

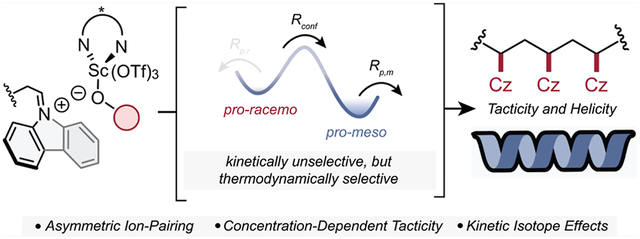
Abstract
The synthesis of stereoregular polymers through ionic mechanisms using asymmetric ion-pairing (AIP) catalysis is emerging as an effective strategy to achieve differentiated material properties from readily available building blocks. Stereoselective cationic polymerization in particular is primed for advancement using AIP by leveraging the breadth of Brønsted and Lewis acid small-molecule catalysis literature; however, mechanistic studies that address polymer-specific phenomena are scarce and, as a result, the lack of mechanistic understanding has limited catalyst design. In a recent study, we demonstrated the only example of a stereoselective and helix-sense-selective cationic vinyl polymerization of N-vinylcarbazole using chiral scandium-bis(oxazoline) Lewis acids. To better understand the mechanism of this highly stereoselective polymerization and elicit design principles for future advances, we present a combined experimental and computational study into the relevant factors that determine tacticity and helicity control. Key mechanistic experiments suggest two competing elementary steps—chain-end conformation equilibration and propagation—whose relative rates can be influenced by monomer concentration, isotope effects, and catalyst design to tune tacticity. In contrast, helicity is influenced by complex relationships between the stereoselectivity of the first monomer propagation and a time-dependent initiator-catalyst mixing time. The more complete understanding of stereoselective cationic polymerization through AIP developed herein provides insights into polymer-specific mechanisms for stereocontrol, which we believe will motivate continued catalyst discovery and development for stereoselective vinyl polymerization.
Keywords: asymmetric ion-pairing, cationic polymerization, tacticity, helix-sense-selectivity, N-vinylcarbazole
Graphical Abstract
Asymmetric ion-pairing (AIP) catalysis is an emerging general conceptual approach to control stereochemistry during cationic polymerizations.1 In contrast to achiral catalysts that rely on solely chain-end control,2–6 the chiral counterion provides control over the main-chain stereochemistry through catalyst-directed stereoinduction by leveraging electrostatic interactions with the growing carbenium chain-end, while also facilitating enantiofacial monomer addition.7,8 AIP cationic polymerization has enabled access to isotactic polymers that were inaccessible through other methods, and it has proven particularly useful for monomers that lack chirality, substantial steric bulk, and/or Lewis basic-directing groups.8–12 Isotactic polymers made through AIP have demonstrated attractive thermomechanical, adhesion, and transport properties that are otherwise inaccessible by their atactic counterparts.2,8,10–15
An additional dimension of stereochemistry accessible to AIP catalysis is control of the stereochemical conformation (i.e., helicity) of the polymer.16,17 While an isotactic vinyl polymer is pseudo-symmetric, the use of an enantioenriched catalyst can result in optically active secondary structures. This approach is known as helix-sense-selective polymerization (HSSP) and was originally developed by Okamoto for the anionic polymerization of bulky triphenyl methyl methacrylates.18 HSSP has been leveraged for the synthesis of commercial, high-value helically chiral materials for chiral separations, chiral recognition, spintronics, and circular polarized luminescence.18–22 However, its translation to cationic polymerization methods is underexplored.
While the emergence of AIP cationic polymerization has resulted in polymers with performance advantaged properties, several polymer-specific phenomena in stereoselective catalysis present notable challenges for mechanistic studies and, thus, hinder mechanism guided catalyst design. For example, stereoselective polymerizations include multiple operative diastereomeric interactions inherent to the chiral chain-end and catalyst,7,23 competing chain-end and catalyst control mechanisms, and a large stereochemical structure space arising from atropoisomeric structures, asymmetric backbones, and non-trivial stereo sequences.8,16,24 These complexities make detailed mechanistic studies challenging, and the resulting lack of mechanistic understanding about the operative interactions that dictate stereoselective monomer addition limits efforts to expand these approaches to other monomer classes and achieve higher selectivities (e.g. > 95% m). The adoption of physical organic tools for mechanistic investigations would prove enabling for advancing the field of AIP polymerizations.1,25
In a recent report, we disclosed the first stereoselective cationic HSSP of a vinyl monomer—N-vinylcarbazole (NVC)—using a scandium-bis(oxazoline) (Sc-BOX) Lewis acid as a catalyst (Figure 1A).15 Backed by a combination of experimental and computational studies, we hypothesized that helicity was correlated to the stereoselectivity of the first monomer addition, whereas the tacticity was governed by the thermodynamics of the chain-end conformation during propagation. These modes of selectivity diverged from traditional rationale for stereoinduction during propagation, which invoke kinetic stereoselectivity arising from a ΔΔG‡ between meso and racemo addition under Curtin–Hammett control.2,7,26–28 To gain a more complete understanding of this stereoselective polymerization, we hypothesized that the use of both traditional and modern tools in physical organic chemistry would uncover key aspects of these two stereo-determining mechanisms and establish principles for the design of AIP catalysts specific for polymerization.
Figure 1.
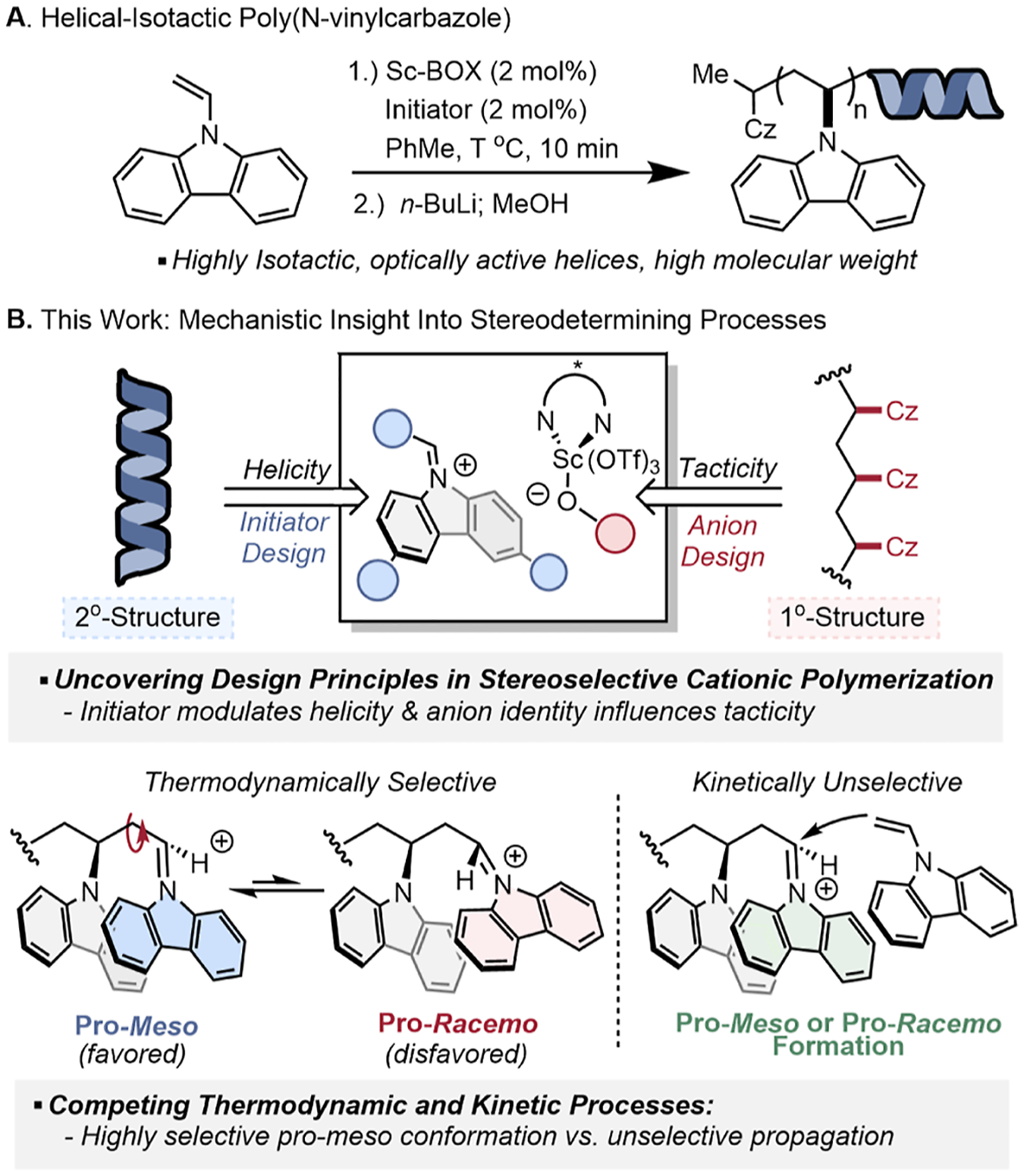
(A) HSSP of N-vinyl carbazole and (B) mechanistic rationale for the stereoselective cationic HSSP of N-vinylcarbazole.
Herein, we report an investigation into the relevant factors that influence stereoselectivity during the cationic HSSP of NVC by using a combination of kinetic investigations, temperature-dependent stereoselectivity analyses, isotope labeling, and initiator derivatization (Figure 1B). By expanding our knowledge of this polymerization mechanism, we demonstrate methods to tune both helicity and tacticity independently and temporally. Key mechanistic insights suggest two competing processes that determine the population of chain-end stereoisomers, whose relative rates determine control of tacticity: chain-end conformer equilibration and monomer propagation. Both processes play a role in the stereochemical outcome and are depicted in Figure 2b (step II and III). Exploration of the structure–selectivity relationships between initiator identity and helix-sense-selectivity elucidated a time-dependent relationship that provides a rationale to tune the magnitude of helicity through control of reaction conditions. The mechanistic insights uncovered through this study shed light on catalyst design principles in AIP that are specific to polymerization, including how chain-end effects dictate tacticity, while providing structure–stereoselectivity relationships that are broadly useful in asymmetric synthesis.
Figure 2.
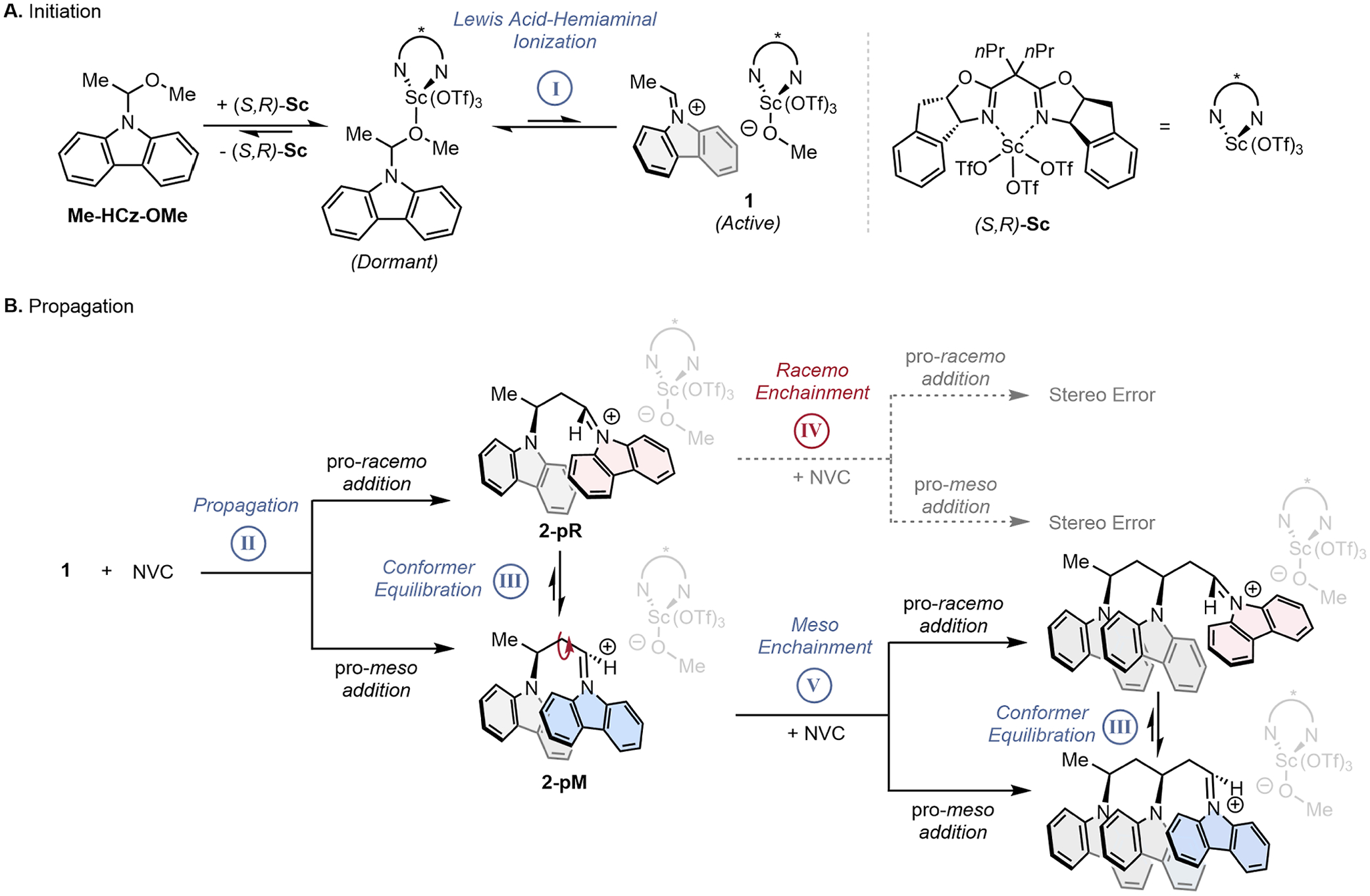
Proposed mechanism of stereocontrol during initiation (A) and polymerization (B) of N-vinylcarbazole.
Identity of the Propagating Chiral Anion.
Control of tacticity of poly(NVC) during polymerization is determined, in part, by the identity of the propagating chiral anion that is generated upon ionization of the hemiaminal initiator Me-HCz-OMe (Figure 2A). Ion-pairing between the chiral anion and iminium chain-end, 1, is inherently non-directional, and previous work has proposed that tight ion-pairing is needed for effective stereoinduction.7 Accordingly, we investigated the effect that steric parameters of the O-alkyl initiator substituent had on the resulting tacticity using the (S,R)-Sc Lewis acid catalyst (Figure 3). Synthesis of hemiaminal initiators allowed the systematic assessment of steric hindrance on isotacticity; increasing sterics from −OMe substitution toward larger −OEt, −OiPr, and −OtBu groups led to a marked decrease in stereocontrol, from 90.6 to 85.3% meso triads (% mm). These data fit our hypothesis of stereoselectivity being driven by tight ion pairing as a larger initiating group presumably results in a more separated ion pair during propagation and, thus, lower tacticity by significantly reducing the ΔG or increasing ΔG‡ for conformer equilibration (Figure S1). Attempts to generate smaller pro-anion groups, such as −Cl, −CN, and −OCF3, were found to be synthetically inaccessible due to the instability of the parent hemiaminal.
Figure 3.
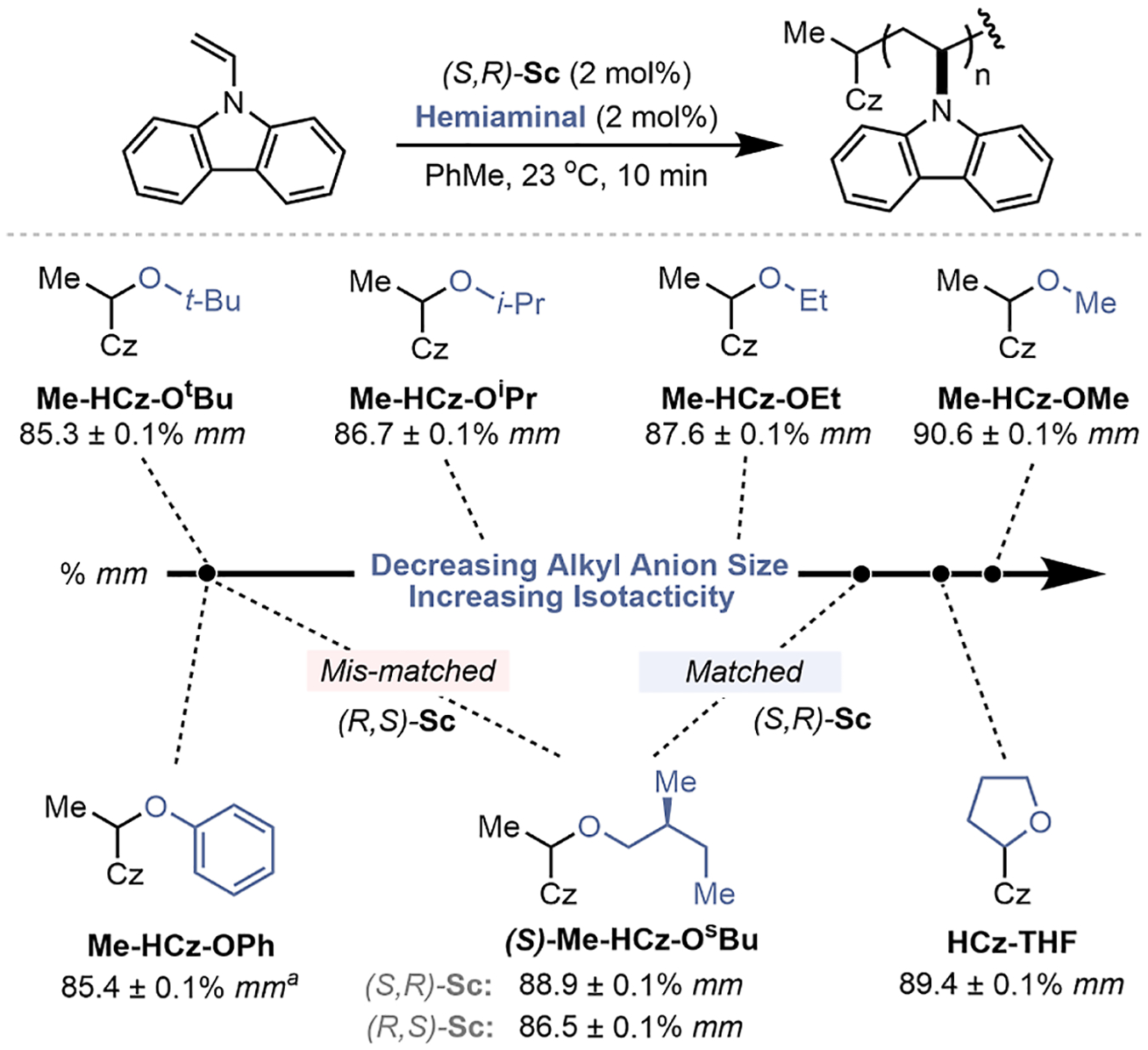
Effect of the alkyl anion structure on poly(NVC) isotacticity reported in % meso triads (% mm) in triplicate with standard error; reaction conditions: [NVC] = 0.12 M, [(S,R)-Sc] = 2.4 mM, [initiator] = 2.4 mM, 10 min, 23 °C, 2.59 mmol scale; Cz = carbazole; athis initiator was generated in situ from phenol.
To further probe the influence of the initiator structure on tacticity, we tested three initiators that generate an anion with specific and complementary chain-end interactions. A cyclic hemiaminal (HCz-THF) that covalently tethers the pro-anion to the initiator was synthesized using a copper-mediated C–H functionalization of tetrahydrofuran29 and provided high stereocontrol when used as an initiator (89.4% mm). The chiral pro-anion derived from (S)-(−)-2-methylbutanol, (S)-Me-HCz-OsBu, displayed a match-mismatch effect that depended on ligand stereochemistry, achieving 88.9% mm with the matched (S,R)-Sc and 86.5% mm with the partially matched (R,S)-Sc.7,14 Lastly, we hypothesized that an electron-rich phenoxy-based anion may create an opportunity for favorable π–π interactions with the electron-poor carbazolium chain-end, but the initiator Me-HCz-OPh yielded poor % mm. Additionally, counter-ion effects were evaluated for the chiral scandium Lewis-acid catalyst. Use of ScCl3 or substituting the triflates for other non-coordinating ions—namely PF6 and BArF24—led to erosion of isotacticity (Table S4), demonstrating the importance of Sc(OTf)3 that is a precursor to 1.
Kinetics of Polymerization.
We next investigated the kinetics of stereoselective NVC polymerization using Me-HCz-OMe as the initiator because it provided the highest isotacticity with (S,R)-Sc. The reaction progression was determined by 1H NMR after quenching with a 0.1 M solution of n-butyl lithium (nBuLi) in hexanes. Pseudo-first-order kinetics were observed with an observed rate (kobs) of 63.0 × 10−3 s−1 at 23 °C, which is consistent with other cationic polymerizations (Figure 4A, Table S2).2,7
Figure 4.
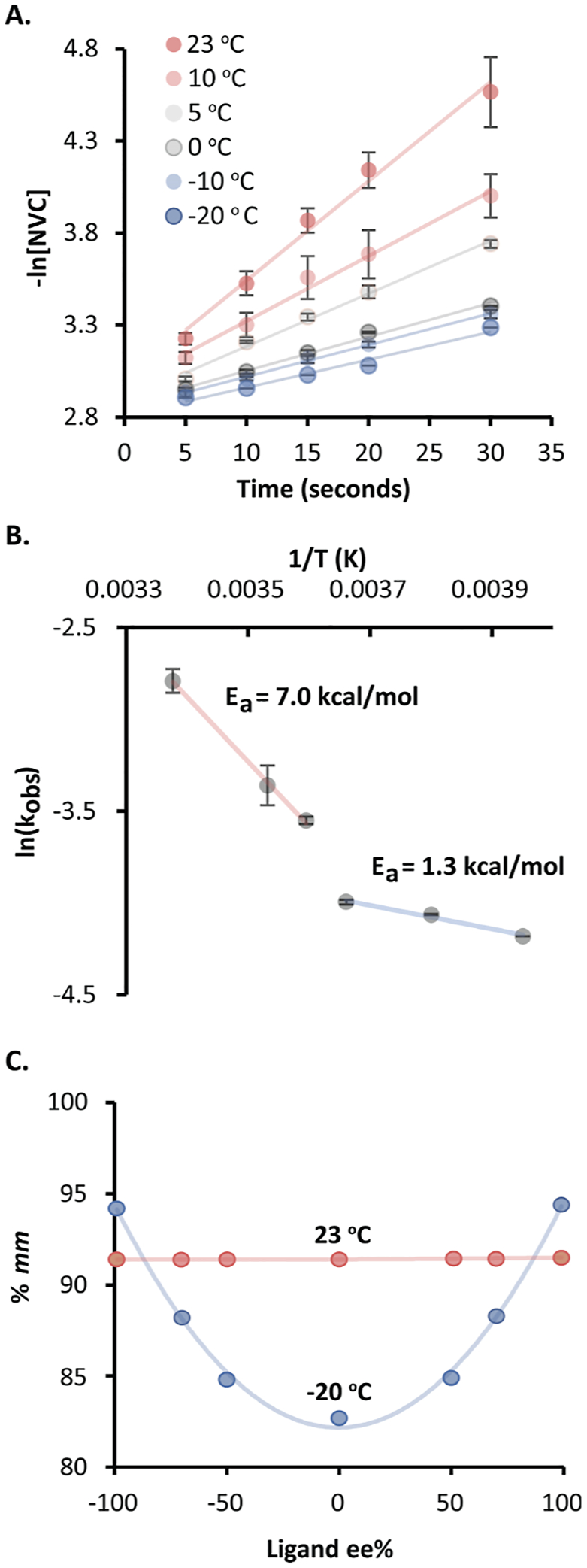
(A) Kinetics of NVC polymerization under optimized conditions ([NVC] = 0.06 M, [(S,R)-Sc] = 1.2 mM, [Me-HCz-OMe] = 1.2 mM). Reported data points are the mean of three individual experiments, and error bars indicate the standard error; (B) Arrhenius analysis of the kinetic data; and (C) non-linear effects of ligand ee %.
Transforming and plotting the logarithm of the kobs from conversion data in Figure 4A against inverse temperature yielded an Arrhenius plot (Figure 4B). Non-Arrhenius behavior was observed over the temperature range analyzed, resulting in two separate linear regimes that correlated to an activation energy (Ea) of 7.0 kcal/mol between 23 and 5 °C and an Ea of 1.3 kcal/mol between 0 and −20 °C. The discontinuity in Arrhenius behavior commonly arises from either a change in the rate-determining step (RDS) of polymerization or a change in the active catalyst structure. Use of a racemic ligand provided the same tacticity at 23 °C as the enantiopure ligand; however, at −20 °C, a decrease in selectivity was observed with scalemic ligand mixtures, indicating a nonlinear effect potentially consistent with catalyst heterochiral clustering but not homochiral clustering (Figure 4C).30 Such clustering is presumably possible with scandium Lewis acids, which have been observed with coordination numbers up to 7 and could accommodate a clustered ML2 structure.31,32 Ligand disproportionation to form an ML2 structure would also form Sc(OTf)3, which is known to be a highly active but unselective polymerization catalyst for NVC and could account for the loss of selectivity and lower Ea. Unfortunately, due to the instability of the iminium (1), attempts to grow single crystals were unsuccessful, and solutions of (S,R)-Sc were significantly convoluted by 1H NMR; thus, preventing direct observation of potential catalyst clustering.
Further experiments led us to the hypothesize that a change in RDS is a more likely explanation of the discontinuity in Arrhenius behavior. An Eyring analysis of stereoselectivity demonstrated a linear relationship between temperature and isotacticity, providing evidence that the catalyst structure remains the same during the stereo-determining step within the temperature range from −20 to 23 °C (Figure 5A). This experiment led us to conclude that catalyst clustering, if it were occurring, could not be solely responsible for the non-Arrhenius behavior. For these reasons, we sought to further explore the potential for a change to the RDS as temperature changes. At higher temperatures, we hypothesized that reversible chain-end ionization (Figure 2A, step I) is the RDS to propagation. At lower temperatures, however, the ionized state 1 is sufficiently stabilized, and we hypothesized that monomer propagation could become the RDS. Supporting evidence for this hypothesis is our observation of a loss of molecular-weight control at lower temperatures,15 potentially due to a lack of reversible chain-end capping.
Figure 5.
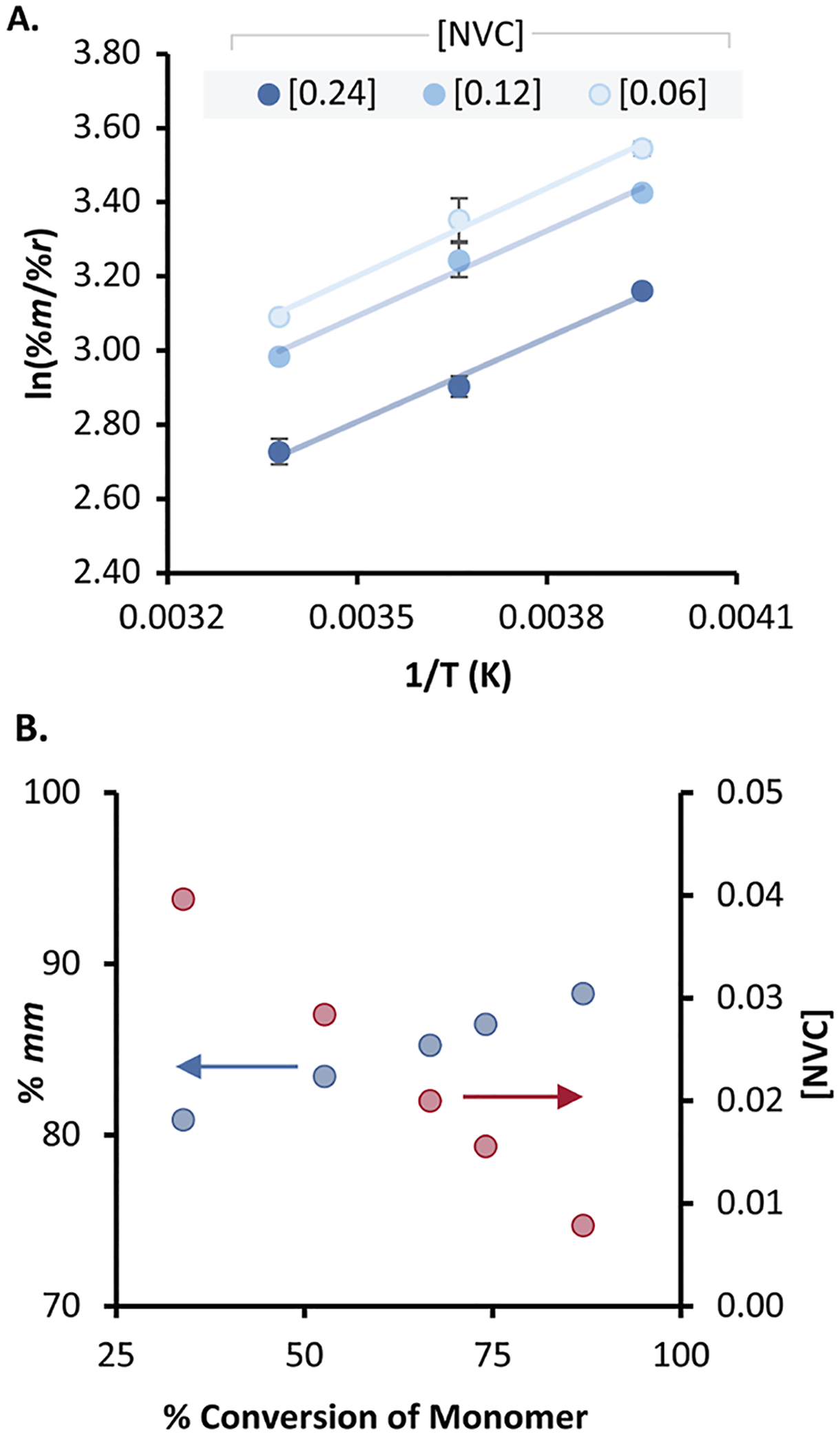
(A) Eyring analysis of NVC polymerization that quantitates tacticity as a function of temperature at different monomer concentrations in triplicate with standard errors and (B) monitoring the change in % mm as a function of monomer conversion; each data point is a separate polymerization.
Stereo-Determining Steps of Tacticity.
While conducting the kinetic study, we observed that the polymers produced were soluble during early stages of the polymerization, but as they approached complete conversion, the polymers became insoluble. Motivated by this observation, we monitored the % mm of the polymerization as a function of conversion and found that the % mm of the resultant polymer increased with increasing conversion (Figure 5B). While some changes in selectivity may be expected in the first few propagation steps, the stereoselectivity of a polymerization is typically not dependent on conversion.7,33 We hypothesized that this behavior was due to concentration-dependent phenomenon, where every monomer enchainment represents a bifurcation point on the potential energy surface.
To further probe this phenomenon, a modified Eyring analysis was carried out, and the ratio of % meso (% m) to % racemo diads (% r) was found by characterizing the % mm with high-temperature 1H NMR in d2-tetrachloroethane at 100 °C and transformed to % m using the Bovey chain-end control relationship.34 Over the temperature range of 23 to −20 °C, a linear relationship between the natural log of selectivity and the reciprocal of temperature is observed (Figure 5A). These data equate to a ΔΔG‡ of 1.50 kcal/mol at a monomer concentration of 0.12 M. Reactions below −20 °C and above room temperature were not analyzed because they quickly become heterogeneous.
To gain additional evidence for this phenomenon, we initiated three different polymerizations at starting monomer concentrations 0.24, 0.12, and 0.06 M NVC and observed that the isotacticity was higher for the polymerizations run at lower concentration. When ln(% m/% r) is plotted against [NVC] for each temperature, a linear relationship is also produced, but the y-intercept of the plot is distinct (Figure S3). Interestingly, similar concentration effects have been observed for the cationic polymerization of benzyl vinyl ether with BF3 etherate but were not investigated.35
To explain the concentration-dependent stereoselectivity, we propose that two competing elementary steps are operative that determine the populations of chain-end stereoisomers and that monomer concentration influences the magnitude to which each are operative (Scheme 1). The first step is a kinetically unselective addition of NVC to the iminium chain-end. This addition occurs regardless of the instantaneous chain-end conformation and, therefore, has the potential to enchain either a meso or racemo diad. The second step is a conformer equilibration (i.e., bond rotation) between the pro-meso and pro-racemo chain-end stereoisomeric conformers. As described in our previous work, the pro-meso conformer is thermodynamically favored due to beneficial orbital overlap between the iminium chain end and the last enchained monomer, the lack of gauche interactions, and presence of the chiral catalyst.
Scheme 1.
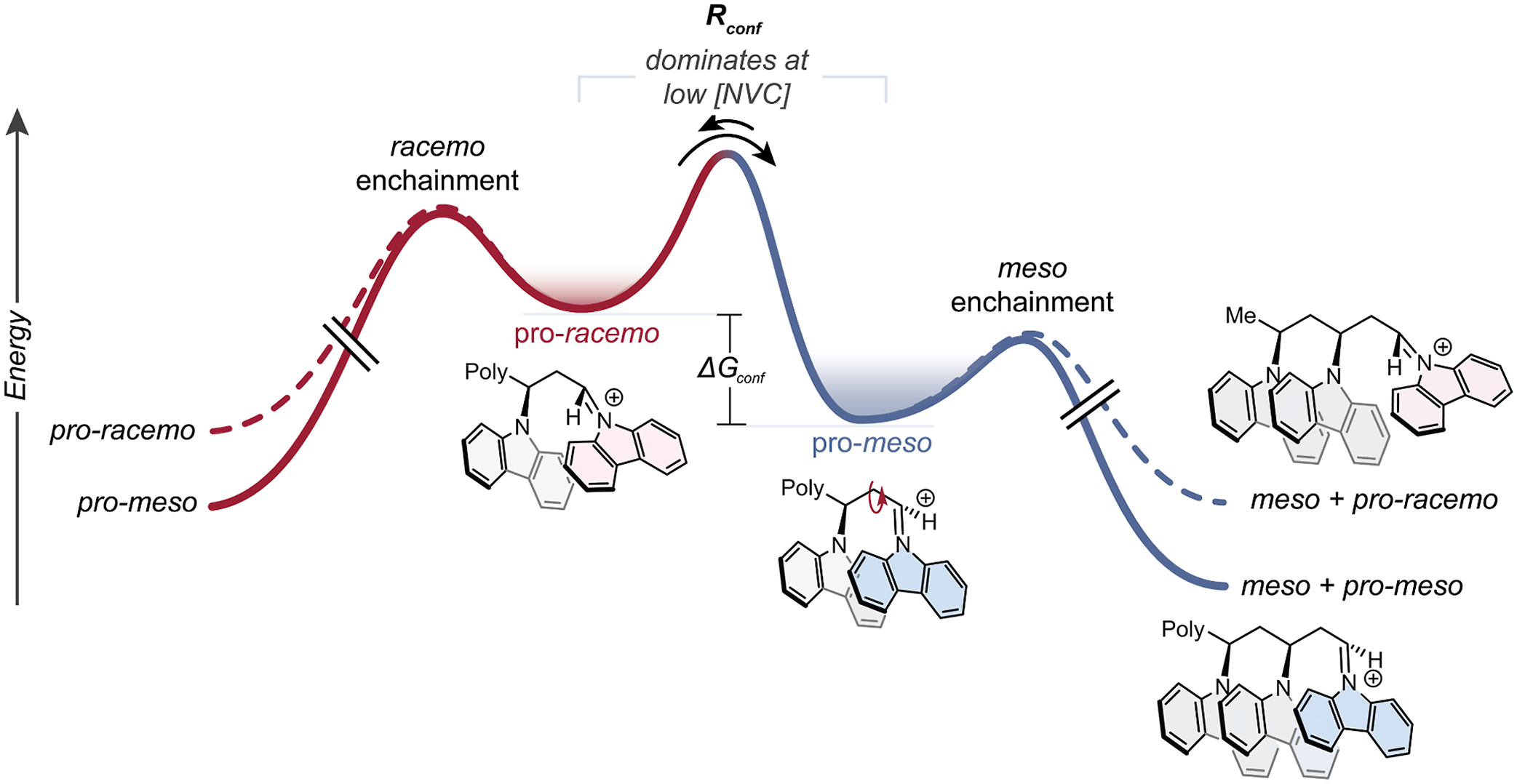
Proposed Reaction Coordinate Diagram of Polymerizationa
aLowering [NVC] or lowering temperature increases the relative rate of conformer equilibration compared to propagation, which drives selective meso enchainment and leads to isotactic polymers.
Competition between these two elementary steps—propagation and chain-end equilibration—dictate the stereochemical outcome of each monomer enchainment. These two rates both appear to be rapid; however, the rate of propagation is of pseudo-first-order in [NVC], whereas conformer equilibration is presumably zero-order in [NVC]. By lowering [NVC], the relative rate of conformer equilibration becomes significantly faster than propagation, which drives selective monomer addition to the pro-meso chain end and leads to isotactic polymers. At higher [NVC], monomer propagation occurs faster than the chain-end equilibration, leading to an erosion of stereoselectivity. Therefore, at low concentration, the system is put into a Curtin–Hammett scenario where thermodynamics of the iminium chain-end conformation determine tacticity. We hypothesize that the modest initial % mm seen during polymerization (Figure 5) arises from the high initial [NVC], where unselective propagation dominates without allowing ample time for conformer equilibration to “correct” to a pro-meso chain-end. To support this hypothesis, addition of monomer over 30 min at room temperature led to a 2% increase in % mm (91 to 93% mm), compared to the same conditions with the instantaneous injection of monomer solution. Attempts at slow monomer addition at colder temperatures and lower effective concentrations yielded polymer that was insoluble to conditions for tacticity assignment, suggesting a % mm greater than 95%.
To gain more evidence for this proposed mechanism, we sought to identify an experiment that changed the rates of conformer equilibration and propagation independently. We hypothesized that trideuteration of the vinyl group of NVC would result in an inverse-secondary kinetic isotope effect (KIE) due to the hybridization of vinylic C–D bonds changing from sp2 to sp3 upon propagation. Up to six deuterium atoms potentially interact in the bond-forming transition state (Figure 6), with three changing from sp2 to sp3 hybridization and two interacting via hyperconjugation to yield three inverse-α and two inverse-β secondary KIEs with a maximum KIE of approximately 0.2 (see Supporting Information Section S2.8).36 To conduct this experiment, a d3-N-vinylcarbazole (d3-NVC) monomer was synthesized in one-step via addition of carbazole to deuterated acetylene using a previously reported procedure by Ledovskaya et al.37
Figure 6.
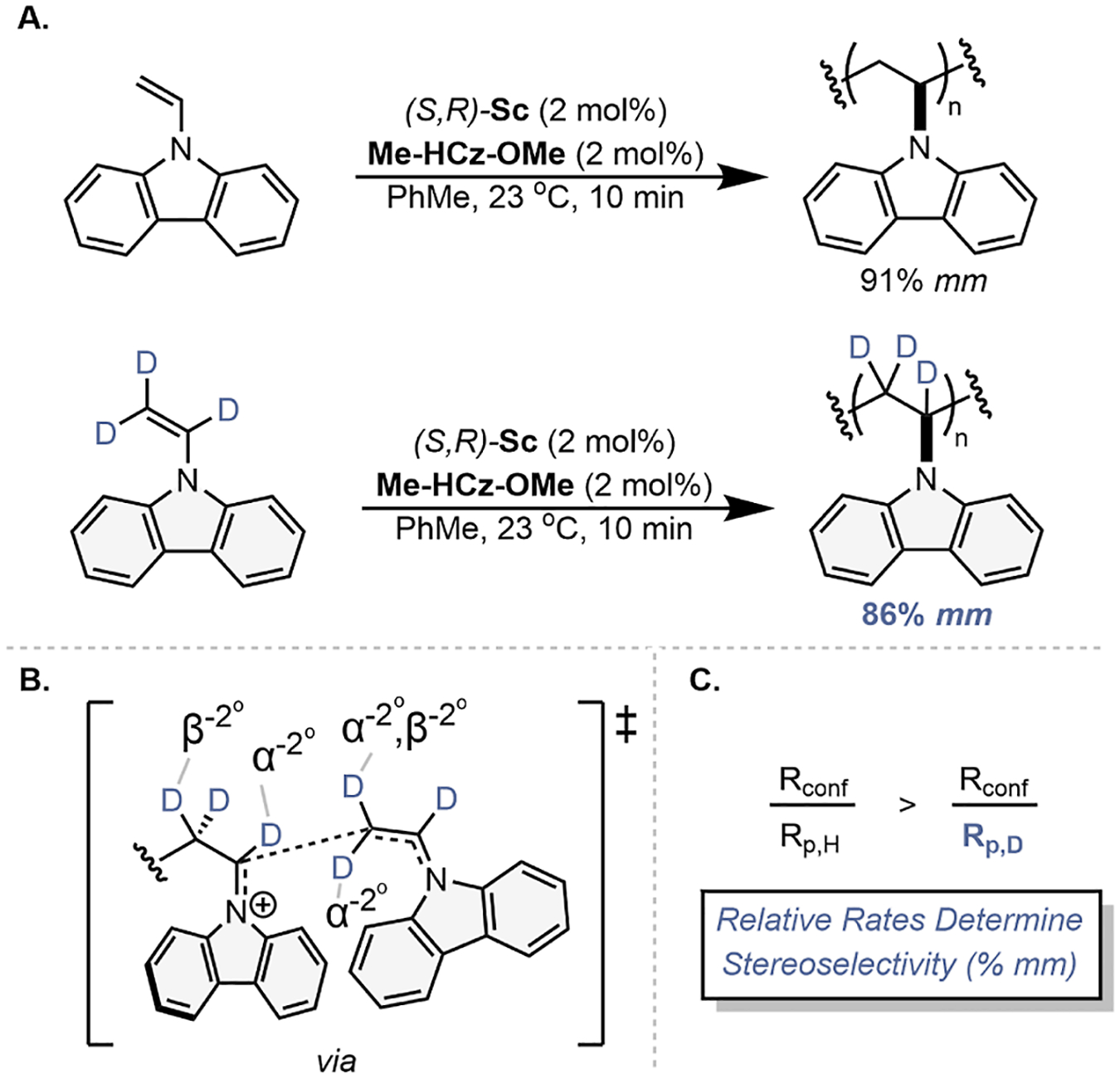
(A) Polymerization of NVC and d3-NVC and their resulting tacticity (% mm) determined by variable temperature 1H NMR and 13C NMR, respectively; (B) proposed KIE of deuterium atoms involved in the transition state of monomer propagation; and (C) relative rates of polymerization to conformer equilibration.
Polymerization of d3-NVC yielded poly(NVC) with a fully deuterated backbone and a tacticity of 86% mm, which is 5% mm lower than polymerization with the non-deuterated monomer (Figure 6A). This observation is consistent with a faster rate of polymerization relative to conformer equilibration for the d3-NVC polymerization, and it supports the proposal that the relative rates of these two elementary steps determines the magnitude of isotacticity.
Kinetic data for the d3-NVC polymerization was unable to be collected as conversion was greater than 90% after 5 s. Based off concentration experiments, a minimum KIE of 0.33 is estimated if pseudo-first-order kinetics are maintained (see Supporting Information Section S2.8 for derivation). Other potential rationalizations for this KIE are that trideuteration affects the rate of conformer equilibration through a steric KIE or changes the location of the equilibrium between pro-meso and pro-racemo chain-end conformations. Steric KIEs are typically significantly smaller in magnitude than the observed KIEs and, if operative, would likely increase the forward rate of conformer equilibration and improve the stereoselectivity due to the proposed sterically demanding transition state. Alternatively, the Cϕ position of imines and oxocarbeniums has been postulated to have strong C–H hydrogen bonding, which results in a positive KIE upon deuteration of this position.38,39 This phenomenon is potentially contributing to the large inverse KIE observed here via hydrogen bonding interactions with the anionic counterion.
The use of conformational locks represents a complementary experiment that changes the rate of conformer equilibration without significantly affecting the rate of propagation. Conformational locks are commonly employed to reduce conformational freedom in geometrically confined systems. For example, adding a tert-butyl group onto a cyclohexane ring effectively locks it into a conformation by increasing the energy required to ring flip. For our (S,R)-Sc catalyst system, we hypothesized that the tert-butyl group on 3,6-di-tert-butyl-N-vinylcarbazole (t-Bu NVC) would function as a conformational lock by increasing the energy required for chain-end conformer equilibration (Figure 2B, step III) and, therefore, resulting in a polymer with lower isotacticity (Figure 7).
Figure 7.
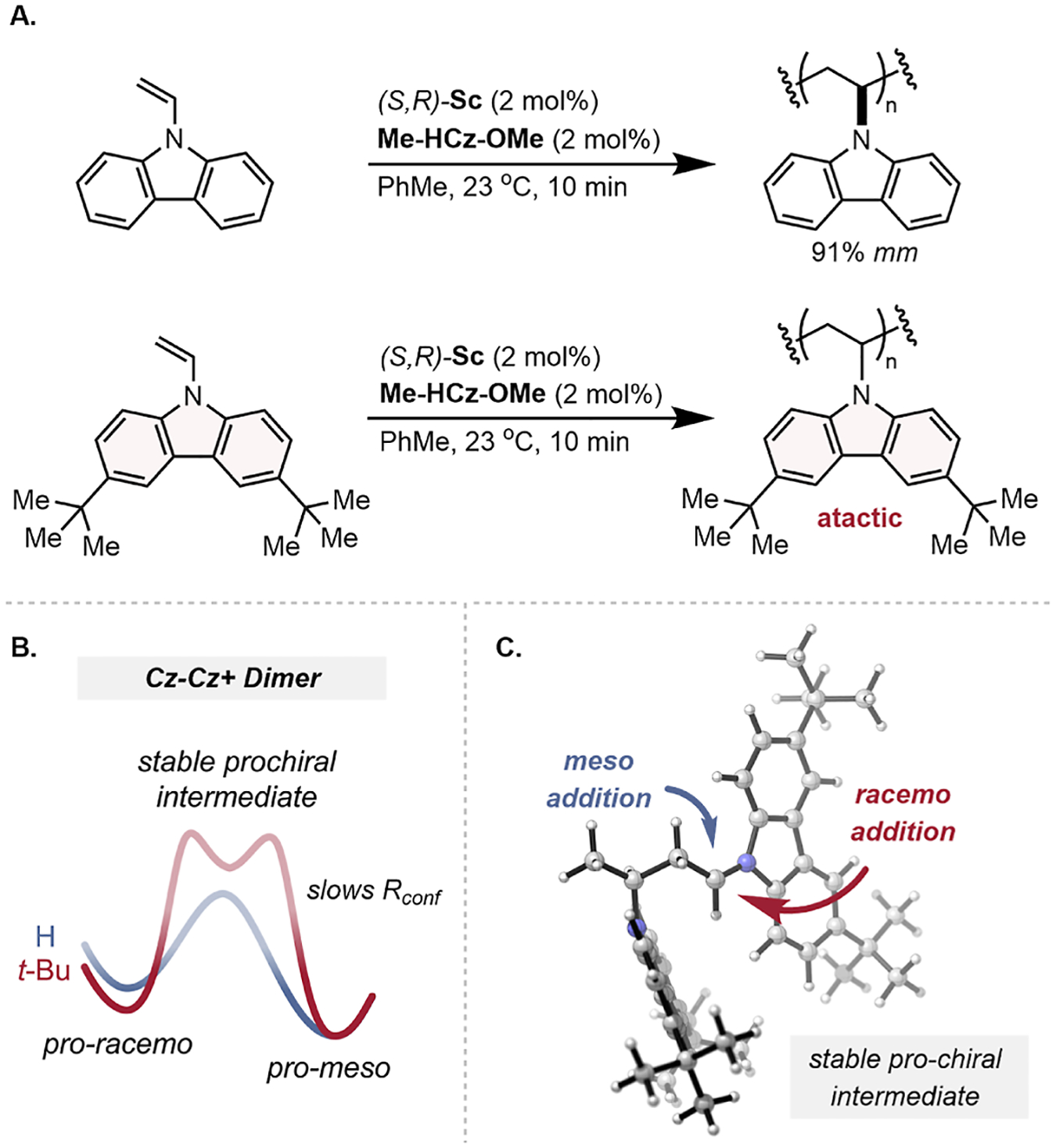
(A) Polymerization of NVC and t-Bu-NVC and their resulting tacticity and (B) reaction coordinate diagram displaying the relative energies between pro-meso and pro-racemo conformers and the presence of a stable intermediate with both faces of the iminium available, whose DFT-optimized structure is shown in (C).
Polymerization of t-Bu NVC under our standard conditions produced an atactic polymer (Figure 7A). In fact, control experiments demonstrated that the use of (S,R)-Sc as a catalyst produced poly(t-Bu NVC) with the same stereo micro-structure as a polymer prepared with TfOH or Sc(OTf)3. A relaxed dihedral scan of the iminium chain end after a single propagation with density functional theory (DFT) provided insights into the relative energies of the chain-end conformers.15 Unlike for NVC, there exists an intermediate structure where the carbazolium chain end sits anti to the last enchained monomer and exposes both the si and re face of the chain end for potential monomer addition (Figures 7B,C, S10). The barrier height is increased roughly from 4 to 8 kcal/mol—suggesting a slower rate of conformer equilibration—and the pro-racemo conformation is disfavored less as seen through a smaller ΔG (Figure S11). This monomer presents a case where chain-end conformer equilibration is slowed compared to the rate of polymerization, resulting in an atactic polymer.
In contrast to our observations, recently reported work by Aoshima and co-workers disclosed a ZnCl2-based catalyst system for the stereoselective living cationic polymerization of t-Bu NVC.27 They observed qualitative isotactic enrichment for this monomer at −78 °C with their catalyst, which was attributed to the increase in ΔΔG‡ between meso and racemo addition under kinetic stereocontrol. This result highlights that the stereo-determining step of this complementary catalytic approach is fundamentally different to the Sc–BOX catalysts described herein.
Initiator Design for Helical Control.
Our previous work demonstrated that the stereoselectivity of the first monomer propagation was a strong determinant in the helicity of the resulting polymer.15 Therein, we showed that use of an enantioenriched initiator increased the magnitude of helicity when matched with the appropriate catalyst enantiomer. To probe helicity control more fully through the initiator design, we hypothesized that the synthesis of a library of sterically and electronically distinct hemiaminal initiators would provide structure–stereoselectivity relationships to better understand the structural factors that dictate helicity (Figure 8). Tacticity was held constant by maintaining the same pro-alkoxide derived from (S,R)-Sc for all species studied. Polymerization was initiated under standard conditions with 2 mol % loading of the designated hemiaminal initiator, isolated via precipitation twice, and analyzed by circular dichroism (CD) spectroscopy in tetrahydrofuran. A positive cotton peak is observed in the CD at 296 nm, which we designated to the helical structure of the polymer backbone. As a reference, the initiator derived from NVC (Me-HCz-OMe) provided a CD response of 790 deg cm2 dmol−1.15
Figure 8.
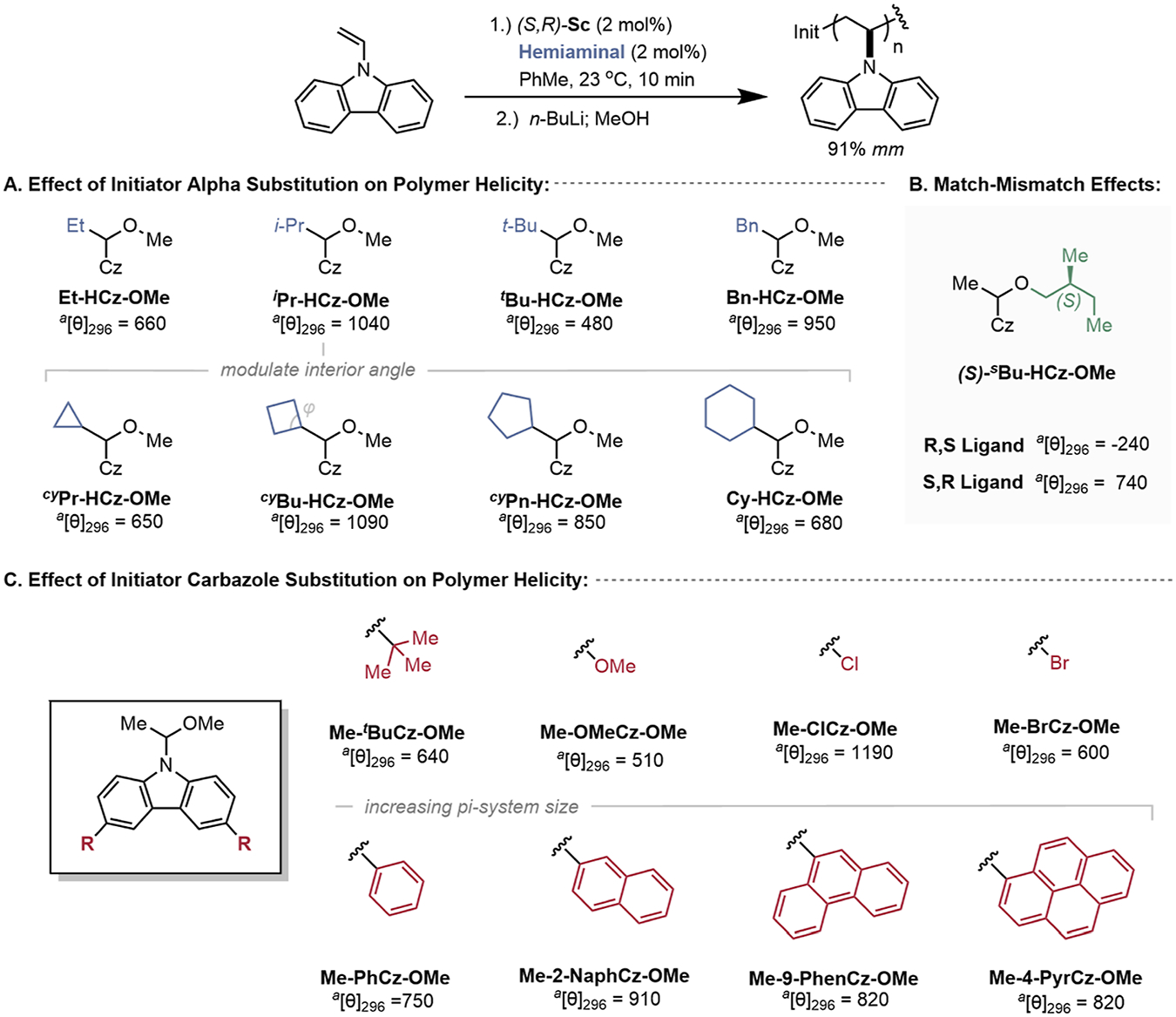
Hemiaminal substituent effects on resulting polymer helical excess measured through CD. Polymerization was carried out at 23 °C under optimized conditions ([NVC] = 0.12 M, [(S,R)-Sc] = 1.2 mM, [hemiaminal] = 1.2 mM) with a 5 min pre-mix between the initiator and the catalyst, then a 10 min polymerization time and subsequent quenching with n-BuLi (0.1 M) solution. aUnits for molar ellipticity are deg cm2 dmol−1.
Different aliphatic-substituted hemiaminal initiators were accessed first by condensing carbazole with various aldehydes or acetals in the presence of MeOH and an acid catalyst. Increasing the steric bulk alpha to the hemiaminal improves the CD response when going from ethyl (Et-HCz-OMe) and isopropyl (iPr-HCz-Ome), with a maximum of 1040 deg cm2 dmol−1 (Figure 8A). We hypothesize that this is due to an increase in the stereoselectivity of the first propagation step due to steric effects of the prochiral iminium initiating species. Further increasing the steric demand to tert-butyl, tBu-HCz-OMe, leads to a significant decrease in response, possibly because tert-butyl prevents intimate ion-pairing between the chiral anion and the iminium formed upon ionization. Cyclic alkyl groups alpha to the hemiaminal show that decreasing the interior angle from cyclohexyl (Cy-HCz-OMe) to cyclobutyl (cyBu-HCz-OMe) improves the magnitude of helicity. A further decrease of the interior angle to cyclopropyl (cyPr-HCz-OMe) is deleterious and may arise from the significant change in electronics of the iminium via p-orbital donation of the cyclopropyl ring.40 Lastly, a benzylic substituent (Bn-HCz-OMe) showed a high CD response with 950 deg cm2 dmol−1, possibly due to interactions of the counteranion with the π-system of the initiator. Diastereomeric initiator (S)-Me-HCz-OsBu demonstrated a large match–mismatch effect for CD response, possibility indicating a complementary approach to control the magnitude of helicity (Figure 8B).
Subsequently, we sought to understand how the electronic properties of the initiating carbazole heterocycle influenced the magnitude of helicity. Carbazole derivatives substituted at the 3- and 6-position were synthesized and used as initiators. Polymerization initiated from substituted carbazole hemiaminals with electron-donating groups such as tert-butyl (Me-tBuCz-OMe) and methoxy (Me-OMeCz-OMe) provided poor CD responses (Figure 8C). Small electron-withdrawing groups such as chlorine (Me-ClCz-OMe) displayed the largest magnitude increase of helicity for all initiators studied, up to 1190 deg cm2 dmol−1, while the sterically larger bromine (Me-BrCz-OMe) resulted in one of the lowest responses with 600 deg cm2 dmol−1. Phenyl substitution and further extensions of the π-system (2-Naph-, 9-phenanthryl, and 4-pyrenyl) showed moderately large responses, but no trends between the size of the π-system and helical excess were observed.
Well-established structure–reactivity trends have been observed with many reactions that invoke the reaction of an iminium with an electron-rich alkene in the presence of chiral anion; therefore, we were intrigued by the lack of the relationships observed by the initiator derivatives tested, as shown in Figure 8. In these reactions, a constant pre-mixing time of the hemiaminal and Lewis acid of 5 min was used. We hypothesized that a dynamic thermodynamic resolution of the racemic hemiaminal may be responsible for the lack of trends in CD data as different substitutions may need to enrich for longer periods of time to reach maximum CD response.
To probe this idea, the mixing time of catalyst (S,R)-Sc and Me-HCz-OMe was changed from 0, 1, 5, 30, and 120 min before the addition of monomer solution, and an enrichment of CD response was observed upon isolation of the resulting polymer, providing a temporal handle for the control of helicity (Figure S7). DFT results at the b3lyp-d3/def2svp//b3lyp-d3/def2tzvp level suggest a sufficient ΔG (0.69 kcal/mol) between the diastereomeric intermediates of the initiator-Lewis-acid complex for a dynamic thermodynamic resolution of the initiator (Table S5). The same mixing time experiment was conducted on Me-BrCz-OMe and iPr-HCz-OMe, and more complex behavior was observed in which an initial increase in CD response was measured, and then, the response decayed at longer mixing times (Figure S7). These data highlight the importance of optimizing reaction conditions for each initiator individually as complex degradation, thermodynamic, and kinetic pathways possibly lead to a large temporal dependence on the pre-mixing phase and helical response.
CONCLUSIONS
Complementary kinetic, computational, and experimental work has provided a mechanistic rationale for the independent control of both tacticity and helicity in the stereoselectivity HSSP of NVC. A systematic investigation into the identity of the propagating counterion provided design principles for the role of the catalyst and initiating species. Temperature-dependent kinetic studies revealed that a change in the reaction mechanism occurs between −20 and 23 °C. Eyring analysis of stereoselectivity, kinetic isotope labeling, and conformational lock experiments implicate possible competing stereo-determining steps whose relative rates are both concentration and temperature dependent. Competition between these two elementary steps—propagation and chain-end equilibration (Figure 2, step II and III)—dictates the stereochemical outcome of each monomer enchainment. Because the rate of propagation is of pseudo-first-order in [NVC], whereas conformer equilibration is presumably of zero-order in [NVC], lowering [NVC] or temperature increases the relative rate of conformer equilibration compared to propagation, which drives selective monomer addition to the pro-meso chain end and leads to isotactic polymers. Lastly, structure–selectivity relationships were established for how initiating species and mixing time influence the magnitude of poly(NVC) helicity, which revealed a large temporal response that is dictated by a complex relationship between thermodynamic and kinetic factors that lead to helicity. This study enabled both a significantly deeper understanding of cationic stereoselective HSSP of NVC and demonstrated the value of using traditional and modern tools of physical-organic chemistry to understand polymer-specific phenomena in stereoselective catalysis. We envision that this work will provide improved approaches to access stereo-controlled materials for applied studies and inform continued catalyst evolution for stereoselective vinyl polymerization through AIP mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the many members of the Leibfarth group who have contributed in various ways. Additionally, the UNC Department of Chemistry’s NMR Core Laboratory provided expertise and instrumentation that enabled this study with support from the National Science Foundation (CHE-1828183 and CHE-0922858). We acknowledge the Macromolecular Interactions Facility (CD; supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086) for assistance with CD instrumentation.
Funding
This work is supported by the National Institute of General Medical Sciences of the National Institute of Health under award number 1-R35-GM142666-01 and the Arnold and Mabel Beckman Foundation under a Beckman Young Investigator Award.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c02165.
Synthesis, experimental information, computational details, and geometries (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.3c02165
Contributor Information
Cole C. Sorensen, Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599, United States
Vittal Bhat, Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
Anthony Y. Bello, Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599, United States
Frank A. Leibfarth, Department of Chemistry, University of North Carolina, Chapel Hill, North Carolina 27599, United States
REFERENCES
- (1).Sorensen CC; Kozuszek CT; Borden MA; Leibfarth FA Asymmetric Ion-Pairing in Stereoselective Vinyl Polymerization. ACS Catal. 2023, 13, 3272–3284. [Google Scholar]
- (2).Watanabe H; Kanazawa A; Aoshima S Stereospecific Living Cationic Polymerization of N-Vinylcarbazole through the Design of ZnCl2-Derived Counteranions. ACS Macro Lett. 2017, 6, 463–467. [DOI] [PubMed] [Google Scholar]
- (3).Hirokawa Y; Higashimura T; Matsuzaki K; Kawamura T; Uryu T Stereochemistry in cationic polymerization of alkenyl ethers. III. Effect of the alkoxy group on the steric structure of polymers. J. Polym. Sci., Part A-1: Polym. Chem 1979, 17, 3923–3931. [Google Scholar]
- (4).Hirokawa Y; Higashimura T; Matsuzaki K; Uryu T Stereochemistry in Cationic Polymerization of Alkenyl Ethers. II. Formation of Poly(B-Substituted Vinyl Ether)s with Erythro-Meso Structures. J. Polym. Sci., Part A: Polym. Chem 1979, 17, 1473–1481. [Google Scholar]
- (5).Yuki H; Hatada K; Ota K; Kinoshita I; Murahashi S; Ono K; Ito Y Stereospecific Polymerization of Benzyl Vinyl Ether by BF3•OEt2. J. Polym. Sci., Part A: Polym. Chem 1969, 7, 1517–1536. [Google Scholar]
- (6).Kamigaito M; Sawamoto M; Higashimura T Alkoxy-Substituted Titanium(IV) Chlorides as Lewis Acid Activators for Living Cationic Polymerization of Isobutyl Vinyl Ether. Control of Lewis Acidity in the Design of Initiating Systems1. Macromolecules 1995, 28, 5671–5675. [Google Scholar]
- (7).Varner TP; Teator AJ; Reddi Y; Jacky PE; Cramer CJ; Leibfarth FA Mechanistic Insight into the Stereoselective Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc 2020, 142, 17175–17186. [DOI] [PubMed] [Google Scholar]
- (8).Uchiyama M; Watanabe D; Tanaka Y; Satoh K; Kamigaito M Asymmetric Cationic Polymerization of Benzofuran through a Reversible Chain-Transfer Mechanism: Optically Active Polybenzofuran with Controlled Molecular Weights. J. Am. Chem. Soc 2022, 144, 10429–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Natta G; Farina M; Peraldo M; Bressan G Asymmetric Synthesis of Optically Active Di-Isotactic Polymers from Cyclic Monomers. Makromol. Chem 1961, 43, 68–75. [Google Scholar]
- (10).Teator AJ; Leibfarth FA Catalyst-Controlled Stereoselective Cationic Polymerization of Vinyl Ethers. Science 2019, 363, 1439–1443. [DOI] [PubMed] [Google Scholar]
- (11).Knutson PC; Teator AJ; Varner TP; Kozuszek CT; Jacky PE; Leibfarth FA Brønsted Acid Catalyzed Stereoselective Polymerization of Vinyl Ethers. J. Am. Chem. Soc 2021, 143, 16388–16393. [DOI] [PubMed] [Google Scholar]
- (12).Zhang X; Yang Z; Jiang Y; Liao S Organocatalytic, Stereoselective, Cationic Reversible Addition–Fragmentation Chain-Transfer Polymerization of Vinyl Ethers. J. Am. Chem. Soc 2022, 144, 679–684. [DOI] [PubMed] [Google Scholar]
- (13).Teator AJ; Varner TP; Jacky PE; Sheyko KA; Leibfarth FA Polar Thermoplastics with Tunable Physical Properties Enabled by the Stereoselective Copolymerization of Vinyl Ethers. ACS Macro Lett. 2019, 8, 1559–1563. [DOI] [PubMed] [Google Scholar]
- (14).Watanabe H; Yamamoto T; Kanazawa A; Aoshima S Stereoselective Cationic Polymerization of Vinyl Ethers by Easily and Finely Tunable Titanium Complexes Prepared from Tartrate-Derived Diols: Isospecific Polymerization and Recognition of Chiral Side Chains. Polym. Chem 2020, 11, 3398–3403. [Google Scholar]
- (15).Sorensen CC; Leibfarth FA Stereoselective Helix-Sense-Selective Cationic Polymerization of N-Vinylcarbazole Using Chiral Lewis Acid Catalysis. J. Am. Chem. Soc 2022, 144, 8487–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Okamoto Y; Nakano T Asymmetric Polymerization. Chem. Rev 1994, 94, 349–372. [Google Scholar]
- (17).Ito S; Nozaki K Asymmetric Polymerization. In Catalytic Asymmetric Synthesis; 3rd Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp 931–985. [Google Scholar]
- (18).Okamoto Y; Suzuki K; Ohta K; Hatada K; Yuki H Optically Active Poly (Triphenylmethyl Methacrylate) with One-Handed Helical Conformation. J. Am. Chem. Soc 1979, 101, 4763–4765. [Google Scholar]
- (19).Watanabe K; Osaka I; Yorozuya S; Akagi K Helically π-Stacked Thiophene-Based Copolymers with Circularly Polarized Fluorescence: High Dissymmetry Factors Enhanced by Self-Ordering in Chiral Nematic Liquid Crystal Phase. Chem. Mater 2012, 24, 1011–1024. [Google Scholar]
- (20).Xu L; Gao BR; Xu XH; Zhou L; Liu N; Wu ZQ Controlled Synthesis of Cyclic-Helical Polymers with Circularly Polarized Luminescence. Angew. Chem. Int. Ed 2022, 61, 61. [DOI] [PubMed] [Google Scholar]
- (21).Maeda K; Nozaki M; Hashimoto K; Shimomura K; Hirose D; Nishimura T; Watanabe G; Yashima E Helix-Sense-Selective Synthesis of Right-and Left-Handed Helical Luminescent Poly-(Diphenylacetylene)s with Memory of the Macromolecular Helicity and Their Helical Structures. J. Am. Chem. Soc 2020, 142, 7668–7682. [DOI] [PubMed] [Google Scholar]
- (22).Song X; Li Y-X; Zhou L; Liu N; Wu Z-Q Controlled Synthesis of One-Handed Helical Polymers Carrying Achiral Organoiodine Pendants for Enantioselective Synthesis of Quaternary All-Carbon Stereogenic Centers. Macromolecules 2022, 55, 4441–4449. [Google Scholar]
- (23).Cram DJ; Kopecky KR Studies in Stereochemistry. XXX. Models for Steric Control of Asymmetric Induction1. J. Am. Chem. Soc 1959, 81, 2748–2755. [Google Scholar]
- (24).Ito S; Nozaki K Asymmetric Polymerization. In Catalytic Asymmetric Synthesis; John Wiley & Sons, Ltd, 2010; pp 931–985. [Google Scholar]
- (25).Dougherty DA; Anslyn EV Modern Physical Organic Chemistry; University Science Books, 2005; pp 421–488. [Google Scholar]
- (26).Nakano T; Okamoto Y; Hatada K Asymmetric Polymerization of Triphenylmethyl Methacrylate Leading to a One-Handed Helical Polymer: Mechanism of Polymerization. J. Am. Chem. Soc 1992, 114, 1318–1329. [Google Scholar]
- (27).Watanabe H; Kanazawa A; Okumoto S; Aoshima S Role of the Counteranion in the Stereospecific Living Cationic Polymerization of N-Vinylcarbazole and Vinyl Ethers: Mechanistic Investigation and Synthesis of Stereo-Designed Polymers. Macromolecules 2022, 55, 4378–4388. [Google Scholar]
- (28).Teator AJ; Varner TP; Knutson PC; Sorensen CC; Leibfarth FA 100th Anniversary of Macromolecular Science Viewpoint: The Past, Present, and Future of Stereocontrolled Vinyl Polymerization. ACS Macro Lett. 2020, 9, 1638–1654. [DOI] [PubMed] [Google Scholar]
- (29).Yang Q; Choy PY; Fu WC; Fan B; Kwong FY Copper-Catalyzed Oxidative C-H Amination of Tetrahydrofuran with Indole/Carbazole Derivatives. J. Org. Chem 2015, 80, 11193–11199. [DOI] [PubMed] [Google Scholar]
- (30).Guillaneux D; Zhao SH; Samuel O; Rainford D; Kagan HB Nonlinear Effects in Asymmetric Catalysis. J. Am. Chem. Soc 1994, 116, 9430–9439. [Google Scholar]
- (31).Evans DA; Sweeney ZK; Rovis T; Tedrow JS Highly Enantioselective Syntheses of Homopropargylic Alcohols and Dihydrofurans Catalyzed by a Bis(oxazolinyl)pyridine–Scandium Triflate Complex. J. Am. Chem. Soc 2001, 123, 12095–12096. [DOI] [PubMed] [Google Scholar]
- (32).Evans DA; Scheidt KA; Fandrick KR; Lam HW; Wu J Enantioselective Indole Friedel-Crafts Alkylations Catalyzed by Bis(Oxazolinyl)Pyridine-Scandium(III) Triflate Complexes. J. Am. Chem. Soc 2003, 125, 10780–10781. [DOI] [PubMed] [Google Scholar]
- (33).Hatada K; Ute K; Tanaka K; Imanari M; Fujii N Two-Dimensional NMR Spectra of Isotactic Poly(methyl methacrylate) Prepared with t-C4H9MgBr and Detailed Examination of Tacticity. Polym. J 1987, 19, 425–436. [Google Scholar]
- (34).Bovey FA; Tiers GVD Polymer NSR Spectroscopy. II. The High Resolution Spectra of Methyl Methacrylate Polymers Prepared with Free Radical and Anionic Initiators. J. Polym. Sci., Part A: Polym. Chem 1996, 34, 711–720. [Google Scholar]
- (35).Yuki H; Hatada K; Ota K; Kinoshita I; Murahashi S; Ono K; Ito Y Stereospecific Polymerization of Benzyl Vinyl Ether by BF3.OEt2. J. Polym. Sci 1969, 7, 1517–1536. [Google Scholar]
- (36).Streitwieser A; Jagow RH; Fahey RC; Suzuki S Kinetic Isotope Effects in the Acetolyses of Deuterated Cyclopentyl Tosylates. J. Am. Chem. Soc 1958, 80, 2326–2332. [Google Scholar]
- (37).Ledovskaya MS; Voronin VV; Rodygin KS; Posvyatenko AV; Egorova KS; Ananikov VP Direct Synthesis of Deuterium-Labeled O -S -N-Vinyl Derivatives from Calcium Carbide. Synthesis 2019, 51, 3001–3013. [Google Scholar]
- (38).Terada M; Tanaka H; Sorimachi K Enantioselective Direct Aldol-Type Reaction of Azlactone via Protonation of Vinyl Ethers by a Chiral Brønsted Acid Catalyst. J. Am. Chem. Soc 2009, 131, 3430–3431. [DOI] [PubMed] [Google Scholar]
- (39).Klausen RS; Kennedy CR; Hyde AM; Jacobsen EN Chiral Thioureas Promote Enantioselective Pictet-Spengler Cyclization by Stabilizing Every Intermediate and Transition State in the Carboxylic Acid-Catalyzed Reaction. J. Am. Chem. Soc 2017, 139, 12299–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fleming I Molecular Orbitals and Organic Chemical Reactions; Student Edition, 3rd ed.; John Wiley & Sons, Inc., 2009; pp 71–72. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.