

Abstract
As new methods to interrogate glycan organization on cells develop, it is important to have a molecular level understanding of how chemical fixation can impact results and interpretations. Site-directed spin labeling technologies are well suited to study how the spin label mobility is impacted by local environmental conditions, such as those imposed by cross-linking effects of paraformaldehyde cell fixation methods. Here, we utilize three different azide-containing sugars for metabolic glycan engineering with HeLa cells to incorporate azido glycans that are modified with a DBCO-based nitroxide moiety via click reaction. Continuous wave X-band electron paramagnetic resonance spectroscopy is employed to characterize how the chronological sequence of chemical fixation and spin labeling impacts the local mobility and accessibility of the nitroxide-labeled glycans in the glycocalyx of HeLa cells. Results demonstrate that chemical fixation with paraformaldehyde can alter local glycan mobility and care should be taken in the analysis of data in any study where chemical fixation and cellular labeling occur.
Keywords: Sialic acid, metabolic glycan engineering, chemical fixation, crosslinking, glycan, organization, electron paramagnetic resonance spectroscopy
1. Introduction
Cells are shielded by a layer of carbohydrates known as the “cell glycocalyx” that plays an important role in various biological events [1]. Cell surface carbohydrates exist mainly as conjugates with lipids and proteins to assist their attachment onto the cell wall or membrane. Changes in the composition and presentation patterns of glycans in the cell glycocalyx are associated with many diseases [2–4]. For example, more than 80% of cell surface glycoconjugates in the vertebrate brain are glycolipids [5]. It has been revealed that glycosphingolipids (GSLs) are the major component of cell surface glycolipids and are involved in many processes such as signal transduction [6], cell recognition, adhesion and apoptosis [7, 8], embryonic and nerve system development [9, 10], and drug resistance [11]. Peculiar GSL expression and presentation are related to various pathologies such as cancer [12, 13], diabetes [14], sclerosis [15], Alzheimer’s disease (AD) [16, 17], and lysosomal storage disorders [18]. Another type of prominent glycolipids is glycosylphosphatidylinositols (GPIs), whose attachment to proteins is one of the most common posttranslational modifications [19]. GPIs help anchor proteins to the extracellular membrane and are related to many biological activities from signal transduction [20] to cancer [21–24], AD [25], etc. It was also demonstrated that GPI-anchored proteins could not function properly without GPI [26, 27] and that GPI deficiency was lethal [28].
It is generally recognized that the structure of glycans is extremely adaptable to enable their broad functioning via interactions with each other and with other molecules; therefore, the presentation form of glycans on the cell surface can have a decisive impact on their biological activity [29]. As such there is a continued desire to develop novel tools for interrogating glycan organization and mobility on cells. Recently, we demonstrated how nitroxide radical spin labeling with electron paramagnetic resonance (EPR) spectroscopy can be applied to the study of glycans on cells [30–32]. Spin labeling has a rich history of being utilized to probe the local environment in macromolecules and macromolecular assemblies such as lipids, proteins, membrane proteins, nucleic acids, materials like polymers [33–38].
Spin-labeling EPR, which can be performed at physiologically relevant temperatures and conditions, when combined with a spin-labeled biological molecule, provides information on local dynamics, conformations, accessibility, organization, and hydration environment at local sites within biological environments [33, 34, 39–41]. Typically, a persistent radical, such as a nitroxide moiety, is incorporated into the chosen macromolecule (protein, nucleic acids, lipids, polymers etc.) at specific sites of interest. Several different types of spin-labeling magnetic resonance (SLMR) experiments can be performed to provide this information. The mobility of the spin probe (described as a correlation time, τcorr, and order parameter, S) [41–43] is determined from lineshape analyses (fitting/simulation) of continuous wave (CW) spectra collected at single or multiple frequencies [44, 45]. Interactions between spin-labeled molecules give rise to changes in lineshapes that arise from dipolar [46] and super-exchange interactions [47, 48], which provide distance information over the range of 6–20 Å from lineshape investigations. Interactions and organization with the membrane and bilayer surface can be measured by power saturation [49–54], and Overhauser dynamic nuclear polarization (ODNP) methods [55–61]. The conformation, dynamics and packing of molecules are reflected quantitatively in these data and can be used to track and quantify changes. As such, SLMR represents an important tool to characterize the molecular environment of glycans in model and cellular membranes that mimic normal or pathological states, as well as to characterize changes that occur upon binding to antibodies or other glycan binding partners.
Spin-labeling strategies for proteins often times chemically modify a unique cysteine moiety that was site-specifically incorporated into the system through bio-enabled templated directed amino acid substitutions [34]. Spin-labeling in nucleic acids proceeds oftentimes by chemical modification of a reactive moiety incorporated into the phosphate backbone, sugar or base that occurs either after bioconjugation or prior to the synthesis of the oligomer [37]. In contrast, fully organic synthetic schemes are more often utilized to incorporate the EPR active radicals into lipids and polymers [62]. Our approach for labeling glycans on cells follows two different chemical biology routes, where we utilize orthogonal reaction of azide containing glycans within the glycocalyx of cells with a click-reactive moiety DBCO-based nitroxide spin label (4). The first approach involves metabolic glycan engineering (MGE) with an azide-modified modified sugar (Fig 1A) [30] [63, 64], whereas the second approach exploited enzymatic glycoengineering (EGE) of cell surface glycans with an azide-modified sugar employing different siayltransferases (Fig 1B) [31, 32].
Figure 1:
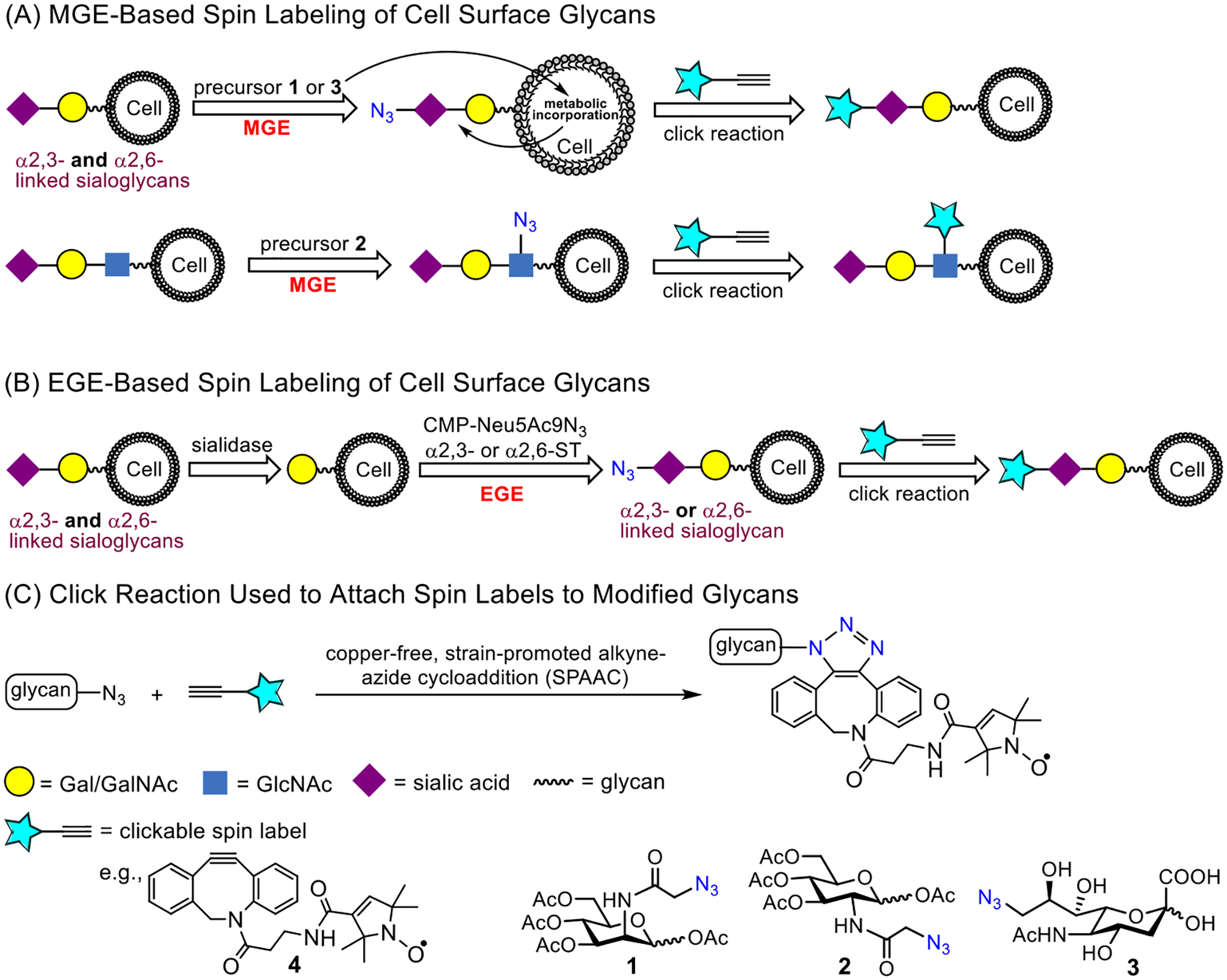
Spin-labelling approach of cell surface sialoglycans via (A) MGE with precursors 1, 2 or 3 and (B) EGE-mediated incorporation of an azide-modified sialic acid (Neu5Ac9N3) residue via either α2,3- or α2,6-siayltransferase (ST) and then a copper-free click reaction (C) with DBCO-SL, 4, to install the nitroxide SL.
In these and numerous other cell-based studies, such as immunostaining and genome editing based-strategies or exogenous expression of a tagged protein as well as transmission electron microscopy, a critical methodological step in the sample preparation of some measurements is the fixation of the cell. It is well accepted that chemical fixation of cells disrupts the “live” cell organization/structure [65–68]. Studies are increasing that characterize the molecular level details that chemical fixation has on protein structural organization, such as DNA-protein interactions, liquid-liquid phase separations and emphasis on biomolecules cell and its organelles [69–72], including AFM based investigations of membrane protein clustering during fixation [73]. Here, we investigated the impact that paraformaldehyde (PFA) fixing of cells has on the mobility and organization of glycans in the glycocalyx with EPR spectroscopy. Specifically, we performed spin-labeling studies using MGE approaches of HeLa cells with three different azide-modified monosaccharides, Ac4ManNAz (1), Ac4GlcNAz (2) and 9-Az-sialic acid (3) (Fig 1), as MGE precursors and varied the order in which cells were spin-labeled and fixed with PFA. EPR spectra were collected and revealed different spectral features that were dependent upon the chronological order of labeling and fixation. Insights into the structural organization giving rise to the different spectral features are inferred from the rates of reduction of the spin label EPR signal by ascorbic acid.
2. Materials and Methods
Materials.
The HeLa cell line was purchased from the American Type Culture Collection (ATCC, United States). The Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin (10000 U/mL), and phosphate-buffered saline (PBS) utilized in the study were purchased from Thermo Fisher Scientific (USA). CMP-Neu5Ac9Az, Ac4ManNAz, and Ac4GlcNAz were synthesized using the previously reported method and verified using NMR and MS spectroscopy [74]. Compound 3 (9-Az-sialic acid) was prepared by our lab according to a reported procedure and its 1H NMR spectra agreed with that in the literature [64, 75]. DBCO-SL 4 was synthesized following our previously reported protocol [32]. The washing buffer contained 1X DPBS with 2% BSA. Buffer 1 contains 1X DPBS with 20 mM MgCl2, 20 mM CaCl2, pH7.5.
Cell culture preparation.
The HeLa cells were maintained in tissue culture flasks under controlled conditions of 37°C, 5% CO2, and 95% air, and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 U per mL of penicillin–streptomycin and 10% fetal bovine serum (FBS). Upon reaching approximately 90% confluency after 3–4 days of culture, the cells were harvested from the culture surface by Gibco™ Trypsin-EDTA (0.25%) and subsequently suspended in a fresh culture medium. An aliquot of cell suspension was used to count cell numbers using a 0.4% trypan blue stain and a hemocytometer. Following that, the required number of cells were transferred to a new culture flask and subjected to the metabolic engineering experiment or directly transferred to a centrifuge tube for chemoenzymatic engineering experiment.
Metabolic engineering-based spin labeling of HeLa cells.
Ac4ManNAz (1) or Ac4GlcNAz (2) stock solution (40 μL, 20 mM stock in ethanol) was transferred to a T-25 tissue culture flask (25 cm2), and the flask was placed in a sterile laminar hood to allow the ethanol to evaporate. Then, 4 mL of DMEM media supplemented with 100 U/mL of penicillin–streptomycin and 10% FBS was added to each flask to reach the desired concentration of 200 μM of 1 or 2. For engineering using 9-Az-sialic acid (3), the required amount (8 μL, 100 mM) of stock in DPBS was directly added to the cell culture media. For negative control, cells were incubated in culture media without 1, 2, or 3. After adding 1.5 million cells to each flask, the cells were cultured for 48 hours under the previously described conditions. The cells were then harvested by adding 750 μL of an EDTA solution (1 mM stock in dd water) in PBS (pH 7.4) to each flask, followed by 8 minutes of centrifugation at 300 rpm and 4 °C to acquire the cell pellet. After that, the cells were washed with washing buffer three times and counted as described above. Finally, the cells were resuspended in a 4% paraformaldehyde (PFA) solution (in PBS, pH 7.5) for 20 min at RT, washed 3 times with washing buffer, and subjected to spin label installation (For cells designated as Fix>SL).
For cells designated as SL>Fix, the DBCO-SL reaction was performed before incubation with 4% PFA, with all the other steps identical. The cell pellet was resuspended in 200 μL of washing buffer containing 100 μM DBCO-SL (4) (20 μL of 1 mM stock in DMSO). The cell suspension was then incubated in a shaker at RT for 1 h with shaking at 300 rpm with occasional pipetting of the air to prevent cells from settling. The reaction was quenched by adding excess ice-chilled washing buffer, followed by centrifugation, and washing three times. Finally, the cells were pelleted down by centrifugation at 6000 rpm (2300 × g) for 1 min, and the cell pellet was subjected to EPR analysis.
MGE-based fluorescent labeling of cells and flow cytometry analysis.
The HeLa cells experienced metabolic engineering through the application of 200 μM of a metabolic reporter molecule 2, following the exact same procedures as described above. Cells were harvested and washed three times with ice-cold washing buffer, then resuspended in 100 μL of washing buffer containing 50 μM of DBCO-FAM (Fluorescein dibenzocyclooctyne, Lumiprobe Corp. Hunt Valley, MD) and kept at RT in the dark for 60 mins with occasional air agitation to the cells. Thereafter following a quick washing with the washing buffer, the cells were incubated in 100 μL of 4% paraformaldehyde solution (in PBS, pH 7.5) for 20 mins at RT. Finally, the cells (designated as Click+ Fix) were washed again three times with the washing buffer before being analyzed on the flow cytometer. An Attune™ Nxt flow cytometer was used with the blue excitation (BL1) laser (Ex: 488 nm, Em: 520 nm), and the data was analyzed using Attune™ Nxt software. For the cells designated as Fix+ Click, the fixing step with 4% PFA was performed prior to the click conjugation process, with all other steps remaining identical. Control samples were subjected to labeling with DBCO-FAM without treatment of 2.
Ascorbic acid quenching of spin labels.
Ascorbic acid quenching of the spin probe was performed following a previously reported method [76]. Using the above-described method, HeLa cells were engineered, then fixed and spin-labeled (or spin-labeled and then fixed). In both the cases, cell pellets were resuspended in 1X DPBS containing 2.30 mM L-ascorbate (9.2 μL from a 50 mM stock in 1X DPBS) in a final volume of 200 μL and then incubated at room temperature (RT) for various time intervals with occasional air agitation in the tube. Samples from different time points were washed three times with an excess ice-cooled washing buffer, and the cell pellet was subjected to EPR analysis.
CW-EPR data collection and analysis of spin-labeled cells.
100–200 G X-band (9.5 GHz) CW-EPR absorption spectra were collected at 30°C using a Magnettech MiniScope MS-5000 bench-top EPR spectrometer with a dielectric resonator. Spectra were reported as an average of 16 scans with 120 mT sweep width, 0.2 mT modulation amplitude, 100 kHz modulation frequency and 1 mW incident microwave power (2 mW incident microwave power on Bruker E500). All of the EPR spectra were area normalized to the cell number, and all spectra were baseline-corrected and processed using the LabVIEW software provided by C. Altenbach and W. Hubbell (https://sites.google.com/site/altenbach/labview-programs).
Lineshape analysis and simulation of EPR spectra.
EPR spectra were simulated using the chili and esfit functions of EasySpin [77] as done in our previous studies using DBCO-SL [30–32]. The A- and g-tensors were previously determined: gxx = 2.0070, gyy = 2.0062, gzz = 2.0033, Axx = 6.7 G, Ayy = 6.7 G, and Azz = 35 G [30]. The other parameters used in EPR lineshape simulations are linewidth, correlation time of motion (τc), and the ordering potential C20. Each EPR spectrum was subjected to 1-, 2-, and 3-component simulations.
3. Results and Discussion
MGE with Ac4ManNAz (1), Ac4GlcNAz (2) or 9-Az-Sialic acid (3) was performed with analogous control samples that did not contain the azide-modified sugars but were subjected to the same procedure of fixing/spin labeling with DBCO-SL (4). Control samples resulted in a background EPR signal of non-specifically interacting labels [30]. As done previously, these cell-count normalized control spectra are subtracted from the cell count normalized labeled spectra. Figure 2 shows the resultant control subtracted EPR spectra for MGE HeLa cells prepared with varied chronological order of labeling and chemical fixation with PFA. Spectra of HeLa cells treated with precursors 1 and 2 that were fixed before labeling are consistent with earlier published data [30]. EasySpin was used to fit the experimental results [77], and spectra are well fit by two components where one spectrum reflects a more mobile lineshape and the other a more immobilized local environment for the spin label (Fig 2 and Fig S1, Table S1). For purposes of comparison within, we follow typical descriptions of nitroxide motional regimes where the boundaries of these motional regimes for nitroxides in solution and attached to biomacromolecules are governed by the anisotropy of the g-tensor and A-tensor magnetic components relative to the resonance field conditions [44]. The X-band isotropic motional regime occurs when the anisotropy of the hyperfine interaction is averaged to zero. The isotropic limit is typically observed for free spin in solution with correlation times (τR) within 5 × 10−11 – 10−9 sec. When the nitroxide is attached to a biomolecule, the correlation time increases. However, an isotropic limit model of analysis of the X-band EPR nitroxide lineshape can still be utilized for correlation times of <1.0 ns τR ≤ 2.0 ns [78–80], described as the “fast limit regime”. As the motion slows, the X-band EPR lineshape is modulated by the time dependent fluctuations of the anisotropic components of the hyperfine tensor for correlation times of τR > 2.0 ns and referred to as the “intermediate regime”. When motion no longer averages the anisoptry of the hyperfine interaction (aka τR > 100 ns), the spectra reflect the rigid limit [45]. Within the 2.0 to 100 ns time regime, we set a boundary at ~ 10 ns to describe a “slow” regime. Results demonstrate that the relative fractions of these components vary depending upon the sugar identity and order of PFA fixation and spin labeling.
Figure 2.
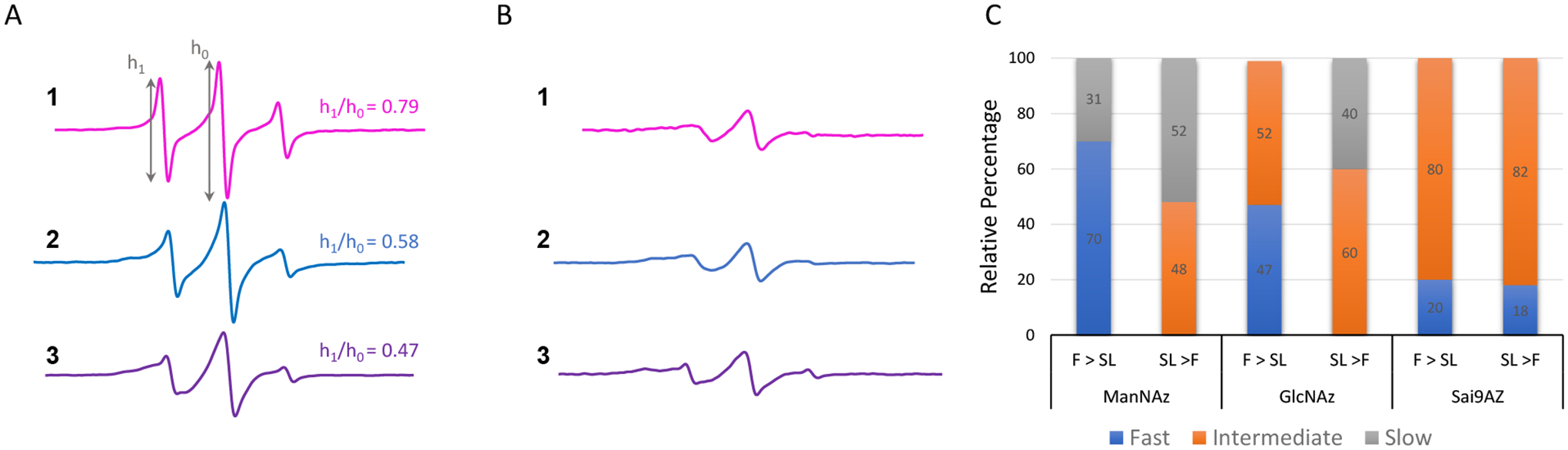
Stack plots of vertically offset 100 G X-band EPR spectra for control-subtracted HeLa cells treated with Ac4ManNAz (1), Ac4GlcNAz (2) or 9-Az-Sialic acid (3) that were (A) fixed with PFA and then reacted with DBCO-SL 4 or (B) reacted with DBCO-SL 4 and then fixed with PFA (dotted lines). Spectra are plotted with normalized integral areas to reflect mobilities. Annotations for the h1 and h0 transition area shown. Values for h1/ho given in the figure are evaluated for spectra from conditions where the cells were fixed and then spin-labeled. (C) Relative results of spectral components from fitting with Easy Spin where τR ≤ 2 ns is designated as “fast”, 2 ns < τR ≤ 10 ns is designated as “intermediate”, and τR > 10 ns is designated as “slow”. Numbers reflect the relative percentages.
Here we report MGE results for the first-time using precursor 3. EPR results for 3 indicate an on average more restricted environment with a complex lineshape of multiple components and only a small fraction of the mobile τR ~ 1 ns component. At first, this result is somewhat surprising given that both 1 and 3 are expected to be incorporated into glycan biosynthesis as sialic acids (Fig 1), and as such we anticipated similar EPR spectra for cells treated with 1 and 3. However, the location of the azide differs in 1 and 3. Furthermore, 1 and 3 experience different metabolic processes, thus there are significant differences between ManNAc with regard to sialic acids regarding their metabolism, incorporation efficiency, selectivity for sialoglycans and chemical modification potential [81]. All these factors may affect the final expression level and pattern of azido sialoglycans on the cell surface, thus the EPR results. Additionally, we recently showed that incorporation of Neu5Ac9N3 with either an α2,3- or α2,6-siayltransferase (ST) resulted in a different resultant EPR lineshape based upon organizational differences that could limit mobility of the rotatable bonds of the spin label, the mobility of the azide-modified sugar, and/or glycosidic bond mobility [32]. The EPR lineshape obtained from MGE with 3 differs from that using 1 or 2 in that they possess a relatively small percentage of a highly mobile site with a more restricted site characterized here as having slow mobility. Irrespective of the complexity, the EPR spectrum of spin-labeled cells treated with 3 reveals the lowest mobility (h1/h0 = 0.47).
For all metabolic precursors used, the lineshape for the EPR spectra that were spin labeled before treatment with PFA (Fig. 2B) reflect lower mobility than when spin-labeled after chemical fixation (Fig 2A). Spectra in Figs 2A and 2B are plotted with signal intensity normalized to area, where broadened spectra have less intensity and reflect overall lowered mobility [33, 34, 41, 82]. More insights into the local mobility of the spin-labeled glycans is afforded from theoretical spectral fitting [77]. For example, with 2, the spectrum resulting from fixing prior to spin labeling contains about 47% of the “fast” mobile component with the remaining signal comprising an intermediate motion component. In contrast, when cells treated with 2 are spin-labeled and then fixed, the EPR results show about 60% of an intermediate component and 40% of a slow component with no spectral components reflective of fast isotropic motion (Fig 2C). Perhaps these results are not surprising. The chemical fixation process with PFA cross-links together proteins on the cell surface and if done after spin labeling it might be expected that the spin-labeled sites are now restricted whereas spin-labeling after cross-linking may only retain sites that are surface exposed leading to a relatively high component of a fast motionally averaged spin-label, aka those sites clearly exposed on the outer sphere of the cell glycocalyx. In contrast, chemical fixation after spin labeling could cluster together the already spin-labeled glycoproteins and entrap labeled glycolipids, giving rise to a higher percentage of restricted glycans. More importantly, spectra obtained when chemical fixation occurred after spin labeling generate very similar spectra irrespective of the identity of the azide-modified sugar used in the MGE treatment (Fig 2B). Thus, differences in the glycan organization, reflected in the EPR spectral line shapes, are only revealed for 1 and 2 when spin labeling occurs after the chemical fixation reactions. Overall, results indicate that insights into glycan mobility can be obscured by chemical fixation.
EPR of spin-labeled cells using 3 for MGE shows slightly different results than obtained for 1 and 2. A small fraction of a fast mobility component is present in the spectrum from treatment with 3 even when SL is performed prior to PFA fixation. The total percentage of the fast mobility components for 3 is ~ 20%. When comparing results obtained when chemical fixation occurs before spin labeling, the relative fraction of the fast mobility component is less than those observed with either 1 (73%) or 2 (47%). Although the presence and relative percentages of fast mobility and intermediate mobility component with MGE treatment of 3 do not differ with the order of chemical fixation, the mobility of the spectral components within a given motional regime differs. The results from simulation of cells treated with 3 with varying order of chemical fixation and labeling are shown in Figure 3. The mobility of both components is slower when fixation occurs after spin labeling (i.e., a larger correlation time means slower motion). As inferred from CW-/X-band EPR lineshape differences, the order of chemical fixation manifests less of an effect on glycan organization and mobility when using 3 compared to 2 or 1 for MGE with HeLa cells.
Figure 3.
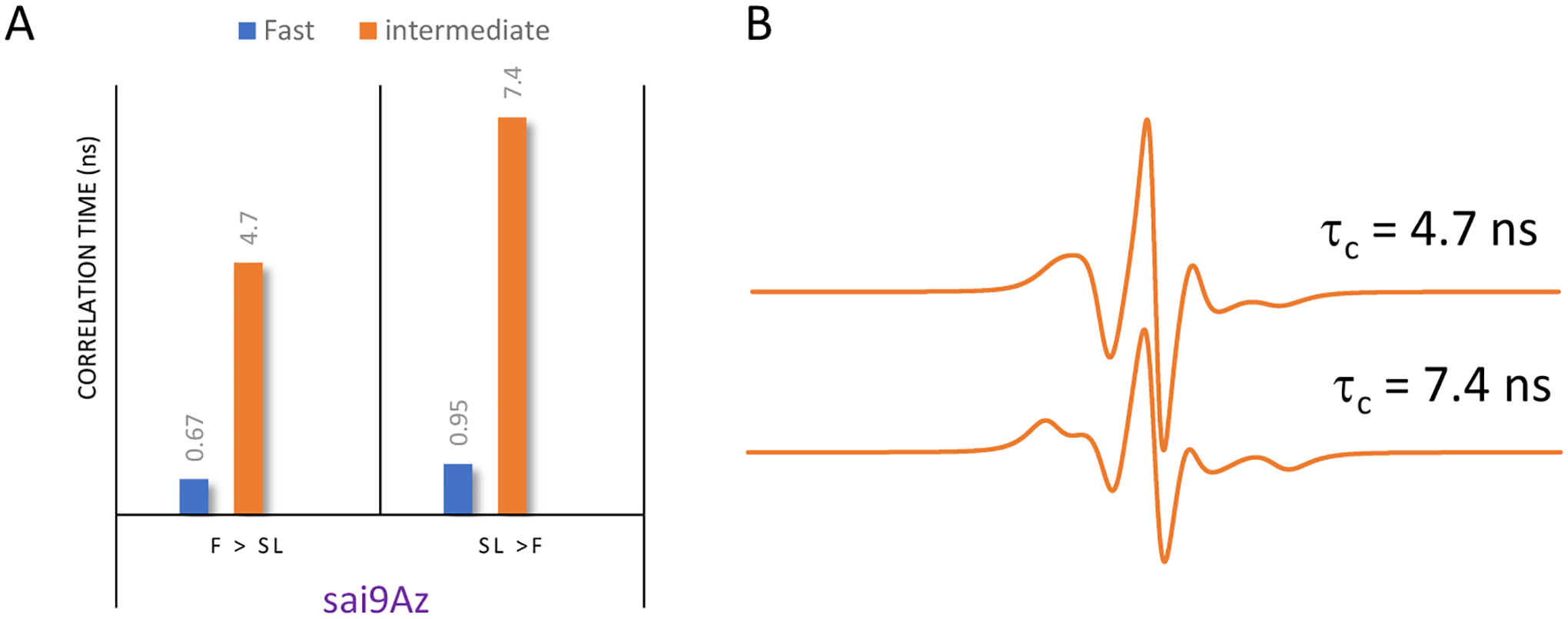
(A) Correlation time analysis from theoretical fitting of MGE of HeLa cells treated with 3 as a function of the order of chemical fixation, with correlation times in units of ns labeled on each bar. (B) X-band intermediate motion EPR component spectra from Easy Spin fitting showing how the EPR lineshape has discernable changes with respect to the intermediate regime correlation times. In both cases the intermediate motional component comprises 80% of the total signal.
It would be interesting to compare the results of fixed and non-fixed cells. Thus, we also tried to obtain EPR spectra/data from the engineered cells without fixing. However, these experiments gave rather flexible results, probably due to the internalization and reduction of spin labels by cells. In this study, we did not optimize the conditions or attempt other methods, e.g., inhibition of internalization, to stabilize spin labels on non-fixed cells, which may be investigated in the future.
To further probe organizational differences that arise from the order of fixation relative to spin labeling, we subjected samples to reduction with ascorbic acid. Ascorbic acid will reduce the nitroxide radical, thus diminishing the intensity of the EPR signal [76]. The ability of ascorbic acid to penetrate the cross-linking extracellular matrix is dependent upon the accessibility and diffusion ability of the small molecule through the crosslinked glycocalyx. This reduction process occurs on the minute time scale, so we can take aliquot samples of cells from a batch reaction, quench with cold water and collect EPR scans. Fig 4 shows a set of spectra obtained for HeLa cells treated with Ac4GlcNAz (2) with different orders of fixing and spin labeling over a 100-minute time span. We note that the total EPR signal from equivalent number of cells that were spin-labeled and then fixed was approximately only ~ 40% total intensity compared to cells that were fixed and then spin-labeled. We attribute this reduction in total EPR signal to chemical reaction of the nitroxide with the chemical reagents formed during PFA cross linking mechanism [83]. Addition of the PFA crosslinking agents to DBCO in solution did not reduce the nitroxide, however, the PFA reaction produced an intermediate Schiff base that can tautomerize to an imine/iminol form [84], which could be capable of reacting with radicals. In contrast, we performed analogous studies with DCBO-fluorophores, which demonstrate a greater increase in fluorescence intensity when labeled prior to cross linking – an opposite trend (Fig 5). As such, we interpret these results to suggest that more of the azide-modified glycans are available for click-reaction when labeling is prior to fixation, but the chemistry of the fixation process can chemically reduce a marked fraction of the total nitroxide radicals incorporated into the glycocalyx.
Figure 4.
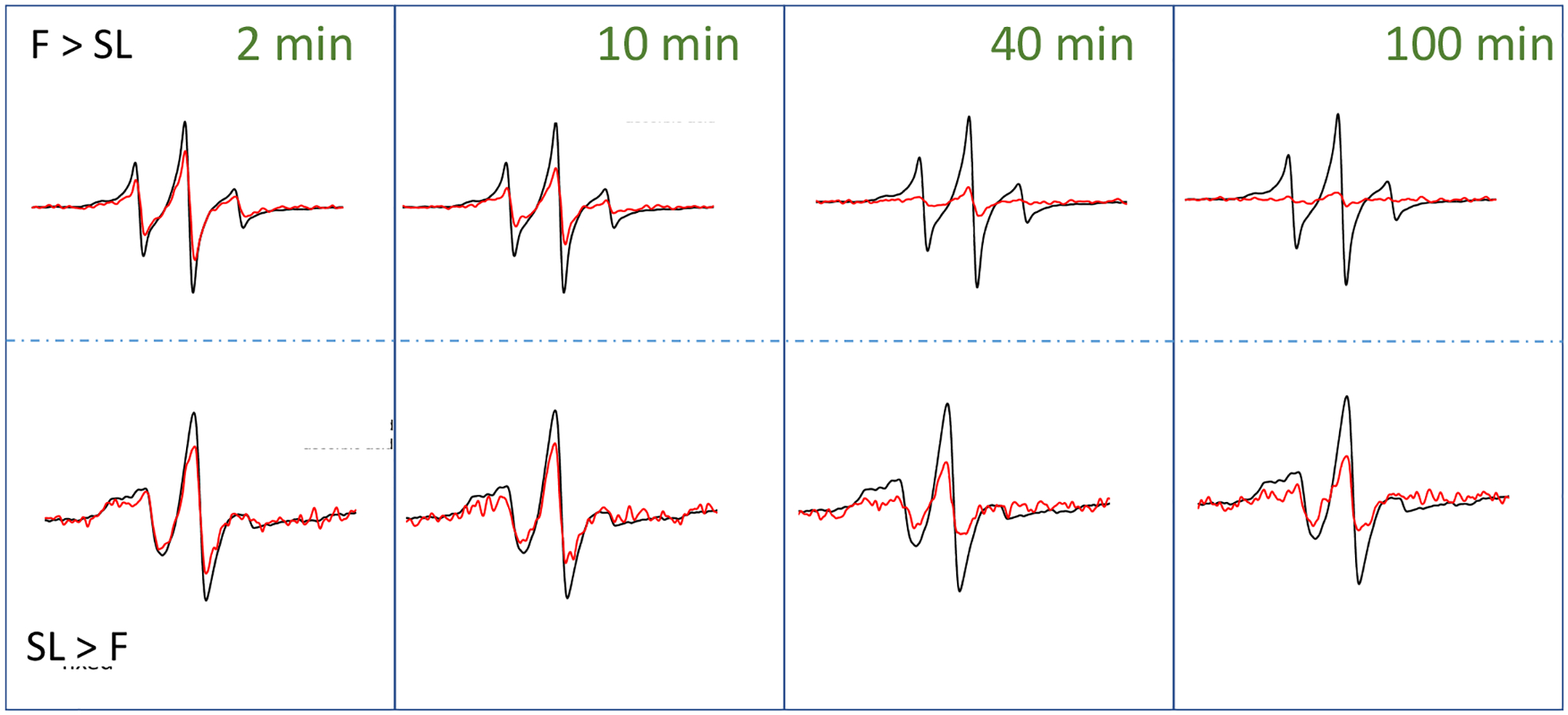
100 G X-band EPR spectra for control subtracted HeLa cells treated with Ac4GlcNAz (2) and DBCO-SL (4) before (black) and after (red) exposure to ascorbic acid over the time course of 100 minutes. The top row presents data for conditions where cells were fixed prior to spin labeling (F > SL), whereas the bottom row show represents data when cells were spin-labeled prior to chemical fixation (SL > F). Time points of 2, 10, 40 and 100 minutes are shown. Time course spectra are given in the supporting information (Figures S2). Spectra after exposure to ascorbic acid represent only 2 scans of signal averaging, whereas the before spectra have better signal-to-noise ratios because they were signal averaged for 16 scans. Spectra are plotted normalized to total cell count, so the decrease in signal is reflective of the decrease in EPR active radical that remains in the sample.
Figure 5.
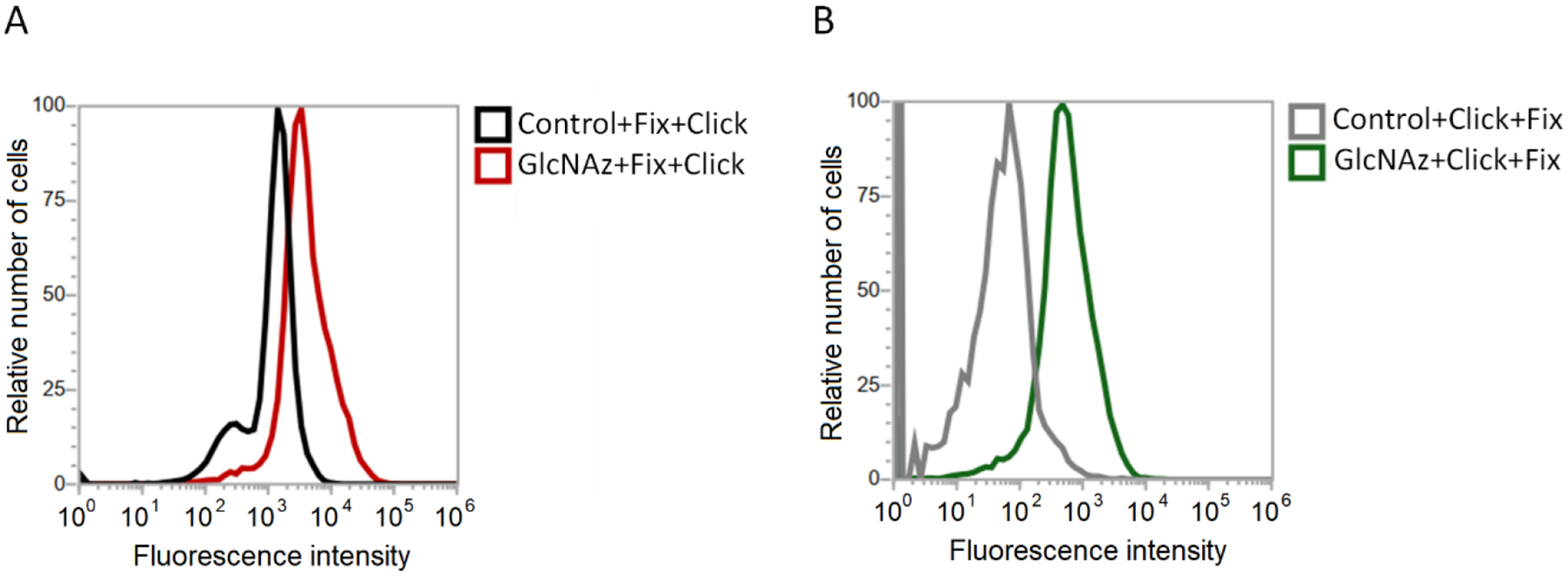
Flow cytometry results demonstrating an increase in fluorescence intensity of HeLa cells treated with 2 and labeled with DBCO-FAM (A) after chemical fixation and (B) prior to chemical fixation, which demonstrate that greater fluorescence is observed when labeled prior to fixing than vice-versa.
Because of the lowered signal-to-noise ratios in the kinetic studies, we did not attempt to simulate the spectra acquired as a function of time in the presence of ascorbic acid. Nevertheless, the spectral lineshape does not appear to change over the time course investigated (Fig S2). Only the total intensity is observed to markedly change over time. Because of this, the kinetic process of ascorbic acid reduction was evaluated by tracking the percentage remaining intensity of the h1 and h0 spectral features, relative to prior to ascorbic acid addition, as a function of time. Fig 6 plots results for ascorbic acid reduction for labeled HeLa cells treated with 2 or 3, which modify GlcNAc and Neu5Ac residues in glycans respectively, with varying order of fixing/spin labeling, where solid lines are fits to the data using a decay function with either one or two independent exponential decay rates,
Where A1 and A2 reflect the relative percentages of each decay component, y0 represents the relative percentage of remaining intensity after 100 minutes of exposure to ascorbic acid and t1 and t2 are the respective decay time constants.
Figure 6.
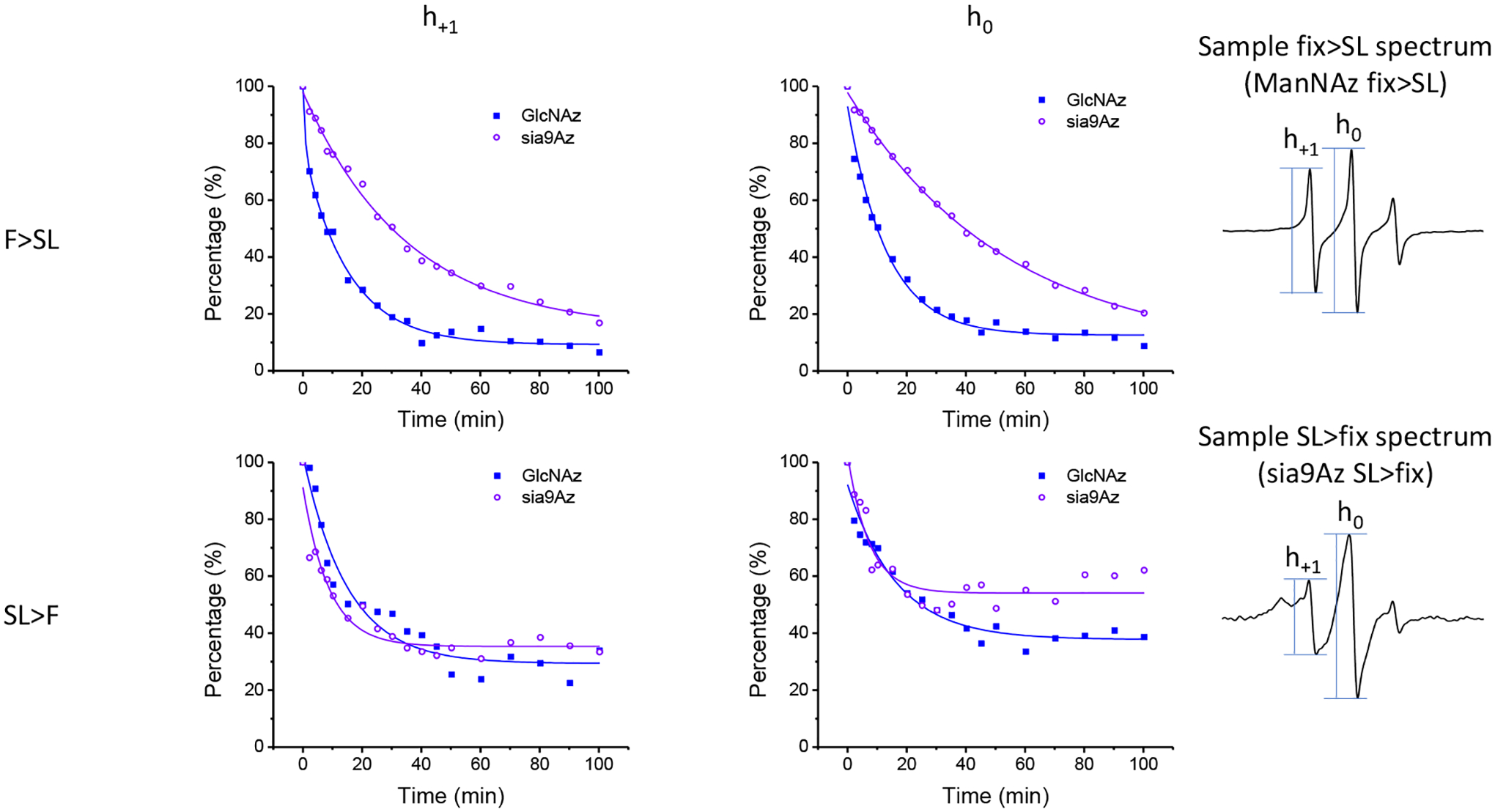
Resultant time course data of ascorbic acid reduction of spin-labeled HeLa cells with Ac4GlcNAz (2, GlcNAz) or 9-Az-Sialic acid (3, sia9Az) and DBCO-SL and evaluated by both the intensities of the h1 (left side plots) and h0 (middle plots) transitions for (top row) chemical fixation before spin-labeling and (bottom row) spin labeling prior to fixation.. Values of h1 and h0 at t=0 were acquired prior to addition of ascorbic acid and were set to 100% after scaling for dilution required by adding ascorbic acid to cells for additional time step measurements. All other intensities were scaled accordingly.
Table 1 summarizes the results from fitting of the decay curves. In most cases a single component exponential fit was only necessary for suitable fitting of the data. In terms of accessibility, results indicate that for both 2 and 3, approximately 90% of the total signal was reduced (i.e.,10% remaining) after 100 minutes of exposure to ascorbic acid when cells were chemically fixed prior to spin labeling, whereas only 60–70% of the signal was reduced (i.e.,30–40% remaining) when spin-labeled prior to chemical fixation, thus, indicating that the crosslinks among biomolecules on cells that lead to broadened EPR spectra also make the SL less accessible to ascorbic acid under these same conditions (Fig. 7A). The results are quite similar for both the h+1 and h0 signals, again supporting that the relative populations of the mobile and immobile components (for spectra Fix prior to SL) do not change significantly throughout the time course of these experiments or that both features have roughly equal accessibility to ascorbic acid. The reasoning follows that the central line has the greatest contributions from both components and the h+1 and h−1 will be more dominated by the relative fraction of the slower component.
Table 1.
Decay rate parameters for ascorbic acid reduction of the nitroxide EPR signal.
| Sugar: | GlcNAz (2) | Sia9Az (3) | ||||||
|---|---|---|---|---|---|---|---|---|
| Order: | SL > Fix | SL > Fix | ||||||
| transition | h0 | h0 | ||||||
| yo | 9.2± 1.1 | 11.5± 0.8 | 29± 2 | 37± 2 | 15± 2 | 7± 2 | 35 ± 2 | 54 ± 2 |
| A1 | 69 ± 3 | 77 ± 2 | 73± 4 | 52± 3 | 84± 2 | 91± 2 | 56 ± 4 | 49 ± 5 |
| t1 | 15 ± 2 | 15 ± 1 | 15± 2 | 19± 2 | 35± 2 | 53± 2 | 9 ± 2 | 7 ± 2 |
| A2 | 22 ± 4 | 12 ± 3 | n.a. | 8 ± 5 | n.a. | n.a. | n.a. | n.a. |
| t2 | 0.07 c | 0.02 c | n.a. | 0.08 c | n.a. | n.a. | n.a. | n.a. |
A second exponential was not needed to adequately fit the data.
These decay components were too fast for accurate measurements given our first time point was longer than this decay time. Nevertheless, this second component was required for adequate fitting of the data.
Figure 7.
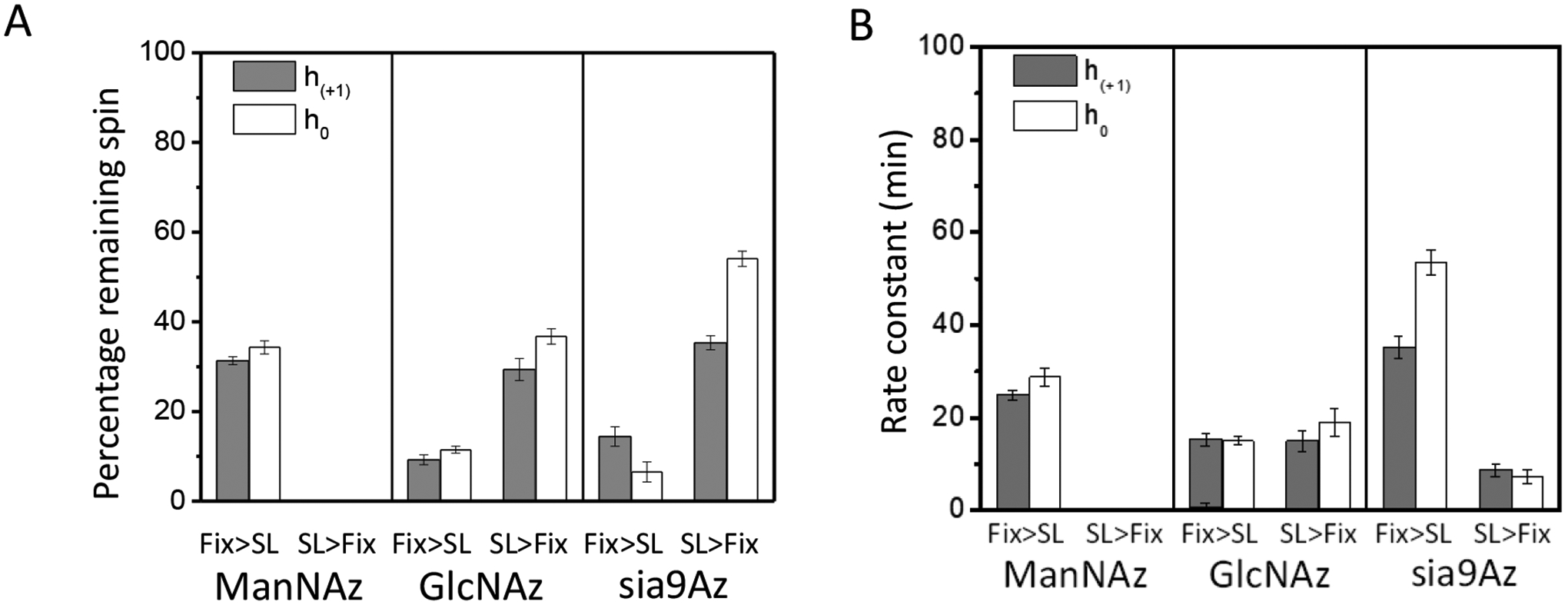
(A) Resultant relative percentage of remaining EPR active spins and (B) kinetic time constants after 100 min exposure to ascorbic acid for HeLa cells treated with Ac4ManNAz (1), Ac4GlcNAz (2) or 9-Az-Sialic acid (3) and DBCO-SL, where the order of fixing and spin-labeling was altered.
In terms of the decay times, the order of reduction in EPR signal followed the trend of 2 was the fastest, followed by 1 and then 3 when cells were chemically fixed and then spin-labeled, where in contrast when the cells were spin-labeled and then chemically fixed (Fig. 7B), 3 now had the fasted reduction time constant. These findings suggest disparate impacts on the organization and accessibility of various types of glycans in the glycocalyx when labeled either prior to or after cell fixation.
Because of this variability, we had since chosen to pursue enzymatic labeling of cells with orthogonal click reactive chemistry where no chemical fixation of cells was utilized in the protocol. However, we were unable to obtain similar high quality reproducible EPR signals from MGE cells that were spin-labeled and not chemically fixed. Through engineering of EPR sample holders that can maintain 4 °C, it may be possible to inhibit endocytosis and reduction of the spin label; yet we want to develop SDSL methods that can be utilized at physiological temperatures. In this case, a less perturbing cell fixation method is desirable. A recent review of the impacts of chemical fixation on the cellular contents of cells has proposed future directions for the development of potentially less perturbing chemical fixatives [69].
4. Conclusions
Taken together, the spin labeling EPR approach is exquisitely well-suited to show how cell fixation impacts the resultant EPR lineshape, which reflects changes in the average mobility and organization of the spin-labeled glycans on the surface of cells. The accessibility measurements also indicate differences in the local environment of the spin labels depending upon the order of treatment with crosslinking agents. These results have clearly demonstrated that the cell fixation protocol can have a significant impact on the organization and mobility of components within the cellular glycocalyx, the accessibility of cell surface glycans for molecular labeling, and the properties and dynamics of labeled glycans. As such, it is suggested that care should be taken when analyzing results of membrane organization obtained from any method where chemical fixation of cells is performed.
Supplementary Material
Funding
This study was supported by NIGMS/NIH (R35GM131686; ZG), NSF (MCB-1715384; GEF), an NIH instrumentation grant (S10 RR031603) for the Bruker E500 spectrometer, and Department of Chemistry, University of Florida for the Magnettech MS-5000 benchtop spectrometer. ZG thanks Steven and Rebecca Scott endowment to support our research.
Footnotes
Competing interests
The authors declare no competing interests.
Availability of data and materials
Ascii files of all EPR and fluorescence data can be obtained by contacting the Corresponding author directly.
References:
- 1.Varki A, Biological roles of glycans. Glycobiology, 2017. 27: p. 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiederschain G, Glycobiology and Human Diseases. 2016, Boca Taton: Taylor & Francis Group. [Google Scholar]
- 3.Zhang X and Kiechle FL, Glycosphingolipids in health and disease. Annu. Clin. Lab. Sci, 2004. 34: p. 3–13. [PubMed] [Google Scholar]
- 4.Rudd PM, Disease Related Glycosylation Changes and Biomarker Discovery: Challenges and Possibilities in an Emerging Field. 2009, Fairfax: IOS Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnaar RL, Suzuki A, and Stanley P, Glycosphingolipids, in Essentials of Glycobiology, Varki A, et al. , Editors. 2008, Cold Spring Harbor Laboratory Press: Cold Spring Harbor: p. 129–142. [Google Scholar]
- 6.Russo D, Parashuraman S, and D’Angelo G, Glycosphingolipid–protein interaction in signal transduction. Int. J. Mol. Sci, 2016. 17: p. 1732 ( 1–23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Ruiz C, Morales A, and Fernandez-Checa JC, Glycosphingolipids and cell death: one aim, many ways. Apoptosis, 2015. 20: p. 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez PHH and Schnaar RL, Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol, 2009. 19: p. 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schengrund C-L, Gangliosides: glycosphingolipids essential for normal neural development and function. Trend. Biochem. Sci, 2015. 40: p. 397–406. [DOI] [PubMed] [Google Scholar]
- 10.Palmano K, et al. , The Role of Gangliosides in Neurodevelopment. Nutrients, 2015. 7: p. 3891–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gouaze-Andersson V and Cabot MC, Glycosphingolipids and drug resistance. Biochim. Biophys. Acta, 2006. 1758: p. 2096–2103. [DOI] [PubMed] [Google Scholar]
- 12.Groux-Degroote S, Guerardel Y, and Delannoy P, Gangliosides: structures, biosynthesis, analysis, and roles in cancer. ChemBioChem, 2017. 18: p. 1146–1154. [DOI] [PubMed] [Google Scholar]
- 13.Qamsari ES, et al. , Ganglioside as a therapy target in various types of cancer. Asian Pac. J. Cancer Prev, 2016. 17: p. 1643–1647. [DOI] [PubMed] [Google Scholar]
- 14.Inokuchi J, GM3 and diabetes. Glycoconj. J, 2014. 31: p. 193–197. [DOI] [PubMed] [Google Scholar]
- 15.Halmer R, Walter S, and Faßbender K, Sphingolipids: Important players in multiple sclerosis. Cell Physiol. Biochem, 2014. 34: p. 111–118. [DOI] [PubMed] [Google Scholar]
- 16.van Echten-Deckert G and Walter J, Sphingolipids: Critical players in Alzheimer’s disease. Prog. Lipid Res, 2012. 51: p. 378–393. [DOI] [PubMed] [Google Scholar]
- 17.Ariga T, Wakade C, and Yu RK, The pathological roles of ganglioside metabolism in Alzheimer’s disease: Effects of gangliosides on neurogenesis. Int. J. Alzh. Dis, 2011. ID 193618: p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginzburg L, Kacher Y, and Futerman KH, The pathogenesis of glycosphingolipid storage disorders. Semin. Cell Dev. Biol, 2004. 15: p. 417–431. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson MAJ and Williams AF, Cell-surface anchoring of proteins via glycosylphosphatidylinositol structure. Annu. Rev. Biochem, 1988. 57: p. 285–320. [DOI] [PubMed] [Google Scholar]
- 20.Robinson PJ, Signal transduction by GPI-anchored membrane proteins. Cell Biol. Intern. Rep, 1991. 15: p. 761–767. [DOI] [PubMed] [Google Scholar]
- 21.Onda H, et al. , A Novel secreted tumor antigen with a glycosylphosphatidylinositol-anchored structure ubiquitously expressed in human cancers. Biochem. Biophys. Res. Commun, 2001. 285(2): p. 235–243. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, et al. , Overexpression of glycosylphosphatidylinositol (GPI) transamidase subunits phosphatidylinositol glycan class T and/or GPI anchor attachment 1 induces tumorigenesis and contributes to invasion in human breast cancer. Cancer Res, 2006. 66(20): p. 9829–9836. [DOI] [PubMed] [Google Scholar]
- 23.Zhao P, et al. , Proteomic identification of glycosylphosphatidylinositol anchor-dependent membrane proteins elevated in breast carcinoma. J. Biol. Chem, 2012. 287: p. 25230–25240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolezala S, et al. , Elevated levels of glycosylphosphatidylinositol (GPI) anchored proteins in plasma from human cancers detected by C. septicum alpha toxin. Cancer Biomark, 2014. 14: p. 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambamurti K, et al. , Glycosylphosphatidylinositol-anchored proteins play an important role in the biogenesis of the Alzheimer’s amyloid beta-protein. J. Biol. Chem, 1999. 274(38): p. 26810–26814. [DOI] [PubMed] [Google Scholar]
- 26.Walter EI, et al. , Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J. Biol. Chem, 1992. 267: p. 1245–1252. [PubMed] [Google Scholar]
- 27.McDowell MA, Ransom DM, and Bangs JD, Glycosylphosphatidylinositol-dependent secretory transport in Trypanosoma brucei. Biochem. J, 1998. 335: p. 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boccuni P, et al. , Glycosyl phosphatidylinositol (GPI)-anchored molecules and the pathogenesis of paroxysmal nocturnal hemoglobinuria. Crit. Rev. Oncol. Hematol, 2000. 33: p. 25–43. [DOI] [PubMed] [Google Scholar]
- 29.Wang B and Boons G-J, Carbohydrate Recognition : Biological Problems, Methods, and Applications. 2011, Hoboken: Wiley. [Google Scholar]
- 30.Jaiswal M, et al. , A metabolically engineered spin-labeling approach for studying glycans on cells. Chem. Sci, 2020. 11: p. 12522–12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaiswal M, et al. , Enzymatic glycoengineering-based spin labelling of cell surface sialoglycans to enable their analysis by electron paramagnetic resonance (EPR) spectroscopy. Analyst, 2022. 147(5): p. 784–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaiswal M, et al. , Different Biophysical Properties of Cell Surface α2,3- and α2,6-Sialoglycans Revealed by Electron Paramagnetic Resonance Spectroscopic Studies. J Phys Chem B, 2023. 127(8): p. 1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubbell WL, Cafiso DS, and Altenbach C, Identifying conformational changes with site-directed spin labeling. Nat Struct Biol, 2000. 7(9): p. 735–9. [DOI] [PubMed] [Google Scholar]
- 34.Hubbell WL, et al. , Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol, 1998. 8(5): p. 649–56. [DOI] [PubMed] [Google Scholar]
- 35.Sahu ID and Lorigan GA, Site-Directed Spin Labeling EPR for Studying Membrane Proteins. Biomed Res Int, 2018. 2018: p. 3248289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen P and Qin PZ, RNA dynamics: perspectives from spin labels. Wiley Interdiscip Rev RNA, 2012. 3(1): p. 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin PZ, et al. , Monitoring RNA base structure and dynamics using site-directed spin labeling. Biochemistry, 2003. 42(22): p. 6772–83. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, et al. , Studying RNA using site-directed spin-labeling and continuous-wave electron paramagnetic resonance spectroscopy. Methods Enzymol, 2009. 469: p. 303–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cafiso DS, Identifying and quantitating conformational exchange in membrane proteins using site-directed spin labeling. Acc Chem Res, 2014. 47(10): p. 3102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fanucci GE and Cafiso DS, Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol, 2006. 16(5): p. 644–53. [DOI] [PubMed] [Google Scholar]
- 41.Columbus L and Hubbell WL, A new spin on protein dynamics. Trends Biochem Sci, 2002. 27(6): p. 288–95. [DOI] [PubMed] [Google Scholar]
- 42.Columbus L and Hubbell WL, Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry, 2004. 43(23): p. 7273–87. [DOI] [PubMed] [Google Scholar]
- 43.Columbus L, et al. , Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry, 2001. 40(13): p. 3828–46. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, et al. , Multifrequency electron spin resonance study of the dynamics of spin labeled T4 lysozyme. J Phys Chem B, 2010. 114(16): p. 5503–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casey TM, et al. , Continuous wave W- and D-Band EPR spectroscopy offer “sweet-spots” for characterizing conformational changes and dynamics in intrinsically disordered proteins. Biochemical and Biophysical Research Communications, 2014. 450(1): p. 723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altenbach C, et al. , Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry, 2001. 40(51): p. 15471–82. [DOI] [PubMed] [Google Scholar]
- 47.Jao CC, et al. , Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A, 2008. 105(50): p. 19666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M, et al. , Investigation of alpha-synuclein fibril structure by site-directed spin labeling. J Biol Chem, 2007. 282(34): p. 24970–9. [DOI] [PubMed] [Google Scholar]
- 49.Pyka J, et al. , Accessibility and dynamics of nitroxide side chains in T4 lysozyme measured by saturation recovery EPR. Biophys J, 2005. 89(3): p. 2059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altenbach C, et al. , Accessibility of nitroxide side chains: absolute Heisenberg exchange rates from power saturation EPR. Biophys J, 2005. 89(3): p. 2103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altenbach C, et al. , A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc Natl Acad Sci U S A, 1994. 91(5): p. 1667–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kirby TL, Karim CB, and Thomas DD, Electron paramagnetic resonance reveals a large-scale conformational change in the cytoplasmic domain of phospholamban upon binding to the sarcoplasmic reticulum Ca-ATPase. Biochemistry, 2004. 43(19): p. 5842–52. [DOI] [PubMed] [Google Scholar]
- 53.Fanucci GE, et al. , Structure and dynamics of the beta-barrel of the membrane transporter BtuB by site-directed spin labeling. Biochemistry, 2002. 41(39): p. 11543–51. [DOI] [PubMed] [Google Scholar]
- 54.Gross A and Hubbell WL, Identification of protein side chains near the membrane-aqueous interface: a site-directed spin labeling study of KcsA. Biochemistry, 2002. 41(4): p. 1123–8. [DOI] [PubMed] [Google Scholar]
- 55.Cheng CY, et al. , Hydration dynamics as an intrinsic ruler for refining protein structure at lipid membrane interfaces. Proc Natl Acad Sci U S A, 2013. 110(42): p. 16838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaminker I, Barnes R, and Han SI, Overhauser Dynamic Nuclear Polarization Studies on Local Water Dynamics. Electron Paramagnetic Resonance Investigations of Biological Systems by Using Spin Labels, Spin Probes, and Intrinsic Metal Ions, Pt B, 2015. 564: p. 457–483. [DOI] [PubMed] [Google Scholar]
- 57.Cheng CY, et al. , Cholesterol enhances surface water diffusion of phospholipid bilayers. Journal of Chemical Physics, 2014. 141(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song J, Allison B, and Han S, Local water diffusivity as a molecular probe of surface hydrophilicity. Mrs Bulletin, 2014. 39(12): p. 1082–1088. [Google Scholar]
- 59.Franck JM, Scott JA, and Han SI, Nonlinear Scaling of Surface Water Diffusion with Bulk Water Viscosity of Crowded Solutions. Journal of the American Chemical Society, 2013. 135(11): p. 4175–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong Y, et al. , Hydrophobicity of arginine leads to reentrant liquid-liquid phase separation behaviors of arginine-rich proteins. Nat Commun, 2022. 13(1): p. 7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moon H, et al. , Evidence for Entropically Controlled Interfacial Hydration in Mesoporous Organosilicas. J Am Chem Soc, 2022. 144(4): p. 1766–1777. [DOI] [PubMed] [Google Scholar]
- 62.Smith IR, et al. , Probing Membrane Hydration at the Interface of Self-Assembled Peptide Amphiphiles Using Electron Paramagnetic Resonance. ACS Macro Lett, 2018. 7(10): p. 1261–1266. [DOI] [PubMed] [Google Scholar]
- 63.Saxon E and Bertozzi CR, Cell surface engineering by a modified Staudinger reaction. Science, 2000. 287(5460): p. 2007–10. [DOI] [PubMed] [Google Scholar]
- 64.Xie R, et al. , In vivo metabolic labeling of sialoglycans in the mouse brain by using a liposome-assisted bioorthogonal reporter strategy. Proc Natl Acad Sci U S A, 2016. 113(19): p. 5173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amato PA, Unanue ER, and Taylor DL, Distribution of actin in spreading macrophages: a comparative study on living and fixed cells. J Cell Biol, 1983. 96(3): p. 750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lonn P and Landegren U, Close Encounters - Probing Proximal Proteins in Live or Fixed Cells. Trends Biochem Sci, 2017. 42(7): p. 504–515. [DOI] [PubMed] [Google Scholar]
- 67.Malinin GI and Malinin TI, Effects of dimethylsulfoxide on the ultrastructure of fixed cells. Biotech Histochem, 2004. 79(2): p. 65–9. [DOI] [PubMed] [Google Scholar]
- 68.Watson P, Jones AT, and Stephens DJ, Intracellular trafficking pathways and drug delivery: fluorescence imaging of living and fixed cells. Adv Drug Deliv Rev, 2005. 57(1): p. 43–61. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida SR, Maity BK, and Chong S, Visualizing Protein Localizations in Fixed Cells: Caveats and the Underlying Mechanisms. J Phys Chem B, 2023. 127(19): p. 4165–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Irgen-Gioro S, et al. , Fixation can change the appearance of phase separation in living cells. Elife, 2022. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kiyoto S, et al. , Improved chemical fixation of lipid-secreting plant cells for transmission electron microscopy. Microscopy (Oxf), 2022. 71(4): p. 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edechi CA, et al. , Comparison of Fixation Methods for the Detection of Claudin 1 and E-Cadherin in Breast Cancer Cell Lines by Immunofluorescence. J Histochem Cytochem, 2022. 70(2): p. 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ichikawa T, et al. , Chemical fixation creates nanoscale clusters on the cell surface by aggregating membrane proteins. Commun Biol, 2022. 5(1): p. 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng B, et al. , 9-Azido Analogues of Three Sialic Acid Forms for Metabolic Remodeling of Cell-Surface Sialoglycans. ACS Chem Biol, 2019. 14(10): p. 2141–2147. [DOI] [PubMed] [Google Scholar]
- 75.Ikeda K, et al. , Chemoenzymatic synthesis of an N-acetylneuraminic acid analogue having a carbamoylmethyl group at C-4 as an inhibitor of sialidase from influenza virus. Carbohydr Res, 1998. 312(4): p. 183–9. [DOI] [PubMed] [Google Scholar]
- 76.Wuebben C, et al. , Site-Directed Spin Labeling of RNA with a Gem-Diethylisoindoline Spin Label: PELDOR, Relaxation, and Reduction Stability. Molecules, 2019. 24(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stoll S and Schweiger A, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. Journal of Magnetic Resonance, 2006. 178(1): p. 42–55. [DOI] [PubMed] [Google Scholar]
- 78.Todd AP and Millhauser GL, Esr-Spectra Reflect Local and Global Mobility in a Short Spin-Labeled Peptide Throughout the Alpha-Helix-]Coil Transition. Biochemistry, 1991. 30(22): p. 5515–5523. [DOI] [PubMed] [Google Scholar]
- 79.Miick SM, Todd AP, and Millhauser GL, Position-Dependent Local Motions in Spin-Labeled Analogs of a Short Alpha-Helical Peptide Determined by Electron-Spin-Resonance. Biochemistry, 1991. 30(39): p. 9498–9503. [DOI] [PubMed] [Google Scholar]
- 80.Pirman NL, et al. , Characterization of the disordered-to-α-helical transition of IA3 by SDSL-EPR spectroscopy. Protein Science, 2011. 20: p. 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moons SJ, et al. , Sialic acid glycoengineering using N-acetylmannosamine and sialic acid analogs. Glycobiology, 2019. 29(6): p. 433–445. [DOI] [PubMed] [Google Scholar]
- 82.Qin PZ, Iseri J, and Oki A, A model system for investigating lineshape/structure correlations in RNA site-directed spin labeling. Biochem Biophys Res Commun, 2006. 343(1): p. 117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffman EA, et al. , Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem, 2015. 290(44): p. 26404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raczuk E, et al. , Different Schiff Bases-Structure, Importance and Classification. Molecules, 2022. 27(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Ascii files of all EPR and fluorescence data can be obtained by contacting the Corresponding author directly.