

Abstract
Monotargeting anticancer agents suffer from resistance and target nonspecificity concerns, which can be tackled with a multitargeting approach. The combined treatment with HDAC inhibitors and PPARγ agonists has displayed potential antitumor effects. Based on these observations, this work involves design and synthesis of molecules that can simultaneously target PPARγ and HDAC. Several out of 25 compounds inhibited HDAC4, and six compounds acted as dual-targeting agents. Compound 7i was the most potent, with activity toward PPARγ EC50 = 0.245 μM and HDAC4 IC50 = 1.1 μM. Additionally, compounds 7c and 7i were cytotoxic to CCRF-CEM cells (CC50 = 2.8 and 9.6 μM, respectively), induced apoptosis, and caused DNA fragmentation. Furthermore, compound 7c modulated the expression of c-Myc, cleaved caspase-3, and caused in vivo tumor regression in CCRF-CEM tumor xenografts. Thus, this study provides a basis for the rational design of dual/multitargeting agents that could be developed further as anticancer therapeutics.
Graphical Abstract
INTRODUCTION
Cancer is the most prevalent disease worldwide and is regulated by genetic and epigenetic mechanisms. The major challenge in anticancer drug discovery is to bring about a therapeutic approach capable of attenuating malignant cells without damaging normal cells. Thus, there exist two fundamentals of a drug, viz., target specificity and potency. The clinical effectiveness of recent monotargeted drugs are transitory because of unexpected resistance and target nonspecificity due to tumor heterogeneity.1,2 To overcome these concerns, an alternative strategy of multitargeting can be applied to hit multiple cancer hallmarks and achieve the desired pharmacological efficiency with reduced detrimental effects such as drug–drug interactions, unforeseen side-effects, and poor patient compliance.3–6 Moreover, in a multitarget approach, the concept of “magic bullets” exist—which are single agents with target specificity and high potency.7
In the development and progression of several malignancies, epigenetics contributes significantly. Tumor cell activation is effectively regulated by epigenetic mechanisms which facilitate escape from chemotherapy; thus, the recent focus of anticancer drug discovery has been directed to epigenetic targets.8,9 There are numerous enzymes involved in epigenetic regulation; for example, histone deacetylases (HDACs) are indispensable in regulation of gene transcription and cellular homeostasis events.10–13 Also, aberrant expression of HDACs has been linked to a variety of solid and hematological malignancies, neurological disorders, and inflammation.14 There are various FDA-approved HDAC inhibitors such as SAHA, belinostat, panobinostat, and romidepsin, as well as various agents under clinical studies.15 However, the use of these inhibitors as a monotherapy against solid tumors has shown limited efficacy; thus, the development of HDAC inhibitors with multitarget specificity has been studied extensively.16–22
PPARs (peroxisome proliferator-activated receptors) are ligand-activated transcription factors that play a key role in the regulation of a large number of genes whose products are directly or indirectly involved in glucose homeostasis and lipid metabolism. The subtype PPARγ is the most widely studied and acts as a key factor in various metabolic processes playing an important role in the regulation of insulin tissue sensitivity and in the management of glucose and lipid uptake and storage.23 PPARγ is the receptor of well-known antidiabetic insulin sensitizer drugs rosiglitazone and pioglitazone, which belong to the thiazolidinedione (TZD) class.24 Apart from its role in the cardiovascular system, PPARγ is vital to cancer cell growth regulation. A large body of evidence shows that agonistic PPARγ ligands exert antitumorigenic effects against a variety of neoplastic cell types, both in vivo and in vitro.25–28 For instance, efatutazone is indicated in thyroid29–31 and nonsmall cell lung cancers;32 additionally, a phase I clinical study demonstrated acceptable tolerance in patients with advanced malignancies,33 and a phase II clinical trial is ongoing.34 Though there are undesirable side effects associated with full agonist activity,35 there exist PPARγ partial agonists endowed with reduced side effects.36–41 For example, in the preclinical setting, PPARγ partial agonist balaglitazone displayed a better safety profile than full agonists.34,42,43 Another partial agonist netoglitazone (MCC-555) has one-tenth the affinity to PPARγ as rosiglitazone, has lesser side effects,44 and is significantly more potent.45 Thus, the search for novel PPARγ partial agonists with structural diversity and safe biological profiles remains an attractive therapeutic strategy to circumvent cancer.
Recent reports suggested that combination treatment with HDAC inhibitors and PPARγ agonists increased cytotoxic effects in a synergistic/additive manner against various cancer cell lines, resulting in proliferation arrest and apoptosis. In some cases, even low doses of a PPARγ ligand in combination with a weak HDAC inhibitor resulted in more profound growth arrest than treatment with either drug alone.46–48
On the basis of this literature, a series of 25 novel TZD-based naphthylidene derivatives were rationally designed and successfully synthesized in pursuit of a multitarget drug approach and simultaneous targeting of HDAC/PPARγ. Biological evaluation of compounds revealed their dual-targeting ability by partial transactivation of PPARγ and selective inhibition of HDAC4. To our knowledge, this is the first ever attempt to develop agents which simultaneously target HDACs and PPARγ, two highly desirable targets in cancer drug discovery. An investigation of the antiproliferative capacity of this series of compounds revealed uncompromised cytotoxicity in a panel of hematological and solid cancer cells, induction of apoptosis, and in vivo tumor regression in CCRF-CEM (T-cell leukemia) xenografts.
RESULTS AND DISCUSSION
Rationale of Designing PPARγ and HDAC Dual-Targeting Analogues.
The main challenge in designing dual HDAC inhibitors and PPARγ agonists was achieving a unique framework which satisfied the binding requirements of both targets. In our previous reports, a distinct pharmacophoric drug-design was used to individually achieve both classes of compounds.49–53 Thus, on the basis of our experience with both targets, a multitargeting strategy was conceptualized in consideration of HDAC and PPARγ pharmacophore properties.
Traditional (full) PPARγ agonists consist of a hydrophobic tail linked to a central aryl ring, which is further linked through another small methylene bridge (linker 2) to the pharmacophoric TZD head (Figure 1). Examples are antidiabetic drugs pioglitazone and rosiglitazone. However, partial PPARγ agonists display a broader range of structural diversity, some being structurally similar to traditional agonists (e.g., netoglitazone), while others have an entirely different structural framework. The major distinguishing feature between full and partial PPARγ agonists is their binding pattern at the active site, which has been reviewed well in the literature. A key structural difference between partial agonist netoglitazone and traditional glitazones is the central naphthalene ring in place of a phenyl ring, suggesting a major contribution of naphthalene in shifting the molecule from full to partial agonist.
Figure 1.
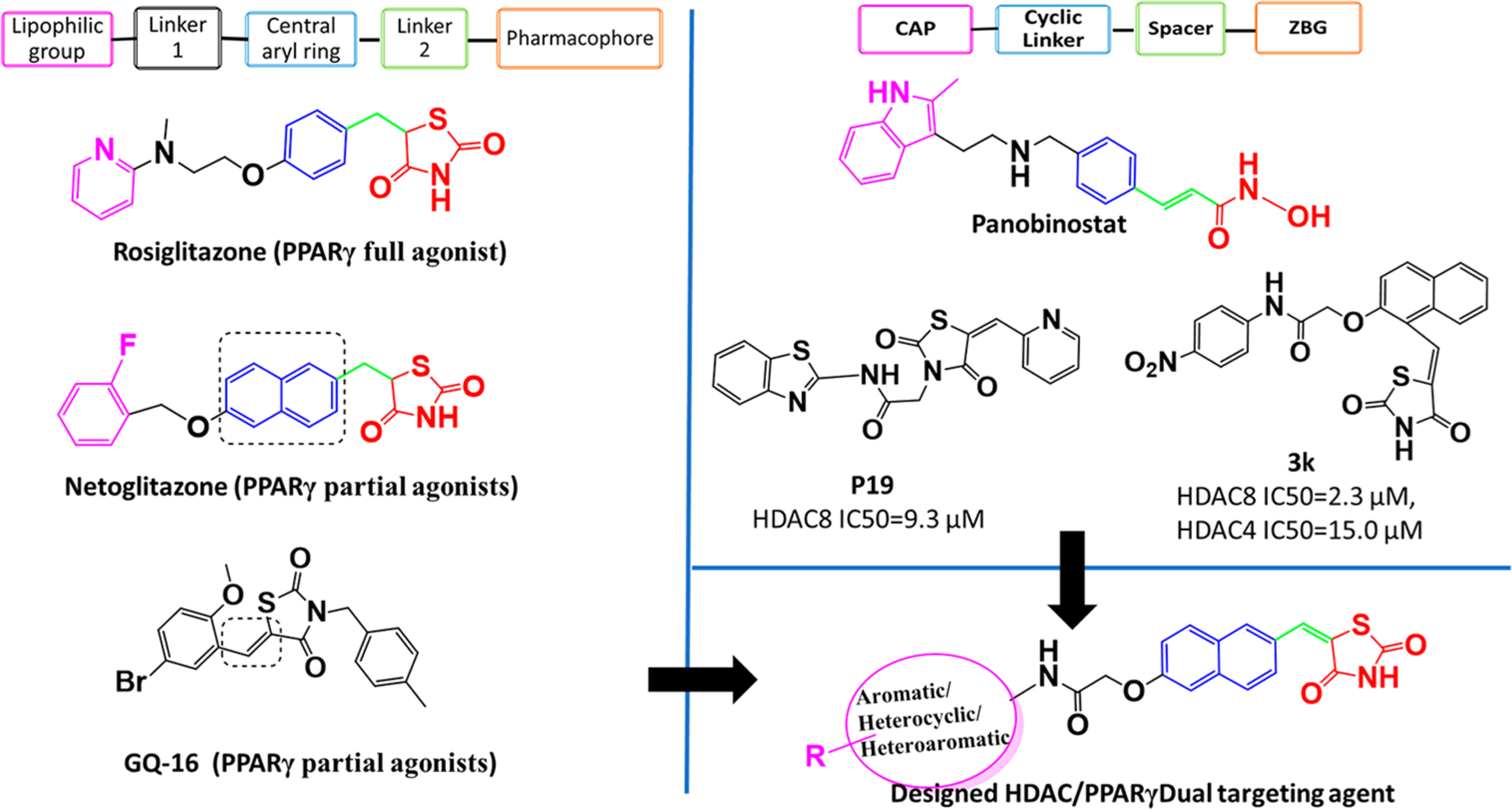
Designing of (partial) PPARγ and HDAC dual targeting agents.
N-Substituted benzylidene TZDs (an aryl ring attached to TZD through –C═CH– instead of –CH–CH2–), including our previously reported compounds and GQ-16, were found to be partial PPARγ agonists.54 Both netoglitazone, with a naphthalene central aryl ring, and GQ-16, with a benzylidene TZD ring, have demonstrated antitumor potential.55 Given the partial PPARγ transactivation and antitumor potential of these compounds, we sought to synthesize compounds with a naphthalene ring attached to TZD ring through a benzylidene like linker (–C═CH–)—thus generating the naphthylidene TZD derivatives which are evaluated herein (designed molecule, Figure 1).
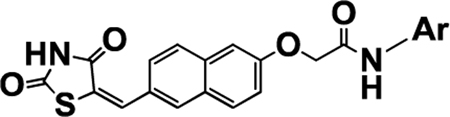
Typical HDAC inhibitors share a broad pharmacophore with a surface recognition cap (SRC, denoted CAP herein) which binds with amino acids of the active site, a hydrophobic linker which occupies the active site channel, and a zinc binding group (ZBG) which chelates the catalytic zinc ion.56 Previously, we developed HDAC inhibitors by placing a TZD ring centrally to obtain N-substituted TZD derivatives like P19 (Figure 1), or terminally to get naphthylidene TZDs like 3k (Figure 1) with good to moderate HDAC inhibitory activity.50,52,53 Both TZD series showed different activity against HDAC4 and HDAC8, which are representative of class IIa and class I human zinc-dependent HDACs. While N-substituted TZD analogues were found to be inactive against HDAC4 with moderate activity against HDAC8 (e.g., P19 IC50 = 9.3 μM), compound 3k with ortho-substituted naphthalene linker showed increased inhibitory activity on both HDAC4(IC50 = 15 μM) and HDAC8 (IC50 = 2.3 μM)52 (Figure 1). Despite significantly different chemical structures of P19 and 3k, their docked binding poses revealed that the carbonyl oxygen of carboxamide binds to the catalytic zinc ion and not the TZD ring. Thus, in the quest of modifying the structure of 3k series compounds to discover more potent HDAC inhibitors, we shifted substitutions at the naphthalene linker in the opposite ends to get a more extended shape than 3k. A primary docking study of these newly designed HDAC inhibitors at the active sites of HDAC4 and HDAC8 revealed that the TZD ring exhibited zinc binding interactions in HDAC4, whereas the carbonyl oxygen of carboxamide was bound to the zinc ion of HDAC8, which was a positive indication that the designed compounds could be HDAC inhibitors with differential potencies and selectivity. Similarly, HDAC inhibitors such as panobinostat have substitutions and a cyclic linker attached ZBG which are opposite to each other (Figure 1). We retained the naphthalene ring as a cyclic linker to help establish the structure–activity relationship (SAR) for HDAC inhibitory activity and partial PPARγ agonistic activity. Although compounds like 3k are structural isomers of the series described herein, the CAP portion have been varied to a great extent—aromatic, heterocyclic, and heteroaryl—to optimize for surface complementarity with HDAC active sites. In addition, the position of the substituents is such that it alters the shape of the molecules, thus orienting them differently from “series 3” and potentially leading to distinct HDAC selectivity and potency.
This rational designing led to oppositely substituted naphthylidene TZD structures in accordance with the pharmacophoric model of both targets (Figure 2). To clarify, the terminal aromatic/heterocyclic/heteroaromatic groups correspond to the lipophilic group of PPARγ agonists and to the CAP for HDAC8 inhibitors. Likewise, the carboxamide alkoxy moiety reflects linker 1 of PPARγ and the connecting unit between CAP and cyclic linker for HDAC4, while the carbonyl of this carboxamide may function as a ZBG for HDAC8. The naphthalene ring serves as the central aryl ring for PPARγ and as cyclic linker for HDACs. The methylene (–C═CH–) group corresponds to linker 2 for PPARγ and spacer for HDACs; and ultimately, the terminal TZD ring is a pharmacophore for PPARγ and ZBG for HDAC4. Therefore, we theorized that these compounds would have the capacity to simultaneously target HDACs and PPARγ.
Figure 2.
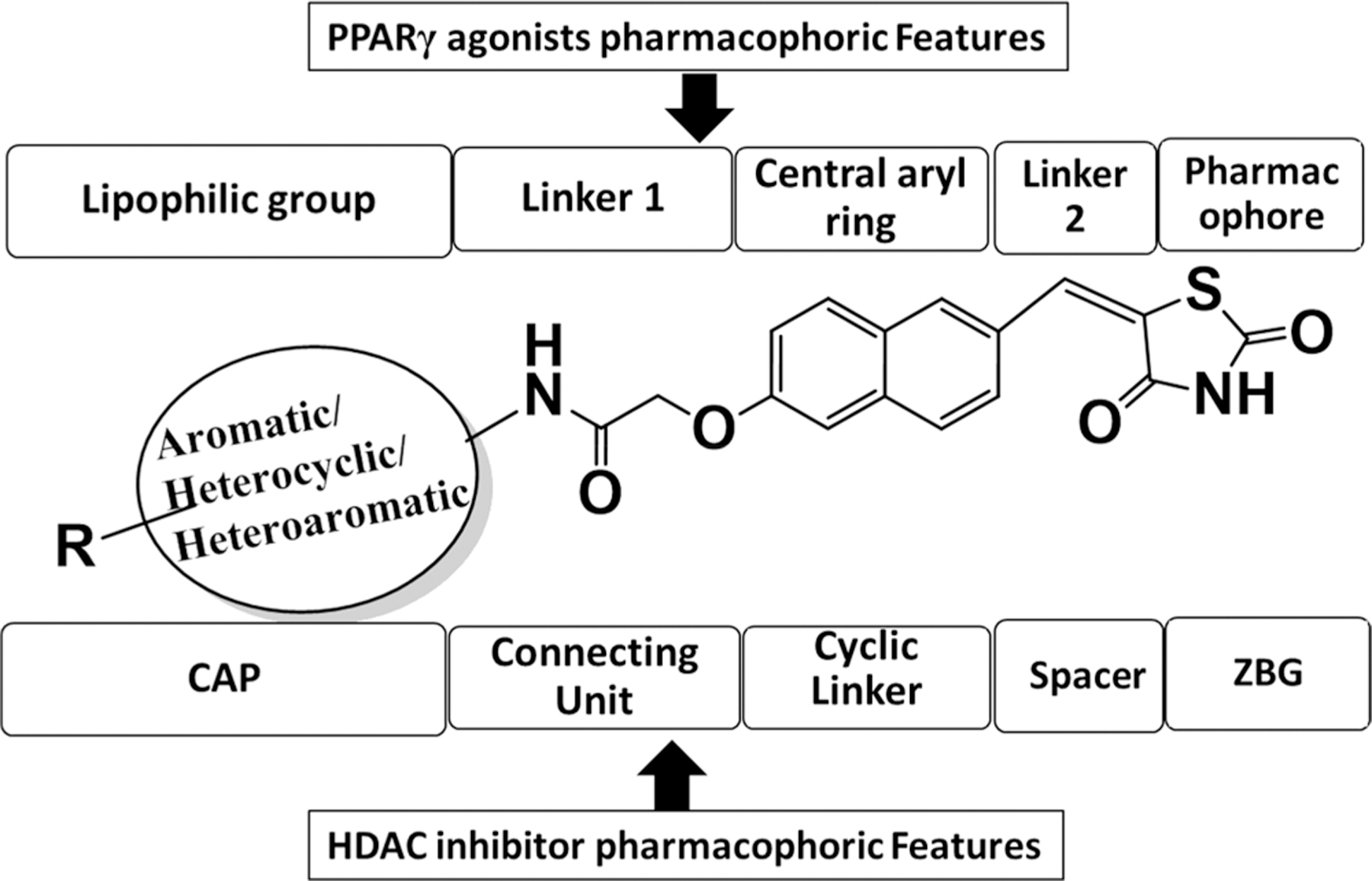
Fitting of designed dual PPARγ and HDAC targeting agents in typical pharmacophoric models of both targets.
Chemistry.
The procedures to synthesize the target compounds with a terminally substituted aromatic ring (7a–7l, 7o–7r, 7u–7v) were outlined in Scheme 1. Commercially available 6-hydroxy-2-naphthaldehyde (3) was reacted with 2,4-TZD (4) under Knoevenagel conditions to afford intermediate 5, which was further condensed with phenyl acetamides (2a–2l, 2o–2r, 2u–2v) under weakly basic conditions. Phenyl acetamides, in turn, were prepared from commercially available aromatic amines (1a–1l, 1o–1r, 1u–1v) as previously reported. Target compounds with a terminally substituted heterocyclic ring 7m, 7n, 7s, and 7t were synthesized using procedures as described in Scheme 2. The phenyl acetamides 2m, 2n, 2s, and 2t were condensed first with 3 and then were reacted by Knoevenagel conditions but using methoxy-ethanol as a solvent rather than traditional knoevenagel solvents. Use of methoxy-ethanol drastically improved the yield and purity of the final compounds. Target compounds 7w–7y were prepared as per the procedure detailed in Scheme 3 in a similar way to heterocyclic derivatives, with the variation of the solvent and reaction conditions.
Scheme 1. Synthesis of Substituted Aromatic Compoundsa.
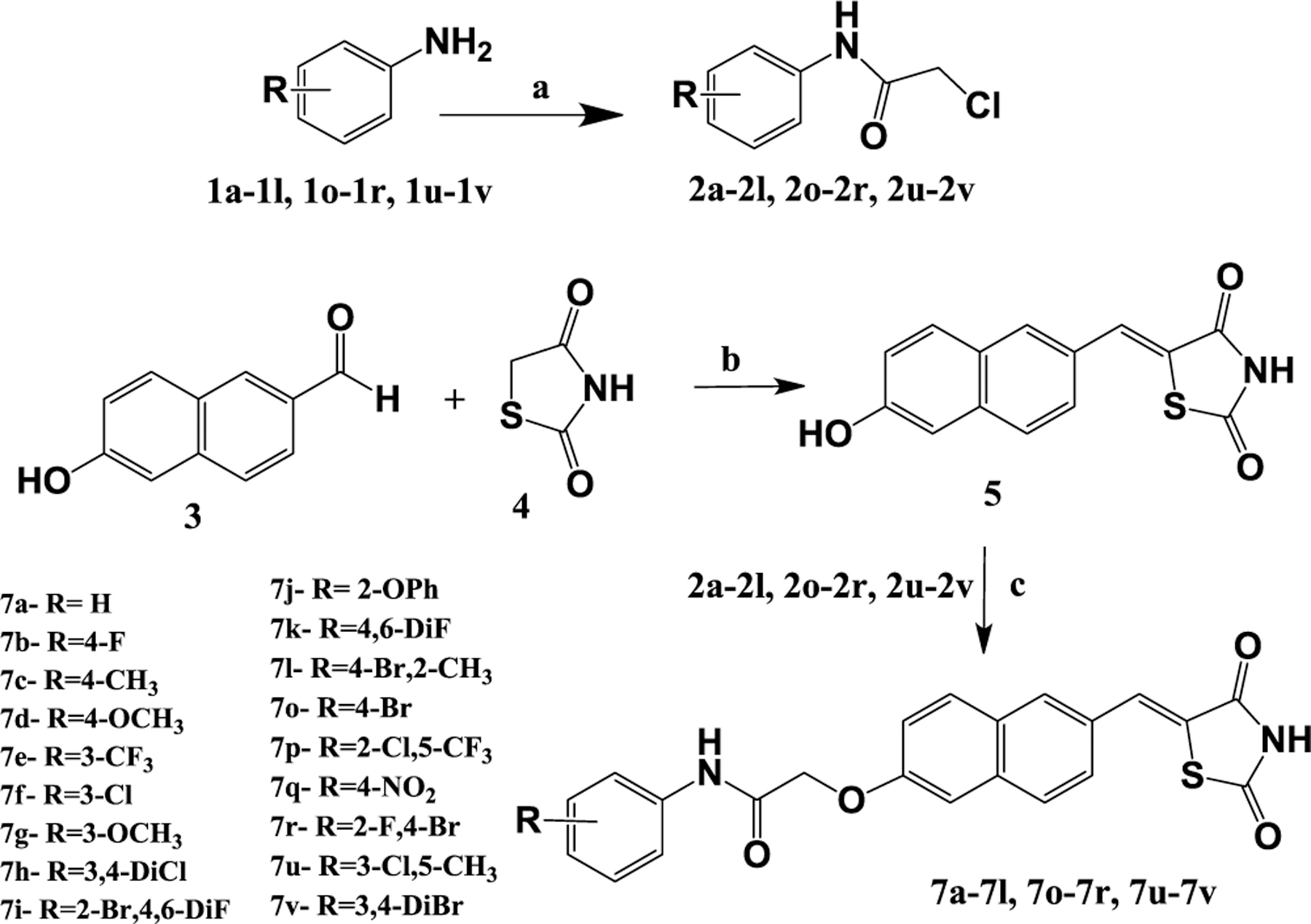
aReagents and conditions: (a) chloroacetyl chloride, K2CO3, DCM, stir, 0–5 °C, 1 h, rt, 3–6 h; (b) piperidinium benzoate, toluene, reflux 3–4 h; (c) DMF, K2CO3, stir 24 h.
Scheme 2. Synthesis of Substituted Heterocyclic Compoundsa.
aReagents and conditions: (a) chloroacetyl chloride, DCM/chloroform, TEA, stir, 0–5 °C, 1 h, rt, 8–12 h; (b) 6-hydroxy naphthaldehyde (3), DMF, K2CO3, stir 36 h; (c) 2,4-thiazolidinedione, methoxy ethanol, piperidine, reflux 3–4 h.
Scheme 3. Synthesis of Substituted Heteroaryl Compoundsa.
aReagents and conditions: (a) chloroacetyl chloride, chloroform, K2CO3, stir, 0–5 °C, 1 h, rt, 24–48 h; (b) 6-hydroxy naphthaldehyde (3), DMF, K2CO3, stir 48 h; (c) 2,4-thiazolidinedione, DMF, piperidine, reflux 3–4 h.
HDAC4/8 Activity Assay.
To determine the effects of synthesized derivatives 7a–7y (Figure 2) on the activity of HDACs and to detect if they exhibit selectivity against class I or class II, all 25 compounds were initially screened at 35 μM concentration on both isoforms, viz., HDAC4 from class II and HDAC8 from class I. The test compounds showed exceptional inhibitory effects on HDAC4, with markedly lower but significant activity on HDAC8 (Figure 3; Table S1 in Supporting Information). On the basis of the results gained in primary screening, compounds inducing a residual enzyme activity of <40% were evaluated at six concentrations to establish a dose–response curve (Figure 2, Table 1). Few compounds were found to be dual HDAC4 and HDAC8 inhibitors. Most of the compounds were more selective and potent on HDAC4 and at least 2 times potent on HDAC4 over HDAC8 at the tested concentrations. The best activities against HDAC4 were comparable to “Cpd 6”57 (Table 1), indicative of the therapeutic potential of this compound series.
Figure 3.
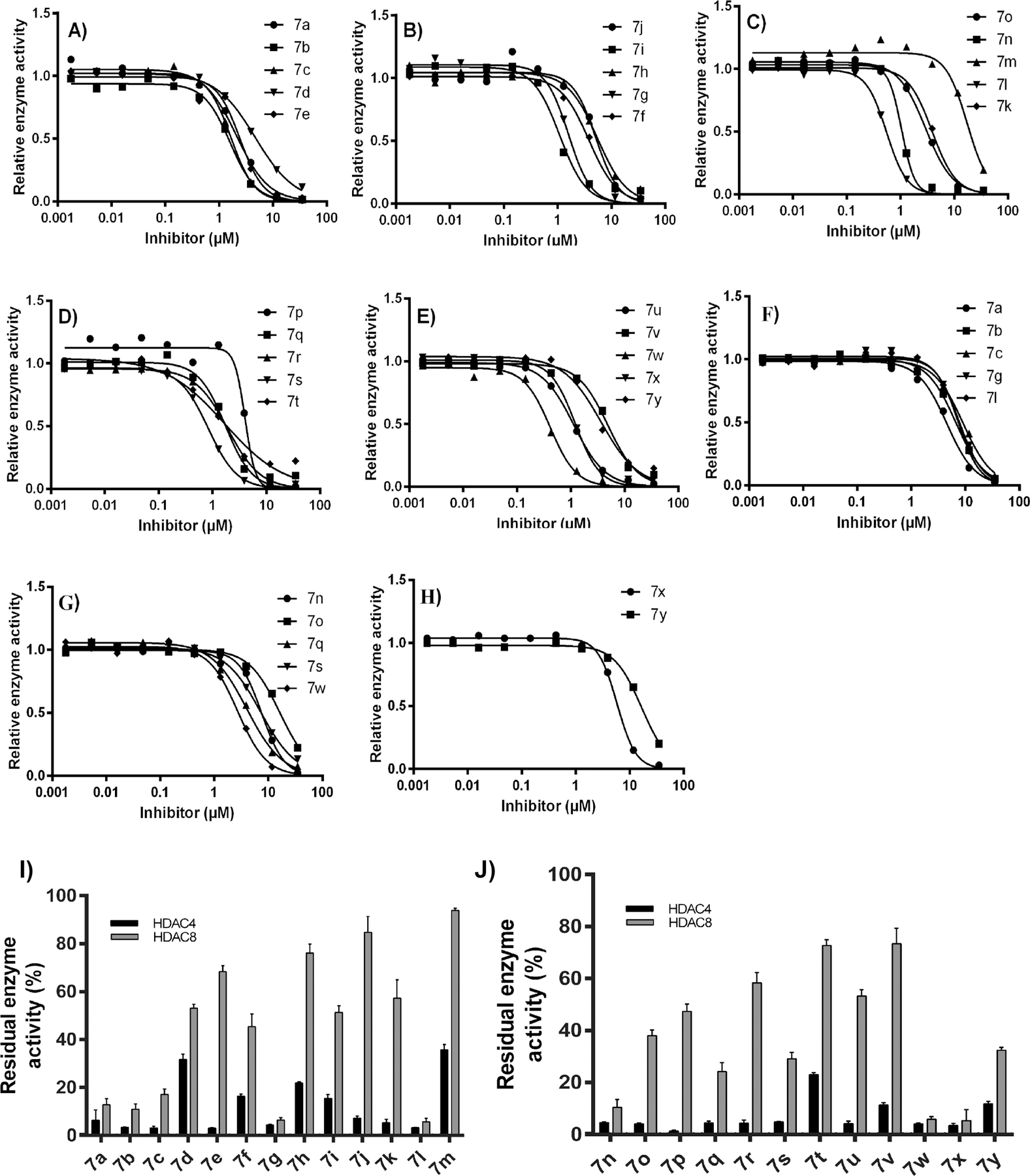
Dose–response curves (DRCs) of indicated inhibitors (A–E) against HDAC4 and (F–H) against HDAC8. Residual enzyme activity of HDAC4 or HDAC8 for compounds 7a–7m (I) and compounds 7n–7y (J) in the presence of 35 μM of the indicated compounds. Data represent means and standard deviations, n = 3. Compounds with less than 40% residual enzyme activity were taken further to determine IC50 values.
Table 1.
IC50 Values of the “Series 7” against HDAC4 and HDAC8 a
| |||
|---|---|---|---|
| compound | Ar group | HDAC4 IC50 (μM) | HDAC8 IC50 (μM) |
| 7a | phenyl | 2.1 ± 0.3 | 4.5 ± 0.2 |
| 7b | 4-fluorophenyl | 1.6 ± 0.3 | 6.6 ± 0.2 |
| 7c | 4-methylphenyl | 1.7 ± 0.3 | 9.0 ± 0.2 |
| 7d | 4-methoxyphenyl | 4.7 ± 0.1 | >50 |
| 7e | 3-trifluoromethylphenyl | 2.5 ± 0.4 | >50 |
| 7f | 3-chlorophenyl | 3.8 ± 0.1 | >50 |
| 7g | 3-methoxyphenyl | 1.6 ± 0.2 | 7.6 ± 0.2 |
| 7h | 3,4-dichlorophenyl | 5.5 ± 0.3 | >50 |
| 7i | 2-bromo-4,6-difluorophenyl | 1.1 ± 1.7 | >50 |
| 7j | 2-phenoxyphenyl | 5.2 ± 0.5 | >50 |
| 7k | 2,4-difluorophenyl | 3.9 ± 0.1 | >50 |
| 7l | 4-bromo-2-methylphenyl | 0.55 ± 0.05 | 7.4 ± 0.3 |
| 7m | pyridin-2-yl | 17 ± 0.7 | >50 |
| 7n | 6-methylpyridin-2-yl | 1.1 ± 1.2 | 7.5 ± 0.2 |
| 7o | 4-Br phenyl | 2.9 ± 0.1 | 16 ± 0.5 |
| 7p | 3-chloro-5 (trifluoromethyl)phenyl | 4.0 ± 0.2 | >50 |
| 7q | 4-nitrophenyl | 1.7 ± 0.4 | 4.0 ± 0.2 |
| 7r | 4-bromo-2-fluorophenyl | 1.9 ± 0.1 | >50 |
| 7s | 6-fluoropyridin-2-yl | 0.84 ± 0.22 | 7.2 ± 0.2 |
| 7t | 5-methylisoxazol-3-yl | 1.7 ± 0.8 | >50 |
| 7u | 3-chloro-4-methylphenyl | 1.1 ± 0.4 | >50 |
| 7v | 3,4-dibromophenyl | 4.9 ± 0.3 | >50 |
| 7w | benzo[d]thiazol-2-yl | 0.42 ± 0.05 | 2.7 ± 0.2 |
| 7x | 4-methylbenzo[d]thiazol-2-yl | 1.2 ± 0.4 | 5.8 ± 0.1 |
| 7y | 6-ethoxybenzo[d]thiazol-2-yl | 3.6 ± 0.3 | 17 ± 0.3 |
| Cpd 6 57 | - | 0.22 | >50 |
| PCI-34051 59 | - | 10 | 0.024 |
Data represent mean ± sd (standard deviations), n = 3. Compounds with less than 40% residual enzyme activity were taken further to determine IC50 values.
Against HDAC8, 12 out of 25 compounds possessed inhibition capacity. The remaining compounds showed no or only moderate activity at tested concentrations. Compounds 7a, 7b, 7c, 7g, 7l, 7n, 7q, 7s, 7w, and 7x exhibited IC50 values of <10 μM, 7o and 7y of < 20 μM. The most potent inhibitor was 7w, which had an IC50 = 2.7 ± 0.2 μM. Among the compounds with a substituted aromatic group (Ar), it was observed that the unsubstituted (7a), monohalo-substituted with F or Br (7b, 7o), and monoalkyl substituted (7c) derivatives retained HDAC8 inhibitory activity. Compounds 7d and 7g are structural isomers with p-OCH3 and m-OCH3 respectively, but only 7g exhibited inhibitory potential, indicating that the point of methoxy group attachment may be important. Compounds with dihalo substitution (7h, 7i, 7k, 7r) were found to be inactive, thus suggesting that dihalo substitution leads to loss of HDAC8 inhibition. Likewise, substitution with Cl (7f), CF3 (7e) or disubstitution with these two groups (7p) did not show HDAC8 inhibition. Additionally, it was observed that compounds with disubstitution at position 3- and 4- of the phenyl ring (7h, 7u, 7v) were not active. When the Ar group is a heterocyclic ring such as pyridinyl, halo-substituted pyridinyl (7s) and methyl substituted pyridinyl (7n) derivatives displayed greater HDAC8 inhibition than the unsubstituted compound (7m). Comparison between benzothiazole containing compounds revealed that an unsubstituted benzothiazole (7w) ring was preferred over alkyl (7x) and alkoxy substitution (7y).
As far as HDAC4 inhibitory activity is concerned, all derivatives exhibited HDAC4 inhibition with IC50 < 6.0 μM, except 7m which had IC50 = 17 ± 0.7 μM. The most potent compounds exhibiting IC50 values in the submicromolar range were 7l, 7s, and 7w (IC50 = 0.55 ± 0.05 μM, 0.84 ± 0.22 μM, and 0.42 ± 0.05 μM respectively). Among aromatic analogues (Ar = substituted phenyl) compounds with no substitution (7a), monohalo-substitution (7b, 7f, 7o), monoalkyl substitution (7c), and monoalkoxy substitution (7d, 7g) were found to be more potent than dihalo substituted compounds (7h, 7k, 7v except 7r). Even, disubstitution with a halogen and methyl group (7l, 7u) was well tolerated with significant activity. In the case of pyridinyl and benzothiazolyl derivatives, the effect of substitution pattern on inhibitory activity was found similar to that of HDAC8. Among pyridinyl derivatives, halogen substitution (7s) and alkyl substitution (7n) led to more potent activity than an unsubstituted pyridine ring (7m), whereas in benzothiazole containing compounds, an unsubstituted benzothiazole ring (7w) was preferred over alkyl (7x) and alkoxy substitution (7y). Since all compounds exhibited HDAC4 inhibition, it can be assumed that an Ar group of all three types, viz., aromatic, heterocyclic, and heteroaromatic, is well tolerated with significant retention of activity.
Compounds 7d, 7e, 7f, 7h, 7i, 7j, 7k, 7m, 7p, 7r, 7t, 7u, and 7v were found more potent on HDAC4 over HDAC8, whereas compounds 7a, 7b, 7c, 7g, 7l, 7n, 7o, 7q, 7s, 7w, 7x, and 7y were found to be dual inhibitors of HDAC4 and HDAC8 with at least 2 times more potency (as reflected from IC50 values) toward HDAC4 than HDAC8.
The difference between our previously reported “series 3” and the current “series 7” is only the position of the TZD ring on the central naphthalene ring. Thus, shifting the point of TZD ring attachment to the opposite side on naphthalene ring leads to an extreme change in HDAC potency and selectivity. In the previously reported “series 3”, the arrangement of the various groups in the molecule was such that they adopted an L-shape which is an ideal fit at the active site of HDAC8 as per the Hou et al. model; instead, in our current “series 7”, the shifting of the TZD ring to the opposite site causes the structure to deviate from an L-shape, which could be the reason why the current series are less selective and potent inhibitors of HDAC8.58 Nevertheless, the common element between our previously reported “series 3” and current “series 7” is the naphthalene linker, thus supporting our hypothesis of incorporating a naphthalene ring as cyclic linker to obtain potent HDAC inhibitors.
Molecular Docking into HDAC4 and HDAC8.
Different TZD scaffolds show different activities against HDAC4 and HDAC8, which are representatives of class IIa and class I of human zinc-dependent HDACs. While N-substituted TZD analogues are generally inactive against HDAC4, there are some examples with moderate activity against HDAC8 (e.g., P19 with an IC50 value of 9.3 μM,53 Figure 4). Docking results suggested that P19 binds to the catalytic zinc ion through its carbonyl oxygen and occupies the lower side pocket which connects the canonic active site pocket and acetate release channel.
Figure 4.
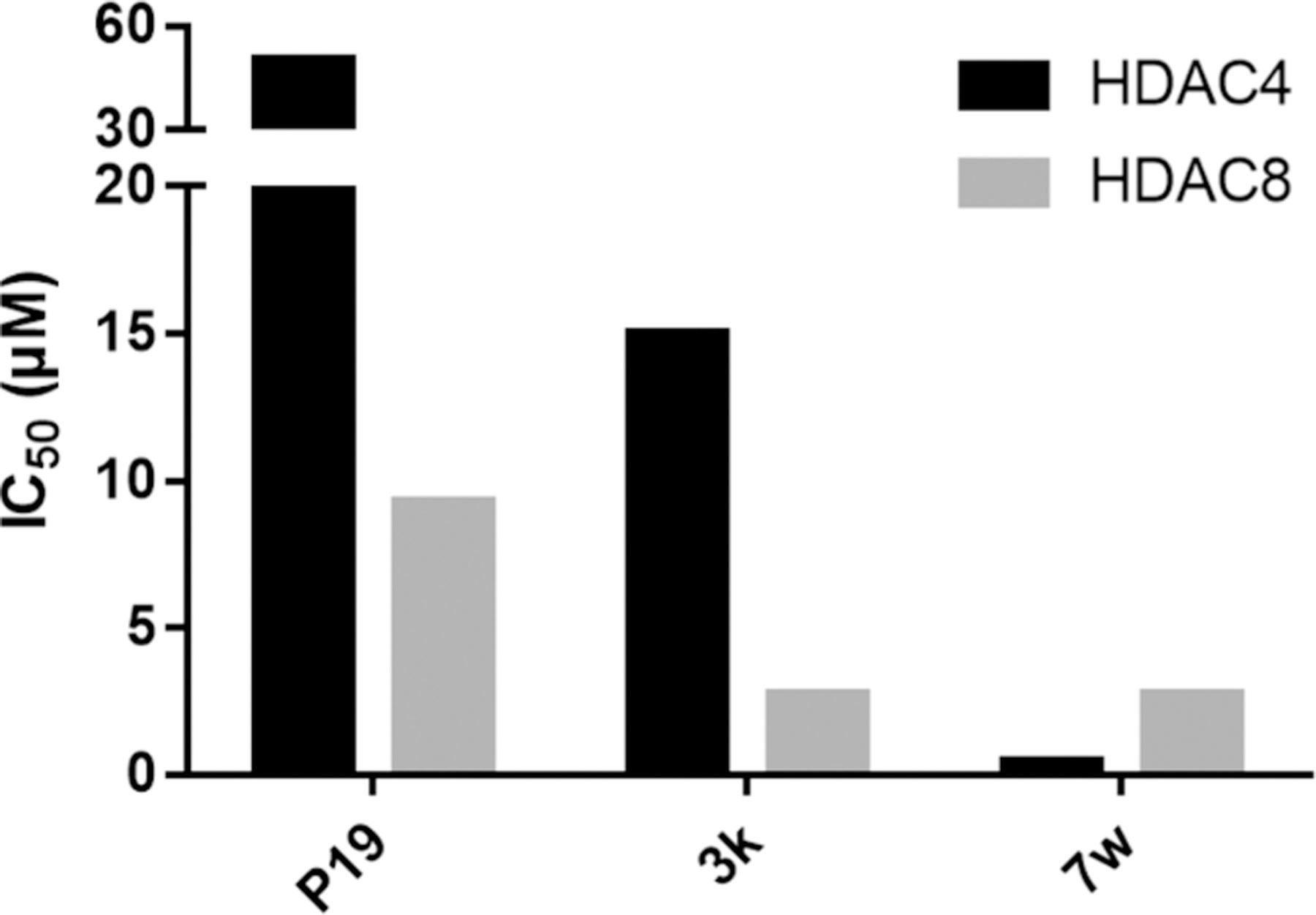
Optimization of dual activity of TZD-analogues against HDAC4 and HDAC8. P19 is a moderate HDAC8 selective inhibitor,53 3k52 shows improved activity on both, HDAC4 and HDAC8, and 7w shows high activity against both HDAC enzymes.
TZD analogue 3k with ortho-substituted naphthalene linker showed increased inhibitory activity on both HDAC4 and HDAC852 (Figure 4). Despite their different chemical structures, in spacer and substitution pattern at the TZD group, P19 and 3k have similar features in their docked binding poses. Namely, the protrusion of the aromatic carboxamide moiety into the transition area between active site and acetate release channel and binding of the carbonyl oxygen to the catalytic zinc ion. “Series 7” differentiate from the branched 3k in that the substitutions at the naphthalene linker are in the opposite position from the TZD group, thus forming a more extended shape. This difference led to a tremendous increase in activity against both HDAC isoenzymes (Figure 4). Compound 7w also showed low micromolar activity against HDAC8 and HDAC4, thus resulting in a dual HDAC4/8 inhibitor with some preference for HDAC4.
To rationalize the observed dual activity of “series 7” analogues against HDAC8 and particularly HDAC4, a comprehensive docking study was performed. At first, the docking routine was validated by redocking of the respective ligands into the used crystal structures of HDAC8- (PDB ID: 3SFF) and HDAC4-ligand complexes (PDB IDs: 2VQJ and 4CBY). The docked and crystallized poses of the ligand within the binding pocket of HDAC8 (PDBID: 3SFF) showed excellent overlap with an RMSD over 26 heavy atoms of 0.2 Å. The thiophene linker and trifluoromethyl warhead of the redocked ligand in HDAC4 (PDB ID: 2VQJ) show a good RMSD (root-mean-square deviation) value of 0.4 Å with respect to the crystal structure (Table S1 in Supporting Information). Since the aromatic headgroup of the trifluoromethyl ketone ligand protrudes into free solution, this part of the molecules is intrinsically flexible and thus not considered for the calculation of RMSD. For HDAC8, the crystal structure with PDB-ID 3SFF, which was previously utilized in docking of 3k, was selected.52 All compounds of “series 7” superposed well and docked smoothly into the binding pocket of HDAC8, showing a very similar binding mode to 3k (Tables S2, S3 in Supporting Information). The aromatic ring of the carboxamide group of 7l fit perfectly into the lower binding pocket adjacent to the catalytic center at the entrance of the acetate release channel, and the carbonyl oxygen was bound to the catalytic zinc ion (Figure 5A). Moreover, the naphthalene linker forms a π-stacking interaction with Phe152 and Phe208 and a π-sulfur interaction with Met274 (Figure 5A,B).
Figure 5.
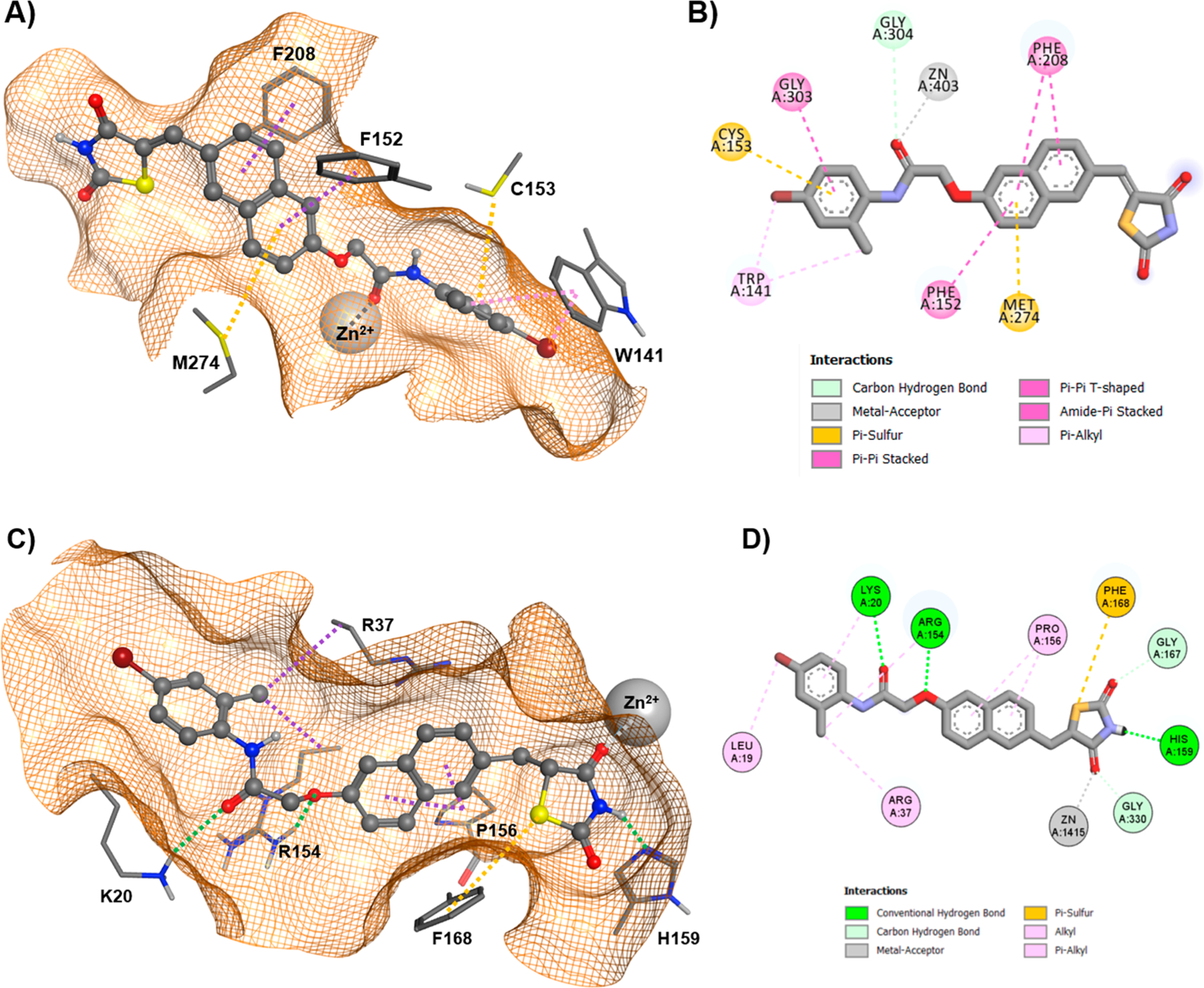
(A) 3D docking pose of 7l within HDAC8, (B) 2D interactions between 7l and HDAC8. (C) Docked 3D binding pose of 7l within the enlarged binding groove of HDAC4o. (D) 2D interactions between 7l and HDAC4o. Conventional hydrogen bonds are indicated by dotted green lines, alkyl and π-alkyl interactions by dotted magenta or pink lines. A dotted gray line denotes metal binding, whereas a dotted orange line highlights a π-sulfur interaction.
The TZD group points out of the binding pocket and is exposed to the outer solvent. The malleability of the HDAC8 binding pocket allows for accommodation of the branched TZD-analogue 3k as well as the more linearly shaped 7l. This exceptional flexibility of HDAC8 has been exploited previously for the design of linkerless or branched isoenzyme selective inhibitors.60,61 However, all of these active compounds contain the potentially problematic hydroxamate group of most canonical HDAC inhibitors. Overall, compounds of “series 7” show good docking scores that are similar to or better than 3k (Table 2).
Table 2.
GBVI/WSA dG Docking Scores for Indicated Molecules and HDAC Isoenzymes
| ID | HDAC4o | HDAC8 | ID | HDAC4o | HDAC8 |
|---|---|---|---|---|---|
| 7a | −8.7 | −9.0 | 7n | −9.2 | −9.6 |
| 7b | −8.7 | −8.8 | 7o | −9.7 | −9.8 |
| 7c | −9.1 | −9.8 | 7p | −9.0 | −10.2 |
| 7d | −9.3 | −10.3 | 7q | −10.3 | −9.9 |
| 7e | −8.9 | −10.0 | 7r | −9.9 | −10.2 |
| 7f | −9.1 | −9.9 | 7s | −8.2 | −10.7 |
| 7g | −10.3 | −9.9 | 7t | −9.5 | −10.5 |
| 7h | −9.1 | −9.8 | 7u | −8.5 | −10.0 |
| 7i | −9.4 | −9.7 | 7v | −8.9 | −10.2 |
| 7j | −10.5 | −10.1 | 7w | −9.6 | −10.2 |
| 7k | −9.0 | −9.8 | 7x | −9.6 | −10.3 |
| 7l | −9.4 | −10.2 | 7y | −10.5 | −11.2 |
| 7m | −9.2 | −9.1 | 3k | −8.7 | −9.5* |
To understand the difference between 3k and 7l in molecular recognition of HDAC4 and HDAC8 appears more complex and challenging. It is of utmost importance to recollect that different types of inhibitors have been shown to stabilize either the open or the closed conformation of the catalytic domain of HDAC4 because of its conformationally flexible second zinc-binding domain.62,63 To explore the impact of open (PDB ID: 2VQJ) and closed (PDB ID: 4CBY) conformations of HDAC4 on the binding affinity of the most active analogues, 7l and 7w (IC50 0.55 ± 0.05 and 0.42 ± 0.05 μM, respectively), docking was performed using both crystal structures. Binding to the enlarged pocket of open conformation HDAC4 (HDAC4o) yielded more favorable binding energies than closed HDAC4 (HDACc) (Table S2 in Supporting Information). Consequently, all TZD analogues have been docked into the large binding groove of HDAC4o, yielding good scores and indicating favorable contacts between protein and ligand (Table 2). A comparison with 3k reveals that the compounds of “series 7” exhibit favorable interactions with both HDAC4o and HDAC8, while 3k shows good affinity toward HDAC8 but is poorly recognized by HDAC4o.52 Thus, docking results are in good agreement with experimental data and may provide insight into important contacts that determine binding mode of action with HDAC4. Docking suggested that “series 7” analogues bind to HDAC4o rather than HDAC4c. The enlarged binding groove of HDAC4o is complementary to the linear shape of the TZD analogues of “series 7”, offering favorable contacts between the amino acids at the surface of the binding pocket and the ligands (Figure 5C). The resulting binding poses were similar among all analogues (Figure S1A,B in Supporting Information). In contrast to the binding mode of the TZD analogues in HDAC8, which is determined by an interaction between the catalytic zinc ion and carbonyl oxygen of the ligand, “series 7” compounds bind the corresponding zinc ion through the TZD group in HDAC4o (Figure 5C,D). A closer look at the distinct contacts between 7l and HDAC4o revealed multiple interactions between the TZD group and binding pocket: one carbonyl oxygen of the TZD ring interacts with the catalytic zinc ion, the amide nitrogen serves as hydrogen donor for His159, and the sulfur atom forms a π-sulfur interaction with the aromatic ring of Phe168 (Figure 5C,D). Moreover, the naphthalene linker is supposed to undergo hydrophobic interactions with Pro156. Two additional charge enhanced hydrogen bonds, between theoxygen next to the naphthalene moiety and Arg154 as well as the distal carbonyl oxygen and Lys20, contribute significantly to the binding of 7l to HDAC4o (Figure 5C,D). Possible hydrophobic interactions between neighboring Arg37, Arg154, and the aromatic head group may also increase affinity depending on the substitution pattern. Altogether, the experimentally demonstrated dual activity of TZD compounds from “series 7” against HDAC4 and HDAC8 agrees with favorable docking scores for both HDAC isoenzymes. Furthermore, the docking results suggest two distinct binding modes of 7l and analogues to HDAC8 and the open conformation of HDAC4 characterized by different complexation of the catalytic zinc ion.
PPARγ Transactivation Assay.
Compounds 7a–7y were analyzed via PPARγ-GAL4 transactivation assay on HepG2 cells in agonist mode at 25, 5, and 1 μM concentrations. Rosiglitazone at a 2 μM concentration was used as a control, while the blank contained 0.1% dimethyl sulfoxide (DMSO). Compounds 7i, 7l, and 7r were the most active and showed efficacy of 20–30% followed by 7c, 7o, and 7q with an efficacy around 10% compared with the control (Table 3) (Figure S4 in Supporting Information). Many compounds showed partial activation of PPARγ, but because Emax was less than 10%, their activity was considered negligible. However, we were successful to determine EC50 (half maximal effective concentration) values for the six compounds listed in Table 3. Thus, our hypothesized design of naphthylidene TZD analogues as partial PPARγ agonists was successful. The most potent partial agonists were 7i and 7l with EC50 values of 0.245 ± 0.006 and 0.359 ± 0.031 μM, respectively. In spite of the small number of PPARγ active compounds, some preliminary remarks can be made about structure–activity relationships. It is very interesting to note that five p-Br (bromo) substituted compounds, 7i, 7l, 7o, 7r, and 7v, displayed PPARγ activity, which indicates that this substitution has the appropriate requisites to elicit the transactivation capacity of these derivatives. For these compounds, other substituents can be tolerated at the ortho position, particularly fluorine (7i, 7r) or methyl (7l), whereas a methyl or a polar nitro group alone at the para position of the phenyl ring led to less potent derivatives (7c, 7q). In contrast, the presence of two bromine atoms, as in the 3,4-disubstituted derivative 7v, led to poor activity. This suggests that an excess steric bulk in this part of the molecule is detrimental for the interaction with the receptor. Given that all heterocyclic and pyridinyl derivatives showed very low activity, it can be assumed that the phenyl ring is more appropriate to achieve significant PPARγ transactivation.
Table 3.
PPARγ Transactivation Assay a
| |||
|---|---|---|---|
| PPARγ |
|||
| compound | Emax%b | EC50 μM | “Ar” group |
| 7c | 13 ± 2 | 1.220 ± 0.260 | 4-methylphenyl |
| 7i | 22.4 ± 1.8 | 0.245 ± 0.006 | 2-bromo-4,6-difluorophenyl |
| 7l | 32 ± 3 | 0.359 ± 0.031 | 4-bromo-2-methylphenyl |
| 7o | 13.0 ± 0.2 | 0.729 ± 0.027 | 4-bromophenyl |
| 7r | 27 ± 6 | 0.791 ± 0.025 | 4-bromo-2-fluorophenyl |
| 7q | 8.5 ± 1.5 | 1.610 ± 0.280 | 4-nitrophenyl |
EC50 values with mean ± SD.
Efficacy values (Emax) were calculated as a percentage of the maximum obtained fold induction with the reference compounds. All transfection experiments were repeated at least twice in triplicate.
These findings illustrate the potential of 7i, 7q, and 7r as dual targeting partial PPARγ agonist and selective HDAC4 inhibitor compounds, and of 7c, 7l, and 7o to target PPARγ, HDAC4, and HDAC8 but with more efficacy (3- to 10-fold) on HDAC4.
Molecular Docking into PPARγ.
To understand the structural basis for the observed partial agonism of 7i, 7l, 7o, 7r, 7c, 7q toward PPARγ, we undertook docking simulations using the Glide module, which is part of the Maestro software suite. Docking experiments were carried out into the X-ray crystal structures of PPARγ ligand binding domain (LBD) complexed to partial agonist CAY10638 (PDB ID: 6DGR).64 This structure was chosen as a template because of the good resolution (2.15 Å) and the similarity of the cocrystallized ligand with the candidate compounds, both containing a TZD headgroup and an unsaturated linker, which restricts the mobility of the TZD group. Analysis of the CAY10638/PPARγ complex revealed that the ligand is H-bonded via two crystallographically observed water molecules to the protein: via HOH708 to L340 (β-sheet) and via HOH706 to S342 (β-sheet). Accordingly, the two water molecules were included in the docking experiments.
The LBD of PPARγ consists of a hydrophobic entrance (arm III) that branches off into two subsites: the polar arm I, which is extended toward H12, and the hydrophobic arm II, which is located between helix H3 and β-sheet.
Compound 7i fit well within the PPARγ LBD, adopting a U-shaped conformation like the known full agonist Rosiglitazone. The interactions between 7i and the PPARγ LBD involved many H-bonds, one halogen bond, and several hydrophobic interactions (Figure 6A,B). The TZD headgroup is located in arm I and establishes a bifurcated H-bond with Nε atom of H323 (d = 2.8 Å) and Oγ atom of S289 (d = 3.2 Å) by one of two carbonyl oxygens. Unlike full agonist Rosiglitazone, 7i shows no direct H-bond with residue Y473 in the activation function domain (AF-2) located on H12. Interestingly, the superimposition of 7i on the cocrystal structure of Rosiglitazone bound to PPARγ LBD (PDB ID: 4EMA) revealed that the TZD headgroup of 7i undergoes a slight shift toward H3, thus preventing the formation of a key H-bond with residue Y473 (Figure 6C). Because stabilization of the AF2 helix in the activated conformation is a key characteristic of full agonism,65 the lack of direct interaction between this domain of the receptor and 7i may explain the observed partial agonist/modulator activities of this class of compounds. The naphthalene moiety is placed in the center of the LBD and makes hydrophobic interactions with C285, I326, Y327, L330, F363, and M364. The phenyl amide tail lies in the subpocket between H3, β-sheet, and Ω-loop (β-sheet subpocket), establishing numerous nonpolar and polar interactions with the participation of HOH708 and HOH706 in setting-up an indirect H-bond network. Specifically, the oxygen atom bound to the naphthalene moiety makes an H-bond with HOH708 (d = 3.1 Å), which in turn interacts with the C═O backbone of L340 (d = 3.1 Å). The bromine atom in the ortho position of the phenyl ring forms a halogen bond with the structural water HOH706 (d = 3.1 Å), that in turn interacts with the NH backbone of S342 (d = 3.1 Å). Hydrophobic contacts are also observed between 7i and the subpocket residues V339, I341, M348 (β-sheet); I281 (H3); and F264 (Ω-loop). These interactions contribute to the stabilization of the H3/β-sheet/Ω-loop region of the LBD, similar to other PPARγ partial agonists such as BVT.13, MRL-24, and nTZDpa.66 It has in fact been demonstrated that the activity of partial agonists, which do not robustly stabilize H12, may stem from the stabilization of the β-sheet subpocket.66–69 In addition, the terminal phenyl ring appears to be optimally oriented for a favorable edge-to-face π-stacking interaction with H266 on the Ω-loop, which contributes to further increase the Ω-loop stabilization. It is thus conceivable to suggest that ligands of PPARγ can afford graded transcriptional responses by employing a compensatory mechanism of β-sheet subpocket interactions to balance the lack of H12 stabilization. However, it is important to note that the Ω-loop is highly unstable and the residues within this loop are quite flexible; thus, the prediction of ligand/Ω-loop interactions might not be accurate and must be interpreted accordingly.67,70
Figure 6.
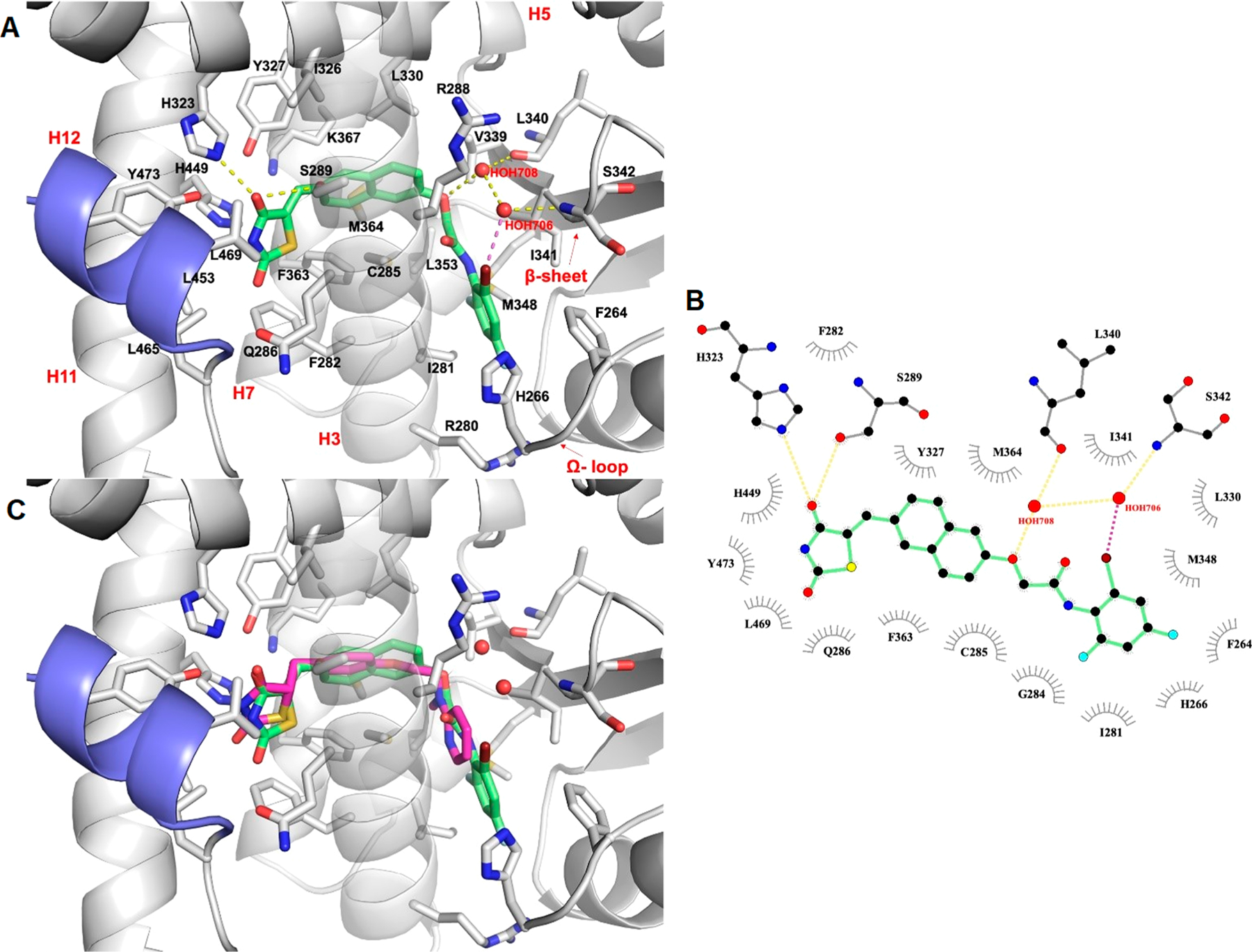
In silico docking of compound 7i into the PPARγ binding pocket. (A) Binding mode of compound 7i (partial agonist, green sticks) into the PPARγ binding site represented as a gray ribbon model. Only amino acids located within 4 Å of the bound ligand are displayed (white sticks) and labeled. H12 is shown in slate. H-bonds discussed in the text are depicted as dashed yellow lines. The halogen bond is shown as dashed magenta lines. The water molecules bridging the interaction of the ligand with the protein are displayed as red spheres. (B) 2D ligand-interaction diagram of 7i. The plot was generated by LigPlot+. H-bonds are shown as dashed yellow lines, while the halogen bond is shown as dashed magenta lines. The water molecules are displayed as red spheres. The spoked arcs represent residues making nonbonded contacts with the ligand. (C) Cα superposition of the complexes of PPARγ with compound 7i and Rosiglitazone (full agonist, magenta sticks, PDB ID: 4EMA).
Docking of 7l, which has a similar potency toward PPARγ as 7i, revealed a binding mode of this compound comparable to that described above for 7i, with the only consistent difference being the lack of the halogen bond, which is outweighed by the formation of a H-bond between the carbonyl oxygen of the phenylacetamide tail and HOH706 (3.5 Å), that in turn interacts with the C═O backbone of S342 (d = 3.1 Å). Moreover, the methyl group at the ortho position on the terminal phenyl ring allows 7l to form additional hydrophobic interaction with L353.
Further docking studies of 7o, 7r, 7c, 7q revealed that these compounds were in the same position as 7i and 7l but were unable to strongly stabilize the β-sheet, thus showing a decrease in potency (Figure S5 in Supporting Information). The low efficacy is due to the lack of the halogen bond with HOH706, since bromine is placed in para position in 7o and 7r, and to steric clashes of methyl and nitro group in 7c and 7q, respectively, with H266 and F264 residues belonging to the Ω-loop.
Cytotoxicity Assessment by DNS Assay.
On the basis of the prior experiments, it was discovered that derivatives 7c, 7i, 7l, 7o, 7r, and 7q were dual HDAC and PPARγ targeting agents; hence, they were considered for further evaluation in regard to their antitumor activity. Compounds 7e, 7f, and 7j were also selected as they were selective for HDAC4, as was 7g for its dual activity against HDAC4 and HDAC8. Thus, a total of 10 compounds were screened via differential nuclear staining (DNS) assay to assess their cytotoxic potential against a panel of seven cell lines. The panel consisted of solid tumor cells: HeLa (cervical adenocarcinoma, epithelial), MDA-MB-231 (triple negative breast, epithelial), and SH-SY5Y (neuroblastoma, epithelial); hematological tumor cells: CCRF-CEM (T-cell leukemia), Ramos (Burkitt’s B-cell lymphoma), and HL60 promyelocytic leukemia, myeloblasts); and noncancerous cells: HS-27 (normal foreskin, fibroblasts). A primary single dose screening of all 10 compounds was performed at a concentration of 10 μM, and those which elicited greater than 40% cell death (Table 4) were analyzed at several concentrations to find their half maximal cytotoxic concentration (CC50). CC50 values indicate the average concentration of compound required to kill 50% of the sample population and is reported as mean ± SD in Table 5.
Table 4.
Primary Single Dose DNS Screening of Selected Compounds for Cytotoxicity in a Panel of Seven Cell Lines at 10 μM Concentration a
| % cell death at 10 μM |
|||||||
|---|---|---|---|---|---|---|---|
| compound | CEM | Ramos | HL60 | HeLa | MDA-MB-231 | SH-SY5Y | HS-27 |
| 7c | >40 | >40 | 20.95 | >40 | 22.80 | >40 | 13 |
| 7e | 25.87 | >40 | 19.55 | 39.09 | 5.96 | 0 | >40 |
| 7f | >40 | >40 | 27.58 | 29.68 | 2.84 | 10.79 | 22.56 |
| 7g | >40 | >40 | 0 | >40 | >40 | 0 | 24.69 |
| 7i | >40 | >40 | 12.43 | >40 | >40 | >40 | >40 |
| 7j | 40.24 | >40 | 0 | >40 | >40 | >40 | 0 |
| 7l | 30.82 | 29.78 | 32.90 | >40 | 31 | >40 | 24.90 |
| 7o | 37.44 | 34.53 | 17.64 | >40 | 0 | >40 | 0 |
| 7q | 42.28 | 35.18 | 12.34 | 38.95 | >40 | 0 | 15.32 |
| 7r | >40 | 3.91 | 0 | 0 | 0 | >40 | >40 |
CEM: T cell leukemia, Ramos: Burkitt’s B cell lymphoma, HL60: promyelocytic leukemia (myeloblasts), HeLa: cervical adenocarcinoma (epithelial), MDA-MB-231: triple negative breast (epithelial), SH-SY5Y: neuroblastoma (epithelial), HS-27: normal foreskin (fibroblasts).
Table 5.
CC50 Values of Selected Compounds (7c, 7e, 7f, 7g, 7i, 7j, 7l, 7o, 7q, 7r) and Standard Drugs (SAHA, Pioglitazone) a
| compounds | cell line | CC50 (μM) | SCI |
|---|---|---|---|
| 7c | CEM | 2.8 ± 0.31 | 14.4 |
| Ramos | 7.0 ± 1.17 | 5.8 | |
| HeLa | 10.3 ± 0.56 | 3.9 | |
| SH-SY5Y | 49.5 ± 1.09 | 0.8 | |
| HS-27 | 40.2 ± 4.75 | - | |
| 7e | Ramos | 6.2 ± 0.28 | >10 |
| 7f | CEM | 10.6 ± 0.22 | >10 |
| Ramos | 9.1 ± 0.08 | >10 | |
| 7g | CEM | 10.7 ± 0.41 | >10 |
| Ramos | 9.3 ± 0.01 | >10 | |
| HeLa | 10.4 ± 0.58 | >10 | |
| MDA-MB-231 | 9.8 ± 0.22 | >10 | |
| 7i | CEM | 9.6 ± 0.15 | 4.2 |
| Ramos | 9.6 ± 0.14 | 4.2 | |
| HeLa | 10.4 ± 0.16 | 3.8 | |
| MDA-MB-231 | 9.8 ± 0.1 | 4.1 | |
| SH-SY5Y | 21.8 ± 0.41 | 1.8 | |
| HS-27 | 39.9 ± 1.95 | - | |
| 7j | Ramos | 10.0 ± 0.39 | >10 |
| HeLa | 10.2 ± 0.52 | >10 | |
| MDA-MB-231 | 10.3 ± 0.12 | >10 | |
| SH-SY5Y | 52.3 ± 3.09 | >10 | |
| 7l | HeLa | 9.9 ± 0.25 | >10 |
| SH-SY5Y | 91.1 ± 4.26 | >10 | |
| 7o | HeLa | 10.1 ± 0.04 | >10 |
| 7q | MDA-MB-231 | 55.8 ± 1.56 | >10 |
| 7r | CEM | 40.9 ± 3.16 | 1.4 |
| SH-SY5Y | 16.0 ± 0.69 | 3.6 | |
| HS-27 | 57.0 ± 8.45 | - | |
| Pioglitazone | CEM | 0% @ 50 μM | - |
| HeLa | 0% @ 10 μM | - | |
| SAHA | CEM | 2.5 ± 0.13 | - |
| HeLa | 2.8 ± 1.47 | - |
CC50 reported as average concentration ± std. deviation (μM); SCI = CC50HS‑27/CC50cancer; Nd*-Not determined
Several experimental compounds were cytotoxic at <10.0 μM (Table 5) on nearly all cancer cell lines except SH-SY5Y; however, compounds 7c and 7i were identified as the most potent candidates from this series. The greatest potency of compound 7c was against CEM (CC50 = 2.8 μM), Ramos (CC50 = 7.0 μM), and HeLa (CC50 = 10.3 μM), whereas 7i was active against CEM (CC50 = 9.6 μM), Ramos (CC50 = 9.6 μM), and MDA-MB-231 (CC50 = 9.8 μM). In comparing the cytotoxicity of “series 7” compounds with existing HDAC or PPARγ-targeted therapeutics, class I and II HDAC inhibitor suberoylanilide hydroxamic acid (SAHA; CEM CC50 = 2.5 μM and HeLa CC50 = 2.8 μM) displayed similar potency to 7c, and highly selective PPARγ agonist pioglitazone had no cytotoxic effect even at 50 μM. Dose–response curves (DRCs) for compounds 7c and 7i against the CEM cell line are shown in Figure 7.
Figure 7.
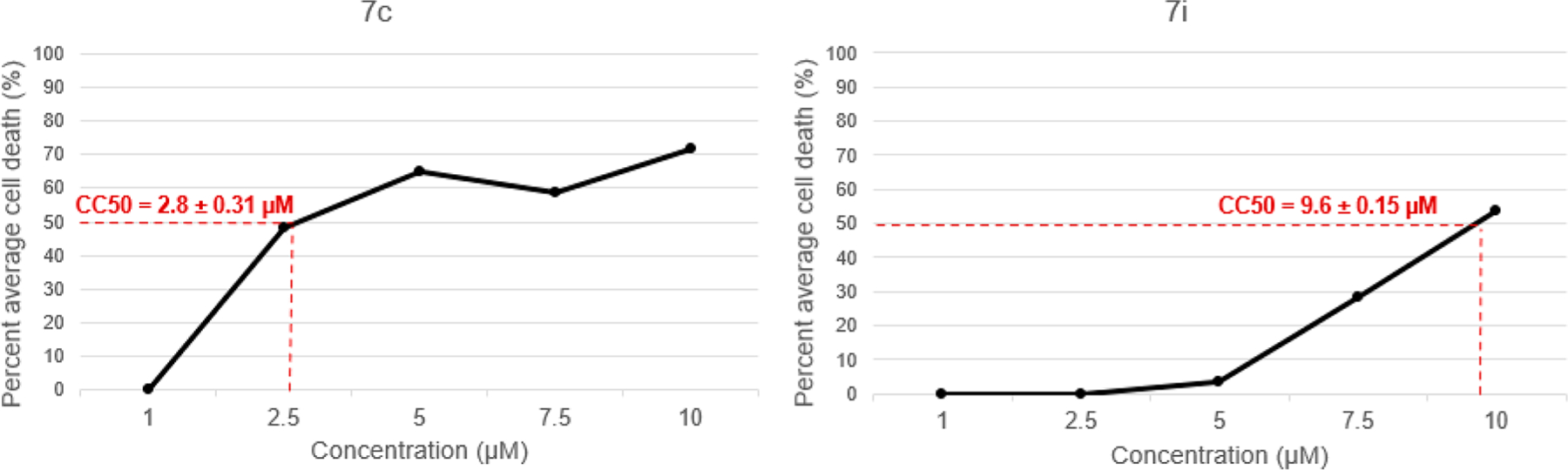
Dose–response curves of compounds 7c and 7i toward CCRF-CEM; n = 3.
Compounds 7c and 7i Exhibit Selectivity toward Tumorigenic Cells.
To compare the cytotoxicity of compounds 7c and 7i between tumorigenic and normal cells, the CC50 value for each compound against HS-27 cells was divided by the CC50 of each cancer cell line to generate selective cytotoxicity index (SCI) values. SCI values greater than 1.0 denote the ability of a given compound to kill cancer cells more effectively than cells of noncancerous origin.
These results revealed substantial selectivity of experimental compounds 7c and 7i toward CEM and other cancer cell lines within our panel. Compound 7c was the most selective, with a 14.4 SCI in the CEM cell line, whereas compound 7i had an SCI value of 4.2. Additional selectivity values for these compounds (and others) are presented in Table 5. Based on the potency and selectivity of 7c and 7i, CEM cells were the chosen model for several experiments that are described herein.
Apoptosis Assay.
To determine whether cell death elicited by experimental compounds 7c and 7i was occurring via apoptosis or necrosis, an Annexin V-fluorescein isothiocyanate/propidium iodide (AnnexinV-FITC/PI) assay was performed via flow cytometry. In this experiment, CEM cells were treated in triplicate with the CC50 or 2xCC50 of 7c, 7i, or SAHA (for comparison) and incubated for 72 h. 4% DMSO (dimethyl sulfoxide), 1 mM H2O2(hydrogen peroxide), and untreated cells were utilized as vehicle, positive, and negative controls respectively (Figure S6 in Supporting Information). Phosphatidylserine (PS) is a phospholipid which flips from the inner to the outer leaflet of the plasma membrane and serves as an “eat me” signal in apoptotic cells. Annexin V is a cellular protein with high affinity for PS that is commonly conjugated to the fluorochrome FITC and used as a probe for PS translocation. Propidium iodide, however, is permeable to all cells with compromised membranes. Apoptotic cell populations in this assay are those which stain positive for AnnexinV-FITC, whereas necrotic cells are those which stain positively for PI but negative for AnnexinV-FITC. This analysis identified significant PS externalization in 7c (21.46 ± 0.33%; p < 0.001) and 7i (26.53 ± 0.96%; p < 0.001) CC50-treated samples when compared with the vehicle control, implying the stimulation of apoptosis by these compounds (Figure 8). Dose (CC50 vs 2×CC50) displayed negligible effect on the proportion of apoptotic cells. Approximately 40 000 events (cells) were analyzed per sample, and statistical significance was determined by Student’s t test (Tables S3–S6 in Supporting Information).
Figure 8.
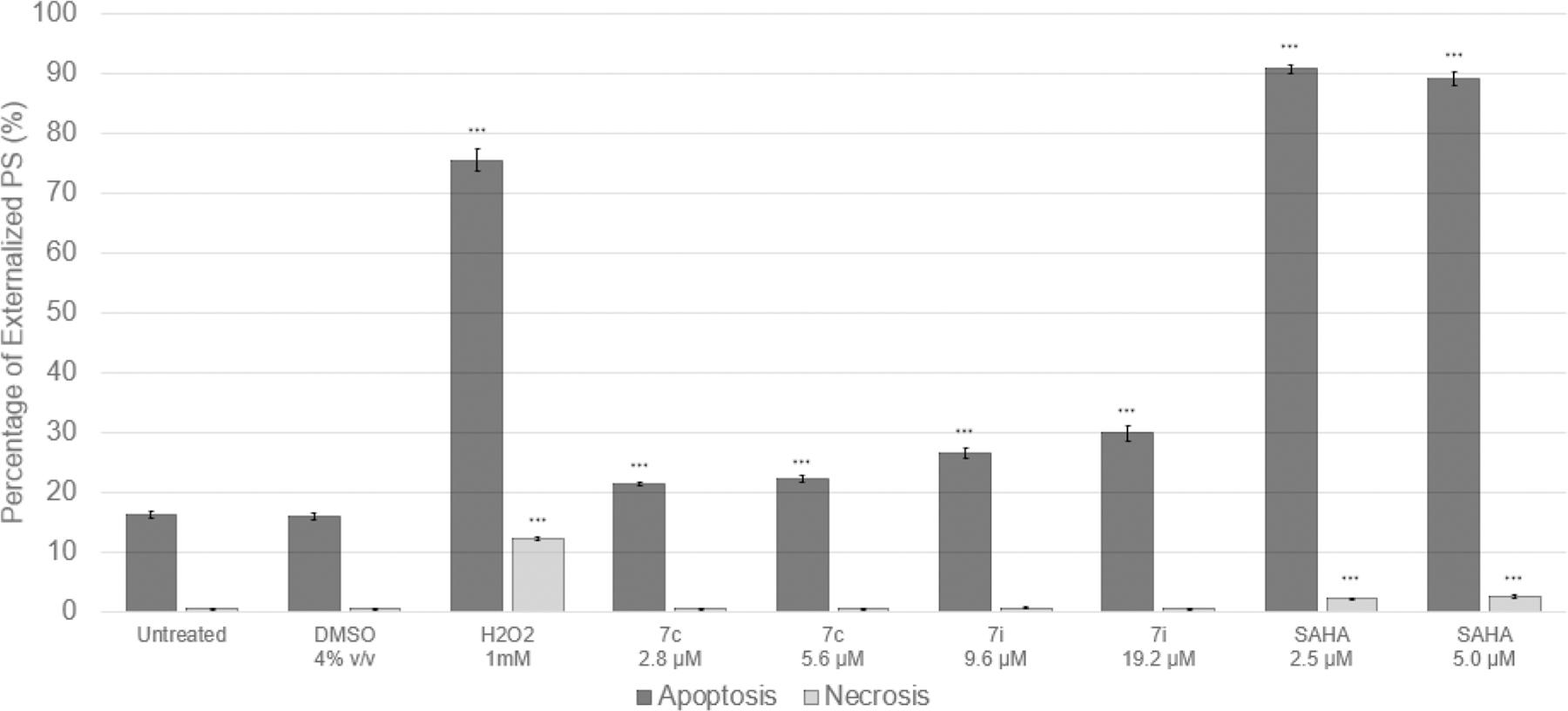
AnnexinV-FITC/PI Assay in CCRF-CEM after 72 h exposure to 7c, 7i, and controls (SAHA, H2O2); n = 3, ***p < 0.001.
Cell Cycle Analysis.
To evaluate the effect of 7c and 7i on the cell cycle, flow cytometry was used to quantify DNA content within each cell sample. CEM cells were exposed, in triplicate, to the CC50 or 2×CC50 of each compound for 72 h. HDAC inhibitor SAHA was likewise included for comparison. 4% v/v DMSO, 1 mM H2O2, and untreated cells were utilized as vehicle, positive, and negative controls repectively. After the incubation period, cells were collected and resuspended in a nuclear isolation medium containing DNA-intercalating dye DAPI (NIM-DAPI) immediately prior to analysis. DAPI fluorescent signal was used to quantify the replication status of cells within each sample, as its intensity is directly proportional to the amount of DNA. Approximately 40 000 events (cells) were analyzed per sample, and statistical significance was determined by Student’s t test (Tables S7–S9 in Supporting Information).
DNA fragmentation, represented by the sub-G0/G1 population, is a key feature of apoptosis. Despite significant apoptosis in our AnnexinV-FITC/PI assay at the same time point (Figure 8), just 6% of sub-G0/G1 accumulation was evident after treatment with 7c and 7i at either concentration. Compound 7c did not have a significant effect on the cell cycle, whereas G0/G1 arrest (p = 0.0103) was evident in 7i CC50-treated samples. Thus, identifying a distinct effect of each compound on the cell cycle which also differs from SAHA (Figure 9).
Figure 9.
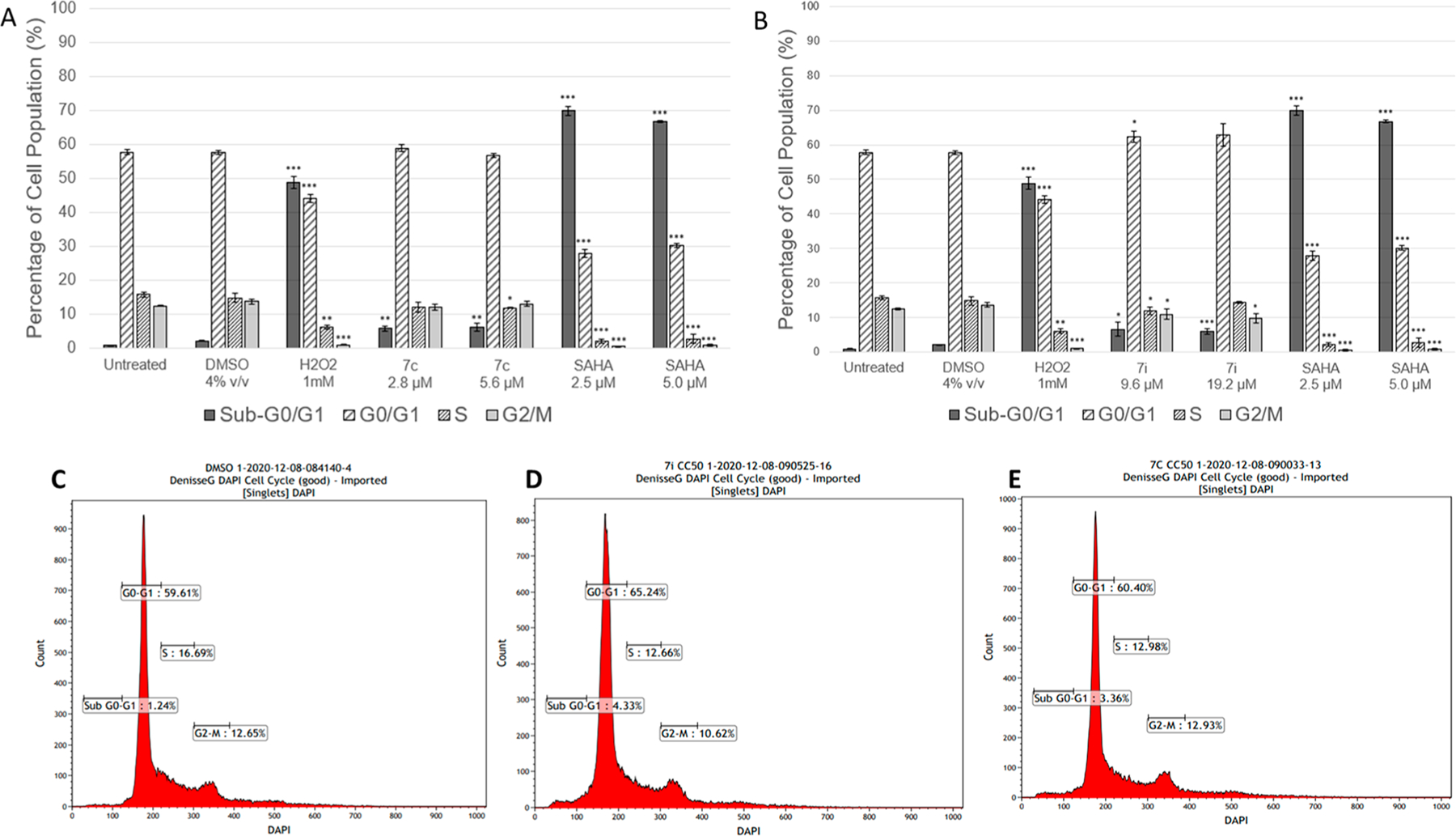
Cell cycle distributions in CCRF-CEM after 72 h exposure to 7c, 7i, and control SAHA (A). Graphical representation of distribution of the cells treated with 7c, untreated control, DMSO, H2O2, and SAHA (B). Graphical representation of distribution of the cells treated with 7i untreated control, DMSO, H2O2, and SAHA. Histograms of vehicle (C), compound 7i (D), compound 7c (E); n = 3, *p < 0.05, **p < 0.01, ***p < 0.001.
Western Blotting.
The c-Myc oncogene is known as a master controller of tumor cell growth regulation and metabolism, which are closely inter-related.71 The metabolic changes driven by c-Myc overexpression, which occurs often in tumor cells, are essential to support the synthesis of nucleic acids, and various proteins/lipids necessary for rapid cell proliferation.72 Thus, downregulation of the c-Myc gene could help to control proliferation of tumor cells. Treatment with both PPARγ agonists and HDAC inhibitors have shown to decrease expression of c-Myc. The PPARγ agonist class glitazones, which include insulin-sensitizing drugs like pioglitazone, have demonstrated an ability to downregulate this gene in various cancer cells.73 When treated with HDAC inhibitor SAHA, c-Myc is acetylated at L-323, and its expression decreases, leading to TRAIL activation and apoptosis.74 Thus, downregulation of c-Myc is an important aspect of antiproliferative effects exerted by these classes of compounds. Often, c-Myc is activated in different types of leukemia and plays a vital role in the induction and progression of leukemogenesis.75–78 To ascertain the effects of PPARγ and HDAC dual targeting compound 7c in CEM (T-cell leukemia) cells, immunoblotting was performed. Western blot analysis of compound 7c in CEM cells revealed downregulation of the c-Myc protein much greater than positive control pioglitazone (Figure 10). To assess if the observed suppression of c-Myc was a cascading effect associated with upregulation of tumor suppressor gene p53,79 this protein was also evaluated; however, no upregulated expression was observed with exposure to compound 7c. This p53-independence suggests that c-Myc suppression could be a feature of the dual HDAC inhibition and PPARγ agonism exerted by compound 7c.
Figure 10.
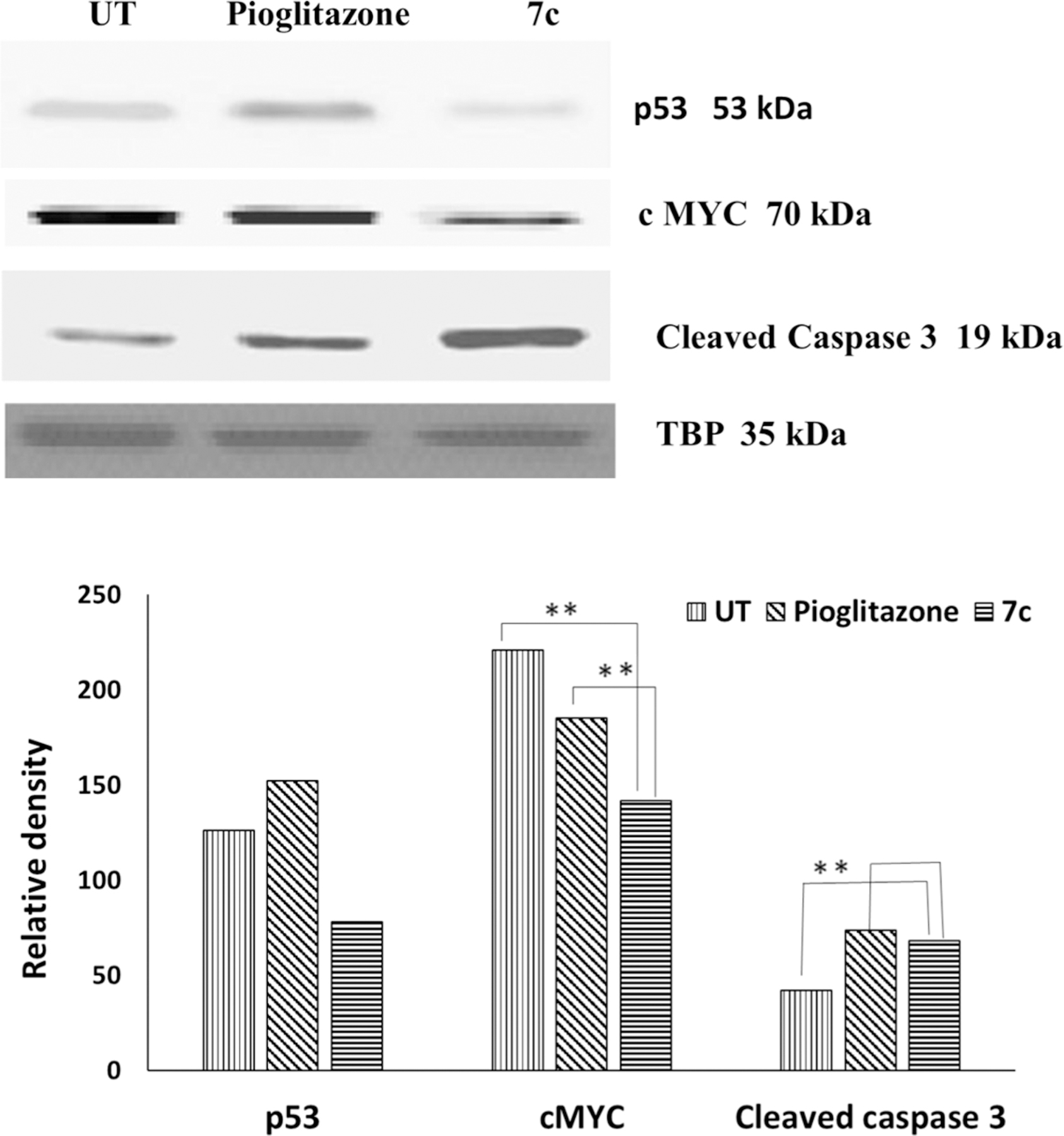
Western blotting of CEM cells incubated with compound 7c (10 μM) and positive control, pioglitazone (10 μM), and untreated cells (UT) for 5 h; n = 2, *p < 0.05, **p < 0.0001. The graph shows the normalized data with respect to TBP band.
Caspases are also known as key mediators of apoptosis,80 and among them, caspase-3 is a frequently activated death protease, which catalyzes the cleavage of many important cellular proteins leading to cell death.81 Both HDAC inhibitors (like SAHA) and PPARγ agonists (like pioglitazone) are linked to enhanced expression of cleaved caspases and apoptotic cell death.82–84 In agreement with these observations, Western blotting revealed an increased expression of cleaved caspase-3 with compound 7c treatment compared with positive control pioglitazone (Figure 10).
The compounds of “series 7” are the very first evidence of molecules simultaneously targeting HDAC and PPARγ. The overall aim of these experiments was an initial evaluation of the antitumor potential of this unique class of molecules. Several analogues, particularly compound 7c, demonstrated effective antitumor activity at micromolar concentrations and substantial selective cytotoxicity. Though further analyses are needed to fully elucidate their mechanism of action, preliminary data on “series 7” strongly supports apoptotic cell death. This is evident by the translocation of PS (AnnexinV-FITC/PI assay), DNA fragmentation (cell cycle analysis), and cleavage of caspase-3 (Western blotting). In addition, c-myc is an oncogene essential for cell cycle progression in human tumor cells, and its suppression has been associated with decreased cell proliferation and arrest, predominantly in G0/G1 phase of the cell cycle.85,86 The downregulation of c-myc that was detected in 7c-treated CEM cells opens the possibility that other analogues in the “series 7” may undergo c-myc dependent exit from the cell cycle and subsequent (apoptotic) death. Such may be the case with compound 7i, for example, considering the observation of G0/G1 phase arrest.
In Vivo Evaluation.
In vivo preclinical tumor growth inhibition has been regarded as an indispensable experiment to understand the expected activity of antitumor agents in humans and thus serves as important milestone in anticancer drug development. The most promising dual targeting derivative 7c, was further evaluated in vivo using a standard subcutaneous xenograft model produced using the CEM cell line. To investigate the capacity of compound 7c to inhibit murine tumor growth, SCID mice were implanted with CEM-derived tumor cells and the tumor allowed to grow to an approximate volume of 100 mm3. The animals were then assigned to three groups (n = 6 mice per treatment group) for treatment with control, Doxorubicin (20 mg/kg), or 7c (50 mg/kg). Treatments were given as described in the methods. As shown in Figure 11, administration of compound 7c significantly retarded tumor growth. This was reflected by reduction in tumor volume by ∼48% compared with the control group (Figure 11A) and a %TGI (tumor growth inhibition) of 24% on day 18. Body weights of the mice were not significantly affected by 7c or doxorubicin administration when compared to the control group, and there were no behavioral changes observed in any of the 7c treated mice. These results suggest that 7c significantly inhibits tumor growth with no signs of severe toxicity.
Figure 11.
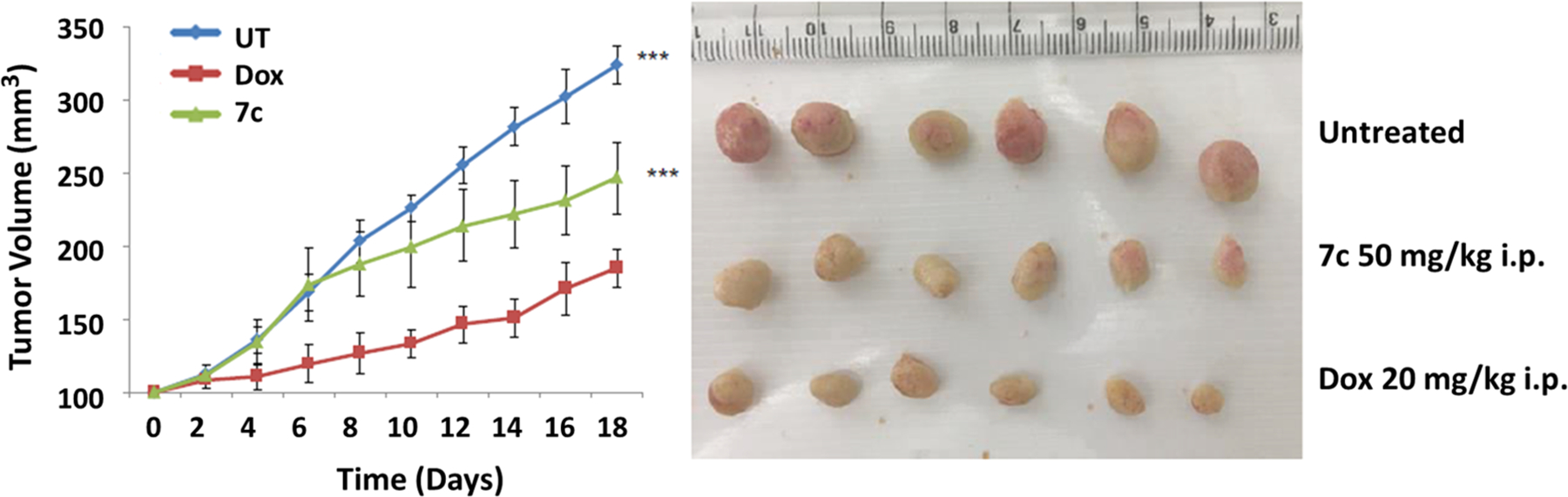
(A) Growth curve of tumor volume vs time in CEM (T cell leukemia) xenografts treated with doxorubicin 20 mg/kg i.p.,7c 50 mg/kg i.p., untreated saline i.p. Error bars represent standard error of the mean (SEM) n = 6. p < 0.001. (B) Excised tumor image of the animals treated with doxorubicin 20 mg/kg i.p., 7c 50 mg/kg i.p., untreated saline i.p.
Many HDAC inhibitors and PPARγ agonists with antitumor capacity have been evaluated for in vivo efficacy, wherein both classes of drugs have showed effective tumor regression at relatively high doses from 50 mg/kg to 400 mg/kg, %TGI ranging from 9% (for low doses) to 40% (at higher doses), with animals showing no sign of severe toxicity. For example, the recently studied HDAC inhibitor belinostat has shown in vivo efficacy in pancreatic cancer at doses of 100 mg/kg in nude mice. Also, its combination with gemcitabine had synergistic effects.87 Similarly, vorinostat was used at a concentration of 150 mg/kg in urothelial carcinoma.88 Several other newer HDAC inhibitors have also been evaluated in vivo in various cancer models.89–94 In the case of in vivo evaluation of PPARγ agonists in various cancer models, a similar trend is observed.95–98 Hence, dual-targeting compound 7c with %TGI of 24% appears more potent than single-targeted agents. Considering its in vivo efficacy at higher doses, 7c analogues and other HDAC inhibitors or PPARγ agonists might be used as an adjuvant to existing chemotherapy to help reduce resistance/toxicity issues and possibly enhance antitumor effects in a synergistic manner. Hence, the in vivo combinatorial effects of compound 7c should be further evaluated.
CONCLUSIONS
The clinical effectiveness of recent single target anticancer drugs has been limited because of unexpected resistance and target nonspecificity. Among the attempts to tackle this concern, a strategy of multitargeting approach could be applied which would be able to hit multiple cancer hallmarks to achieve the desired pharmacological effectiveness with reduced detrimental effects. The combination treatment of HDAC inhibitors and PPARγ agonists increased the cytotoxic effects in a synergistic/additive manner against different cancer cell lines resulting in the arrest of proliferation and increased apoptosis. On the basis of this literature evidence, in an attempt to bring about a multitarget drug approach and simultaneously target HDAC/PPARγ, a series of 25 novel TZD-based naphthylidene derivatives were rationally designed and successfully synthesized. Complete biological evaluation of all compounds revealed their dual targeting ability by partial activation of PPARγ and selective inhibition of HDAC4. In our knowledge, this is the first ever attempt to develop agents that simultaneously target HDAC and PPARγ, two potential targets in cancer drug discovery. Out of 25 compounds synthesized several were found to inhibit HDAC4 effectively and Six compounds showed dual targeting activity with submicromolar potencies; the most potent dual targeting agent 7i had PPARγ EC50 = 0.245 μM and HDAC4 IC50 = 1.1 μM. Out of these six compounds, 7c and 7i were also found to exhibit antiproliferative effects against CEM cells with CC50 values of 2.8 and 9.6 μM, respectively. Both of them also induced apoptosis and caused a significant DNA fragmentation in cell cycle analysis. Further, compound 7c was found to modulate the expression of c-Myc and cleave caspase-3 in Western blotting experiments. These in vitro antiproliferative effects were supported by in vivo tumor regression by compound 7c in CCRF-CEM tumor xenografts. Taken together, this study provides a theoretical basis for rational design of dual/multitargeting agents that could be developed further as anticancer therapeutics.
EXPERIMENTAL SECTION
Chemistry.
Commercial grade reagents of make S D Fine, Sigma-Aldrich or Research Lab were acquired from dealers in India. Thin-layer chromatography (TLC) was done on precoated Merck Silica Gel 60 F254. Melting points were determined by thermo-analytical technique using DSC (Differential scanning calorimetry) device II Nanotechnology (SEIKO) exstar DSC 6220. Infrared (FTIR) spectra were found using Schimadzu FT/IR-8400S with use of direct sampling procedure. 1H and 13C NMR spectra were noted on a Bruker instrument at 400 MHz, and chemical shift values (δ) are described in ppm. J values (Coupling constants) are stated in hertz (Hz). Abbreviations in NMR interpretation are, s- singlet, d-doublet, dd - doublet of doublet, t-triplet, m- multiplet, bs- broad singlet and q - quartet. Mass spectra were recorded with LC-MS Agilent Technologies 1260 Infinity instrument. The conditions of HPLC chromatography: column - Hemochrome C18 (4.6*250 mm), detector- PDA detector, detection wavelength - up to 400 nm, flow rate - 1 mL/min, sample concentration - 10 ppm, oven temperature - 30 °C; technique - gradient elution with a run time of 15 min, mobile phase - methanol:formic acid (0.1%) in 70:30 ratio, mass range-100 to 650 m/z. All final compounds achieved a minimum of 95% purity and molecular ions (of fraction of HPLC elute sent in mass spectrometer) detected in MS of was corresponding to the mass of the individual compounds.
Synthesis and Structural Characterization Details.
The phenyl acetamide intermediates, 2a–2y, were prepared as per the reported procedures and the details of the spectral data and structural characterization could be found in our previous reports.99–101
Synthesis of 5-((6-Hydroxynaphthalen-2-yl)methylene)-thiazolidine-2,4-dione (5).
Compound 5 was synthesized as previously described.52 In brief, toluene (10 mL), commercially available 6-hydroxy-2-naphthaldehyde (3) (6.88 g, 0.04 mmol), and 2,4-thiazolidinedione (4) (4.68 g, 0.04 mmol) was mixed together. To this mixture, was added a catalytic amount of piperidinium benzoate and refluxed in Dean stark apparatus. The reaction was monitored for completion by TLC, after 4–5 h reaction was stopped and mixture was cooled to RT (room temperature). Solid precipitated was collected by filtering under vacuum and washed with water to obtained intermediate 5 (Scheme 1) in crystalline form, which was further recrystallized from methanol. Yellow shiny crystals. Yield 8.8 g (87%). M.P. charred at 300 °C. FTIR (cm−1) 3390, 3119, 1672, 1662, 1587. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.16–7.19 (m, 2H), 7.55 (d, J = 8.6 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.88–7.90 (m, 2H), 8.04 (d, J = 4.0 Hz, 1H), 10.21 (s, 1H), 12.59 (bs, 1H).
Synthesis of Compounds Containing “Ar” as Substituted Aromatic Ring (7a–7l, 7o–7r, 7u–7v).
To the stirred solution of 2a–2l, 7o–7r, and 7u–7v (0.004 mol) in dimethylformamide (DMF, 10 mL) along with potassium carbonate (0.007 mol) was added, 5-((6-hydroxynaphthalen-2-yl)methylene)thiazolidine-2,4-dione, (5) (0.008 mol) and stirred for 24–48 h at RT. The reaction was immobilized with addition of 20 mL of water to get precipitates, which were separated by vacuum filtration, washed several times with water to remove solvent, and purified by TLC column chromatography using ethyl acetate:hexane (30:70) as mobile phase and F254 silica gel as stationary phase by the continuous elution method.
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-phenylacetamide (7a).
Yellow solid; Yield 49.5%; M.P. (DSC) 330.3 °C; IR (cm−1) 3377, 1737, 1687, 1330; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.48 (s, 2H), 7.10 (t, J = 7.4 Hz, 1H), 7.34 (t, J = 8.0 Hz, 2H), 7.40–7.41 (m, 2H), 7.65 (d, J = 4.0 Hz, 3H), 7.91–7.92 (m, 1H), 7.92–7.95 (m, 1H), 8.02 (d, J = 8.0 Hz, 1H), 8.13 (s, 1H), 10.16 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 67.173 (−CH2), 107.325–138.282 (Aromatic), 157.408 (C═C, benzylidene), 166.135, 167.978 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.81%, 3.60 min, 355, 354 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(4-fluorophenyl)acetamide (7b).
Yellow solid; Yield 55.7%; M.P. (DSC) 340.9 °C; IR (cm−1) 3032, 1737, 1680, 1329; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.86 (s, 2H), 7.18 (t, J = 8.0 Hz, 2H), 7.39–7.41 (m, 2H), 7.63–7.69 (m, 3H), 7.90–7.95 (m, 1H), 8.01–8.03 (m, 1H), 8.13 (s, 1H), 10.23 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 67.132 (−CH2), 107.343–134.687 (Aromatic), 157.347 (C═C, benzylidene), 166.118, 167.364, 167.968 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 96.19%, 12.36 min, 422.07, 421.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(p-tolyl)acetamide (7c).
Yellow solid; Yield 52.6%; M.P. (DSC) 298.7 °C; IR (cm−1) 3074, 1737, 1693, 1349; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.26 (s, 3H), 4.85 (s, 2H), 7.14–7.15 (m, 2H), 7.40–7.41 (m, 2H), 7.53–7.55 (m, 3H), 7.91–7.94 (m, 3H), 8.13 (s, 1H), 10.09 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 20.420 (−CH3), 67.168 (−CH2), 107.308–135.727 (Aromatic), 157.398 (C═C, benzylidene), 165.912, 167.372, 167.991 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.73%, 6.70 min, 418.1, 417.1 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(4-methoxyphenyl)acetamide (7d).
Yellow solid; Yield 53.6%; M.P. (DSC) 290.6 °C; IR (cm−1) 3041, 1739, 1683, 1327; 1H NMR (400 MHz, DMSO-d6, δ ppm) 3.72 (s, 3H), 4.83 (s, 2H), 6.89–6.94 (m, 2H), 7.33–7.41 (m, 2H), 7.55–7.57 (m, 2H), 7.63–7.65 (m, 1H), 7.90–7.94 (m, 2H), 8.00–8.03 (m, 1H), 8.13 (s, 1H), 10.02 (s, 1H), 12.68 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 54.947 (−OCH3), 67.182 (−CH2), 105.153–139.947 (Aromatic), 157.549 (C═C, benzylidene), 166.856, 167.420, 167.526 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 99.40%, 5.71 min, 434.09, 433.1 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(3-(trifluoromethyl)phenyl) acetamide (7e).
Yellow solid; Yield 59.2%; M.P. (DSC) 292.8 °C; IR (cm−1) 3055, 1749, 1687, 1332; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.91 (s, 2H), 7.39–7.46 (m, 3H), 7.57–7.61 (m, 1H), 7.64–7.66 (m, 1H), 7.90–7.95 (m, 3H), 8.02 (d, J = 8.0 Hz, 1H), 8.14–8.15 (m, 2H), 10.16 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.670 (−CH2), 107.065–135.851 (Aromatic), 157.501 (C═C, benzylidene), 166.506, 166.562, 167.868 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 95.54%, 7.37 min, 472.1, 471.1 [(M−H)+, 100%].
N-(3-Chlorophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7f).
Yellow solid; Yield 42.4%; M.P. (DSC) 322.5 °C; IR (cm−1) 3093, 1737, 1679, 1330, 677; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.89 (s, 2H), 7.14–7.17 (m, 1H), 7.35–7.41 (m, 3H), 7.55–7.58 (m, 1H), 7.63–7.66 (m, 1H), 7.85–7.86 (m, 2H), 7.90–7.94 (m, 1H), 8.00–8.03 (m, 1H), 8.13 (s, 1H), 10.35 (s, 1H), 12.62 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.981 (−CH2), 107.151–134.680 (Aromatic), 157.875 (C═C, benzylidene), 166.565, 167.553, 167.863 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.98%, 8.54 min, 438.04, 437.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(3-methoxyphenyl)acetamide (7g).
Yellow solid; Yield 48.0%; M.P. (DSC) 277.6 °C; IR (cm−1) 3016, 1743, 1681, 1334; 1H NMR (400 MHz, DMSO-d6, δ ppm) 3.74 (s, 3H), 4.86 (s, 2H), 6.69–6.69 (m, 1H), 7.23–7.23 (m, 2H), 7.36–7.40 (m, 3H), 7.63–7.65 (m, 1H), 7.90–8.02 (m, 3H), 8.00–8.03 (m, 1H), 8.12 (s, 1H), 10.16 (s, 1H), 12.62 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.981 54.988 (−OCH3), 67.153 (−CH2), 105.423–139.477 (Aromatic), 157.384 (C═C, benzylidene), 159.482, 166.182, 167.321, 167.947 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 95.83%, 6.16 min, 434.09, 433.1 [(M−H)+, 100%].
N-(3,4-Dichlorophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7h).
Yellow solid; Yield 36.7%; M.P. (DSC) 321.5 °C; IR (cm−1) 3076, 1735, 1685, 1325; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.90 (s, 2H), 7.39–7.41 (m, 1H), 7.61–7.66 (m, 3H), 7.90–7.95 (m, 3H), 8.01–8.04 (m, 2H), 8.14 (s, 1H), 10.46 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.505 (−CH2), 104.987–134.507 (Aromatic), 157.790 (C═C, benzylidene), 166.630, 167.630 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 100.0%, 14.14 min, 471.01, 471.1 [(M−H)+, 100%].
N-(2-Bromo-4,6-difluorophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)oxy)acetamide (7i).
Yellow solid; Yield 25.6%; M.P. (DSC) 265.8 °C; IR (cm−1) 3230, 1735, 1685, 1348; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.94 (s, 2H), 7.43–7.49 (m, 3H), 7.62–7.67 (m, 2H), 7.92–8.04 (m, 3H), 8.14 (s, 1H), 10.08 (s, 1H), 12.60 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.790 (−CH2), 104.491–134.630 (Aromatic), 157.138 (C═C, benzylidene), 166.987, 167.987 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 95.72%, 5.94 min, 517.97, 518.9 [(M+H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(2-phenoxyphenyl)acetamide (7j).
Yellow solid; Yield 49.0%; M.P. (DSC). 260.0 °C; IR (cm−1) 3074, 1737, 1680, 1330; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.87 (s, 2H), 6.95–7.00 (m, 3H), 7.12–7.20 (m, 3H), 7.24 (d, J = 8.0 Hz, 1H), 7.34–7.39 (m, 3H), 7.63 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.90 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 8.1 (m, 2H), 10.16 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.790 (−CH2), 104.491–134.630 (Aromatic), 157.138 (C═C, benzylidene), 166.987, 167.987 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 95.72%, 5.94 min, 517.97, 518.9 [(M+H)+, 100%].
N-(2,4-Difluorophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7k).
Yellow solid; Yield 50.3%; M.P. (DSC). 306.1 °C; IR (cm−1) 3290, 1755, 1685, 1329; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.92 (s, 2H), 7.10 (m, 1H), 7.34–7.41 (m, 3H), 7.65–7.67 (m, 1H), 7.73–7.75 (m, 1H), 7.89–7.94 (m, 2H), 8.02 (d, J = 8 Hz, 1H), 8.13 (s, 1H), 10.02 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.987 (−CH2), 107.621–134.680 (Aromatic), 157.875 (C═C, benzylidene), 166.565, 167.553, 167.863 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 99.00%, 6.44 min, 440.06, 439.1 [(M−H)+, 100%].
N-(4-Bromo-2-methylphenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)oxy)acetamide (7l).
Yellow solid; Yield 55.9%; M.P. (DSC) 301.7 °C; IR (cm−1) 3288, 1732, 1687, 1348; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.18 (s. 3H), 4.90 (s, 2H), 7.38–7.46 (m, 5H), 7.64–7.67 (m, 1H), 7.90–7.94 (m, 2H), 8.01 (d, J = 12 Hz, 1H), 8.13 (s, 1H), 9.65 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 19.096 (−CH3), 79.658 (−CH2), 114.429–134.589 (Aromatic), 157.952 (C═C, benzylidene), 162.611 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.07%, 10.63 min, 496.01, 497.0 [(M+H)+, 100%].
N-(4-Bromophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7o).
Yellow solid; Yield 51.2%; M.P. (DSC) 316.3 °C; IR (cm−1) 3028, 1730, 1691, 1352; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.87 (s, 2H), 7.38–7.41 (m, 2H), 7.51–7.53 (m, 2H), 7.64–7.66 (m, 3H), 7.91–7.94 (m, 2H), 8.02 (d, J = 12 Hz, 1H), 8.13 (s, 1H), 10.31 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 67.159 (−CH2), 107.355–137.682 (Aromatic), 157.338 (C═C, benzylidene), 166.364, 167.306, 167.928 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.84%, 10.55 min, 481.99, 482.9 [(M+H)+, 100%].
N-(3-Chloro-5-(trifluoromethyl)phenyl)-2-((6-((2,4-dioxothiazoli-din-5-ylidene)methyl)naphthalen-2-yl)oxy)acetamide (7p).
Yellow solid; Yield 44.4%; M.P. (DSC) 313.9 °C; IR (cm−1) 2968, 1726, 1691, 1327; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.97 (s, 2H), 7.36–7.38 (m, 2H), 7.64 (t, J = 4.0 Hz, 3H), 7.91–7.94 (m, 2H), 8.01 (d, J = 8.0 Hz, 2H), 8.30 (s, 1H), 11.11 (s, 1H), 12.68 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 67.851 (−CH2), 107.132–134.554 (Aromatic), 157.521 (C═C, benzylidene), 166.029, 167.687, 167.347 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 99.44%, 9.38 min, 506.03, 505.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(4-nitrophenyl)acetamide (7q).
Yellow solid; Yield 39.0%; M.P. (DSC) 332.0 °C; IR (cm−1) 3010, 1734, 1689, 1329; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.96 (s, 2H), 7.39–7.42 (m, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.92–7.95 (m, 3H), 8.03 (d, J = 8.0 Hz, 1H), 8.14 (s, 1H), 8.25–8.27 (m, 2H), 10.80 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 67.073 (−CH2), 119.389–131.760 (Aromatic), 143.684 (C-NO2 str.), 157.338 (C═C, benzylidene), 162.367 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.10%, 8.60 min, 449.07, 448.0 [(M−H)+, 100%].
N-(4-Bromo-2-fluorophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)oxy)acetamide (7r).
Yellow solid; Yield 32.6%; M.P. (DSC) 314.6 °C; IR (cm−1) 3317, 1732, 1687, 1319; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.95 (s, 2H), 7.37–7.43 (m, 3H), 7.63–7.66 (m, 2H), 7.79–7.83 (m, 1H), 7.91–7.94 (m, 2H), 8.02 (d, J = 8 Hz, 1H), 8.14 (s, 1H), 10.16 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.670 (−CH2), 107.236–135.584 (Aromatic), 157.433 (C═C, benzylidene), 166.622, 166.710, 167.921 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 99.23%, 11.00 min, 499.98, 500.9[(M+H)+, 100%].
N-(3-Chloro-4-methylphenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)oxy)acetamide (7u).
Yellow solid; Yield 44.4%; M.P. (DSC) 304.0 °C; IR (cm−1) 3223, 1737, 1678, 1323; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.28 (s, 3H), 4.86 (s, 2H), 7.30–7.32 (m, 1H), 7.39–7.41 (m, 2H), 7.47–7.49 (m, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.84 (s, 1H), 7.91–7.95 (m, 2H), 8.02 (d, J = 8.0 Hz, 1H), 8.29 (s, 1H), 10.24 (s, 1H), 12.63 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 19.658 (−CH3), 79.096 (−CH2), 114.294–134.611 (Aromatic), 157.333 (C═C, benzylidene), 162.429 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 100.0%, 11.92 min, 452.06, 451.1 [(M−H)+, 100%].
N-(3,4-Dibromophenyl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7v).
Yellow solid; Yield 51.9%; M.P. (DSC) 318.4 °C; IR (cm−1) 3061, 1735, 1680, 1325; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.90 (s, 2H), 7.40–7.42 (m, 2H), 7.61–7.67 (m, 3H), 7.91–7.95 (m, 3H), 8.02–8.05 (m, 1H), 8.30 (s, 1H), 10.45 (s, 1H), 12.62 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 79.120 (−CH2), 117.474–134.671 (Aromatic), 157.429 (C═C, benzylidene), 162.290, 167.063 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 100.0%, 13.32 min, 559.9, 558.9 [(M−H)+, 100%].
Synthesis of Intermediate 6m, 6n, 6s, 6t, 6w–6y.
To the stirred solution of 6-hydroxynaphthaldehyde (3) (0.008 mol) along with potassium carbonate (0.007 mol) in DMF was added the appropriate phenyl acetamides (2m, 2n, 2s, 2t, 2w–2y) (0.004 mol), and the reaction mixture was stirred on magnetic stirrer for 36 h for 6m, 6n, 6s, 6t and for 48 h for 6w–6y. After completion of the reaction water was added to terminate the reaction and generate solid. The solid obtained was washed with water (3 × 50 mL) and dried. It was purified by recrystallization from the appropriate solvents.
2-((6-Formylnaphthalen-2-yl)oxy)-N-(pyridin-2-yl)acetamide (6m).
Brown solid; Yield 63.3%; M.P. 188.9 °C; IR (cm−1) 2918, 1722, 1678, 1301; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.92 (s, 2H), 7.02 (d, J = 16.0 Hz, 2H), 7.41–7.44 (m, 2H), 7.67–7.70 (m, 1H), 7.83–7.86 (m, 3H), 7.91–7.94 (m, 1H), 8.10–8.12 (m, 1H), 8.53 (s, 1H), 10.06 (s, 1H), 10.62 (s, 1H).
2-((6-Formylnaphthalen-2-yl)oxy)-N-(6-methylpyridin-2-yl)-acetamide (6n).
Brown solid; Yield 75.6%; M.P. 148.9 °C; IR (cm−1) 2922, 1722, 1678, 1303; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.43 (s, 3H), 4.98 (s, 2H), 7.01 (d, J = 8.0 Hz, 2H), 7.40–7.43 (m, 2H), 7.66–7.70 (m, 1H), 7.84–7.88 (m, 2H), 7.92–7.94 (m, 1H), 8.11–8.13 (m, 1H), 8.51 (s, 1H), 10.08 (s, 1H), 10.59 (s, 1H).
N-(6-Fluoropyridin-2-yl)-2-((6-formylnaphthalen-2-yl)oxy)-acetamide (6s).
Brown solid; Yield 56.4%; M.P. 202.9 °C; IR (cm−1) 3066, 1721, 1678, 1334; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.99 (s, 2H), 7.01–7.03 (m, 2H), 7.41–7.43 (m, 2H), 7.69–7.72 (m, 1H), 7.84–7.86 (m, 2H), 7.92–7.94 (m, 1H), 8.11–8.13 (m, 1H), 8.51 (s, 1H), 10.06 (s, 1H), 10.58 (s, 1H).
2-((6-Formylnaphthalen-2-yl)oxy)-N-(5-methylisoxazol-3-yl)-acetamide (6t).
White solid; Yield 56.1%; M.P. 213.5 °C; IR (cm−1) 3300, 1722, 1678, 1334; 1H NMR (400 MHz, DMSO-d6, δ ppm)2.26(s, 3H), 4.85 (m, 1H), 7.13 (d, J = 8.0 Hz,1H), 7.43–7.44(m, 1H), 7.53–7.57 (m, 1H), 7.85–7.87 (m, 1H), 7.93–7.95(m, 1H), 8.13(d, J = 8.0 Hz, 1H), 8.51 (s, 1H), 10.04 (s, 1H), 10.08 (s, 1H).
N-(Benzo[d]thiazol-2-yl)-2-((6-formylnaphthalen-2-yl)oxy)-acetamide (6w).
Brownish solid; Yield 41.1%; M.P. 285.2 °C; IR (cm−1) 3383, 1726, 1680, 1334; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.82 (s, 2H), 7.02–7.05 (m, 1H), 7.20–7.23 (m, 1H), 7.36–7.38 (m, 2H), 7.47–7.48 (d, J = 4.0 Hz,1H), 7.67–7.69 (d, J = 8.0 Hz, 1H), 7.80 (m, 1H), 7.82 (m, 1H), 7.88–7.89 (m, 1H), 8.08–8.10 (d, J = 8.0 Hz, 1H), 8.49 (s, 1H), 10.06 (s, 1H).
2-((6-Formylnaphthalen-2-yl)oxy)-N-(4-methylbenzo[d]thiazol-2-yl)acetamide (6x).
Brownish solid; Yield 28.4%; M.P. 243.1 °C; IR (cm−1)3161, 1693, 1658, 1332; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.60 (s, 3H), 5.09 (s, 2H), 7.31 (t, J = 8.0 Hz,1H), 7.34 (m, 1H), 7.45 (t, J = 8.0 Hz,1H), 7.61 (d, J = 4.0 Hz, 1H),), 7.78 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.91–9.32 (m, 1H), 7.97–8.02 (m, 2H), 8.11 (s, 1H), 10.57 (s, 1H).
N-(6-Ethoxybenzo[d]thiazol-2-yl)-2-((6-formylnaphthalen-2-yl)-oxy)acetamide (6y).
Brownish solid; Yield 33.2%; M.P. 263.8 °C; IR (cm−1) 3342, 1680, 1384; 1H NMR (400 MHz, DMSO-d6, δ ppm) 1.30–1.33 (m, 3H), 3.98–4.07 (m, 2H), 4.79 (s, 2H), 6.80–6.82 (m, 1H), 7.26 (m, 1H), 7.35–7.37 (m, 3H), 7.80–7.82 (m, 1H), 7.87–7.89 (m, 1H), 8.07–8.09 (m, 1H), 8.48 (s, 1H), 10.06 (s, 1H).
Synthesis of Compounds Containing “Ar” as Substituted Heterocyclic Ring: 7m, 7n, 7s, and 7t.
Final compound 7m was synthesized by adopting Scheme 2, wherein 2,4-thiazolidinedione (3) (0.005 mol) was dissolved in 2-methoxyethanol (10 mL). To this solution was added piperidine (0.4 mL) dropwise followed by addition of intermediate, 6m (0.002 mol). The reaction mixture was refluxed for 3–5 h and monitored by TLC, after which the reaction was stopped, cooled, and the precipitated solid was collected by filtration and washed with water (3 × 50 mL) followed by methanol (3 × 10 mL). The crude product thus obtained was purified by TLC column chromatography by using hexane:ethyl acetate (50:50 to 25:75 ratio) to give the final products 7m. The other products 7n, 7s, and 7t were obtained by following the same procedure.
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(pyridin-2-yl)acetamide (7m).
Light brown solid; Yield 46.2%; M.P. (DSC) 324.8 °C; IR (cm−1) 3078, 1718, 1681, 1301; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.97 (s, 2H), 7.14–7.17 (m, 1H), 7.36–7.38 (m, 2H), 7.64 (d, J = 8.0 Hz, 1H), 7.81 (t, J = 8.0 Hz, 1H), 7.90–7.93 (m, 2H), 8.05–8.07 (m, 2H), 8.13 (s, 1H), 8.35–8.36 (m, 1H), 10.57 (s, 1H), 12.64 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 71.033 (−CH2), 107.295–138.393 (Aromatic), 148.148 (C═C, benzylidene), 167.001 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 97.19%, 5.78 min, 405.08, 404.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(6-methylpyridin-2-yl)acetamide (7n).
Light brown solid; Yield 42.2%; M.P. (DSC) 274.3 °C; IR (cm−1) 3030, 1726, 1689, 1348; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.43 (s, 3H), 4.95 (s, 2H), 7.00 (d, J = 4.0 Hz, 1H), 7.35–7.37 (m, 2H), 7.62–7.65 (m, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.90–7.92 (m, 3H), 7.99–8.01 (m, 1), 8.13 (s, 1H), 10.56 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 22.497 (−CH3), 71.033 (−CH2), 107.393–138.952 (Aromatic), 148.001 (C═C, benzylidene), 167.148 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 97.41%, 7.88 min, 419.09, 418.1 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(6-fluoropyridin-2-yl)acetamide (7s).
Yellow solid; Yield 43.9%; M.P. (DSC) 307.6 °C; IR (cm−1) 3115, 1712, 1680, 1352; 1H NMR (400 MHz, DMSO-d6, δ ppm) 4.96 (s, 2H), 7.36–7.38 (m, 2H), 7.63–7.65 (m, 1H), 7.71–7.81 (m, 1H), 7.90–7.93 (m, 2H), 8.00 (d, J = 8.0 Hz, 1H), 8.10–8.12 (m, 2H), 8.37(s, 1H), 10.74 (s, 1H), 12.62 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.981 (−CH2), 107.372–134.692 (Aromatic), 147.009 (C N str.), 157.448 (C═C, benzylidene), 166.364, 167.306, 167.928 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 97.77%, 6.02 min, 423.07, 422.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(5-methylisoxazol-3-yl)acetamide (7t).
Yellow solid; Yield 36.7%; M.P. (DSC) 292.4 °C; IR (cm−1) 3010, 1743, 1676, 1350; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.38 (s, 3H), 4.93 (s, 2H), 6.64 (s, 1H), 7.36–7.38 (m, 2H), 7.63–7.66 (m, 1H), 7.91–7.94 (m, 2H), 8.01 (d, J = 8.0 Hz, 1H), 8.3 (s, 1H), 11.18 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 17.898 (−CH3), 66.196 (−CH2), 107.415–134.649 (Aromatic), 157.332 (C═C, benzylidene), 167.333, 168.172 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 98.32%, 5.28 min, 409.07, 408.0 [(M−H)+, 100%].
Synthesis of Compounds Containing “Ar” as Substituted Heteroaryl Ring: 7w, 7x, and 7y.
Final compound 7w was synthesized by the route depicted in Scheme 3, wherein 2,4-thiazolidinedione (3) (0.005 mol) was dissolved in DMF (10 mL). To this solution, was added piperidine (0.8 mL) dropwise followed by addition of intermediate, 6w (0.002 mol). The reaction mixture was refluxed for 3–5 h and monitored by TLC, after which the reaction was stopped, cooled, and 3.0 mL of 30% acetic acid was added to generate the solid which was collected by filtration and washed with water (3 × 50 mL) followed by methanol (3 × 10 mL). The crude product thus obtained was purified by TLC column chromatography by using hexane:ethyl acetate (50:50 to 25:75 ratio) to give the final products 7w.
The other products 7x and 7y were obtained with a similar procedure.
N-(Benzo[d]thiazol-2-yl)-2-((6-((2,4-dioxothiazolidin-5-ylidene)-methyl)naphthalen-2-yl)oxy)acetamide (7w).
Yellow solid; Yield 25.5%; M.P. (DSC) 342.0 °C; IR (cm−1) 3171, 1735, 1685, 1325; 1H NMR (400 MHz, DMSO-d6, δ ppm) 5.10 (s, 2H), 7.34 (t, J = 6.0 Hz, 1H), 7.40–7.42 (m, 2H), 7.47 (t, J = 8.0 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.91–7.95 (m, 2H), 7.99–8.04 (m, 2H), 8.14 (s, 1H), 10.24 (s, 1H), 12.61 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 66.245 (−CH2), 107.235–134.605 (Aromatic), 148.279 (C N str.), 157.429 (C═C, benzylidene), 162.331, 167.485, 168.136 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 99.27%, 9.19 min, 461.05, 460.0 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(4-methylbenzo[d]thiazol-2-yl)acetamide (7x).
Yellow solid; Yield 20.5%; M.P. (DSC) 331.2 °C; IR (cm−1) 3113, 1737, 1689, 1325; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.60 (s, 3H), 4.87 (s, 2H), 7.21–7.24 (m, 2H), 7.27–7.29 (m, 2H), 7.65 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.91–7.95 (m, 2H), 8.03 (d, J = 8.0 Hz, 1H), 8.14 (s, 1H), 10.24 (s, 1H), 12.78 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 17.898 (−CH3), 66.196 (−CH2), 107.415–134.649 (Aromatic), 157.332 (C═C, benzylidene), 167.333, 168.172 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 100.0%, 8.58 min, 475.07, 474.1 [(M−H)+, 100%].
2-((6-((2,4-Dioxothiazolidin-5-ylidene)methyl)naphthalen-2-yl)-oxy)-N-(6-ethoxybenzo[d]thiazol-2-yl)acetamide (7y).
Yellow solid; Yield 20.7%; M.P. (DSC). 347.5 °C; IR (cm−1) 3288, 1732, 1681, 1332; 1H NMR (400 MHz, DMSO-d6, δ ppm) 2.83–2.88 (m, 3H), 4.05–4.06 (m, 2H), 5.06 (s, 2H), 7.03–7.04 (m, 1H), 7.39–7.46 (m, 2H), 7.55 (s, 1H), 7.62–7.69 (m, 1H), 7.86–7.94 (m, 2H), 7.99–8.01 (m, 1H), 8.11–8.14 (m, 1H), 8.51 (s, 1H), 10.08 (s, 1H), 12.54 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 14.624 (−CH3), 64.552 (O−CH2−CH3), 67.347 (−CH2), 107.398–132.003 (Aromatic), 157.302 (C═C, benzylidene), 192.785 (C═O); LC-MS (% Area, RT, Theoretical mass, m/z, I%) 100.0%, 13.36 min, 505.08, 504.1 [(M−H)+, 100%].
HDAC4/8 Activity Assay.
HDAC8 was produced by recombinant technique as described recently.52,102 Recombinant cHDAC4 was expressed using a pET14b vector (Novagen, EMD Millipore) containing the codon-optimized catalytic domain of human HDAC4. The assay was performed as described recently.52 Consecutive dilutions of test compounds in a buffer (75 mM KCl, 25 mM Tris-HCl, pH 8.0, 0.001% Pluronic F-127) were made. Test compound solutions were added and allowed to incubate for 1 h with HDAC8 and HDAC4 isoforms, in a 96-well black microtiter half a rea plate (Greiner) at 30 °C. Then the reaction was started by adding 20 μM of Boc-Lys(trifluoroacetyl)-AMC (Bachem) as a substrate for HDAC4 and HDAC8. After incubation for 60 min at 30 °C, the reaction was terminated by adding 1.7 μM SATFMK for both HDAC4 and HDAC8. The deacetylated substrate was transformed into a fluorescent product by adding 0.4 mg/mL trypsin (Applichem). The release of AMC was traced in a microplate reader (PheraStar Plus, BMG Labtech) at 450 nm (λEx = 350 nm) and were correlated to the activity of enzyme. Dose–response curves (DRCs) were plotted, and IC50 values were calculated.
Docking Method for HDAC4 and HDAC8.
Modeling, preparation, and visualization of structural data as well as molecular docking was performed using MOE 2019 software (Chemical Computing Group ULC, Canada). Three crystal structures, PDB IDs 3SFF, 4CBY and 2VQJ, were obtained from RCSB protein data bank and subjected to the Quickprep procedure of MOE 2019 including 3D protonation for subsequent docking. The partial charges of all protein and ligand atoms were calculated using the implemented Amber14 force field. The docking site was defined by the ligand within the binding pocket of the respective crystal structure. This approach was not permissible for the open conformation of HDAC4 (PDB ID: 2VQJ) since the ligand covered only part of the significantly enlarged binding groove. In this special case, the binding site was analyzed using the Computed Atlas of Surface Topography of proteins (CASTp) (http://sts.bioe.uic.edu/castp/index.html?201l).103 The largest identified pocket with an estimated volume of 1019,7 Å3 was identical with the widely opened binding groove of HDAC4o. The flanking amino acids of this pocket (G36, R37, G331, G330, H198, H158, H159, F227, P156, P155, F168, S123, R154) were used to define the binding site of HDAC4o for the subsequent docking procedure. Molecular docking was performed choosing the triangle matcher for placement of the ligand in the binding site and ranked with the London dG scoring function. The best 50 poses were passed to the refinement and energy minimization in the pocket using the induced fit method, and the 10 best poses were rescored using the GBVI/WSA dG scoring function.
PPARγ Transactivation Assay.
Plasmids.
The expression vector expressing the chimeric receptor containing the yeast Gal4 DNA binding domain fused to the human PPARγ ligand binding domain (LBD), and the reporter plasmid for this Gal4 chimeric receptor (pGal5TKpGL3) containing five repeats of the Gal4 response elements upstream of a minimal thymidine kinase promoter that is adjacent to the luciferase gene were described previously.104
Cell Culture and Transfections.
Human hepatoblastoma cell line HepG2 (Interlab Cell Line Collection, Genoa, Italy) was cultured in minimum essential medium (MEM) containing 10% heat-inactivated fetal bovine serum, penicillin G (100 U mL−1), and streptomycin sulfate (100 μg mL−1) at 37 °C in a humidified atmosphere of 5% CO2. For transactivation assays, 105 cells per well were seeded in a 24-well plate, and transfections were performed after 24 h with CAPHOS, a calcium-phosphate method, according to the manufacturer’s guidelines. Cells were transfected with expression plasmids encoding the fusion protein Gal4–PPARγ LBD (30 ng), pGal5TKpGL3 (100 ng), pCMVβgal (250 ng). Four hours after transfection, cells were treated for 20 h with the indicated ligands in triplicate. Luciferase activity in cell extracts was then determined by a luminometer (VICTOR3 V Multilabel Plate Reader, PerkinElmer). β-Galactosidase activity was determined using ortho-nitrophenyl-β-D-galactopyranoside as described previously.105,106 All transfection experiments were repeated at least twice. Reference compounds, media, and other cell culture reagents were purchased from Sigma-Aldrich (Milan, Italy).
Docking Method for PPARγ.
Computational Chemistry.
Molecular modeling and graphics manipulations were performed using Maestro 11.0 (Schrödinger, LLC, New York, NY, 2020)107 and UCSF-Chimera 1.14 (http://www.cgl.ucsf.edu/chimera) software packages108 running on a E4 Computer Engineering E1080 workstation provided of a Intel Core i7–930 Quad-Core processor. Figures were generated using Pymol 2.4 (Schrödinger, LLC, New York, NY, 2020). 2D ligand-interaction diagram was made using LigPlot+ v.2.2 (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/).109,110
Protein and Ligand Preparation.
The starting coordinates of PPARγ in complex with CAY10638 (PDB ID: 6DGR),64 retrieved from Brookhaven Protein Database, were employed for the docking calculations. The protein was processed with the Protein Preparation Wizard implemented in Maestro. The appropriate bond orders as well as charges and atom types were assigned. Hydrogen atoms were added to the protein consistent with the neutral physiologic pH. The guanidine and ammonium groups of arginine and lysine side chains were considered cationic, whereas the carboxylate groups of the aspartic and glutamic residues were considered anionic. Two well-defined water molecules, HOH708 and HOH706, bridging between CAY10638 and the β-sheet were retained and included in the docking experiments, while the remaining crystallographic water molecules were deleted. The H-bond network was optimized by exhaustive sampling of rotamers, tautomers, and protonation states of titratable amino acids at neutral pH. Imidazole rings H323 and H449 into PPARγ were set in their Nε 2-H (N tau-H) tautomeric state. Finally, the protein hydrogens only were minimized using the Impref module of Impact with the OPLS_2005 force field.
The core structure of compounds 7i, 7l, 7o, 7r, 7c, 7q were sketched using the Molecular Builder module in Maestro. The ligands were then preprocessed with LigPrep 3.3 (Schrödinger, LLC, New York, NY, 2020) and optimized by means of Macromodel 11.5 (Schrödinger, LLC, New York, NY, 2020), employing the MMFFs force field with 1000 steps of steepest descent; the resulting molecules were then submitted to 500 steps of truncated Newton conjugate gradient method. Partial atomic charges were assigned using the OPLS-AA force field.
Docking Calculations.
Docking studies were performed using the Schrödinger Glide algorithm111,112 implemented in Maestro. The docking site was defined as a 10 × 13 × 18 Å inner box centered on the average of coordinates of the native ligand present in the PPARγ crystal structure (PDB ID: 6DGR).64 A scaling factor of 0.8 was set for van der Waals radii of receptor atoms. Ligand sampling was allowed to be flexible. Default docking parameters were used, and no constraints were included. Redocking experiments were run in order to validate the accuracy of Glide at reproducing the position of CAY10638 in the active site. On comparing the conformation of the cocrystallized ligand with the docked poses, it was observed that the SP (standard precision) mode reproduced the bioactive conformation of the cognate ligand with RMSD less than 2 Å. Thus, further molecular docking studies were performed at the SP level. Ligand sampling was allowed to be flexible. At most 10 docking ligand poses were retained per run and ranked using the GlideScore function.111,112 The pose results were analyzed for accuracy through the RMSD of the common core to the original cocrystal ligand core. Binding poses were selected on the basis of the scoring, the similarity to the cocrystallized ligand binding mode, and the consistency of protein–ligand interactions with the experimental data.
Cytotoxicity Assessment by DNS Assay.
Cell Lines and Culture Conditions.
In this study, seven human cell lines were utilized: three leukemia/lymphoma, CCRF-CEM, Ramos, HL-60; one cervical, HeLa; one triple negative breast, MDA-MB-231; one neuroblastoma, SH-SY5Y; and one noncancerous foreskin fibroblast for comparison, HS-27. Cell lines CCRF-CEM, Ramos, and HL-60 were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (HI FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin–except for HL-60, which required 20% HI FBS. Similarly, cell lines MDA-MB-231, HeLa, and HS-27 were cultured in DMEM (dulbecco’s modified eagle medium) medium, and SH-SY5Y in DMEM/F12, supplemented with antibiotics and 10% HI FBS as described above. Cells were continuously maintained in a 37 °C with 5% CO2 humidified atmosphere.
Assessment of cell viability and seeding into multiwell plates was performed when cultures reached 60–75% confluence in the exponential growth phase. Adherent cell lines (HeLa, MDA-MB-231, SH-SY5Y, HS-27) were detached from their vessel by trypsinization. Cell viability was examined by trypan blue exclusion, and only cultures with ≥90% viable cells were seeded for experimentation.
Preparation of Compounds for In Vitro Assays.
To prepare stock solutions, aliquots of dried experimental or reference compound were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM and stored at −20 °C prior to use. For experiments, fresh aliquots of compounds stocks were thawed and diluted with DMSO to a 100× or 250× working concentration to minimize dose volume and possible vehicle-induced cytotoxicity.
Cytotoxicity Screening via the Differential Nuclear Staining (DNS) Assay.
To evaluate the cytotoxicity of “series 7” compounds (7c, 7e, 7f, 7g, 7i, 7j, 7l, 7o, 7q, 7r) the live cell imaging-based differential nuclear staining (DNS) assay was utilized.113 For this experiment, cells were seeded in 96-well plates at a density of 5000 cells/well, incubated overnight, and exposed to experimental or control treatments in quadruplicate for 72 h. In each plate, 1% DMSO, 1 mM H2O2, and untreated cells were utilized as controls. One hour prior to imaging, a mixture of fluorescent nuclear dyes Hoechst 33342 and propidium iodide (PI) were added to wells at a concentration of 1 μg/mL each. Cytotoxicity was determined as the percentage of Hoechst 33342+/PI+ cells. For each cell-containing well, a 2 × 2 montage of images were captured and segmented using a GE InCell Analyzer 2000 high-content analysis system and associated software. Additionally, for comparative purposes, the cytotoxicity of reference compounds SAHA and pioglitazone were assessed in an identical manner.
Calculation of CC50 and SCI Values.
Individual data points were normalized by subtracting the average percentage of cytotoxicity in vehicle-treated cells from experimental values. Cell death was then reported as a cytotoxicity percentage after 10 μM treatment or CC50 value for each individual compound. The concentration of experimental compound which kills 50% of the cell population (cytotoxic concentration 50%; CC50), was calculated by linear interpolation of normalized data points using the two concentrations nearest 50% cytotoxicity and reported as an average of 3–4 replicates. Selective cytotoxicity index (SCI) values were calculated by dividing the CC50 of noncancerous HS-27 cells by the CC50 of individual cancer cell lines.
Apoptosis Assay.
To evaluate the type of cell death (apoptosis or necrosis) elicited by experimental compounds 7c, and 7i, the Annexin V-FITC/PI assay was performed. In this experiment, CCRF-CEM cells were seeded in a 24-well plate at a density of 50 000 cells/well in 1 mL of complete RPMI-1640 culture medium. Cells were then treated in triplicate with the CC50 or 2×CC50 of compounds 7c, 7i, or HDAC inhibitor SAHA for comparison and incubated for 72 h. 4% DMSO, 1 mM H2O2, and untreated cells were utilized as vehicle, positive, and negative controls respectively. Following treatment, cells were collected and centrifuged at 500g for 5 min at 4 °C. Meanwhile, reagents were collected and prepared on ice. The supernatant of each sample was discarded, cell pellets resuspended in 103.5 μL of a AnnexinV-FITC/PI/binding buffer mixture and incubated (covered) on ice for 30 min. Afterward, an additional 300 μL of ice-cold binding buffer was added and samples were analyzed by beckman coulter cytomics FC 500 flow cytometer and associated kaluza software. Approximately 40 000 events (cells) were analyzed per sample. Total apoptosis was calculated by summing early (AnnexinV+/PI−) and late (AnnexinV+/PI+) apoptotic events. Statistical significance was assessed by comparison of vehicle control samples to other treatments via independent samples t-test (p < 0.05).
Cell Cycle Analysis.
To evaluate the effect of experimental compounds 7c and 7i on cell cycle progression, CCRF-CEM cells were seeded in 24-well plates at a density of 50 000 cells/well in 1 mL of complete culture medium. Cells were then treated in triplicate with the CC50 or 2×CC50 of compounds 7c, 7i, or HDAC inhibitor SAHA for comparison and incubated for 72 h. 4% DMSO, 1 mM H2O2, and untreated cells were utilized as vehicle, positive, and negative controls. Following treatment, cells were collected and centrifuged at 262 g for 5 min at 20 °C. For each sample, the supernatant was discarded, and cells resuspended in 100 μL of PBS and 200 μL of nuclear isolation medium (NIM)-DAPI solution to permeabilize cell membranes and stain DNA. Then they were immediately analyzed by beckman coulter cytomics FC 500 flow cytometer and associated kaluza software. Approximately 40 000 events (cells) were analyzed per sample. Statistical significance was assessed by comparison of vehicle control samples to other treatments via independent samples t-test (p < 0.05).
Western Blotting.
CCRF-CEM Cells were grown in multiwell plate which were washed twice with cold PBS, lysed in Laemmli buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS) supplemented with Complete protease inhibitors (Roche), sonicated before protein quantification (DC BioRad Protein Assay cat no. 500–0114). Samples with equal protein quantity were supplemented with 5% β-mercaptoethanol, heated (95 °C; 12 min), size fractionated on a 9% SDS-PAGE gel, and transferred to nitrocellulose membranes. All Blue Precision Plus Protein Standard (Bio-Rad cat. no. 161–0373) was used as protein size marker. Membranes were blocked for 45 min in PBS-T-milk (0.05% Tween, 5% dried fat-free milk); incubated with primary antibody (3 h; RT; di luted in PBS-T milk), washed (PBS-T) and incubated with secondary antibodies (1 h; RT; diluted in PBS-T milk). After removal of unbound secondary antibodies, signals were revealed using Super signal west femto maximum sensitivity substrate (Pierce). Antibodies were purchased from cell signaling (p53 no. 9282, cMYC no. 9402, Cleaved caspase no. 9661, Ac Tub no. 5335). All the bands were quantified and normalized with respect to TBP band by using ImageJ software, version 1.2.
In Vivo Evaluation.
In vivo tumor growth inhibition has served as demonstrative model of achieving expected activity of antitumor agents in human. Hence, the most promising dual targeting compound 7c was further evaluated in vivo using standard subcutaneous xenograft model produced using CEM cell line. All the experiments were approved by the concerned IAEC (institutional animal ethics committee, Approval No. CPCSEA/IAEC/KT/05/20/5). 8–10 weeks old SCID mice were housed individually under pathogen-free conditions in ventilated cage system with a 12 h light/dark cycle. The area was controlled for noise and humidity. Animals were fed autoclaved commercial pellets and water ad libitum and were handled in a laminar air flow during the experimentation. Primary dose escalation was performed with 5 animals which were administered 500 mg/kg and 1000 mg/g single dose of 7c i.p. Animal receiving 1000 mg/kg displayed symptoms of distress and mortality in 2 animals. Animals getting 500 mg/kg did not exhibit any mortality, and clinical distress signs were recovered within 6 h. Hence 1/10th of 500 mg/kg dose was selected for efficacy study. Briefly CEM, ATCC (T cell leukemia) cells (1 × 105) suspended in serum free medium were injected on the back of the mice and allowed to form palpable tumors. Tumors were minced and regrafted in experimental animals. Administration of test sample were done when the tumors attained a volume of nearly 100 mm3, which were measured using digital vernier calipers (Mitutoyo JAPAN) and computed by utilizing the formula: volume = [(length × width2)/2]. Animals were randomly divided in to three groups based on body weight and tumor volume and allocated to treatments: vehicle, 7c (50 mg/kg i.p.) and standard clinical agent doxorubicin (20 mg/kg i. p.). Treatments were administered on days 1–5, 8–12, 15–18. Tumor volume was calculated by formula: volume = (width)2 × length/2. At the end of the experiment, the animals were sacrificed by cervical dislocation. The animals were dissected, and tumors were excised and immediately imaged.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Indo-Poland joint research program grant from Department of Science and Technology (DST) with reference number DST/INT/Pol/P-27/2016 (to CSR). Part of the Funding for this work was provided by the Research Centers in Minority Institutions (RCMI) program grants 5G12MD007592 and 5U54MD007592 to the Border Biomedical Research Center (BBRC) at UTEP from the National Institute on Minority Health and Health Disparities, a component of NIH. The authors thank the personnel of the Genomic Analysis and the Cell Characterization and Biorepository Core Facilities at the University of Texas at El Paso (UTEP). We also acknowledge funding through the LOEWE priority program TRABITA, State of Hesse, Germany.
ABBREVIATIONS USED
- TZD
2,4-thiazolidine dione
- HDAC
histone deacetylase
- PPARγ
peroxisome proliferator-activated receptor gamma
- ZBG
zinc binding group
- SRC
surface recognition cap
- SAR
structure activity relationship
- DMF
dimethyl formamide
- DCM
dichloromethane
- TEA
triethanolamine
- LBD
ligand binding domain
- PDB
protein data bank
- DNS
differential nuclear staining
- SAHA
suberoylanilide hydroxamic acid
- CC50
half maximal cytotoxic concentration
- IC50
half maximal inhibitory concentration
- EC50
half maximal effective concentration
- DMEM
dulbecco’s modified eagle medium
- HI FBS
heat-inactivated fetal bovine serum
- SCI
selective cytotoxicity index
- AnnexinV-FITC/PI
annexin V-fluorescein isothiocyanate/propidium iodide
- PS
phosphatidylserine
- DMSO
dimethyl sulfoxide
- NIM-DAPI
nuclear isolation medium containing DNA intercalating dye DAPI
- DNA
deoxyribonucleic acid
- TGI
total growth inhibition
- TLC
thin layer chromatography
- FTIR
Fourier transform infrared spectroscopy
- NMR
nuclear magnetic resonance
- PDA
photodiode array detector
- DSC
differential scanning calorimetry
- HPLC
high performance liquid chromatography
- MP
melting point
- DRC
dose–response curve
- CASTp
computed atlas of surface topography of proteins
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00491.
Additional docking figures of HDAC and PPARγ docking studies; PPARγ activation assay images; apoptosis histograms, value tables, and statistical data; cell cycle value tables and statistical data; HDAC4 and HDAC8 residual activity values tables; compounds spectral details such as 1H NMR and 13C NMR, IR spectra, DSC graph, LCMS PDA chromatograms, and mass spectra of all target compounds (PDF)
Molecular formula strings and some data (CSV)
3D structure graphics (ZIP)
3D structure graphics (ZIP)
The authors declare no competing financial interest.
Contributor Information
Kalpana Tilekar, Department of Pharmaceutical Chemistry, Bharati Vidyapeeth’s College of Pharmacy, Navi Mumbai 400614, India.
Jessica D. Hess, Cellular Characterization and Biorepository Core Facility, Border Biomedical Research Center, Department of Biological Sciences, The University of Texas at El Paso, El Paso, Texas 79968, United States
Neha Upadhyay, Department of Pharmaceutical Chemistry, Bharati Vidyapeeth’s College of Pharmacy, Navi Mumbai 400614, India.
Alessandra Lo Bianco, Department of Pharmacy, “Drug Discovery” Laboratory, University of Napoli “Federico II”, 80131 Napoli, Italy.
Markus Schweipert, Department of Chemical Engineering and Biotechnology, University of Applied Science, 64295 Darmstadt, Germany.
Antonio Laghezza, Department of Pharmacy-Drug Sciences, University of Bari “Aldo Moro”, 70126 Bari, Italy.
Fulvio Loiodice, Department of Pharmacy-Drug Sciences, University of Bari “Aldo Moro”, 70126 Bari, Italy.
Franz-Josef Meyer-Almes, Department of Chemical Engineering and Biotechnology, University of Applied Science, 64295 Darmstadt, Germany.
Renato J. Aguilera, Cellular Characterization and Biorepository Core Facility, Border Biomedical Research Center, Department of Biological Sciences, The University of Texas at El Paso, El Paso, Texas 79968, United States
Antonio Lavecchia, Department of Pharmacy, “Drug Discovery” Laboratory, University of Napoli “Federico II”, 80131 Napoli, Italy.
C S Ramaa, Department of Pharmaceutical Chemistry, Bharati Vidyapeeth’s College of Pharmacy, Navi Mumbai 400614, India.
REFERENCES
- (1).Raghavendra NM; Pingili D; Kadasi S; Mettu A; Prasad SVUM Dual or Multi-Targeting Inhibitors: The next Generation Anticancer Agents. Eur. J. Med. Chem 2018, 143, 1277–1300. [DOI] [PubMed] [Google Scholar]
- (2).Fu R; Sun Y; Sheng W; Liao D Designing Multi-Targeted Agents: An Emerging Anticancer Drug Discovery Paradigm. Eur. J. Med. Chem 2017, 136, 195–211. [DOI] [PubMed] [Google Scholar]
- (3).Csermely P; Agoston V; Pongor S The Efficiency of Multi-Target Drugs: The Network Approach Might Help Drug Design. Trends Pharmacol. Sci 2005, 26 (4), 178–182. [DOI] [PubMed] [Google Scholar]
- (4).O’Boyle NM; Meegan MJ Designed Multiple Ligands for Cancer Therapy. Curr. Med. Chem 2011, 18 (31), 4722–4737. [DOI] [PubMed] [Google Scholar]
- (5).Morphy R; Rankovic Z Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem 2005, 48 (21), 6523–6543. [DOI] [PubMed] [Google Scholar]
- (6).Peters J-U Polypharmacology–Foe or Friend? J. Med. Chem 2013, 56 (22), 8955–8971. [DOI] [PubMed] [Google Scholar]
- (7).Anighoro A; Bajorath J; Rastelli G Polypharmacology: Challenges and Opportunities in Drug Discovery: Miniperspective. J. Med. Chem 2014, 57 (19), 7874–7887. [DOI] [PubMed] [Google Scholar]
- (8).Jones PA; Issa J-PJ; Baylin S Targeting the Cancer Epigenome for Therapy. Nat. Rev. Genet 2016, 17 (10), 630. [DOI] [PubMed] [Google Scholar]
- (9).Kelly TK; De Carvalho DD; Jones PA Epigenetic Modifications as Therapeutic Targets. Nat. Biotechnol 2010, 28 (10), 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Falkenberg KJ; Johnstone RW Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat. Rev. Drug Discovery 2014, 13 (9), 673. [DOI] [PubMed] [Google Scholar]
- (11).Lin H-Y; Chen C-S; Lin S-P; Weng J-R; Chen C-S Targeting Histone Deacetylase in Cancer Therapy. Med. Res. Rev 2006, 26 (4), 397–413. [DOI] [PubMed] [Google Scholar]
- (12).Mai A Histone Deacetylase Inhibitors: Updated Studies in Various Epigenetic-Related Diseases. Journal of Clinical Epigenetics 2016, 2 (1), 7. [Google Scholar]
- (13).Minucci S; Pelicci PG Histone Deacetylase Inhibitors and the Promise of Epigenetic (and More) Treatments for Cancer. Nat. Rev. Cancer 2006, 6 (1), 38–51. [DOI] [PubMed] [Google Scholar]
- (14).Eckschlager T; Plch J; Stiborova M; Hrabeta J Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci 2017, 18 (7), 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tan J; Cang S; Ma Y; Petrillo RL; Liu D Novel Histone Deacetylase Inhibitors in Clinical Trials as Anti-Cancer Agents. J. Hematol. Oncol 2010, 3 (1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dong G; Chen W; Wang X; Yang X; Xu T; Wang P; Zhang W; Rao Y; Miao C; Sheng C Small Molecule Inhibitors Simultaneously Targeting Cancer Metabolism and Epigenetics: Discovery of Novel Nicotinamide Phosphoribosyltransferase (NAMPT) and Histone Deacetylase (HDAC) Dual Inhibitors. J. Med. Chem 2017, 60 (19), 7965–7983. [DOI] [PubMed] [Google Scholar]
- (17).Ling Y; Xu C; Luo L; Cao J; Feng J; Xue Y; Zhu Q; Ju C; Li F; Zhang Y; et al. Novel β-Carboline/Hydroxamic Acid Hybrids Targeting Both Histone Deacetylase and DNA Display High Anticancer Activity via Regulation of the P53 Signaling Pathway. Journal of medicinal chemistry 2015, 58 (23), 9214–9227. [DOI] [PubMed] [Google Scholar]
- (18).Liu C; Ding H; Li X; Pallasch CP; Hong L; Guo D; Chen Y; Wang D; Wang W; Wang Y; Hemann MT; Jiang H A DNA/HDAC Dual-Targeting Drug CY190602 with Significantly Enhanced Anticancer Potency. EMBO Mol. Med 2015, 7 (4), 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tang C; Li C; Zhang S; Hu Z; Wu J; Dong C; Huang J; Zhou H-B Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. J. Med. Chem 2015, 58 (11), 4550–4572. [DOI] [PubMed] [Google Scholar]
- (20).Yang EG; Mustafa N; Tan EC; Poulsen A; Ramanujulu PM; Chng WJ; Yen JJ; Dymock BW Design and Synthesis of Janus Kinase 2 (JAK2) and Histone Deacetlyase (HDAC) Bispecific Inhibitors Based on Pacritinib and Evidence of Dual Pathway Inhibition in Hematological Cell Lines. J. Med. Chem 2016, 59 (18), 8233–8262. [DOI] [PubMed] [Google Scholar]
- (21).Yao L; Mustafa N; Tan EC; Poulsen A; Singh P; Duong-Thi M-D; Lee JX; Ramanujulu PM; Chng WJ; Yen JJ; et al. Design and Synthesis of Ligand Efficient Dual Inhibitors of Janus Kinase (JAK) and Histone Deacetylase (HDAC) Based on Ruxolitinib and Vorinostat. Journal of medicinal chemistry 2017, 60 (20), 8336–8357. [DOI] [PubMed] [Google Scholar]
- (22).Zang J; Liang X; Huang Y; Jia Y; Li X; Xu W; Chou CJ; Zhang Y Discovery of Novel Pazopanib-Based HDAC and VEGFR Dual Inhibitors Targeting Cancer Epigenetics and Angiogenesis Simultaneously. J. Med. Chem 2018, 61 (12), 5304–5322. [DOI] [PubMed] [Google Scholar]
- (23).Tontonoz P; Spiegelman BM Fat and Beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem 2008, 77 (1), 289–312. [DOI] [PubMed] [Google Scholar]
- (24).Campbell I The Clinical Significance of PPAR Gamma Agonism. Curr. Mol. Med 2005, 5 (3), 349–363. [DOI] [PubMed] [Google Scholar]
- (25).Grommes C; Landreth GE; Heneka MT Antineoplastic Effects of Peroxisome Proliferatoractivated Receptor γ Agonists. Lancet Oncol 2004, 5 (7), 419–429. [DOI] [PubMed] [Google Scholar]
- (26).Piemontese L; Cerchia C; Laghezza A; Ziccardi P; Sblano S; Tortorella P; Iacobazzi V; Infantino V; Convertini P; Dal Piaz F; Lupo A; Colantuoni V; Lavecchia A; Loiodice F New Diphenylmethane Derivatives as Peroxisome Proliferator-Activated Receptor Alpha/Gamma Dual Agonists Endowed with Anti-Proliferative Effects and Mitochondrial Activity. Eur. J. Med. Chem 2017, 127, 379–397. [DOI] [PubMed] [Google Scholar]
- (27).Theocharis S; Margeli A; Vielh P; Kouraklis G Peroxisome Proliferator-Activated Receptor-Gamma Ligands as Cell-Cycle Modulators. Cancer Treat. Rev 2004, 30 (6), 545–554. [DOI] [PubMed] [Google Scholar]
- (28).Youssef J; Badr M Peroxisome Proliferator-Activated Receptors and Cancer: Challenges and Opportunities: PPARs and Cancer. Br. J. Pharmacol 2011, 164 (1), 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shimazaki N; Togashi N; Hanai M; Isoyama T; Wada K; Fujita T; Fujiwara K; Kurakata S Anti-Tumour Activity of CS-7017, a Selective Peroxisome Proliferator-Activated Receptor Gamma Agonist of Thiazolidinedione Class, in Human Tumour Xenografts and a Syngeneic Tumour Implant Model. Eur. J. Cancer 2008, 44 (12), 1734–1743. [DOI] [PubMed] [Google Scholar]
- (30).Smallridge RC; Copland JA; Brose MS; Wadsworth JT; Houvras Y; Menefee ME; Bible KC; Shah MH; Gramza AW; Klopper JP; et al. Efatutazone, an Oral PPAR-γ Agonist, in Combination with Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. Journal of Clinical Endocrinology & Metabolism 2013, 98 (6), 2392–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tachibana K; Yamasaki D; Ishimoto K; Doi T The Role of PPARs in Cancer. PPAR research 2008, 2008, 102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Serizawa M; Murakami H; Watanabe M; Takahashi T; Yamamoto N; Koh Y Peroxisome Proliferator-Activated Receptor γ Agonist Efatutazone Impairs Transforming Growth Factor B2-Induced Motility of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistant Lung Cancer Cells. Cancer Sci 2014, 105 (6), 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nemenoff RA; Weiser-Evans M; Winn RA Activation and Molecular Targets of Peroxisome Proliferator-Activated Receptor-γ Ligands in Lung Cancer. PPAR Res 2008, 2008, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hong F; Xu P; Zhai Y The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci 2018, 19 (8), 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shi GQ; Dropinski JF; McKeever BM; Xu S; Becker JW; Berger JP; MacNaul KL; Elbrecht A; Zhou G; Doebber TW; Wang P; Chao Y-S; Forrest M; Heck JV; M?ller DE; Jones AB Design and Synthesis of α-Aryloxyphenylacetic Acid Derivatives: A Novel Class of PPARα/γ Dual Agonists with Potent Antihyperglycemic and Lipid Modulating Activity. J. Med. Chem 2005, 48 (13), 4457–4468. [DOI] [PubMed] [Google Scholar]
- (36).Burgermeister E; Schnoebelen A; Flament A; Benz J; Stihle M; Gsell B; Rufer A; Ruf A; Kuhn B; Märki HP; Mizrahi J; Sebokova E; Niesor E; Meyer M A Novel Partial Agonist of Peroxisome Proliferator-Activated Receptor-γ (PPARγ) Recruits PPARγ-Coactivator-1α, Prevents Triglyceride Accumulation, and Potentiates Insulin Signaling in Vitro. Mol. Endocrinol 2006, 20 (4), 809–830. [DOI] [PubMed] [Google Scholar]
- (37).Kroker AJ; Bruning JB Review of the Structural and Dynamic Mechanisms of PPAR γ Partial Agonism. PPAR Res 2015, 2015, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lu I-L; Huang C-F; Peng Y-H; Lin Y-T; Hsieh H-P; Chen C-T; Lien T-W; Lee H-J; Mahindroo N; Prakash E; Yueh A; Chen H-Y; Goparaju CMV; Chen X; Liao C-C; Chao Y-S; Hsu JT-A; Wu S-Y Structure-Based Drug Design of a Novel Family of PPARγ Partial Agonists: Virtual Screening, X-Ray Crystallography, and in Vitro/in Vivo Biological Activities. J. Med. Chem 2006, 49 (9), 2703–2712. [DOI] [PubMed] [Google Scholar]
- (39).Rizos C; Elisaf M; Mikhailidis D; Liberopoulos E How Safe Is the Use of Thiazolidinediones in Clinical Practice? Expert Opin. Drug Saf 2009, 8 (1), 15–32. [DOI] [PubMed] [Google Scholar]
- (40).Sabatino L; Ziccardi P; Cerchia C; Muccillo L; Piemontese L; Loiodice F; Colantuoni V; Lupo A; Lavecchia A Chiral Phenoxyacetic Acid Analogues Inhibit Colon Cancer Cell Proliferation Acting as PpARγ Partial Agonists. Sci. Rep 2019, 9 (1), 5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wright MB; Bortolini M; Tadayyon M; Bopst M Minireview: Challenges and Opportunities in Development of PPAR Agonists. Mol. Endocrinol 2014, 28 (11), 1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Agrawal R; Jain P; Dikshit SN Balaglitazone: A Second Generation Peroxisome Proliferator-Activated Receptor (PPAR) Gamma (γ) Agonist. MRMC 2012, 12 (2), 87–97. [DOI] [PubMed] [Google Scholar]
- (43).Henriksen K; Byrjalsen I; Qvist P; Beck-Nielsen H; Hansen G; Riis BJ; Perrild H; Svendsen OL; Gram J; Karsdal MA; Christiansen C Efficacy and Safety of the PPARγ Partial Agonist Balaglitazone Compared with Pioglitazone and Placebo: A Phase III Randomized, Parallel-Group Study in Patients with Type 2 Diabetes on Stable Insulin Therapy. Diabetes/metabolism research and reviews 2011, 27 (4), 392–401. [DOI] [PubMed] [Google Scholar]
- (44).Sun N; Lu G; Lin M; Fan G; Wu Y Subchronic Toxicity and Toxicokinetics of MCC-555, a Novel Thiazolidinedione, after 270-Day Repeated Oral Administration in Dogs. Environ. Toxicol. Pharmacol 2009, 27 (2), 237–246. [DOI] [PubMed] [Google Scholar]
- (45).Yamaguchi K; Cekanova M; McEntee MF; Yoon J-H; Fischer SM; Renes IB; Van Seuningen I; Baek SJ Peroxisome Proliferator-Activated Receptor Ligand MCC-555 Suppresses Intestinal Polyps in ApcMin/+ Mice via Extracellular Signal-Regulated Kinase and Peroxisome Proliferator-Activated Receptor-Dependent Pathways. Mol. Cancer Ther 2008, 7 (9), 2779–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Annicotte J-S; Iankova I; Miard S; Fritz V; Sarruf D; Abella A; Berthe M-L; Noël D; Pillon A; Iborra F; Dubus P; Maudelonde T; Culine S; Fajas L Peroxisome Proliferator-Activated Receptor γ Regulates E-Cadherin Expression and Inhibits Growth and Invasion of Prostate Cancer. Mol. Cell. Biol 2006, 26 (20), 7561–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Aouali N; Palissot V; El-Khoury V; Moussay E; Janji B; Pierson S; Brons NHC; Kellner L; Bosseler M; Van Moer K; Berchem G Peroxisome Proliferator-Activated Receptor γ Agonists Potentiate the Cytotoxic Effect of Valproic Acid in Multiple Myeloma Cells: PPARγ and Valproic Acid in Multiple Myeloma Cells. Br. J. Haematol 2009, 147 (5), 662–671. [DOI] [PubMed] [Google Scholar]
- (48).Chang T-H; Szabo E Enhanced Growth Inhibition by Combination Differentiation Therapy with Ligands of Peroxisome Proliferator-Activated Receptor-γ and Inhibitors of Histone Deacetylase in Adenocarcinoma of the Lung. Clin. Cancer Res 2002, 8 (4), 1206–1212. [PubMed] [Google Scholar]
- (49).Joshi H; Marulkar K; Gota V; C S R Hydroxy Cinnamic Acid Derivatives as Partial PPAR γ Agonists: In Silico Studies, Synthesis and Biological Characterization Against Chronic Myeloid Leukemia Cell Line (K562). Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 2017, 17 (4), 524–541. [DOI] [PubMed] [Google Scholar]
- (50).Mohan R; Sharma AK; Gupta S; Ramaa CS Design, Synthesis, and Biological Evaluation of Novel 2, 4-Thiazolidinedione Derivatives as Histone Deacetylase Inhibitors Targeting Liver Cancer Cell Line. Med. Chem. Res 2012, 21 (7), 1156–1165. [Google Scholar]
- (51).Pal T; Joshi H; Ramaa CS Design and Development of Oxazol-5-Ones as Potential Partial PPAR-γ Agonist against Cancer Cell Lines. Anti-Cancer Agents Med. Chem 2014, 14 (6), 872–883. [DOI] [PubMed] [Google Scholar]
- (52).Tilekar K; Upadhyay N; Jänsch N; Schweipert M; Mrowka P; Meyer-Almes FJ; Ramaa CS Discovery of 5-Naphthylidene-2,4-Thiazolidinedione Derivatives as Selective HDAC8 Inhibitors and Evaluation of Their Cytotoxic Effects in Leukemic Cell Lines. Bioorg. Chem 2020, 95, 103522. [DOI] [PubMed] [Google Scholar]
- (53).Upadhyay N; Tilekar K; Jänsch N; Schweipert M; Hess JD; Henze Macias L; Mrowka P; Aguilera RJ; Choe J; Meyer-Almes F-J; C S R Discovery of Novel N-Substituted Thiazolidinediones (TZDs) as HDAC8 Inhibitors: In-Silico Studies, Synthesis, and Biological Evaluation. Bioorganic Chemistry 2020, 100, 103934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Amato A; Rajagopalan S; Lin J; Carvalho B; Figueira A; Lu J; Ayers S; Mottin M; Silveira R; Telles de Souza PC; Mourão R; Saad M; Togashi M; Simeoni L; Abdalla D; Skaf M; Polikparpov I; Lima M; Galdino S; Neves F; et al. GQ-16, a Novel Peroxisome Proliferator-Activated Receptor (PPAR) Ligand, Promotes Insulin Sensitization without Weight Gain. J. Biol. Chem 2012, 287, 28169–28179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ferreira APB; Coelho MS; Amato AA; Neves F; de AR; Rodrigues IC; Royer C Effect of PPAR Partial Agonist, GQ-16, on Viability of Breast Cancer Cells in Culture. FASEB J 2017, 31 (S1), 876.5–876.5. [Google Scholar]
- (56).Miller TA; Witter DJ; Belvedere S Histone Deacetylase Inhibitors. J. Med. Chem 2003, 46 (24), 5097–5116. [DOI] [PubMed] [Google Scholar]
- (57).Tessier P; Smil DV; Wahhab A; Leit S; Rahil J; Li Z; Déziel R; Besterman JM Diphenylmethylene Hydroxamic Acids as Selective Class IIa Histone Deacetylase Inhibitors. Bioorg. Med. Chem. Lett 2009, 19 (19), 5684–5688. [DOI] [PubMed] [Google Scholar]
- (58).Hou X; Du J; Liu R; Zhou Y; Li M; Xu W; Fang H Enhancing the Sensitivity of Pharmacophore-Based Virtual Screening by Incorporating Customized ZBG Features: A Case Study Using Histone Deacetylase 8. J. Chem. Inf. Model 2015, 55 (4), 861–871. [DOI] [PubMed] [Google Scholar]
- (59).Kleinschek A; Meyners C; Digiorgio E; Brancolini C; Meyer-Almes F-J Potent and Selective Non-Hydroxamate Histone Deacetylase 8 Inhibitors. ChemMedChem 2016, 11 (23), 2598–2606. [DOI] [PubMed] [Google Scholar]
- (60).Huang W-J; Wang Y-C; Chao S-W; Yang C-Y; Chen L-C; Lin M-H; Hou W-C; Chen M-Y; Lee T-L; Yang P; Chang C-I Synthesis and Biological Evaluation of Ortho -Aryl N-Hydroxycinnamides as Potent Histone Deacetylase (HDAC) 8 Isoform-Selective Inhibitors. ChemMedChem 2012, 7 (10), 1815–1824. [DOI] [PubMed] [Google Scholar]
- (61).KrennHrubec K; Marshall BL; Hedglin M; Verdin E; Ulrich SM Design and Evaluation of ‘Linkerless’ Hydroxamic Acids as Selective HDAC8 Inhibitors. Bioorg. Med. Chem. Lett 2007, 17 (10), 2874–2878. [DOI] [PubMed] [Google Scholar]
- (62).Bottomley MJ; Lo Surdo P; Di Giovine P; Cirillo A; Scarpelli R; Ferrigno F; Jones P; Neddermann P; De Francesco R; Steinkühler C; Gallinari P; Carfí A Structural and Functional Analysis of the Human HDAC4 Catalytic Domain Reveals a Regulatory Structural Zinc-Binding Domain. J. Biol. Chem 2008, 283 (39), 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bürli RW; Luckhurst CA; Aziz O; Matthews KL; Yates D; Lyons KA; Beconi M; McAllister G; Breccia P; Stott AJ; Penrose SD; Wall M; Lamers M; Leonard P; Müller I; Richardson CM; Jarvis R; Stones L; Hughes S; Wishart G; Haughan AF; O’Connell C; Mead T; McNeil H; Vann J; Mangette J; Maillard M; Beaumont V; Munoz-Sanjuan I; Dominguez C Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J. Med. Chem 2013, 56 (24), 9934–9954. [DOI] [PubMed] [Google Scholar]
- (64).Shang J; Brust R; Griffin PR; Kamenecka TM; Kojetin DJ Quantitative Structural Assessment of Graded Receptor Agonism. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (44), 22179–22188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nolte RT; Wisely GB; Westin S; Cobb JE; Lambert MH; Kurokawa R; Rosenfeld MG; Willson TM; Glass CK; Milburn MV Ligand Binding and Co-Activator Assembly of the Peroxisome Proliferator-Activated Receptor-γ. Nature 1998, 395 (6698), 137–143. [DOI] [PubMed] [Google Scholar]
- (66).Bruning JB; Chalmers MJ; Prasad S; Busby SA; Kamenecka TM; He Y; Nettles KW; Griffin PR Partial Agonists Activate PPARγ Using a Helix 12 Independent Mechanism. Structure 2007, 15 (10), 1258–1271. [DOI] [PubMed] [Google Scholar]
- (67).Vasaturo M; Fiengo L; De Tommasi N; Sabatino L; Ziccardi P; Colantuoni V; Bruno M; Cerchia C; Novellino E; Lupo A; Lavecchia A; Piaz FD A Compound-Based Proteomic Approach Discloses 15-Ketoatractyligenin Methyl Ester as a New PPARγ Partial Agonist with Anti-Proliferative Ability. Sci. Rep 2017, 7, 41273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kroker AJ; Bruning JB Review of the Structural and Dynamic Mechanisms of PPAR γ Partial Agonism. PPAR Res 2015, 2015, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Liberato MV; Nascimento AS; Ayers SD; Lin JZ; Cvoro A; Silveira RL; Martinez L; Souza PCT; Saidemberg D; Deng T; Amato AA; Togashi M; Hsueh WA; Phillips K; et al. Medium Chain Fatty Acids Are Selective Peroxisome Proliferator Activated Receptor (PPAR) c Activators and Pan-PPAR Partial Agonists. PLoS One 2012, 7 (5), e36297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Jang JY; Bae H; Lee YJ; Choi YI; Kim H-J; Park SB; Suh SW; Kim SW; Han BW Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARγ. Sci. Rep 2018, 8 (1), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hsieh AL; Walton ZE; Altman BJ; Stine ZE; Dang CV MYC and Metabolism on the Path to Cancer. Semin. Cell Dev. Biol 2015, 43, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Miller DM; Thomas SD; Islam A; Muench D; Sedoris K C-Myc and Cancer Metabolism. Clin. Cancer Res 2012, 18 (20), 5546–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Elrod HA; Sun S-Y PPAR γ and Apoptosis in Cancer. PPAR Res 2008, 2008, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Nebbioso A; Carafa V; Conte M; Tambaro FP; Abbondanza C; Martens J; Nees M; Benedetti R; Pallavicini I; Minucci S; Garcia-Manero G; Iovino F; Lania G; Ingenito C; Belsito Petrizzi V; Stunnenberg HG; Altucci L C-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res 2017, 23 (10), 2542–2555. [DOI] [PubMed] [Google Scholar]
- (75).Hoffman B; Amanullah A; Shafarenko M; Liebermann DA The Proto-Oncogene c-Myc in Hematopoietic Development and Leukemogenesis. Oncogene 2002, 21 (21), 3414–3421. [DOI] [PubMed] [Google Scholar]
- (76).Renneville A; Roumier C; Biggio V; Nibourel O; Boissel N; Fenaux P; Preudhomme C Cooperating Gene Mutations in Acute Myeloid Leukemia: A Review of the Literature. Leukemia 2008, 22 (5), 915–931. [DOI] [PubMed] [Google Scholar]
- (77).Salvatori B; Iosue I; Djodji Damas N; Mangiavacchi A; Chiaretti S; Messina M; Padula F; Guarini A; Bozzoni I; Fazi F; Fatica A Critical Role of C-Myc in Acute Myeloid Leukemia Involving Direct Regulation of MiR-26a and Histone Methyltransferase EZH2. Genes Cancer 2011, 2 (5), 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Slack GW; Gascoyne RD MYC and Aggressive B-Cell Lymphomas. Adv. Anat. Pathol 2011, 18 (3), 219–228. [DOI] [PubMed] [Google Scholar]
- (79).Yu L; Yu T-T; Young KH Cross-Talk between Myc and P53 in B-Cell Lymphomas. Chronic Diseases and Translational Medicine 2019, 5 (3), 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Li J; Yuan J Caspases in Apoptosis and Beyond. Oncogene 2008, 27 (48), 6194–6206. [DOI] [PubMed] [Google Scholar]
- (81).Porter AG; Jänicke RU Emerging Roles of Caspase-3 in Apoptosis. Cell Death Differ 1999, 6 (2), 99–104. [DOI] [PubMed] [Google Scholar]
- (82).Lee YJ; Won AJ; Lee J; Jung JH; Yoon S; Lee BM; Kim HS Molecular Mechanism of SAHA on Regulation of Autophagic Cell Death in Tamoxifen-Resistant MCF-7 Breast Cancer Cells. Int. J. Med. Sci 2012, 9 (10), 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Shao Y; Gao Z; Marks PA; Jiang X Apoptotic and Autophagic Cell Death Induced by Histone Deacetylase Inhibitors. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (52), 18030–18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Szoka L; Palka J Capsaicin Up-Regulates pro-Apoptotic Activity of Thiazolidinediones in Glioblastoma Cell Line. Biomed. Pharmacother 2020, 132, 110741. [DOI] [PubMed] [Google Scholar]
- (85).Berns K; Hijmans EM; Bernards R Repression of C-Myc Responsive Genes in Cycling Cells Causes G 1 Arrest through Reduction of Cyclin E/CDK2 Kinase Activity. Oncogene 1997, 15 (11), 1347–1356. [DOI] [PubMed] [Google Scholar]
- (86).Wang H; Mannava S; Grachtchouk V; Zhuang D; Soengas M; Gudkov A; Prochownik E; Nikiforov M C-Myc Depletion Inhibits Proliferation of Human Tumor Cells at Various Stages of the Cell Cycle. Oncogene 2008, 27 (13), 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Dovzhanskiy DI; Arnold SM; Hackert T; Oehme I; Witt O; Felix K; Giese N; Werner J Experimental in Vivo and in Vitro Treatment with a New Histone Deacetylase Inhibitor Belinostat Inhibits the Growth of Pancreatic Cancer. BMC Cancer 2012, 12 (1), 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Eto S; Saeki K; Yoshitake R; Yoshimoto S; Shinada M; Ikeda N; Kamoto S; Tanaka Y; Kato D; Maeda S; Tsuboi M; Chambers J; Uchida K; Nishimura R; Nakagawa T Anti-Tumor Effects of the Histone Deacetylase Inhibitor Vorinostat on Canine Urothelial Carcinoma Cells. PLoS One 2019, 14 (6), No. e0218382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Chan CT; Qi J; Smith W; Paranol R; Mazitschek R; West N; Reeves R; Chiosis G; Schreiber SL; Bradner JE; Paulmurugan R; Gambhir SS Syntheses and Discovery of a Novel Class of Cinnamic Hydroxamates as Histone Deacetylase Inhibitors by Multimodality Molecular Imaging in Living Subjects. Cancer Res 2014, 74 (24), 7475–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Chen C-H; Lee C-H; Liou J-P; Teng C-M; Pan S-L Molecular Mechanisms Underlying the Antitumor Activity of (E)-N-Hydroxy-3-(1-(4-Methoxyphenylsulfonyl)-1,2,3,4-Tetrahydroquinolin-6-Yl)Acrylamide in Human Colorectal Cancer Cells in Vitro and in Vivo. Oncotarget 2015, 6 (34), 35991–36002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Huang H-L; Peng C-Y; Lai M-J; Chen C-H; Lee H-Y; Wang J-C; Liou J-P; Pan S-L; Teng C-M Novel Oral Histone Deacetylase Inhibitor, MPT0E028, Displays Potent Growth-Inhibitory Activity against Human B-Cell Lymphoma in Vitro and in Vivo. Oncotarget 2015, 6 (7), 4976–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Rettig I; Koeneke E; Trippel F; Mueller WC; Burhenne J; Kopp-Schneider A; Fabian J; Schober A; Fernekorn U; von Deimling A; Deubzer HE; Milde T; Witt O; Oehme I Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth in Vitro and in Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis 2015, 6 (2), No. e1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Yang Z; Wang T; Wang F; Niu T; Liu Z; Chen X; Long C; Tang M; Cao D; Wang X; Xiang W; Yi Y; Ma L; You J; Chen L Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem 2016, 59 (4), 1455–1470. [DOI] [PubMed] [Google Scholar]
- (94).Yao Y; Tu Z; Liao C; Wang Z; Li S; Yao H; Li Z; Jiang S Discovery of Novel Class I Histone Deacetylase Inhibitors with Promising in Vitro and in Vivo Antitumor Activities. J. Med. Chem 2015, 58 (19), 7672–7680. [DOI] [PubMed] [Google Scholar]
- (95).Cellai I; Petrangolini G; Tortoreto M; Pratesi G; Luciani P; Deledda C; Benvenuti S; Ricordati C; Gelmini S; Ceni E; Galli A; Balzi M; Faraoni P; Serio M; Peri A In Vivo Effects of Rosiglitazone in a Human Neuroblastoma Xenograft. Br. J. Cancer 2010, 102 (4), 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Fujita M; Hasegawa A; Yamamori M; Okamura N In Vitro and in Vivo Cytotoxicity of Troglitazone in Pancreatic Cancer. J. Exp. Clin. Cancer Res 2017, 36 (1), 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Grommes C; Karlo JC; Caprariello A; Blankenship D; DeChant A; Landreth GE The PPARγ Agonist Pioglitazone Crosses the Blood–Brain Barrier and Reduces Tumor Growth in a Human Xenograft Model. Cancer Chemother. Pharmacol 2013, 71 (4), 929–936. [DOI] [PubMed] [Google Scholar]
- (98).Higuchi T; Sugisawa N; Miyake K; Oshiro H; Yamamoto N; Hayashi K; Kimura H; Miwa S; Igarashi K; Kline Z; Bouvet M; Singh SR; Tsuchiya H; Hoffman RM Pioglitazone, an Agonist of PPARγ, Reverses Doxorubicin-Resistance in an Osteosarcoma Patient-Derived Orthotopic Xenograft Model by Downregulating P-Glycoprotein Expression. Biomed. Pharmacother 2019, 118, 109356. [DOI] [PubMed] [Google Scholar]
- (99).Kabir A; Tilekar K; Upadhyay N; Ramaa CS Novel Anthraquinone Derivatives as Dual Inhibitors of Topoisomerase 2 and Casein Kinase 2: In Silico Studies, Synthesis and Biological Evaluation on Leukemic Cell Lines. Anti-Cancer Agents Med. Chem 2019, 18 (11), 1551–1562. [DOI] [PubMed] [Google Scholar]
- (100).Patil V; Tilekar K; Mehendale-Munj S; Mohan R; Ramaa CS Synthesis and Primary Cytotoxicity Evaluation of New 5-Benzylidene-2,4-Thiazolidinedione Derivatives. Eur. J. Med. Chem 2010, 45 (10), 4539–4544. [DOI] [PubMed] [Google Scholar]
- (101).Vora D; Upadhyay N; Tilekar K; Jain V; C S R Development of 1,2,4-Triazole-5-Thione Derivatives as Potential Inhibitors of Enoyl Acyl Carrier Protein Reductase (InhA) in Tuberculosis. IJPR 2019, 18 (4), 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Jänsch N; Meyners C; Muth M; Kopranovic A; Witt O; Oehme I; Meyer-Almes F-J The Enzyme Activity of Histone Deacetylase 8 Is Modulated by a Redox-Switch. Redox Biol 2019, 20, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Tian W; Chen C; Lei X; Zhao J; Liang J CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res 2018, 46 (W1), W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Raspé E; Madsen L; Lefebvre AM; Leitersdorf I; Gelman L; Peinado-Onsurbe J; Dallongeville J; Fruchart JC; Berge R; Staels B Modulation of Rat Liver Apolipoprotein Gene Expression and Serum Lipid Levels by Tetradecylthioacetic Acid (TTA) via PPARalpha Activation. J. Lipid Res 1999, 40 (11), 2099–2110. [PubMed] [Google Scholar]
- (105).Pinelli A; Godio C; Laghezza A; Mitro N; Fracchiolla G; Tortorella V; Lavecchia A; Novellino E; Fruchart J-C; Staels B; Crestani M; Loiodice F Synthesis, Biological Evaluation, and Molecular Modeling Investigation of New Chiral Fibrates with PPARα and PPARγ Agonist Activity. J. Med. Chem 2005, 48 (17), 5509–5519. [DOI] [PubMed] [Google Scholar]
- (106).Porcelli L; Gilardi F; Laghezza A; Piemontese L; Mitro N; Azzariti A; Altieri F; Cervoni L; Fracchiolla G; Giudici M; Guerrini U; Lavecchia A; Montanari R; Di Giovanni C; Paradiso A; Pochetti G; Simone GM; Tortorella P; Crestani M; Loiodice F Synthesis, Characterization and Biological Evaluation of Ureidofibrate-Like Derivatives Endowed with Peroxisome Proliferator-Activated Receptor Activity. J. Med. Chem 2012, 55 (1), 37–54. [DOI] [PubMed] [Google Scholar]
- (107).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (108).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- (109).Wallace AC; Laskowski RA; Thornton JM LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng., Des. Sel 1995, 8 (2), 127–134. [DOI] [PubMed] [Google Scholar]
- (110).Laskowski RA; Swindells MB LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model 2011, 51 (10), 2778–2786. [DOI] [PubMed] [Google Scholar]
- (111).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47 (7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- (112).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47 (7), 1750–1759. [DOI] [PubMed] [Google Scholar]
- (113).Lema C; Varela-Ramirez A; Aguilera RJ Differential Nuclear Staining Assay for High-Throughput Screening to Identify Cytotoxic Compounds. Curr. Cell Biochem 2011, 1 (1), 1–14. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.