studies will match patients with DS only and patients with DS and CHD to evaluate the excess risk of mortality.
Our observation (Figure S2) that patients with CHD‐DS with complex malformations had lower mortality compared with patients with CHD without DS reflects the shortcoming of grouping cardiac malformations. Patients with CHD‐DS commonly have tetralogy of Fallot or atrioventricular septal defects, which are defects with a favorable prognosis once they are repaired. However, in the classification in this study, these patients were grouped and compared with those with more complex lesions, such as hypoplastic left heart syndrome or single ventricles, which are relatively more frequent among patients with complex CHD but rare in patients with CHD‐DS.
Patients with DS can be affected by many different medical conditions, and a CHD diagnosis is not the only factor that may affect long‐term outcomes and survival. A recent consensus article by Dimopoulos et al describes the diagnosis and management of cardiovascular disease in patients with DS. 9 It would be of interest in future studies to determine the main causes of comorbidities and death in surgically corrected patients with CHD‐DS and whether this group of patients requires better follow‐up.
Strengths and Limitations
This study had several strengths. First, the data were from nationwide registers, and each patient with a CHD‐DS diagnosis was matched for sex and birth year with 8 control subjects. Second, the registers used in this study, the Swedish National Patient Register and Cause of Death Register, require mandatory participation and long‐term follow‐up, which makes these registers suitable for population‐based studies. Health care in Sweden is also accessible for all citizens and mainly government funded. Therefore, the data are considered representative for Sweden and may be applicable to other countries with similar organization of the health care system. Furthermore, the study design made follow‐up with minimal loss possible.
Administrative data from outpatient clinics were first included in the Swedish National Patient Register in 2001; data before this year and from primary care sources were unavailable, which is a limitation of the study. As the National Patient Register was not nationwide until 1987 for inpatient care, there is a risk of underreported CHD cases (most likely among the less complex lesion groups) during the first birth period of 1970 to 1989. Another limitation is that the patients with CHD without DS were unmatched when compared with the patients with CHD‐DS, which may have led to underestimation of the increased risk of mortality in patients with CHD‐DS. Moreover, although the diagnoses from the Swedish National Register for inpatient care have been found to have high validity (85%–95% positive predictive value for most cardiovascular diagnoses), 19 there is no published formal validation of the CHD and DS diagnostic codes. However, a previous study of patients with CHD with myocardial infarction showed only slightly lower validity of the CHD diagnosis compared with that in the previous study. 20
Conclusions
The risk of mortality among patients with CHD‐DS was 25 times higher compared with matched controls, and it was 2 times higher compared with patients with CHD without DS. However, the survival rate increased in patients with CHD‐DS born after 1990 compared with those born before 1990, coinciding with the modern era of congenital heart care.
Sources of Funding
This work was funded by the Swedish state under an agreement between the Swedish government and county councils (the ALF agreement, grants 236611 and 917361), the Swedish Heart‐Lung Foundation (grant 20180644), and the Swedish Research Council (VRREG 2019‐00193 and VR 2018‐02527).
Disclosures
None.
Supporting information
Tables S1–S6
Figures S1–S3
Acknowledgments
We thank Jane Charbonneau, DVM, from Edanz (www.edanz.com/ac) for editing a draft of this article.
This article was sent to John L. Jefferies, MD, MPH, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.031392
For Sources of Funding and Disclosures, see page 8.
See Editorial by Ware.
References
- 1. van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos‐Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta‐analysis. J Am Coll Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025 [DOI] [PubMed] [Google Scholar]
- 2. Giang KW, Mandalenakis Z, Fedchenko M, Eriksson P, Rosengren A, Norman M, Hanseus K, Dellborg M. Congenital heart disease: changes in recorded birth prevalence and cardiac interventions over the past half‐century in Sweden. Eur J Prev Cardiol. 2023;30:169–176. doi: 10.1093/eurjpc/zwac227 [DOI] [PubMed] [Google Scholar]
- 3. Versacci P, Di Carlo D, Digilio MC, Marino B. Cardiovascular disease in down syndrome. Curr Opin Pediatr. 2018;30:616–622. doi: 10.1097/MOP.0000000000000661 [DOI] [PubMed] [Google Scholar]
- 4. Morris JK, Garne E, Wellesley D, Addor MC, Arriola L, Barisic I, Beres J, Bianchi F, Budd J, Dias CM, et al. Major congenital anomalies in babies born with Down syndrome: a EUROCAT population‐based registry study. Am J Med Genet A. 2014;164A:2979–2986. doi: 10.1002/ajmg.a.36780 [DOI] [PubMed] [Google Scholar]
- 5. Bull MJ. Down syndrome. N Engl J Med. 2020;382:2344–2352. doi: 10.1056/NEJMra1706537 [DOI] [PubMed] [Google Scholar]
- 6. Yuan SM, Jing H. Palliative procedures for congenital heart defects. Arch Cardiovasc Dis. 2009;102:549–557. doi: 10.1016/j.acvd.2009.04.011 [DOI] [PubMed] [Google Scholar]
- 7. Amark K, Sunnegardh J. The effect of changing attitudes to Down's syndrome in the management of complete atrioventricular septal defects. Arch Dis Child. 1999;81:151–154. doi: 10.1136/adc.81.2.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Korten MA, Helm PC, Abdul‐Khaliq H, Baumgartner H, Kececioglu D, Schlensak C, Bauer UM, Diller GP; Competence Network for Congenital Heart Defects Investigators . Eisenmenger syndrome and long‐term survival in patients with Down syndrome and congenital heart disease. Heart. 2016;102:1552–1557. doi: 10.1136/heartjnl-2016-309437 [DOI] [PubMed] [Google Scholar]
- 9. Dimopoulos K, Constantine A, Clift P, Condliffe R, Moledina S, Jansen K, Inuzuka R, Veldtman GR, Cua CL, Tay ELW, et al. Cardiovascular complications of Down syndrome: scoping review and expert consensus. Circulation. 2023;147:425–441. doi: 10.1161/CIRCULATIONAHA.122.059706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Botto LD, Lin AE, Riehle‐Colarusso T, Malik S, Correa A; National Birth Defects Prevention Study . Seeking causes: classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol. 2007;79:714–727. doi: 10.1002/bdra.20403 [DOI] [PubMed] [Google Scholar]
- 11. Irving CA, Chaudhari MP. Cardiovascular abnormalities in Down's syndrome: spectrum, management and survival over 22 years. Arch Dis Child. 2012;97:326–330. doi: 10.1136/adc.2010.210534 [DOI] [PubMed] [Google Scholar]
- 12. Zhu JL, Hasle H, Correa A, Schendel D, Friedman JM, Olsen J, Rasmussen SA. Survival among people with Down syndrome: a nationwide population‐based study in Denmark. Genet Med. 2013;15:64–69. doi: 10.1038/gim.2012.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peterson JK, Kochilas LK, Knight J, McCracken C, Thomas AS, Moller JH, Setty SP. Long‐term survival and causes of death in children with trisomy 21 after congenital heart surgery. J Pediatr. 2021;231:246–253.e3. doi: 10.1016/j.jpeds.2020.12.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tararbit K, Bui TT, Lelong N, Thieulin AC, Goffinet F, Khoshnood B. Clinical and socioeconomic predictors of pregnancy termination for fetuses with congenital heart defects: a population‐based evaluation. Prenat Diagn. 2013;33:179–186. doi: 10.1002/pd.4043 [DOI] [PubMed] [Google Scholar]
- 15. Bergstrom S, Carr H, Petersson G, Stephansson O, Bonamy AK, Dahlstrom A, Halvorsen CP, Johansson S. Trends in congenital heart defects in infants with Down syndrome. Pediatrics. 2016;138:138. doi: 10.1542/peds.2016-0123 [DOI] [PubMed] [Google Scholar]
- 16. Aasen HM, Solberg B, Stangenes KM, Nohr EA, Eggebo TM. Trisomy 21–incidence, diagnostics and pregnancy terminations 1999‐2018. Tidsskr Nor Laegeforen. 2021;141:141. doi: 10.4045/tidsskr.21.0221 [DOI] [PubMed] [Google Scholar]
- 17. Lou S, Petersen OB, Jorgensen FS, Lund ICB, Kjaergaard S; Danish Cytogenetic Central Registry Study G roup; Vogel I. National screening guidelines and developments in prenatal diagnoses and live births of Down syndrome in 1973‐2016 in Denmark. Acta Obstet Gynecol Scand. 2018;97:195–203. doi: 10.1111/aogs.13273 [DOI] [PubMed] [Google Scholar]
- 18. Brodwall K, Greve G, Leirgul E, Klungsoyr K, Holmstrom H, Vollset SE, Oyen N. The five‐year survival of children with Down syndrome in Norway 1994‐2009 differed by associated congenital heart defects and extracardiac malformations. Acta Paediatr. 2018;107:845–853. doi: 10.1111/apa.14223 [DOI] [PubMed] [Google Scholar]
- 19. Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, Heurgren M, Olausson PO. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450. doi: 10.1186/1471-2458-11-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fedchenko M, Mandalenakis Z, Hultsberg‐Olsson G, Dellborg H, Eriksson P, Dellborg M. Validation of myocardial infarction diagnosis in patients with congenital heart disease in Sweden. BMC Cardiovasc Disord. 2020;20:460. doi: 10.1186/s12872-020-01737-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S6
Figures S1–S3