

Abstract
Background
Obesity is accompanied by dysregulated inflammation, which can contribute to vasculometabolic complications including metabolic syndrome and atherosclerosis. Recently, clonal hematopoiesis of indeterminate potential (CHIP) has emerged as a risk factor for cardiovascular diseases. We aimed to determine how CHIP is related to immune cell function, systemic inflammation, and vasculometabolic complications in obese individuals.
Methods and Results
Two hundred ninety‐seven individuals with overweight and obesity, between the ages of 54 and 81 years, were recruited in a cross‐sectional study. Clonal hematopoiesis driver mutations (CHDMs) were identified with an ultrasensitive targeted assay. Assessment of carotid artery atherosclerosis was performed with ultrasound. Detailed immunological parameters, including cytokine production capacity of peripheral blood mononuclear cells, and targeted plasma proteomics analysis, were studied. Adipose tissue inflammation was determined in subcutaneous fat biopsies. Individuals with CHIP had higher concentrations of circulating IL (interleukin)‐6. Total number of leukocytes and neutrophils were higher in individuals with CHIP. In contrast, ex vivo cytokine production capacity of peripheral blood mononuclear cells was significantly lower in individuals with CHIP. Sex‐stratified analysis showed that men with CHDMs had significantly higher leukocyte and neutrophil counts, and ex vivo cytokine production capacity was lower in women with CHDMs. Surprisingly, the presence of atherosclerotic plaques was significantly lower in individuals with CHDMs. There was no relation between CHIP and metabolic syndrome.
Conclusions
In individuals with overweight or obesity, CHDMs are not associated with vasculometabolic complications, but rather with a lower presence of carotid plaques. CHDMs associate with increased circulating inflammatory markers and leukocyte numbers, but a lower peripheral blood mononuclear cell cytokine production capacity.
Keywords: atherosclerosis, clonal hematopoiesis, immunological parameters, obesity
Subject Categories: Atherosclerosis
Nonstandard Abbreviations and Acronyms
- CHDM
clonal hematopoiesis driver mutation
- CHIP
clonal hematopoiesis of indeterminate potential
- DNMT3A
DNA methyltransferase 3a
- TET2
tet methylcytosine dioxygenase 2
- VAF
variant allele frequency
Research Perspective.
What New Question Does This Study Raise?
In a cohort of subjects with overweight or obesity, using an ultrasensitive targeted assay, we detected clonal hematopoiesis driver mutations (CHDMs) in 28% of subjects and clonal hematopoiesis of indeterminate potential in 11%.
The presence of CHDMs was associated with higher circulating leukocytes, neutrophils, and interleukin‐6, but with a lower ex vivo peripheral blood mononuclear cell cytokine production capacity.
CHDM presence was not associated with metabolic syndrome or insulin resistance and was associated with a lower presence of carotid atherosclerotic plaques.
What Questions Should Be Addressed Next?
To investigate how CHDM presence affects monocyte versus macrophage cytokine production, both in the unstimulated and stimulated conditions.
To investigate in detail how CHDMs differentially affect the initiation, progression, and destabilization of atherosclerotic plaques, in particular in the presence of obesity.
The prevalence of overweight and obesity has increased globally in the past decades. This poses a great risk for cardiometabolic diseases and a significant burden on the health care system. 1 Obesity is associated with the development of atherosclerosis, the leading cause of cardiovascular diseases (CVD), via metabolic dysregulation and chronic low‐grade inflammation. Furthermore, obesity can induce activation of innate immune cells, such as monocytes, which are critical in the development of atherosclerotic CVD. 2 In patients with established atherosclerotic CVD, isolated circulating monocytes are characterized by a hyperresponsive phenotype. 3 , 4 , 5 In the arterial wall, monocyte‐derived macrophages play a key role in the pathophysiology of atherosclerosis. 6 Similarly, monocyte‐derived macrophages in the adipose tissue are important regulators of inflammation and insulin resistance. 7 , 8
Innate immune cell function is influenced by many factors, and a recently described mechanism is clonal hematopoiesis. Somatic mutations are common in highly proliferative tissues, including the hematopoietic stem and progenitor cells in the bone marrow. If, by chance, 1 of these mutations confers a selective survival or fitness advantage, it leads to the clonal expansion of that cell. 9 Recent advances in sequencing technologies demonstrated that clonal hematopoiesis is common among the aging population. 10 Clonal hematopoiesis driver mutations (CHDMs) are linked with an increased risk for hematological malignancies, 11 , 12 but multiple epidemiological studies also show that presence of CHDMs is associated with an increased risk of atherosclerotic CVD. 13
Common somatically mutated genes include DNMT3A (DNA methyltransferase 3a), TET2 (tet methylcytosine dioxygenase 2), ASXL1 (ASXL transcriptional regulator 1), JAK2 (Janus kinase 2), and TP53 (tumor protein p53). 9 Clonal hematopoiesis of indeterminate potential (CHIP) is defined as the presence of CHDMs with a variant allele frequency (VAF) ≥2% without clinical diagnosis of a hematological malignancy. 11 This cutoff is predominantly determined by the limitations of standard sequencing methods, including whole‐exome sequencing, frequently used to identify CHDMs. Recently, the 2% cutoff has been challenged, underscoring the potential clinical relevance of driver mutations with lower VAFs. 14
A few experimental preclinical studies suggest that CHDMs are involved in the metabolic and cardiovascular complications of obesity. For example, experimental studies in mice showed that TET2‐driven clonal hematopoiesis is causal to age‐ and obesity‐related insulin resistance. This is mediated by the activation of innate immune cells, in particular plaque macrophages, via NLRP3‐inflammasome driven IL (interleukin)‐1β production. 15 , 16 Furthermore, single‐cell RNA sequencing of unstimulated human monocytes from patients with heart failure revealed higher expression of inflammatory genes in individuals harboring DNMT3A mutations. In the general population, the presence of CHDM is associated with higher waist‐to‐hip ratio. 17 There are also indications for reverse causality, with atherosclerosis‐associated inflammation accelerating clonal hematopoiesis in experimental mouse models. 18 Using the exact same sequencing technique and gene panel as in our study, Andersson‐Assarsson et al recently reported an increase in time in obese individuals, but not in those who underwent bariatric surgery. 19
Here, we propose that clonal hematopoiesis is an important driver of inflammation in patients with obesity, predominantly associated with older age, and of the subsequent development of metabolic syndrome and atherosclerosis. We hypothesize that this is mediated by hyperinflammatory responsiveness of monocyte‐derived macrophages with CHDMs in the arterial wall and in the adipose tissue, which is also reflected by hyperresponsiveness in circulating monocytes. To this end, we identified a panel of candidate CHDMs with an ultrasensitive assay in a cohort of 297 human subjects with overweight and obesity. We associated CHDMs to parameters of metabolic and atherosclerotic complications, to parameters of systemic inflammation, as well as to the innate immune cell phenotype and function and adipose tissue inflammation. Because the regulation of inflammation in relation to metabolic syndrome is highly sex specific, we performed all analyses stratified by sex. 20 Lastly, we performed a DNMT3A (DNA methyltransferase 3a)‐specific analysis, because this gene harbored the most CHDMs in our cohort, and it was shown to play a causal role in driving atherosclerosis in murine models. 21
Methods
Study Subjects and Clinical Measurements
This cross‐sectional single center cohort study was part of the Human Functional Genomics Project, a large scale project including various cohorts designed to understand the effects of human genetic variation and diversity of gut microbiome on the immune system during health and disease. The 300 Obesity cohort is 1 of the cohorts belonging to the Human Functional Genomics Project, specifically aimed at understanding the role of inflammation in individuals with obesity. A cohort of 302 individuals (the 300 Obesity cohort) with a body mass index >27 kg/m2, between the ages of 54 and 82 years, consisting mostly of Western European ancestry, was recruited in the Radboud University Medical Center from 2014 to 2016. The research protocol was approved by the Radboud University Ethical Committee (nr. 46846.091.13), and all subjects gave written informed consent. The study protocol was performed in accordance with the 1975 Declaration of Helsinki.
For an extensive description of the cohort and measurements, please see ter Horst et al. 20
Lipid‐lowering medication was stopped 4 weeks before measurements. Blood samples were drawn between 8:30 am and 12:00 pm in the morning after an overnight fast. The blood samples were collected into EDTA vacutainers, and the following laboratory procedures took place immediately after collection. Total blood cell counts were performed in fresh EDTA blood samples using a Sysmex Hematoanalyzer XE5000. Plasma samples were stored at −80 °C until further measurement. Blood glucose, triglycerides, total cholesterol, high‐density lipoprotein cholesterol, and low‐density lipoprotein cholesterol were determined by standard laboratory protocols. Systolic and diastolic blood pressure were measured after 30 minutes of supine rest.
Metabolic syndrome was defined according to the National Cholesterol Education Program ATP III criteria. 22 Participants having at least 3 of the following characteristics were determined to have metabolic syndrome:
Abdominal obesity: waist circumference ≥102 cm in men or ≥88 cm in women.
Triglycerides: serum triglycerides ≥150 mg/dL (1.7 mmol/L) or treatment with lipid‐lowering drugs.
High‐density lipoprotein cholesterol: serum high‐density lipoprotein cholesterol <40 mg/dL (1 mmol/L) in men or <50 mg/dL (1.3 mmol/L) in women.
Blood pressure: ≥130/85 mm Hg or treatment for hypertension.
Fasting plasma glucose: ≥100 mg/dL (5.6 mmol/L) or treatment for elevated blood glucose.
Insulin resistance was determined according to the Homeostasis Model Assessment model. 23
Cardiovascular Measurements
Carotid artery measurements were performed by ultrasound, as described previously. 24 Carotid intima‐media thickness (IMT) was determined in the proximal 1‐cm straight portion of the carotid artery at 90o, 120o, and 180o angles for 6 heart beats. The presence and thickness of plaques were assessed in the common carotid, internal carotid, or external carotid artery or at the carotid bulbus. A focal thickening of the carotid wall of at least 1.5× IMT or an IMT >1.5 mm was termed as plaque presence. Pulse wave velocity and augmentation index adjusted for heart rate were measured to assess arterial stiffness by SphygmoCor (ATCOR Medical) under standard operating protocol.
Adipose Tissue Analysis
Detailed methodology on adipose tissue measurements was previously described. 20 Briefly, subcutaneous abdominal adipose tissue samples were obtained by needle biopsies under local anesthesia (biopsy needle 14 gauge, 100 Sterican, B/Braun, Ref: 4665473). The biopsies were performed 6‐ to 10‐cm lateral to the umbilicus in the right lower quadrant.
Adipocyte size, presence of macrophages, and crown‐like structures were determined by fluorescent microscopy (Zeiss Axiphot) using hematoxylin and eosin in combination with CD68 staining. From 4 microscopic fields of view, adipocyte cell diameters were measured and indicated as area and Feret minimal diameter (the minimal diameter of each cell). Adipose tissue sections were stained with a CD68‐monoclonal antibody (Serotec; Oxford) to identify macrophages. The percentage of macrophages was determined as the total number of macrophages divided by the total number of adipocytes from 15 random microscopic fields of view. A crown‐like structure was defined as an adipocyte surrounded by at least 3 macrophages. 25 , 26
Additionally, total RNA was isolated from adipose tissue samples by Trizol (Invitrogen) extraction, followed by cDNA library preparation (iScript cDNA synthesis kit; Bio‐Rad). mRNA expression levels were determined by real‐time polymerase chain reaction and normalized to housekeeping gene (RPL37A [ribosomal protein L37a]) expression. Primer sequences can be found in Table S1.
Identification of CHDMs
CHDMs were identified in the whole blood by an ultrasensitive assay, as previously described. 19 , 27 In short, 300 single‐molecule molecular inversion probes were designed against a selection of well‐known hot spots of a panel of 24 clonal hematopoiesis driver genes (Table S2). The DNMT3A gene, which contains the most driver mutations, was entirely covered. 28 We used the single‐molecule molecular inversion probe panel designed in 2017, 29 which was also used in recent studies 19 , 27 (Table S3). For each sample, 2 technical polymerase chain reaction replicates were run; thereafter, 2 independent data processing strategies and a quality control step were performed. Variant allele frequencies were calculated using samtools mpileup 30 (Table S4).
All identified CHDMs were validated in publicly available data sets. For individuals with >1 CHDM, the CHDM with the highest VAF was included in the analysis. We divided all identified CHDMs into: low VAF for CHDMs with a VAF <2% and high VAF for CHDMs with a VAF ≥2% in accordance with the current CHIP cutoff.
From the 302 individuals of the cohort, whole blood samples of 3 individuals could not be obtained, and the samples from 2 individuals did not pass the quality control for sequencing. Therefore, 5 individuals were completely excluded from this study, corresponding to 297 individuals for the final analysis.
Peripheral Blood Mononuclear Cell Isolation and Ex Vivo Stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated with differential density centrifugation over Ficoll‐Paque (GE Healthcare). PBMCs were then washed 3 times by centrifugation with phosphate saline buffer. Isolated PBMCs were resuspended in Dutch modified Roswell Park Memorial Institute 1640 medium (Invitrogen) supplemented with 50 μg/mL gentamicin (Centrafarm), 2 mmol/L GlutaMAX, and 1 mmol/L pyruvate (Life Technologies). There were 0.5×106 cells per well stimulated for 24 hours in 96‐well round‐bottom plates (Greiner) at 37 °C and 5% CO2. Stimuli used include Roswell Park Memorial Institute medium as the negative control, lipopolysaccharide (Sigma‐Aldrich, Escherichia coli serotype 055:B5, further purified as described 31 ) at low (1 ng/mL) and high concentrations (100 ng/mL), and Pam3Cys (1 μg/mL) (EMC Microcollections, L2000). Supernatants were collected after 24 hours and stored at −20 °C until measurements were performed.
Lipopolysaccharide and Pam3Cys are agonists of TLR4 (toll‐like receptor 4) and TLR2 (toll‐like receptor 2), respectively. These TLRs are relevant receptors in the context of atherosclerosis and adipose tissue inflammation, because several damage‐associated molecular patterns and pathogen‐associated molecular patterns in these microenvironments stimulate these receptors. 32 In addition, these TLR agonists are chosen based on our previous work, in which we showed that lipopolysaccharide and Pam3Cys stimulation of PBMCs can accurately identify monocytes with a hyperinflammatory (trained) phenotype. 33 , 34 We capture the response of monocytes by measuring monocyte‐specific cytokines that can be released upon 24‐hour stimulation when adaptive immune response cannot be initiated yet.
Cytokine Measurements
Cytokine concentrations upon stimulation of PBMCs and in plasma were measured with commercially available ELISA kits according to instructions supplied by the manufacturer. For detailed information on the ELISA kits used, please refer to Table S5.
Proteomic Profiling
The concentrations of 177 inflammatory proteins from the Olink Cardiovascular II and Inflammatory panels were measured using the previously described proximity extension assays technology (Olink Bioscience AB, Uppsala, Sweden). 35 Quality control was ensured using internal extension control and interplate control. The data are presented in Normalized Protein eXpression values, an arbitrary unit on log2 scale.
Statistical Analysis
Distribution of data was assessed with the Shapiro‐Wilk test. Normally distributed data are shown as mean±SD. If the data did not follow a normal distribution, they are shown as median and interquartile range. Each group (all CHDM, high VAF, and low VAF) was independently compared with the subjects without CHDM (no‐CHDM group) with the Student t test or Mann‐Whitney U test when appropriate. Spearman correlation was used to determine the association between VAF and various clinical and immunological parameters. The same statistical methodology was applied for the sex‐specific analyses. P<0.05 is considered statistically significant and is indicated with an asterisk in tables. All statistical analyses were performed in R version 4.1.1 (R Core Team). Part of the data is publicly available on the website of Human Functional Genomics Project (bbmri.nl), and the rest is available upon reasonable request to the Human Functional Genomics Project committee.
Results
The study population included 297 individuals with a median age of 67 years (interquartile 1 and 3, 63–71 years) and body mass index of 30 kg/m2 (28–32 kg/m2). Men represented 55% of the cohort. Fifty‐four percent of the cohort met the National Cholesterol Education Program Adult Treatment Panel III criteria of metabolic syndrome, and approximately half of the participants had carotid atherosclerotic plaques as measured by ultrasound. Hypertension was diagnosed in 60% of the study population, whereas diabetes was present in 12%. Median total cholesterol concentration was 6.3 mmol/L and triglyceride concentration was 1.61 mmol/L. Twenty‐seven percent of the cohort used lipid‐lowering drugs. Detailed baseline characteristics of the study population are presented in Table 1.
Table 1.
Baseline Characteristics of the Study Cohort (n=297)
| Parameter | Median (IQR1–IQR3) or % |
|---|---|
| Clinical data | |
| Sex, men, % | 55% |
| Age, y | 67 (63–71) |
| BMI, kg/m2 | 30 (28–32) |
| Waist‐to‐hip ratio | 1 (0.9–1) |
| Systolic blood pressure, mm Hg | 130 (119–139) |
| Diastolic blood pressure, mm Hg | 80 (74–86) |
| Heart rate, bpm | 61 (55–67) |
| Hypertension, % | 60% |
| Plaque presence, % | 53% |
| Diabetes, % | 12% |
| Metabolic syndrome, NCEP ATP III criteria, % | 54% |
| Pack‐years | 16.1 (8.5–31) |
| Laboratory data | |
| Creatinine, μmol/L | 80 (67–91) |
| Glucose, mmol/L | 5.4 (5–6) |
| Urate, mmol/L | 0.4 (0.3–0.4) |
| Total cholesterol, mmol/L | 6.3 (5.6–7) |
| Triglycerides, mmol/L | 1.6 (1.3–2.1) |
| HDL cholesterol, mmol/L | 1.3 (1.1–1.5) |
| Non‐HDL cholesterol, mmol/L | 4.9 (4.2–5.7) |
| LDL cholesterol, mmol/L | 4.1 (3.5–4.7) |
| Concomitant medication | |
| Antihypertensive drugs, % | 45% |
| Antidiabetic drugs, % | 9% |
| Lipid lowering drugs, % | 27% |
Data are shown as median (IQR1–IQR3) and percentage where appropriate. BMI indicates body mass index; HDL, high‐density lipoprotein; IQR, interquartile range; LDL, low‐density lipoprotein cholesterol; and NCEP ATP III, National Cholesterol Education Program Adult Treatment Panel III.
CHDM Prevalence and Characteristics
One hundred ten candidate CHDMs were identified in 85 individuals; 62 individuals (21% of the cohort) carried a single CHDM, and 23 individuals (8%) had >1 CHDM (Figure [A]). The VAF of all CHDMs ranged from 0.01% to 34.5%, with a mean of 3.3% and median of 1.1%.
Figure . Characterization of candidate CHDMs identified in our study.
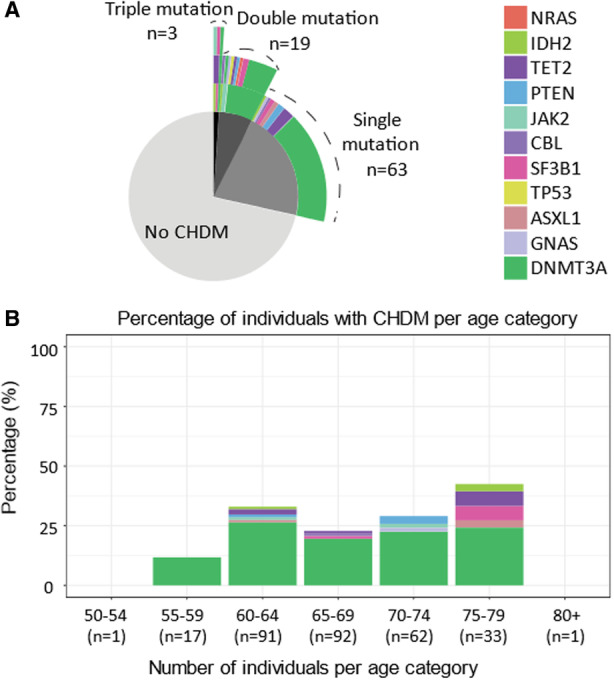
A, Pie chart indicating number of individuals without CHDMs, with a single mutation, double mutations, and triple mutations. Top rings indicate genes affected from the mutations. For >1 mutation, carrying individuals second and third mutations are displayed as multiple rings on top of each other. B, Percentage of individuals with CHDMs per age category and gene affected. Age range of the entire cohort is depicted, and n refers to total number of individuals in that age category. CHDM indicates clonal hematopoiesis driver mutation.
In 33 individuals, we identified CHIP (ie, CHDM with a VAF ≥2%), and 52 individuals had CHDMs with VAF <2%. We identified CHDMs in 11 individual genes. Mutations in DNMT3A (73%) and TET2 (7%) genes were the most common in the entire cohort (Figure [A]). Additionally, DNMT3A mutations were the most common across all age groups (Figure [B]).
Relationship Between CHDM and Clinical Characteristics
We did not observe statistically significant differences in baseline characteristics between subjects with and without CHDMs, apart from the finding that individuals with CHDMs with VAF <2% presented with higher heart rate (Table 2). Although there was no significant association between sex and presence of CHDMs, we observed a trend toward more women in the all‐CHDM group (P=0.054). Sex‐specific analysis revealed that men with CHIP were significantly older than those without CHDMs, and men with CHDMs with VAF <2% had a significantly higher heart rate (Table S6). In women, there were no differences in baseline characteristics in any of the groups (Table S7). We did not observe a correlation between presence or size of the clones and any of the other baseline parameters. When restricting the analyses only to CDHMs in DNMT3A, we observed significantly more women among the carriers of CHDMs in this gene (data not shown).
Table 2.
Baseline Characteristics According to CHDM Status
| Characteristic | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| Age, y | 66 (63–70) | 68 (63–72) | 67 (63–74) | 68 (63–71) | 0.1 |
| Sex, men, % | 58 | 46 | 45 | 46 | |
| Weight, kg | 88 (82–97) | 89 (81–99) | 89 (80–98) | 87 (82–99) | −0.1 |
| Height, cm | 172 (165–178) | 171 (164–176) | 172 (164–176) | 171 (164–177) | −0.1 |
| BMI, kg/m2 | 30 (28–32) | 30 (28–32) | 30 (28–32) | 30 (28–32) | 0 |
| Creatinine, μmol/L | 81 (67–92) | 77 (68–89) | 76 (70–88) | 78 (67–89) | −0.2 |
| Glucose, mmol/L | 5.4 (5–6) | 5.4 (5–6) | 5 (5–6) | 5.4 (5–6) | 0.004 |
| Total cholesterol, mmol/L | 6 (6–7) | 6 (5–7) | 6 (5–7) | 6 (6–7) | −0.1 |
| Triglycerides, mmol/L | 1.6 (1.3–2.2) | 1.5 (1.3–2) | 1.5 (1.3–2) | 1.5 (1.3–2) | −0.023 |
| Heart rate, bpm | 61 (55–67) | 64 (58–71) | 62 (59–67) | 66 (57–73) * | −0.2 |
| Antihypertensives, % | 47 | 40 | 52 | 33 | |
| Lipid lowering drugs, % | 28 | 25 | 18 | 29 | |
| Antidiabetic drugs, % | 9 | 7 | 6 | 8 |
All data are given as median (interquartile ranges 1–3) unless indicated otherwise. BMI indicates body mass index; CHDM, clonal hematopoiesis driver mutation; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
Association of CHDM With Parameters of Metabolic Dysregulation
We investigated the association between presence of CHDMs and presence of metabolic syndrome and its individual components, and parameters related to insulin resistance and diabetes type 2. We did not observe a higher prevalence of metabolic syndrome, its individual components, or diabetes in individuals with clonal hematopoiesis (Table 3).
Table 3.
Parameters of Metabolic Dysregulation (Metabolic Syndrome and Diabetes) According to CHDM Status
| Parameter | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| Diabetes, % | 14 | 9 | 9 | 10 | |
| Metabolic syndrome, % | 55 | 52 | 55 | 50 | |
| Liver fat, mg/cm3 | 0.058 (0.024–0.12) | 0.062 (0.029–0.16) | 0.059 (0.034–0.14) | 0.066 (0.017–0.17) | −0.01 |
| Waist circumference, cm | 106 (100–110) | 105 (100–112) | 106 (100–110) | 104 (100–113) | −0.09 |
| Hip circumference, cm | 110 (106–114) | 111 (107–114) | 109 (106–114) | 111 (109–114) | −0.09 |
| HDL cholesterol, mmol/L | 1.27 (1.1–1.51) | 1.32 (1.14–1.51) | 1.32 (1.11–1.51) | 1.33 (1.14–1.52) | 0.02 |
| LDL cholesterol, mmol/L | 4.15 (3.57–4.72) | 4.06 (3.29–4.64) | 4.04 (3.18–4.58) | 4.07 (3.36–4.81) | −0.07 |
| Systolic blood pressure, mm Hg | 129 (118–138) | 130 (123–140) | 129 (123–140) | 131 (123–140) | −0.05 |
| Diastolic blood pressure, mm Hg | 80 (74–85) | 80 (72–87) | 76 (72–87) | 80 (72–87) | −0.08 |
| HOMA‐IR | 7 (4–11) | 7 (4–16) | 5 (4–12) | 8 (4–16) | ‐0.13 |
All data are given as median (interquartile ranges 1–3) unless indicated otherwise. CHDM indicates clonal hematopoiesis driver mutation; HDL, high‐density lipoprotein; HOMA‐IR, Homeostatic Model Assessment of Insulin Resistance; LDL, low‐density lipoprotein; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
Association Between Adipose Tissue Inflammation and Presence of CHDMs
Adipose tissue is an important site for generation of various cytokines and adipokines and is strongly associated with the development of cardiometabolic complications of obesity. Therefore, we characterized adipose tissue biopsies by immunohistochemistry and quantitative polymerase chain reaction and explored whether adipose tissue inflammation is more severe in individuals with CHDMs.
Although we did not identify a higher number of CD68+ macrophages by immunohistochemistry, we observed significantly higher expression of CD68, adiponectin, and tumor necrosis factor in individuals with CHDMs with VAF <2% measured with quantitative polymerase chain reaction (Table S8).
Relationship Between CHDM and Parameters of Atherosclerosis
To explore whether the presence of CHDM correlates with the presence of atherosclerosis in subjects with obesity, we investigated IMT and carotid plaque presence and characteristics in relation to CHDM. We found that the presence of carotid plaques was lower in individuals with any CHDM and CHDMs with VAF <2% compared with individuals without CHDMs. The same results were obtained for only the CHDMs in the DNMT3A gene (data not shown). Individuals with CHIP mutations had a significantly higher augmentation index, suggestive of an increased systemic arterial stiffness (Table 4).
Table 4.
cIMT and Parameters of Carotid Plaques and Measures of Arterial Stiffness According to CHDM Status of the Participants
| Parameter | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| cIMT, μm | 791 (703–884) | 753 (691–859) | 815 (713–882) | 745 (692–832) | 0.11 |
| Carotid plaque presence, % | 59 | 38* | 48 | 31* | |
| No. of plaques | 1 (1–2) | 1 (1–2) | 2 (1–2) | 1 (1–2) | 0.16 |
| Maximum plaque thickness, mm | 2.2 (1.8–2.8) | 2.65 (1.98–3.2) | 2.75 (2–3.2) | 2.55 (1.87–2.97) | 0.17 |
| Maximum stenosis, % | 0.29 (0.23–0.39) | 0.3 (0.25–0.39) | 0.37 (0.27–0.4) | 0.29 (0.22–0.34) | 0.25 |
| PWV, m/s | 9.2 (8.4–10.7) | 9.4 (8.1–10.4) | 9.9 (8.2–10.4) | 9.3 (8.1–10.9) | −0.03 |
| Augmentation index, % | 25.7 (20.5–30.3) | 27.5 (21.6–33.3) | 29.9 (24.6–34.8)* | 25.3 (19–32.1) | 0.31 |
| History of CVD, % | 16 | 14 | 15 | 13 |
All data are given as median (interquartile ranges 1–3) unless indicated otherwise. CHDM indicates clonal hematopoiesis driver mutation; cIMT, carotid intima‐media thickness; CVD, cardiovascular disease; PWV, pulse wave velocity; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
Leukocyte Number and Function and Presence of CHDMs
The absolute number of leukocytes and thrombocytes was measured in whole blood. We identified a significantly higher number of total leukocytes, and specifically neutrophils, in individuals with CHDMs and in CHIP carriers. DNMT3A CHDM carriers also had significantly higher number of total leukocytes (data not shown). There were no differences in monocyte or lymphocyte counts (Table 5). Sex‐specific analysis revealed men with CHIP had significantly higher neutrophil‐to‐lymphocyte ratio (Table S9). In contrast, there was no difference in any leukocyte count in women (Table S10).
Table 5.
Leukocyte Numbers and Differentiation, and Thrombocyte Numbers Separated According to CHDM Status of the Participants
| Variable | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| Leukocytes 109/L | 5.7 (5–6.6) | 6.1 (5.3–7.4)* | 6.2 (5.5–7.6)* | 5.9 (5.3–7.4) | 0.15 |
| Neutrophils 109/L | 3.2 (2.6–3.7) | 3.4 (2.7–4.4)* | 3.6 (2.9–4.5)* | 3.2 (2.7–4.3) | 0.13 |
| Lymphocytes 109/L | 1.8 (1.5–2.2) | 1.9 (1.6–2.3) | 1.9 (1.6–2.2) | 1.9 (1.6–2.3) | 0.1 |
| Monocytes 109/L | 0.5 (0.4–0.6) | 0.5 (0.4–0.6) | 0.5 (0.4–0.6) | 0.5 (0.4–0.6) | 0.1 |
| Eosinophils 109/L | 0.16 (0.09–0.21) | 0.15 (0.1–0.29) | 0.13 (0.09–0.29) | 0.16 (0.11–0.22) | 0.002 |
| Basophils 109/L | 0.03 (0.02–0.04) | 0.03 (0.02–0.04) | 0.03 (0.02–0.05) | 0.03 (0.02–0.04) | ‐0.03 |
| Thrombocytes 109/L | 225 (194–262) | 231 (196–266) | 226 (203–281) | 231 (196–264) | 0.18 |
| NLR | 1.72 (1.31–2.18) | 1.8 (1.41–2.25) | 1.81 (1.5–2.42) | 1.78 (1.32–2.08) | 0.021 |
All data are given as median (interquartile ranges 1–3). CHDM indicates clonal hematopoiesis driver mutation; NLR, neutrophil‐to‐lymphocyte ratio; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
Ex vivo cytokine production capacity of PBMCs can be used as a measure of inflammatory responsiveness, and this has been shown to be higher in patients with established coronary heart disease. 3 , 5 Therefore, we characterized proinflammatory cytokine production capacity of PBMCs upon stimulation with Pam3Cys and lipopolysaccharide at different concentrations. Interestingly, we observed significantly lower production of IL‐1β upon Pam3Cys stimulation in individuals with all CHDMs and CHDMs with VAF <2. Likewise, stimulation with lipopolysaccharide (both concentrations) led to significantly lower production of IL‐6 in individuals with CHDMs, and CHIP carriers had significantly lower IL‐6 upon stimulation with high concentration of lipopolysaccharide. Lastly, stimulation with Pam3Cys resulted in significantly less IL‐6 production in all groups compared with individuals without any CHDMs (Table 6). Interestingly, the lower production of these cytokines in individuals with CHDMs was solely seen in women and not in men (Tables S11 and S12). When restricting the analyses to CHDMs in DNMT3A, we observed similarly lower ex vivo cytokine production capacity seen in the entire cohort (data not shown).
Table 6.
Ex Vivo Cytokine Production Capacity of Peripheral Blood Mononuclear Cells Separated According to CHDM Status of the Participants
| Variable | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| Lipopolysaccharide (1 ng/mL) IL‐1β (pg/mL) | 1075 (599–1936) | 998 (564–1710) | 1036 (564–2013) | 905 (561–1399) | 0.05 |
| Lipopolysaccharide (100 ng/mL) IL‐1β (pg/mL) | 2476 (1544–3713) | 2210 (1245–3375) | 2703 (1280–3274) | 1823 (1240–3396) | 0.01 |
| Pam3Cys (1 μg/mL) IL‐1β (pg/mL) | 280 (111–640) | 174 (77–474)* | 163 (53–569) | 180 (82–424)* | −0.004 |
| Lipopolysaccharide (1 ng/mL) IL‐6 (pg/mL) | 5750 (3278–8968) | 4850 (2969–6547)* | 4464 (2989–6547) | 5318 (2963–6449) | −0.06 |
| Lipopolysaccharide (100 ng/mL) IL‐6 (pg/mL) | 8322 (5206–12 536) | 7167 (4279–10 797)* | 6062 (3606–9402)* | 7410 (4611–11 114) | −0.09 |
| Pam3Cys (1 μg/mL) IL‐6 (pg/mL) | 3975 (1759–6545) | 2010 (1021–4548) * | 2010 (811–4548)* | 2034 (1364–4627)* | −0.05 |
All data are given as median (interquartile ranges 1–3). CHDM indicates clonal hematopoiesis driver mutation; IL, interleukin; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
Relationship Between CHDMs and Circulating Cytokines and Adipokines
To determine the association between systemic inflammation and presence of CHDMs, we measured a selection of circulating cytokines and adipokines in plasma. We observed significantly higher concentrations of circulating IL‐6 in individuals with CHIP. We identified a trend toward higher circulating IL‐1β concentration in individuals with CHDMs with VAF <2%, although this did not reach statistical significance (P=0.053). We did not observe statistically significant differences in high‐sensitivity C‐reactive protein concentrations. Additionally, individuals with CHDMs with VAF <2% had significantly higher concentrations of resistin in circulation (Table 7). We did not observe associations for any other circulating marker. There was no sex‐specific association between circulating markers and the presence of CHDMs (Tables S13 and S14).
Table 7.
Circulating Cytokine and Adipokine Concentrations Separated According to CHDM Status of the Participants
| Variable | No CHDM (n=212) | All CHDM (n=85) | High VAF (n=33) | Low VAF (n=52) | Correlation with VAF |
|---|---|---|---|---|---|
| IL‐6, pg/mL | 2.4 (1.7–3.4) | 2.6 (1.7–4.1) | 2.9 (1.9–4.4) * | 2.4 (1.6–3.4) | 0.18 |
| IL‐1β, pg/mL | 0.06 (0.06–0.11) | 0.06 (0.06–0.12) | 0.06 (0.06–0.13) | 0.07 (0.06–0.11)† | ‐0.048 |
| IL‐18, pg/mL | 304 (227–490) | 288 (216–484) | 320 (221–455) | 285 (210–505) | 0.024 |
| IL‐18bp, ng/mL | 17.2 (14.1–21) | 16.1 (13.3–19.2) | 16 (13–17.8) | 16.2 (14.1–20) | ‐0.14 |
| hsCRP, μg/mL | 1.9 (0.9–3.3) | 2 (1.2–3.3) | 1.9 (0.9–3.9) | 2 (1.4–3.3) | 0.083 |
| AAT, mg/mL | 0.9 (0.6–1.7) | 0.9 (0.6–1.4) | 0.9 (0.6–1.4) | 0.9 (0.6–1.4) | 0.019 |
| Resistin, ng/mL | 10.6 (8.2–13.5) | 11.5 (9.2–15) | 11.6 (8.7–13.1) | 11.4 (9.5–16.2)* | ‐0.095 |
| Leptin, ng/mL | 15.8 (9.7–29.5) | 18.1 (11.1–35.5) | 17.2 (9.3–25.5) | 19.1 (11.6–36.6) | ‐0.028 |
| Adiponectin, μg/mL | 4.3 (2.8–6) | 4.3 (3.1–5.7) | 4.3 (3.4–5.2) | 4.3 (3.1–6.4) | 0.15 |
All data are given as median (interquartile ranges 1–3). AAT indicates α‐1 antitrypsin; CHDM, clonal hematopoiesis driver mutation; hsCRP, high sensitivity C‐reactive protein; IL, interleukin; IL‐18bp, IL‐18 binding protein; and VAF, variant allele frequency.
Indicates P<0.05 compared with the no‐CHDM group. Correlation is indicated with Spearman correlation coefficient.
P=0.053.
To further investigate the association between circulating proteins and CHDM status, we performed a targeted proteomics approach with Olink panels Cardiovascular II and Inflammation. Table S15 shows the list of proteins that were significantly different in individuals with CHDMs. However, significance was lost after correction for multiple testing. Figure S1 shows volcano plots indicating all proteins that were significantly higher or lower in the participants with CHDMs.
Discussion
In this cross‐sectional study of older individuals with overweight or obesity, we investigated the presence of clonal hematopoiesis driver mutations in relation to a wide range of clinical and immunological parameters. We hypothesized that the presence of CHDMs predisposes to the development of metabolic syndrome and atherosclerosis and that this is mediated by immune cell activation and systemic inflammation. Approximately 28% of the individuals had a CHDM. We showed that individuals with CHDMs had higher numbers of leukocytes and neutrophils and a higher circulating IL‐6 concentration. In contrast, the ex vivo cytokine production capacity of PBMCs from individuals with CHDMs appeared to be lower compared with those without. On a clinical level, the presence of CHDMs of CHIP was not associated with the presence of metabolic syndrome or atherosclerosis. Rather, the presence of carotid plaques was significantly lower in individuals with CHDMs. Lastly, we identified that several of the associations with CHDMs were sex specific.
Acquisition of somatic mutations is a hallmark of aging, and obesity is known to accelerate the pace of aging. 36 Systemic inflammation and activation of the immune system drives the development of metabolic and atherosclerotic complications in obesity. 7 Thus, clonal hematopoiesis may be pivotal in the association between obesity and atherosclerotic cardiovascular disease in aging individuals.
Several epidemiological studies identified the most common clonal hematopoiesis driver mutations to be in DNMT3A and TET2 genes. 10 , 13 Although a VAF ≥2% has been used traditionally to distinguish CHIP, recent advances in sequencing methodologies with superior sequencing depth have revealed that smaller clone sizes can also be associated with cardiovascular disease. 37 Recent work by Assmus et al argued for mutation‐specific cutoff values, 1.15% for DNMT3A and 0.73% for TET2, to predict all‐cause mortality in patients with heart failure. 14 Given the relevance of clones with smaller sizes, we included them in our analysis.
We observed a trend toward more women having CHDMs in this population. This finding prompted us to perform a sex‐specific analysis. Men with CHIP were significantly older than those without CHDMs. However, women with or without CHIP were the same age. Because women on average live longer than men, the clone growth might be delayed. Additionally, it has been shown that men generally have shorter telomere lengths, a trait associated with clonal hematopoiesis. 38 , 39
We hypothesized that in individuals with overweight and obesity, CHDMs would predispose to metabolic syndrome and to atherosclerosis due to increased monocytes responsiveness and increased systemic inflammation. We observed higher leukocyte and neutrophil numbers in individuals with CHDMs, in particular when the VAF was ≥2% (ie, in the presence of CHIP). In addition, the circulating IL‐6 concentration was higher in individuals with CHIP, confirming previous findings. 40 A previous genetic study showed that the increased CVD risk associated with CHIP is abrogated in individuals with a genetically determined reduced IL‐6 signaling, 41 although this was not confirmed in a larger study. 28 In contrast to the higher circulating IL‐6, we did not find a higher cytokine production capacity in PBMCs of individuals with CHDMs; rather, we observed a lower production capacity for IL‐6 and IL‐1β in women with CHDMs. This contrasts the experimental finding that TET2‐deficient mouse macrophages had increased cytokine production capacity. 16 Furthermore, single‐cell RNA sequencing of unstimulated human monocytes from patients with heart failure revealed increased expression of inflammatory genes in individuals harboring DNMT3A mutations. 37 These discrepant findings might indicate a differential effect of CHDMs on the inflammatory phenotype of monocytes versus macrophages or on baseline inflammatory gene expression versus protein production upon stimulation. A comparable differential inflammatory phenotype was observed between monocytes and macrophages of patients with primary aldosteronism, with a higher stimulated cytokine response in macrophages but not in monocytes. 24 One potential explanation is that the circulating PBMCs might have developed tolerance by the continuous exposure to higher concentrations of cytokines, thus impairing the ex vivo cytokine production capacity upon stimulation. Interestingly, the lower cytokine production capacity in the presence of CHDMs is exclusively observed in women in the sex‐stratified analysis.
In contrast to our hypothesis, we did not observe more atherosclerosis in individuals with CHDMs, as assessed with carotid ultrasound measuring to IMT and atherosclerotic plaques. Surprisingly, the presence of carotid plaques was even significantly lower in these individuals. Of note, the effect of CHDMs or CHIP on nonsymptomatic atherosclerotic plaques has never been studied before. Various studies showed a strong association of CHIP with the occurrence of cardiovascular events, which are mostly triggered by destabilization of atherosclerotic plaques and subsequent thrombus formation. 10 , 14 Thus, our data suggest that clonal hematopoiesis does not facilitate the initial development of atherosclerotic plaques, but rather the destabilization of these plaques. In line with this, atherosclerotic plaque development starts as early as in the second or third decade of life, whereas clonal hematopoiesis is primarily seen in individuals >55 years of age. 6 , 11
Finally, there was no association between CHDMs or CHIP and markers of metabolic complications of obesity, including metabolic syndrome and its individual components, liver fat, and insulin resistance. Similarly, we did not observe increased adipose tissue inflammation in individuals with CHDMs or CHIP.
DNMT3A mutations are unequivocally the most common drivers of clonal hematopoiesis identified in several cohorts including ours. Recently, mechanistic studies in mice identified DNMT3A mutations to be causally linked to atherosclerosis. 21 Therefore, we also performed all analyses restricted to the CHDMs in DNMT3A. We recapitulated the majority of our findings from the main cohort, with an even stronger lower ex vivo cytokine production capacity, as well as lower carotid plaque presence. There were significantly more women among DNMT3A carriers, as previously observed. 42 Absolute number of leukocytes was significantly higher in DNMT3A carriers.
A limitation of our study is the limited sample size of our cohort. This prevented us from studying the effects of individual genes harboring clonal hematopoiesis driver mutations. A second potential limitation is the sequencing approach used in identification of CHDMs. Although the use of single‐molecule molecular inversion probes allowed for ultrasensitive detection of small clones, the probes were designed against the majority of well‐known clonal hematopoiesis hotspots, except for DNMT3A, which was covered entirely. Thus, we cannot exclude the possibility that unknown drivers may be located outside the targets included in our assay. Furthermore, our cohort mainly consists of individuals with Western European origin. Therefore, our findings might not be generalized to diverse ethnic backgrounds.
A significant advantage of our study is that we performed extensive phenotyping of metabolic and atherosclerotic clinical parameters, as well as inflammatory and immune parameters. To the best of our knowledge, this study is the first to explore the association between CHDMs and the presence of nonsymptomatic atherosclerotic plaques and immune cell function in individuals with overweight or obesity. In addition, the use of single‐molecule molecular inversion probes allowed us to identify CHDMs with a low variant allele frequency.
In conclusion, we showed that in individuals with overweight or obesity, the presence of CHDMs is associated with higher circulating leukocyte and neutrophil numbers, and higher IL‐6 concentration, yet with an impaired cytokine production capacity of isolated PBMCs in women. Because multiple factors are analyzed in each section, we cannot entirely eliminate potential false‐positive findings. Therefore, our findings need to be validated in independent cohorts, preferentially with increased cohort size. Additionally, studies combining the differential assessment of monocyte and macrophage phenotypes, at both unstimulated and stimulated states, are needed to understand our observation of lower PBMC‐cytokine production capacity in CHDM carriers. Furthermore, we found no association between CHDMs and the presence of metabolic syndrome or carotid atherosclerotic plaques, supporting the concept that clonal hematopoiesis does not affect atherosclerosis formation per se, but might trigger plaque destabilization and the subsequent occurrence of cardiovascular events.
Sources of Funding
N.P.R., M.G.N., and L.A.B.J. received a CVON grant from the Dutch Cardiovascular Alliance and Dutch Heart Foundation (CVON2018‐27; IN CONTROL II). N.P.R. was the recipient of a grant of the ERA‐CVD Joint Transnational Call 2018 supported by the Dutch Heart Foundation (JTC2018, project MEMORY; 2018T093). S.B. is supported by the Dutch Heart Foundation (2018T028). M.G.N. was supported by an ERC Advanced Grant (#833247) and a Spinoza grant of the Netherlands Organization for Scientific Research.
Disclosures
None.
Supporting information
Data S1
Tables S2–S4
Acknowledgments
The authors thank the investigators (I.C.L. van den Munckhof, R. ter Horst, K. Schraa, M. Jaeger, H. Lemmers, and H. Dijkstra) who performed the inclusion of the study participants and executed laboratory work.
Preprint posted on MedRxiv July 08, 2023. doi: https://doi.org/10.1101/2023.07.07.23292396.
This article was sent to Tiffany M. Powell‐Wiley, MD, MPH, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.031665
For Sources of Funding and Disclosures, see page 10.
References
- 1. González‐Muniesa P, Mártinez‐González M‐A, Hu FB, Després J‐P, Matsuzawa Y, Loos RJF, Moreno LA, Bray GA, Martinez JA. Obesity. Nat Rev Dis Primers. 2017;3:17034. doi: 10.1038/nrdp.2017.34 [DOI] [PubMed] [Google Scholar]
- 2. Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel‐Latif A, Smyth SS, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835. doi: 10.1016/j.cmet.2014.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bekkering S, van den Munckhof I, Nielen T, Lamfers E, Dinarello C, Rutten J, de Graaf J, Joosten LA, Netea MG, Gomes ME, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. 2016;254:228–236. doi: 10.1016/j.atherosclerosis.2016.10.019 [DOI] [PubMed] [Google Scholar]
- 4. Noz MP, Bekkering S, Groh L, Nielen TM, Lamfers EJ, Schlitzer A, El Messaoudi S, van Royen N, Huys EH, Preijers FW, et al. Reprogramming of bone marrow myeloid progenitor cells in patients with severe coronary artery disease. elife. 2020;9:e60939. doi: 10.7554/eLife.60939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354. doi: 10.1084/jem.20150900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–533. doi: 10.1038/s41586-021-03392-8 [DOI] [PubMed] [Google Scholar]
- 7. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55:31–55. doi: 10.1016/j.immuni.2021.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. 2020;126:1549–1564. doi: 10.1161/circresaha.119.315896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jan M, Ebert BL, Jaiswal S. Clonal hematopoiesis. Semin Hematol. 2017;54:43–50. doi: 10.1053/j.seminhematol.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sano S, Wang Y, Walsh K. Clonal hematopoiesis and its impact on cardiovascular disease. Circ J. 2018;83:2–11. doi: 10.1253/circj.CJ-18-0871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17:137–144. doi: 10.1038/s41569-019-0247-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, Rasper T, Abou‐El‐Ardat K, Herrmann E, Berkowitsch A, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A‐ and TET2‐driver gene mutations. Eur Heart J. 2020;42:257–265. doi: 10.1093/eurheartj/ehaa845 [DOI] [PubMed] [Google Scholar]
- 15. Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana‐Huete V, Ferrer‐Pérez A, Matesanz N, Herrero‐Cervera A, Sano S, et al. TET2‐loss‐of‐function‐driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. 2020;33:108326. doi: 10.1016/j.celrep.2020.108326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pasupuleti SK, Ramdas B, Burns SS, Palam LR, Kanumuri R, Kumar R, Pandhiri TR, Dave UP, Yellapu NK, Zhou X, et al. Obesity‐induced inflammation exacerbates clonal hematopoiesis. J Clin Invest. 2023;133:e163968. doi: 10.1172/JCI163968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, Cheek D, Iwamoto Y, Schloss MJ, Vandoorne K, et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. 2021;184:1348–1361.e1322. doi: 10.1016/j.cell.2021.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andersson‐Assarsson JC, van Deuren RC, Kristensson FM, Steehouwer M, Sjöholm K, Svensson PA, Pieterse M, Gilissen C, Taube M, Jacobson P, et al. Evolution of age‐related mutation‐driven clonal haematopoiesis over 20 years is associated with metabolic dysfunction in obesity. EBioMedicine. 2023;92:104621. doi: 10.1016/j.ebiom.2023.104621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rt H, ICLvd M, Schraa K, Aguirre‐Gamboa R, Jaeger M, Smeekens SP, Brand T, Lemmers H, Dijkstra H, Galesloot TE, et al. Sex‐specific regulation of inflammation and metabolic syndrome in obesity. Arterioscler Thromb Vasc Biol. 2020;40:1787–1800. doi: 10.1161/ATVBAHA.120.314508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rauch PJ, Gopakumar J, Silver AJ, Nachun D, Ahmad H, McConkey M, Nakao T, Bosse M, Rentz T, Vivanco Gonzalez N, et al. Loss‐of‐function mutations in Dnmt3a and Tet2 lead to accelerated atherosclerosis and concordant macrophage phenotypes. Nat Cardiovasc Res. 2023;2:805–818. doi: 10.1038/s44161-023-00326-7 [DOI] [PubMed] [Google Scholar]
- 22. Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation. 2002;106:3143–3421. doi: 10.1161/circ.106.25.3143 [DOI] [PubMed] [Google Scholar]
- 23. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/bf00280883 [DOI] [PubMed] [Google Scholar]
- 24. van der Heijden C, Ter Horst R, van den Munckhof ICL, Schraa K, de Graaf J, Joosten LAB, Danser AHJ, Netea MG, Deinum J, Rutten J, et al. Vasculometabolic and inflammatory effects of aldosterone in obesity. J Clin Endocrinol Metab. 2020;105:2719–2731. doi: 10.1210/clinem/dgaa356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cossins BC, van den Munckhof I, Rutten JHW, van der Graaf M, Stienstra R, Joosten LAB, Netea MG, Li Y, Riksen NP. Sex‐specific association between adipose tissue inflammation and vascular and metabolic complications of obesity. J Clin Endocrinol Metabol. 2023;108:2537–2549. doi: 10.1210/clinem/dgad193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Geng J, Zhang X, Prabhu S, Shahoei SH, Nelson ER, Swanson KS, Anastasio MA, Smith AM. 3D microscopy and deep learning reveal the heterogeneity of crown‐like structure microenvironments in intact adipose tissue. Sci Adv. 2021;7:eabe2480. doi: 10.1126/sciadv.abe2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Heijden WA, van Deuren RC, van de Wijer L, van den Munckhof ICL, Steehouwer M, Riksen NP, Netea MG, de Mast Q, Vandekerckhove L, de Voer RM, et al. Clonal hematopoiesis is associated with low CD4 nadir and increased residual HIV transcriptional activity in virally suppressed individuals with HIV. J Infect Dis. 2021;225:1339–1347. doi: 10.1093/infdis/jiab419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kessler MD, Damask A, O'Keeffe S, Banerjee N, Li D, Watanabe K, Marketta A, Van Meter M, Semrau S, Horowitz J, et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature. 2022;612:301–309. doi: 10.1038/s41586-022-05448-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Acuna‐Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney L, Veltman JA, Gilissen C, Hoischen A. Ultra‐sensitive sequencing identifies high prevalence of clonal hematopoiesis‐associated mutations throughout adult life. Am J Hum Genet. 2017;101:50–64. doi: 10.1016/j.ajhg.2017.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deuren RCV. CHMIP‐RsCh‐PIPELINE. Github; 2021. [Google Scholar]
- 31. Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll‐like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618 [DOI] [PubMed] [Google Scholar]
- 32. Zimmer S, Grebe A, Latz E. Danger signaling in atherosclerosis. Circ Res. 2015;116:323–340. doi: 10.1161/circresaha.116.301135 [DOI] [PubMed] [Google Scholar]
- 33. Bekkering S, Stiekema LCA, Bernelot Moens S, Verweij SL, Novakovic B, Prange K, Versloot M, Roeters van Lennep JE, Stunnenberg H, de Winther M, et al. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. 2019;30:1–2. doi: 10.1016/j.cmet.2019.05.014 [DOI] [PubMed] [Google Scholar]
- 34. van der Heijden C, Groh L, Keating ST, Kaffa C, Noz MP, Kersten S, van Herwaarden AE, Hoischen A, Joosten LA, Timmers HJ, et al. Catecholamines induce trained immunity in monocytes in vitro and in vivo. Circ Res. 2020;127:269–283. doi: 10.1161/circresaha.119.315800 [DOI] [PubMed] [Google Scholar]
- 35. Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, et al. Homogenous 96‐Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9:e95192. doi: 10.1371/journal.pone.0095192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carlsson LMS, Sjöholm K, Jacobson P, Andersson‐Assarsson JC, Svensson P‐A, Taube M, Carlsson B, Peltonen M. Life expectancy after bariatric surgery in the Swedish obese subjects study. N Engl J Med. 2020;383:1535–1543. doi: 10.1056/NEJMoa2002449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abplanalp WT, Cremer S, John D, Hoffmann J, Schuhmacher B, Merten M, Rieger MA, Vasa‐Nicotera M, Zeiher AM, Dimmeler S. Clonal hematopoiesis–driver DNMT3A mutations alter immune cells in heart failure. Circ Res. 2021;128:216–228. doi: 10.1161/CIRCRESAHA.120.317104 [DOI] [PubMed] [Google Scholar]
- 38. Aviv A, Levy D. Hemothelium, clonal hematopoiesis of indeterminate potential, and atherosclerosis. Circulation. 2019;139:7–9. doi: 10.1161/circulationaha.118.038434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andreu‐Sánchez S, Aubert G, Ripoll‐Cladellas A, Henkelman S, Zhernakova DV, Sinha T, Kurilshikov A, Cenit MC, Jan Bonder M, Franke L, et al. Genetic, parental and lifestyle factors influence telomere length. Commun Biol. 2022;5:565. doi: 10.1038/s42003-022-03521-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cook EK, Izukawa T, Young S, Rosen G, Jamali M, Zhang L, Johnson D, Bain E, Hilland J, Ferrone CK, et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019;3:2482–2486. doi: 10.1182/bloodadvances.2018024729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131. doi: 10.1161/circulationaha.119.044362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamphuis P, van Zeventer IA, de Graaf AO, Salzbrunn JB, van Bergen M, Dinmohamed AG, van der Reijden BA, Schuringa JJ, Jansen JH, Huls G. Sex differences in the spectrum of clonal hematopoiesis. Hemasphere. 2023;7:e832. doi: 10.1097/hs9.0000000000000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S2–S4