

Abstract
Some G protein–coupled receptors (GPCRs) demonstrate biased signaling, such that ligands of the same receptor exclusively or preferentially activate certain downstream signaling pathways over others. This phenomenon may result from ligand-specific receptor phosphorylation by GPCR kinases (GRKs). GPCR signaling can also exhibit location bias because GPCRs traffic to and signal from subcellular compartments in addition to the plasma membrane. Here, we investigated whether GRKs contributed to location bias in GPCR signaling. GRKs translocated to endosomes following stimulation of the chemokine receptor CXCR3 or other GPCRs in cultured cells. GRK2, GRK3, GRK5, and GRK6 showed distinct patterns of recruitment to the plasma membrane and to endosomes, depending on the identity of the biased ligand used to activate CXCR3. Analysis of engineered forms of GRKs that localized to either the plasma membrane or endosomes demonstrated that biased CXCR3 ligands elicited different signaling profiles that depended upon the subcellular location of the GRK. Each GRK exerted a distinct effect on the regulation of CXCR3 engagement of β-arrestin, internalization, and activation of the downstream effector kinase ERK. Our work highlights a role for GRKs in location-biased GPCR signaling and demonstrates the complex interactions between ligands, GRKs, and cellular location that contribute to biased signaling.
INTRODUCTION
G protein–coupled receptors (GPCRs) represent the largest and most versatile class of receptors in humans and can elicit cellular responses to various stimuli, including photons, odorants, peptides, lipids, and small molecules (1). Due to the extensive involvement of GPCRs in normal physiology and pathophysiology, approximately 35% of all U. S. Food and Drug Administration–approved drugs target one or more of the 800 human GPCRs (2, 3). GPCRs engage an array of signaling effectors to actuate a cellular response, such as G proteins, GPCR kinases (GRKs), and β-arrestins (4). Ligand binding to plasma membrane–embedded GPCRs promotes a conformational change in the receptor that induces the activation of G proteins and subsequent G protein–dependent signaling (5, 6). The GRKs and other kinases then phosphorylate the intracellular surface of the receptor, promoting the binding of β-arrestins, which can desensitize receptor coupling to G proteins, induce receptor internalization into clathrin-coated pits, and act as scaffolds for other signaling effectors, including mitogen activated protein kinases (MAPKs) and E3 ubiquitin ligases (7, 8). Some ligand:GPCR complexes preferentially activate distinct signaling pathways over others, a phenomenon referred to as biased signaling (9–11).
One proposed mechanism for biased signaling, the phosphorylation barcode hypothesis, posits that unique ligand-induced GPCR phosphorylation patterns promote selective engagement with specific signaling effectors, thereby producing functionally selective outputs (12–14). Many studies demonstrate the critical role the phosphorylation barcode plays in driving specific β-arrestin recruitment and conformational signatures, ultimately promoting unique downstream signaling (15–18). Despite the critical function of GRKs in receptor phosphorylation, the role of the GRKs in promoting biased signaling has not been widely studied. The seven identified mammalian GRKs (1–7) are divided into three subfamilies according to sequence and structure (19), wherein GRKs 1 and 7 comprise the GRK1 subfamily, GRKs 2 and 3 the GRK2 subfamily, and GRKs 4, 5, and 6 the GRK4 subfamily (20, 21). GRKs 2, 3, 5, and 6 are ubiquitously expressed in human tissues (22), and it has been demonstrated that each of these GRKs performs a unique signaling role at GPCRs (23). Although no clear consensus target sequence has been identified for specific GRKs (24), GRKs have a strong preference for nearby phosphorylated residues, and GPCR phosphorylation may ultimately be determined by other serine, threonine, and tyrosine kinases that initially phosphorylate the receptor and subsequent phosphorylation by different GRKs (25). GRKs 2 and 3 are localized to the cytoplasm, and their ligand-induced engagement with a GPCR depends upon interaction of the GRK pleckstrin homology (PH) domain with free Gβγ subunit (26), whereas GRKs 5 and 6 are constitutively present on the plasma membrane (27, 28). GPCRs demonstrate heterogeneity in the putative phosphorylation patterns present on intracellular loop 3 (ICL3) and at the C-terminus (29), and it has been difficult to experimentally delineate the specific contributions of individual GRKs to the generation of unique GPCR phosphorylation patterns and subsequent receptor signaling (30). Certain residues on numerous GPCRs are specifically targeted by individual GRKs, but other sites are phosphorylated by multiple GRKs (13, 31, 32). Differences in the relative amounts of GRKs in various tissues has also been proposed as a means of attaining GRK-specific functionality. For example, GRK5 and GRK6 are present in similar amounts in B- and T-cells, yet GRK5 is the predominant family member present in cardiac tissue (24). Although distinct structural elements, phosphosite targets, and tissue-specific differences in abundance have helped understand GRK specificity, further studies are needed to fully resolve this complex system.
It has been demonstrated that GPCRs can signal from numerous subcellular locations beyond the plasma membrane, including endosomes, mitochondria, the Golgi apparatus, and the endoplasmic reticulum (33–44). We previously demonstrated that biased ligands of the chemokine receptor CXCR3 induce distinct patterns of G protein activation and β-arrestin recruitment at endosomes compared to the plasma membrane, and CXCR3 internalization contributes to the overall biased cellular output of this receptor (45). Given the critical role of receptor phosphorylation in biased GPCR signaling and previous evidence that GRKs can exist in specific subcellular compartments (46–50), it is likely that the GRKs play a substantial role in generating functionally selective responses in different cellular locations. Yet, to our knowledge, few experiments have assessed the specific effects of GRK engagement with GPCRs in endosomes on biased signaling.
Here, we show that biased CXCR3 agonists differentially promoted GRK engagement with CXCR3 at the plasma membrane. We also provide evidence demonstrating that some ligands promoted GRK translocation to endosomes in a pattern entirely distinct from that observed at the plasma membrane. We further show that individual GRKs exhibited distinct effects on CXCR3 signaling depending on the biased ligands used to activate the receptor. We demonstrated that the association of GRKs 2 and 3 with CXCR3 was largely dependent upon the activation of G proteins, but some ligands enabled G protein–independent recruitment. Each GRK demonstrated unique roles in regulating the receptor’s ability to engage β-arrestins, become internalized, and activate extracellular signal–regulated kinase (ERK). Using engineered forms of GRKs that localize to specific subcellular locations, we showed that GRK identity and subcellular localization modulated GPCR signaling depending on the ligand used to activate the receptor. Lastly, we demonstrated that GRK recruitment to endosomes was not unique to CXCR3 but was observed for a panel of other therapeutically relevant, well-studied GPCRs, suggesting that location-specific GRK activity occurs across the GPCR superfamily. These findings suggest that subcellular engagement of GRKs with GPCRs may facilitate location-specific signaling responses and that the role of individual GRKs is differentially directed by specific ligands. These data demonstrate a complex interaction between GRK family members, ligands, and cellular locations in driving a GPCR’s overall biased signaling outputs.
RESULTS
Biased ligands elicit different GRK recruitment patterns to the plasma membrane and to CXCR3
CXCR3 is a chemokine receptor with endogenous ligands that exhibit biased signaling in various forms, such as in their differential formation of Gαi:β-arrestin complexes and markedly different abilities to induce G protein– or β-arrestin–mediated signaling (51–54). CXCR3 is primarily found on activated CD8+ T cells and natural killer cells, directs cellular functions like chemotaxis and T cell polarization, and is implicated in inflammatory conditions such as cancer, atherosclerosis, autoimmunity, and allergic contact dermatitis (11). We first determined if activation of CXCR3 using biased ligands promoted distinct patterns of GRK recruitment to the plasma membrane in HEK293 cells. Using a previously validated NanoLuc binary technology (nanoBiT) complementation assay based on the generation of active luciferase by the association of small BiT (SmBiT) and large BiT (LgBiT) subunits with one another (55), we monitored the interaction between the plasma membrane marker CD8α-SmBiT and GRK2-, GRK3-, GRK5-, or GRK6-LgBiT following stimulation of untagged CXCR3 (Fig. 1A). Using confocal microscopy in HEK293T cells, we demonstrated colocalization of CD8α-mCherry and CXCR3-mCerulean, consistent with CD8α and CXCR3 residing in the same subdomains within the plasma membrane (fig. S1). We used three endogenous, biased CXCR3 agonists, CXCL9, CXC10, and CXCL11, and two synthetic CXCR3 agonists, VUF10661 and VUF11418, for the nanoBiT complementation experiments (56). Previous publications measuring relative amounts of G protein signaling and β-arrestin recruitment demonstrate that CXCL11 is relatively β-arrestin–biased, whereas CXCL10, VUF10661, and VUF11418 are relatively G protein–biased. CXCL9 acts as a β-arrestin–biased partial agonist (52, 57).
Fig. 1. GRK recruitment to the plasma membrane and to CXCR3.
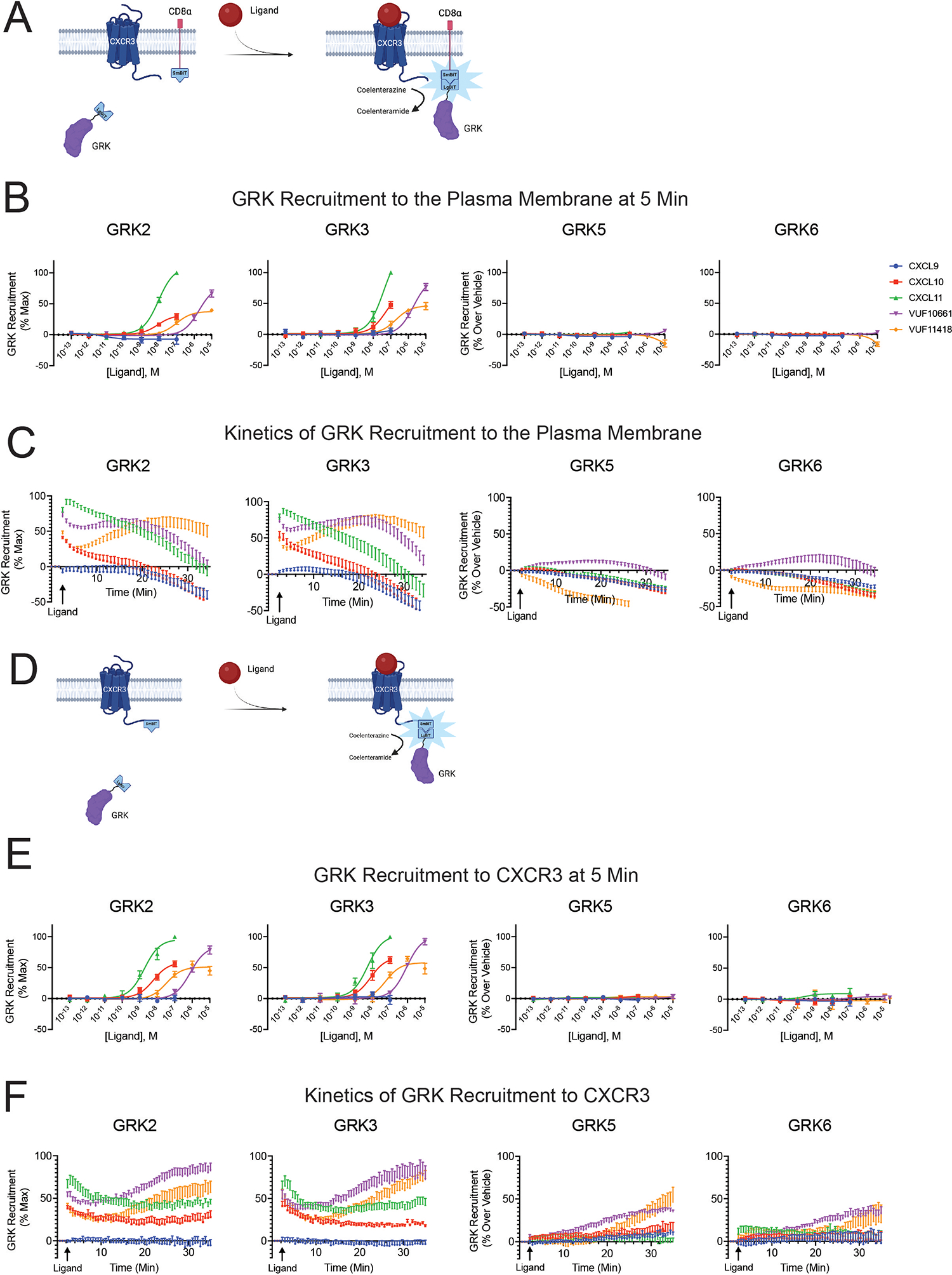
(A) Schematic representation of a NanoBiT complementation assay detecting GRK recruitment to the plasma membrane using GRK-LgBiT and CD8α-SmBiT in HEK293 cells stimulated with CXCR3 agonists. Upon recruitment of GRK to the plasma membrane, the smBiT and LgBiT generate a functional luminescent signal as a readout of GRK. (B) Agonist concentration-response at five minutes and (C) kinetic time-course of GRK2, GRK3, GRK5, and GRK6 recruitment to the plasma membrane in response to the indicated ligands. (D) Schematic representation of a NanoBiT complementation assay detecting GRK recruitment to CXCR3 using GRK-LgBiT and CXCR3-SmBiT in HEK293 cells stimulated with CXCR3 agonists. (E) Agonist concentration-response at five minutes and (F) kinetic time-course of GRK2, GRK3, GRK5, and GRK6 recruitment to CXCR3. All experiments were performed following stimulation with 100nM CXCL9, CXCL10, or CXCL11 or 10μM VUF10661 or VUF11418. Data shown are the mean ± SEM of n=5 independent experiments. Concentration-response curves and kinetic data for GRK2 and GRK3 are normalized to maximum observed signal across all ligands. Concentration-response curves and kinetic data for GRK5 and GRK6 are shown as luminescence change over vehicle for each ligand. GRK2 and GRK3 both demonstrate maximum recruitment patterns of CXCL11 > VUF10661 > (CXCL10=VUF11418) > CXCL9 as assessed using one-way ANOVAs and Tukey’s post hoc testing on the luminescence values at maximum dose.
After 5 minutes, the recruitment patterns of GRK2 and GRK3 were similar to each other, with differences in efficacy and potency between the biased ligands (Fig. 1B). Specifically, CXCL11 and VUF10661 were the most efficacious agonists followed by CXCL10 and VUF11418. CXCL9 evoked little to no detectable recruitment of GRK2 or GRK3 at physiologic concentrations of the chemokine. GRK2 and GRK3 are known to localize differently from GRK5 and GRK6 in the cell at baseline, and we performed confocal microscopy using YFP-tagged GRKs and CXCR3-mCerulean to confirm cytosolic localization of GRK2 and GRK3 and constitutive membrane localization of GRK5 and GRK6 (58–60) (fig. S2). Thus, ligand-induced recruitment to the plasma membrane was not necessarily an illustrative readout of GRK5 and GRK6 activity, as evinced by the lack of detectable additional recruitment of these GRKs over baseline by physiological chemokine concentrations in our assay (Fig. 1B).
In analyzing the kinetic data of GRK recruitment to the plasma membrane in response to 100nM CXCL9, CXCL10, or CXCL11 and 10μM VUF10661 or VUF11418, we observed significant differences across ligands. All ligands, except CXCL9, elicited rapid recruitment of GRK2 and GRK3 to the plasma membrane upon receptor activation (Fig. 1C). However, at 30 minutes, all ligands except VUF11418, show a marked decrease in luminescence. Given that most ligands induced maximum GRK recruitment to the plasma membrane at 5 minutes, we elected to show the concentration-response data at this time point (Figs. 1B). These data demonstrate that the ligands not only promoted different magnitudes of GRK2 and GRK3 translocation but also produced unique kinetic signatures for the interaction of individual GRKs with the plasma membrane. Differential GRK recruitment magnitude and kinetics may contribute to the generation of specific phosphorylation barcodes and biased signaling.
When the same experiment was performed using CXCR3-SmBiT to assess GRK recruitment to the receptor, we observed similar concentration-response curves to those seen for plasma membrane recruitment for all GRKs at 5 minutes after stimulation (Fig. 1, D and E). Unexpectedly, when analyzing GRK recruitment over time, we observed an increase in the luminescence signal at 30 minutes for all four GRKs in response to the synthetic ligands but not to the endogenous chemokines (Fig. 1F). For GRK2 and GRK3, following an initial robust phase of GRK recruitment induced by the synthetic ligands, there was a slight decrease in luminescence at 10 minutes, followed by an increase in luminescence at 30 minutes (Fig. 1F). CXCL10 and CXCL11 elicited sustained interaction of GRKs with the receptor even at 30 minutes (Fig. 1F), whereas the luminescence signal returned to baseline at this time when measuring GRK recruitment to the plasma membrane (Fig. 1C). By contrast, GRK5 and GRK6 showed little initial detectable increase in luminescence signal following stimulation with VUF10661 and VUF11418 but demonstrated a very slow and late increase after 10 minutes (Figs. 1F).
GRK recruitment to endosomes differs from recruitment to the plasma membrane
Based on the unexpected kinetic tracings of GRK recruitment to CXCR3 compared to the plasma membrane, we hypothesized this data may reflect GRKs interacting with the receptor at subcellular locations in addition to or other than the plasma membrane. To evaluate the potential recruitment of the GRKs to endosomes, we repeated the NanoBiT complementation assays using the GRK-LgBiT constructs, wild-type (WT) CXCR3, and the previously validated early endosome marker 2x-Fyve tagged with SmBiT (61, 62) (Fig. 2A). We observed that GRK2, GRK3, and GRK5 were recruited to endosomes, with VUF10661 exhibiting the most robust recruitment of these GRKs (Fig. 2B). We also observed detectable recruitment of GRK3 to endosomes using CXCL11, VUF11418, and CXCL10. The overall pattern of ligand-induced GRK recruitment to endosomes substantially differed from the recruitment pattern observed at the plasma membrane. GRK recruitment to endosomes was significantly slower, consistent with previously reported kinetic data on GPCR internalization (63, 64).
Fig. 2. CXCR3-mediated GRK recruitment to endosomes.
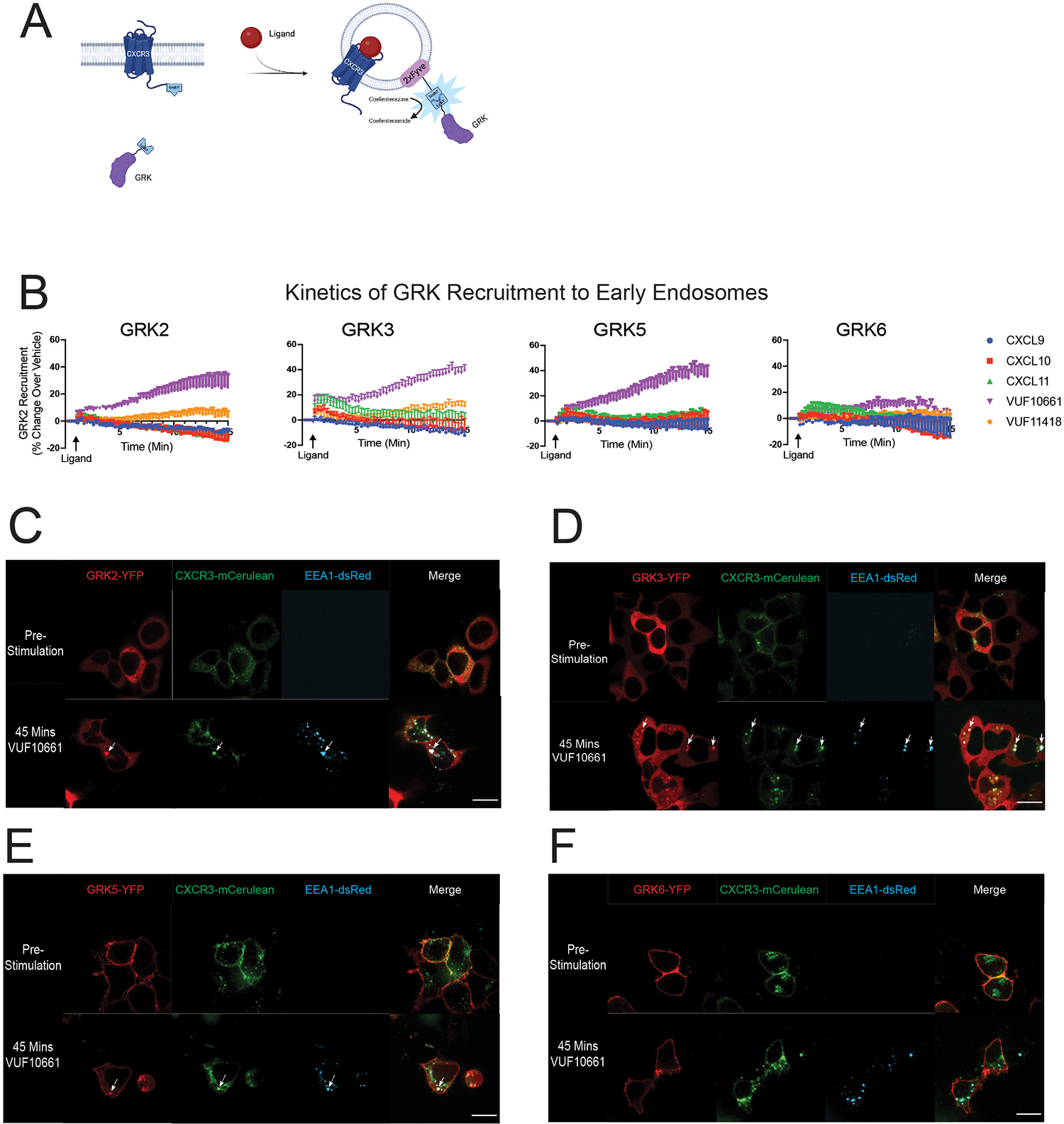
(A) Schematic representation of the NanoBiT complementation assay used to detect GRK recruitment to endosomes using wild-type CXCR3, LgBiT-tagged GRK, and 2xFyve-SmBiT in HEK293 cells stimulated with CXCR3 agonists. Following activation of the receptor and recruitment of GRK to endosomes, the smBiT and LgBiT undergo complementation and generate a functional luminescent signal as a readout of GRK endosomal recruitment. (B) Kinetic data for GRK2, GRK3, GRK5, and GRK6 recruitment to early endosomes upon stimulation with 100nM CXCL9, CXCL10, or CXCL11 or 10μM VUF10661 or VUF11418. Kinetic tracings are shown as luminescence change over vehicle. (C to F) Confocal microscopy images of HEK293 cells transfected with CXCR3-mCerulean and EEA1-dsRed plus GRK2-YFP (C), GRK3-YFP (D), GRK5-YFP (E), or GRK6-YFP (F). Images were taken before or 45 minutes after stimulation with 10μM of VUF10661. Scale bar, 21 μm. Plate-based experiments show the mean ± SEM of n=5 independent experiments. Confocal microscopy images are representative of n=3 independent experiments. Using one-way ANOVA with Tukey’s post hoc testing, the time to maximum GRK2 and GRK3 recruitment to the plasma membrane (~2 minutes) was significantly different than the time to maximum recruitment to the endosome (~13 minutes) at a p-value of <0.05.
To support these findings using confocal microscopy, we transfected HEK293 cells with the GRK-YFP and CXCR3-mCerulean and stimulated cells with agonist for 45 minutes (figs. S3, A and B; S4, A and B; S5, A to E). The 45-minute time point was chosen because this is when maximal CXCR3 internalization was observed using confocal microscopy; however, CXCR3 and GRKs were detected in intracellular puncta as early as 15 minutes following agonist stimulation, consistent with our luminescence data (fig. S6, A and B). These data are consistent with many other reports that demonstrate GPCR localization to early endosomes is dynamic and can occur within a few minutes following agonist stimulation but may last for up to 90 minutes (63, 65–67). Although all ligands promoted receptor internalization, we only observed the formation of intracellular puncta that were both YFP+ and mCerulean+, as supported by a line scan analysis, suggestive of colocalized receptor and GRK in endosomes, for some ligand:GRK combinations, consistent with our luminescence data (figs. S3, A and B; S4, A and B; S5, A to E). Of note, we observed colocalization of GRK3-YFP and CXCR3-mCerulean following treatment with CXCL11 but not with CXCL10 nor CXCL9 (fig. S3B).
In contrast, following treatment with VUF10661, we observed robust colocalization of CXCR3-mCerulean with GRK2-YFP, GRK3-YFP, and to a lesser extent GRK5-YFP. To more rigorously demonstrate colocalization of GRKs and CXCR3 in endosomes, we repeated the confocal experiment with co-transfection of the endosomal marker EEA1-dsRed following VUF10661 treatment (Fig. 2, C to F). Consistent with our previous confocal experiment, we observed colocalization of GRK2-YFP, GRK3-YFP, and GRK5-YFP with CXCR3-mCerulean and EEA1-dsRed. These findings suggest that ligands may direct specific activities of the individual GRKs through selective trafficking of certain GRKs to endosomes.
GRK2 and GRK3 recruitment to CXCR3 largely depends on G protein activation for some, but not all ligands
A primary feature distinguishing the GRK2 and GRK4 subfamilies is that GRK2 and GRK3 recruitment to a GPCR rely on interactions of their PH domain with free Gβγ following heterotrimeric G protein dissociation (68, 69). Although studies have shown that mutations in the PH domain of GRK2 abolish its ability to interact with free Gβγ and phosphatidylinositol 4,5-bisphosphate (70), previous work also suggests G protein–independent mechanisms of GRK recruitment and functionality (71). Seminal work using purified GRK1, GRK2, GRK5, and the GPCRs β2 adrenergic receptor (β2AR) and rhodopsin demonstrated the ability of GRKs to phosphorylate activated receptors in the absence of G proteins (72–76). Additionally, this work showed that GRK2, but not GRK5, kinase activity is greatly augmented in the presence of Gβγ (73, 76). Another study demonstrated a role for G protein–independent GRK2 recruitment to the dopamine D2 receptor (D2R) in facilitating β-arrestin engagement in cells (77). However, whether biased ligands of CXCR3 can elicit distinct GRK recruitment mechanisms is unclear.
To evaluate the necessity of G protein activation in GRK2 and GRK3 recruitment to CXCR3, we assessed GRK recruitment to this predominantly Gαi-coupled receptor (78) in the absence of Gαi using ΔG6 cells, a previously validated HEK293 cell line devoid of all G proteins except Gαi/o family members (79). Specifically, these cells lack GNAS, GNAL, GNA11, GNA12, GNA13, and GNAQ. Using these cells, we performed the NanoBiT complementation assay using GRK2- or GRK3-LgBiT and CXCR3-SmBiT with and without the addition of pertussis toxin (PTX) to inhibit Gαi/o activation (80, 81) (Fig. 3, A and B). The kinetic tracings for GRK recruitment to CXCR3 in ΔG6 HEK293 cells (fig. S7, A and B) were overall similar to those observed in WT HEK293 cells (Fig. 1F). Unexpectedly, we were able to detect GRK2 and GRK3 recruitment after CXCL9 stimulation in the ΔG6 cells, a process we did not observe in WT HEK293 cells. We found that PTX treatment significantly reduced GRK2 and GRK3 recruitment, although there was incomplete inhibition of GRK2 recruitment induced by the endogenous chemokines (Fig. 3A) and GRK3 recruitment by all ligands (Fig. 3B). It is possible that these findings are due to incomplete inhibition of Gαi/o activation with PTX. However, we observed near complete inhibition of GRK2 recruitment with VUF10661 and VUF11418 but relatively little change with CXCL9, suggesting that PTX treatment completely inhibited Gαi/o activation in these assays (Fig. 3A). These data validate previous reports suggesting that recruitment of GRK2 family proteins is largely, but not completely, dependent on G protein activation, with some contribution of G protein–independent mechanisms for GRK2 and GRK3 recruitment.
Fig. 3. GRK recruitment to CXCR3 in G protein–deficient cells.
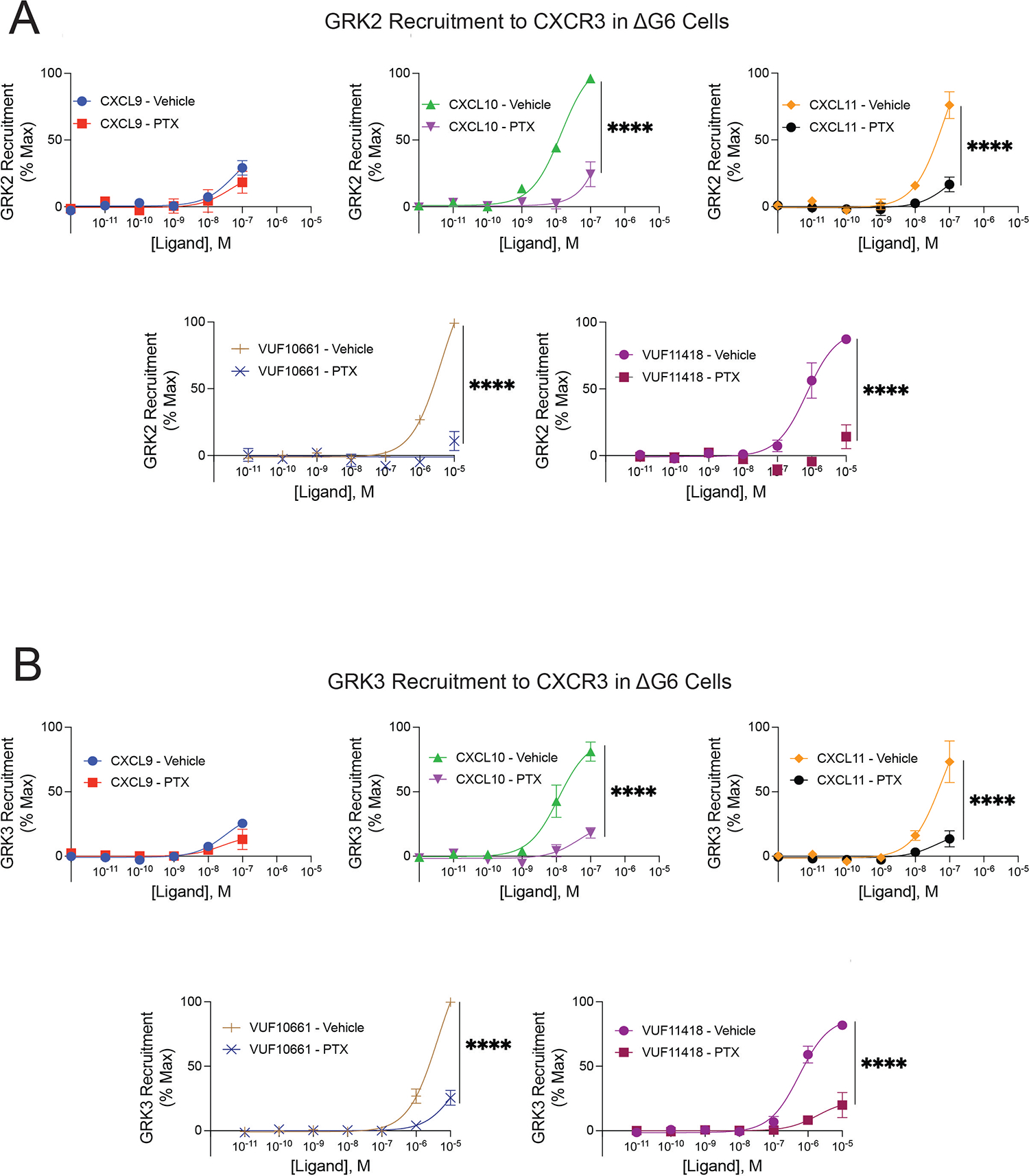
Agonist concentration-response of (A) GRK2 and (B) GRK3 recruitment to CXCR3 with and without pretreatment with 200ng/mL pertussis toxin (PTX) in ΔG6 knockout cells treated with CXCL9, CXCL10, CXCL11, VUF10661 or VUF11418. GRK recruitment was measured using a split luciferase assay involving LgBiT-tagged GRK2 or GRK3 and CXCR3-SmBiT. Data shown are the mean ± SEM of n=3 independent experiments, significance testing by one-way ANOVA with Šidák post hoc testing comparing luminescent signal at maximum dose between treatment conditions (vehicle vs. PTX). Concentration-response curves are normalized to maximum signal observed across all ligands. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001.
To further corroborate these findings, we repeated these experiments in HEK293 cells devoid of all Gα proteins (ΔG7 cells), specifically GNAS, GNAL, GNAQ, GNA11, GNA12, GNA13, GNAI1, GNAI2, GNAI3, GNAO1, GNAZ, GNAT1, and GNAT2 (82). Upon rescued expression of Gαi1, we observed a significant increase in GRK2 recruitment with CXCL11 and VUF10661, and in GRK3 recruitment with VUF10661 (fig. S8, A to D). All ligands except CXCL9 demonstrated an increase, albeit not to statistically significant extents, in GRK2 or GRK3 recruitment following rescue of Gαi1. Even in the absence of Gαi activity, VUF10661 maintained a partial ability to recruit GRK2, whereas CXCL10, CXCL11, VUF10661, and VUF11418 all partially recruited GRK3. Although G protein–dependent mechanisms are responsible for the majority of GRK2 and GRK3 recruitment to CXCR3 in response to stimulation by different ligands, these findings demonstrate an additional aspect of GRK specificity, whereby different ligands can sometimes elicit G protein–independent mechanisms to promote GRK2/3 recruitment.
GRK subcellular localization differentially affects β-arrestin 2 recruitment and CXCR3 internalization
Given that biased CXCR3 agonists promoted differential GRK recruitment across subcellular locations, we next determined if the cellular localization of a GRK could influence signaling. We therefore generated location-restricted GRK constructs to localize GRKs to either the plasma membrane or endosomes. We used a membrane targeting sequence from Lyn kinase to create plasma membrane–tethered forms (Mem-GRK) of GRK2 and GRK3, and a 2x-Fyve early endosome tag to develop endosome-tethered forms (Endo-GRK) of GRK2, GRK3, GRK5, and GRK6. The constructs were validated through confocal microscopy (Fig. 4, A and B) and evaluated further using co-expression with the membrane marker CD8α and the endosomal marker EEA1 to confirm proper localization (fig. S9, A and B). Endo-GRK5 and Endo-GRK6 demonstrated both plasma membrane and endosome localization upon addition of the 2x-Fyve tag given their baseline localization to the plasma membrane.
Fig. 4. β-arrestin recruitment and CXCR3 internalization in cells expressing location-specific forms of GRK2, GRK3, or GRK6.
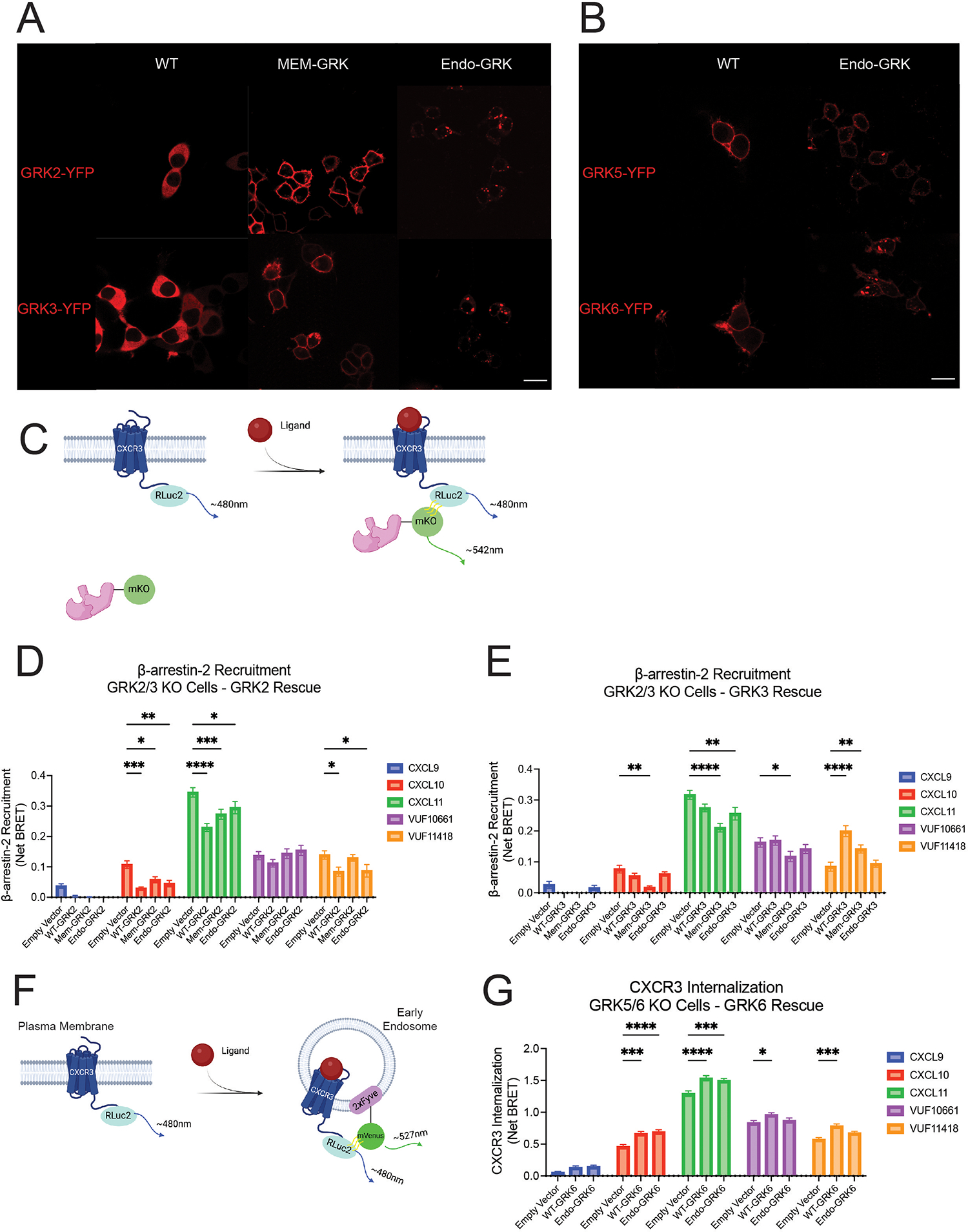
(A and B) Confocal microscopy images of HEK293T cells transfected with plasma membrane–localized (Mem) or endosome-localized (Endo) forms of GRK2 or GRK3 (A) and GRK5 or GRK6 (B) tagged with YFP (false red coloring for clarity). Scale bars, 26μm. (C) Schematic representation of β-arrestin 2-mKO recruitment to CXCR3-RLuc2 using bioluminescence resonance energy transfer (BRET). (D and E) BRET experiments were performed in ΔGRK2/3 cells transfected with 50ng of the indicated GRK2 constructs (D), 25ng of the indicated GRK3 constructs (E), or empty vector. (F and G) Schematic representation of BRET-based assay (F) to assess CXCR3 internalization in ΔGRK5/6 cells transfected with 50ng of the indicated GRK6 constructs or empty vector (G). Cells were stimulated with 100nM CXCL9, CXCL10, or CXCL11 or 10μM VUF10661 or VUF11418 in all experiments. Data shown are the mean AUC ± SEM of n=5 independent experiments,, significance testing by two-way ANOVA with Tukey post hoc testing conducted between empty vector and other transfections conditions. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001.
Because the GRK N-terminus is involved in recognition of and docking to active GPCRs, it is possible that the N-terminal placement of the localization tags on the GRKs could interfere with their function (83, 84). To determine if the location-specific GRK constructs could interact with CXCR3, we performed Biolumescence Resonance Energy Transfer (BRET) assays using CXCR3 tagged with Renilla Luciferase II (RLucII) and YFP-tagged GRK constructs. Endo-GRK2 and Mem-GRK3 interacted with the receptor following treatment with VUF10661, providing evidence that these tags did not significantly disrupt GRK:GPCR interactions (figs. S9, C and D). We additionally observed that the Mem-GRK and Endo-GRK constructs had higher basal BRET values as compared to the WT-GRK constructs, demonstrating that the location-specific GRK constructs were in close proximity to both plasma membrane and endosomal CXCR3, even in the absence of ligand stimulation. Given these higher basal BRET values, likely representing increased constitutive receptor association, we expected and observed a smaller ligand-induced effect using Mem-GRK-YFP and Endo-GRK-YFP as compared to WT-GRK-YFP (figs. S9, C and D).
To evaluate the signaling consequences of these location-specific forms of GRK, we used two previously validated CRISPR/Cas9–edited HEK293 cell lines, one devoid of GRK2 and GRK3 (ΔGRK2/3) and the other devoid of GRK5 and GRK6 (ΔGRK5/6) (85, 86), rather than siRNA or pharmacologic strategies, to ensure complete elimination of endogenous GRK kinase activity. We performed immunoblotting on WT, ΔGRK2/3, and ΔGRK5/6 HEK293 cells to determine the amount of each GRK to transfect based on WT expression amounts (fig. S10, A to D). We then used BRET to monitor ligand-induced recruitment of fluorescently tagged β-arrestin 2 (β-arrestin 2-mKO) recruitment to CXCR3-RLucII in either ΔGRK2/3 or ΔGRK5/6 cells transfected with individual WT GRK constructs (Fig. 4C). For CXCL10, CXCL11, and VUF11418 treatment, we observed that expression of WT-GRK2 decreased β-arrestin 2 recruitment, whereas VUF10661 stimulation had no significant effect on WT-GRK2–mediated β-arrestin 2 recruitment (Fig. 4D). Further, expression of WT-GRK3 enhanced β-arrestin 2 recruitment for VUF11418 treatment but had no significant effect for other ligand conditions (Fig. 4E). By contrast, WT-GRK5 and WT-GRK6 had minimal effect on β-arrestin 2 recruitment for all but one ligand condition (figs. S11, A and B). These findings suggest that the WT GRKs have distinct effects on CXCR3 engagement with β-arrestin 2. These effects were conserved across ligands for some GRKs but agonist-dependent for others, indicative of the ability of different ligands to confer unique properties to GRK family members.
Furthermore, the effects of the location-specific GRKs from WT-GRK were ligand-specific. With CXCL11, both Mem-GRK2 and Endo-GRK2 partially reduced the WT effect on β-arrestin 2 recruitment (Fig. 4D). However, upon VUF11418 stimulation, Mem-GRK2, but not Endo-GRK2, altered the WT phenotype. A different profile was observed for GRK3. Although β-arrestin 2 recruitment increased following expression of WT-GRK3 with VUF11418 treatment, this effect was reduced when GRK3 was localized to the plasma membrane and lost when it was bound to endosomes (Fig. 4E). Although WT-GRK3 had no detectable effect on β-arrestin 2 recruitment for CXCL10, CXCL11, or VUF10661, Mem-GRK3 decreased β-arrestin 2 recruitment for all three ligands. Whereas β-arrestin 2 recruitment was susceptible to alterations in GRK2 and GRK3 cellular localization, GRK5 and GRK6 demonstrated little effect (fig. S11, A and B). These data suggest a role of location-dependent activities of the GRKs on β-arrestin 2 recruitment, where the relative direction and magnitude of change is GRK-, ligand-, and location-dependent.
Given the role of β-arrestins in promoting receptor internalization, we next assessed if individual GRKs had distinct effects on CXCR3 trafficking to endosomes. We expressed individual GRKs in ΔGRK2/3 or ΔGRK5/6 cells transfected with CXCR3-RLuc2 and 2x-Fyve-mVenus to assess CXCR3 proximity to early endosomes following ligand stimulation (Fig. 4F). Whereas WT-GRK2 significantly decreased β-arrestin 2 engagement of CXCR3 for CXCL10, CXCL11 and VUF11418 stimulation, expression of WT-GRK2 unexpectedly elicited minimal changes to CXCR3 internalization across most ligands (fig. S11C). Likewise, although we observed a robust increase in β-arrestin 2 recruitment by WT-GRK3 upon VUF11418 stimulation, there was no effect on CXCR3 internalization (fig. S11D). By contrast, even though WT-GRK6 showed little activity in affecting β-arrestin 2, expression of WT-GRK6 enhanced CXCR3 internalization across all ligands except CXCL9 (Fig. 4G). Expression of WT-GRK5 also showed a minimal effect on CXCR3 internalization for all ligands apart from VUF10661 (fig. S11E). These findings are indicative of specific roles for distinct GRKs in driving different GPCR signaling events. Whereas our data suggest a prominent function of GRK2 and GRK3 in regulating β-arrestin 2 recruitment, we found a substantial role for GRK6 in mediating receptor internalization.
Subcellular localization modulates the distinct effects of GRKs on cytosolic and nuclear ERK1/2 activity in a ligand-dependent fashion
We next investigated the activation of the kinases ERK1 and ERK2 (ERK1/2) as a marker of mitogen-activated protein kinase (MAPK) signaling, a prototypical GPCR downstream signaling pathway that can be activated by both G proteins and β-arrestins (87, 88). We transfected a previously developed BRET-based biosensor that detects ERK kinase activity in the nucleus or cytoplasm (45, 89) into the ΔGRK2/3 or ΔGRK5/6 cell lines. We did not observe any differences in basal ERK activity following expression of any of the GRK constructs (fig. S12, A to D).
WT-GRK2 had little effect on cytosolic ERK activity, with minor decreases in ERK activity with VUF10661 (Fig. 5A). However, WT-GRK2 significantly inhibited nuclear ERK activity across all ligands (Fig. 5B). In contrast, WT-GRK3 was inhibitory to both cytosolic and nuclear ERK activation for most ligands tested (Fig. 5, C and D). WT-GRK5 had an entirely different profile, wherein it diminished cytosolic ERK activity upon CXCL10 and VUF11418 stimulation but had no significant effect on nuclear ERK activity with these ligands (Fig. 5, E and F). CXCL11 also demonstrated reduced cytosolic ERK activity with WT-GRK5 but enhanced nuclear ERK activation. These findings support distinct roles of individual GRKs in regulating the activation of cytosolic and nuclear pools of ERK. Consistent with our previous assays examining upstream signaling events, activities of GRK subtypes were specific for each ligand.
Fig. 5. Cytoplasmic and nuclear ERK activation by GRKs localized to distinct cellular compartments.
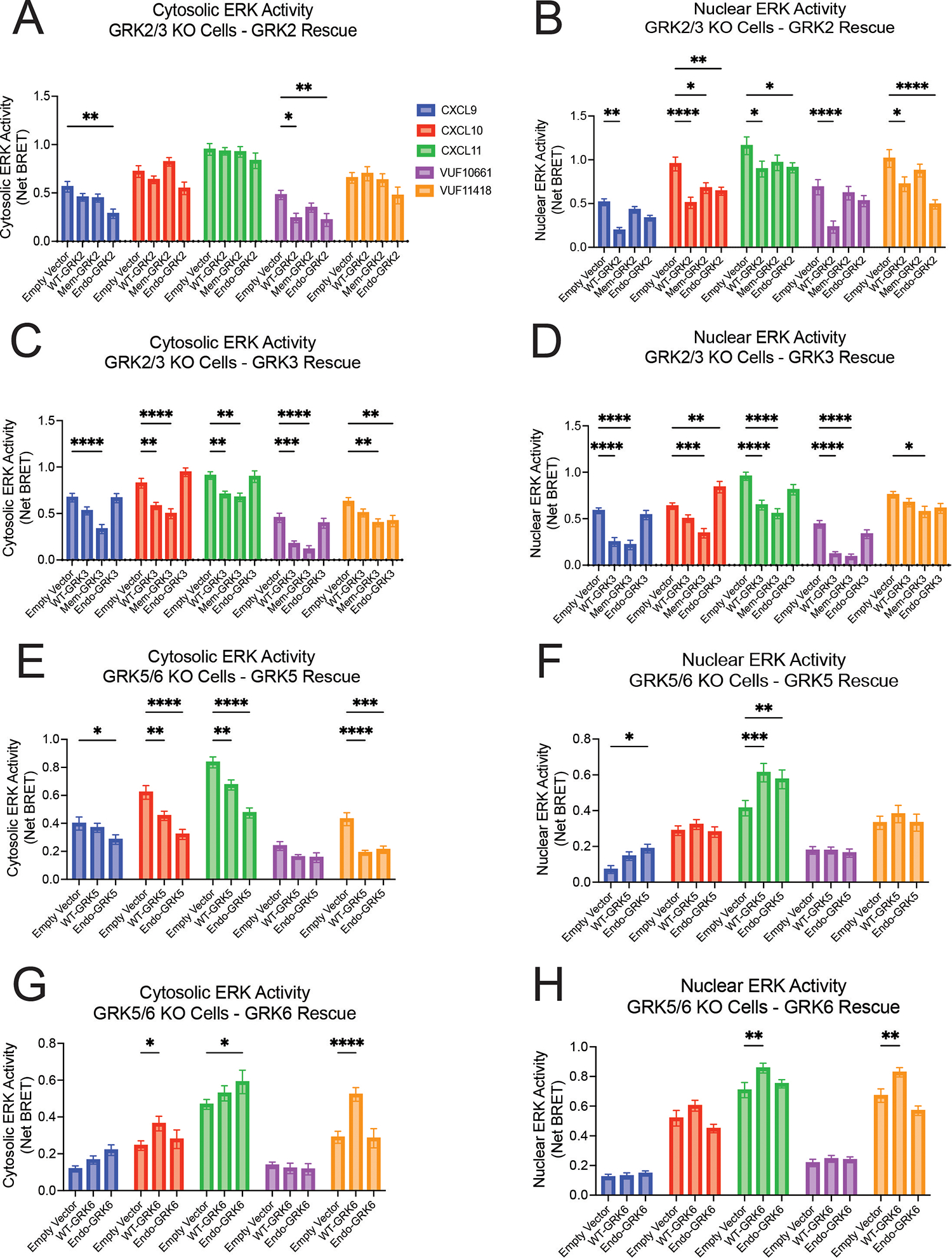
(A to D) Cytoplasmic and nuclear ERK activity in ΔGRK2/3 cells expressing the indicated GRK2 (A and B) or GRK3 (C and D) constructs or empty vector. Cytoplasmic and nuclear ERK activity in ΔGRK5/6 cells expressing the indicated GRK5 (E and F) or GRK6 (G and H) constructs or empty vector. 50 ng GRK2 and GRK6 constructs and 25 ng GRK3 constructs were transfected into cells. 100nM CXCL9, CXCL10, or CXCL11 or 10μM VUF10661 or VUF11418 were used in all experiments. Data shown are the mean AUC ± SEM of n=5 independent experiments,, significance testing by two-way ANOVA with Tukey post hoc testing conducted between empty vector and other transfections conditions. *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001.
When using the location-specific forms of GRK, Endo-GRK2 inhibited nuclear ERK activity similarly to WT-GRK2, except with CXCL9 and VUF10661 (Fig. 5B). For most ligands, WT-GRK3 and Mem-GRK3 significantly reduced both nuclear and cytosolic ERK activation, whereas Endo-GRK3 had little effect on ERK activity as compared to empty vector (Fig. 5, C and D). Further, although WT-GRK5 and Endo-GRK5 decreased cytosolic ERK activation following treatment with CXCL10, CXCL11, and VUF11418, we observed a substantial increase in nuclear ERK activation with CXCL11 treatment (Fig. 5, E and F). Endo-GRK6 demonstrated little effect in modulating nuclear or cytosolic ERK activity as compared to empty vector, whereas WT-GRK6 increased ERK activation in both the nucleus and cytosol for some ligands (Fig. 5, G and H). These data demonstrated that altering GRK localization had differential effects on the activation of nuclear and cytoplasmic ERK. By modulating the ligand, the GRK, and the subcellular location of the GRK, it was possible to generate numerous and highly specific patterns of ERK activity, highlighting the complex interaction between GRKs and a GPCR activated by a single ligand.
Other GPCRs recruit GRKs to endosomes
Given our finding that biased CXCR3 agonists promoted differential GRK recruitment to the plasma membrane and endosomes, we wondered if this phenomenon occurred at other GPCRs. We selected an array of GPCRs, including β2AR, μ-opioid receptor (MOR), angiotensin II type I receptor (AT1R), V2 vasopressin receptor (V2R), and atypical chemokine receptor 3 (ACKR3). We then probed GRK2, GRK3, GRK5, and GRK6 recruitment to the plasma membrane and endosomes following ligand stimulation using the Nano-BiT complementation assay. Cells were transfected with WT receptor, GRK-LgBiT, and either CD8-SmBiT for the plasma membrane or 2xFyve-SmBiT for the endosome.
We found that these receptors differed in their ability to induce GRK recruitment to the plasma membrane. For example, stimulation of the V2R produced the most robust recruitment of GRK2 and GRK3 to the plasma membrane, whereas stimulation of the β2AR induced the greatest recruitment of GRK5 and GRK6 to the plasma membrane (Fig. 6A). Although GRK5 and GRK6 are constitutively membrane-localized, this apparent increase may represent a change in the membrane distribution of these GRKs because CD8α can be found in specific membrane microdomains (90, 91). We observed substantial recruitment of the GRKs to endosomes for many of the receptors tested, with distinct patterns when compared to plasma membrane recruitment (Fig. 6, A and B). GRK2 and GRK3 were robustly recruited to endosomes upon activation of the AT1R (Fig. 6B). The β2AR, MOR, and V2R all demonstrated the ability to recruit GRK2 and/or GRK3 to endosomes, albeit to different extents and with distinct kinetic tracings. GRK5 and GRK6 similarly demonstrated recruitment to endosomes across many receptors in a pattern that was different than that observed at the plasma membrane. (Fig. 6, A and B). We further validated these findings with the AT1R using confocal microscopy. Following treatment with angiotensin II (AngII), we observed colocalization of GRK3-YFP and GRK6-YFP with AT1R-mKO in endosomes (Fig. 6, C and D). These data demonstrated that GRK trafficking to endosomes is a conserved signaling mechanism for many GPCRs and that distinct patterns of GRK engagement with GPCRs at the plasma membrane and endosomes may facilitate signaling specificity and promote biased responses.
Fig. 6. GRK recruitment to plasma membrane and endosomes at other GPCRs.
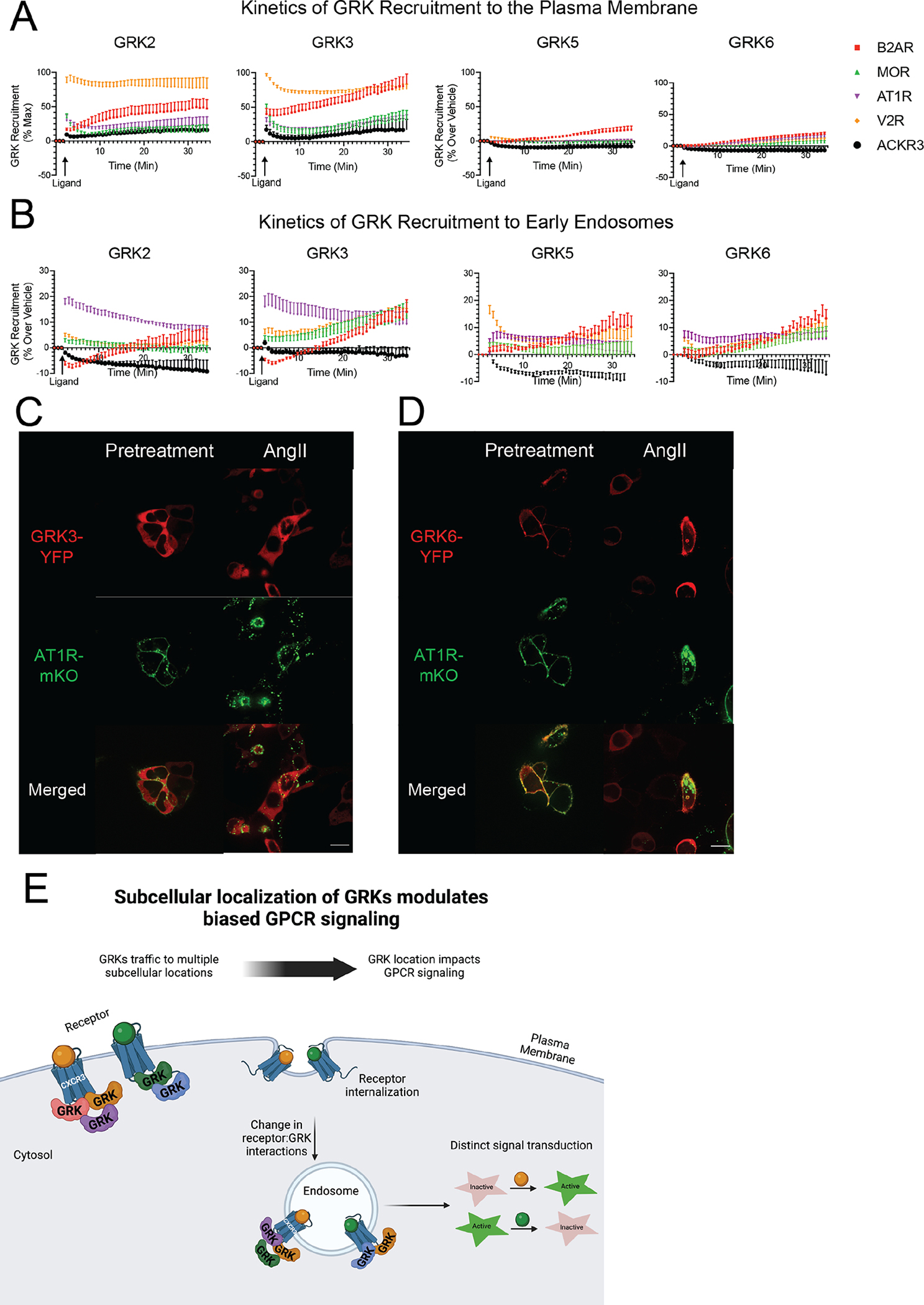
(A) Recruitment of GRK2, GRK3, GRK5, or GRK6 to the plasma membrane in HEK293 cells upon stimulation of each specified receptor with a single application of the corresponding agonist. (B) Recruitment of GRK2, GRK3, GRK5, and GRK6 to the endosome in HEK293 cells upon stimulation of each specified receptor with a single application of the corresponding agonist. (C and D) Confocal microscopy of mKO-tagged angiotensin II type I receptor (AT1R-mKO) and either GRK3-YFP (C) or GRK6-YFP (D) 45 minutes after addition of vehicle or 10μM angiotensin II (AngII). Scale bars, 16μm. The β2-adrenergic receptor (B2AR) was stimulated with 10μM isoproterenol, the μ-opioid receptor (MOR) was stimulated with 10μM DAMGO, the angiotensin II type I receptor (AT1R) was stimulated with 10μM angiotensin II, the V2 vasopressin receptor (V2R) was stimulated with 1μM arginine vasopressin, and the atypical chemokine receptor 3 (ACKR3) was stimulated with 10μM of WW36. Data shown are the mean ± SEM of n=5 independent experiments, and are normalized luminescence change over vehicle. Confocal microscopy images are representative of n=3 independent experiments. (E) Working model showing how the subcellular location of GRKs influences GPCR signaling induced by biased ligand. Biased ligands (orange and green) activate GPCRs, such as CXCR3, at the plasma membrane and engage distinct GRK subtypes. Following receptor internalization, the GRK subtypes interacting with each receptor can change, demonstrating spatially-dependent GPCR signaling. These unique ligand:receptor:GRK complexes at different cellular locations drive distinct signaling outputs.
DISCUSSION
Nearly all GPCRs are known to interact with G proteins, GRKs, and β-arrestins, but these interactions have primarily been studied at the plasma membrane. Published studies demonstrate that G proteins and β-arrestins can signal from endosomes (92–94). Here, we provide evidence that GRKs can traffic to endosomes following agonist stimulation of multiple GPCRs (Fig. 6E). Additionally, using both endogenous and synthetic CXCR3 agonists, we showed that the pattern of GRK recruitment to the plasma membrane differed substantially from that observed at endosomes. For example, whereas CXCL11 promoted robust GRK2 recruitment to CXCR3 at the plasma membrane, it did not produce any detectable recruitment of GRK2 at endosomes. Similarly, stimulation of the V2R promoted more GRK2 and GRK3 recruitment to the plasma membrane than did stimulation of the AT1R, but this pattern was reversed in endosomes. It is likely that the subcellular localization of the GRKs contributes to the ability of these GPCRs to initiate spatially distinct signaling patterns. Elucidation of the unique roles of individual GRK subtypes on overall GPCR signaling has received appreciable attention, in large measure due to the potential contribution of single GRKs to a wide range of pathophysiological processes. The dysregulation of GRK2 has been linked to the progression of mitochondrial lesions in Alzheimer’s disease, cardiovascular dysfunction, pathological angiogenesis, and chronic inflammation, among others (95–99).
Using biased ligands, we observed that G protein activation was a critical mechanism underlying the biased recruitment of GRKs to CXCR3 at the plasma membrane. These findings are consistent with previous reports that have demonstrated that GRK2 and GRK3 are recruited to GPCRs through their PH domain recognizing free Gβγ following G protein activation (26, 68, 74). However, using two different CRISPR/Cas9 GRK knockout cell lines, we found that GRK2 and GRK3 could be recruited to CXCR3 independently of G protein activation. Furthermore, the relative amount of G protein–independent GRK recruitment observed was dependent on the ligand, the specific GRK, and the cell type. It is possible that these observations are a consequence of using genetically modified HEK293 cells, but other work has demonstrated a similar phenomenon for the D2R, which can recruit GRK2 in the absence of G protein activation (77). We build upon this previous work, because our data suggest that a single GPCR may use both G protein–dependent and –independent mechanisms to recruit GRKs to the plasma membrane, depending on the specific ligand that activates the receptor.
This observation of G protein–independent GRK recruitment raises the question of whether this phenomenon has physiologic relevance. Although our data demonstrated that GRK recruitment robustly increased in the presence of cognate G proteins, we were still able to detect GRK recruitment using cells devoid of G proteins. Changes in the abundances of GRKs and G proteins are associated with some disease states. For example, GRK2 is present in almost three-fold higher amounts in the failing heart compared to the healthy heart, leading to enhanced activity in adrenergic receptor desensitization (100). Similarly, there are reports of decreased and increased heterotrimeric G proteins in disease states like asthma, myocardial infarction, and breast cancer (101). It is possible that G protein–independent mechanisms of GRK recruitment are present in both physiologic and pathophysiologic states, depending on the specific context of the cellular system. Further work is warranted to better understand these findings.
We explored the contributions of GRK subcellular localization to biased GPCR signaling by developing location-specific forms of GRKs and characterizing CXCR3 signaling using GRK knockout cell lines. We demonstrated that modifications to GRK cellular localization could alter the receptor’s ability to recruit β-arrestin 2, internalize, and activate ERK, depending on the specific ligand and GRK. Thus, GRK subtype functionality appears to be not only driven by distinctions between the proteins themselves, but also by unique properties conferred to each GRK by different GPCR ligands. We observed that expression of GRK2 and GRK3 in ΔGRK2/3 cells led to decreased β-arrestin 2 recruitment. These findings were unexpected considering canonical GPCR signaling suggests receptor phosphorylation by GRKs leads to increased β-arrestin recruitment. It is possible that GRK2 and GRK3 phosphorylate sites on CXCR3 that are inhibitory to β-arrestin recruitment or that β-arrestin recruitment to CXCR3 is primarily mediated through a GRK-independent process. Similarly, GRK5 and GRK6 (which are still present in ΔGRK2/3 cells) may promote arrestin recruitment for some agonist conditions, and GRK2 and GRK3 may compete with these kinases for the receptor. There is evidence that different GRKs can serve unique functions at a single GPCR (102, 103), and the specific role of GRKs 2, 3, 5 and 6 at CXCR3 similarly seem to be nonredundant.
We also observed an increase in CXCR3 internalization following expression of GRK6 in ΔGRK5/6 cells at most ligands (Fig. 4G). However, we did not observe any change in β-arrestin 2 recruitment under similar transfection conditions (fig. S11B). There are many possible explanations for these findings. Although β-arrestins are known to play a role in GPCR internalization through interactions with AP-2 and clathrin, there are β-arrestin–independent mechanisms of internalization as well (104–106). It is therefore likely that GPCR internalization is not always directly correlated with β-arrestin recruitment. GRKs could regulate GPCR internalization through mechanisms independent of β-arrestin recruitment, possibly by modulating β-arrestin conformation or receptor interactions with other effector proteins (107, 108). Previous work has demonstrated that different ligands can cause the same receptor to differentially engage the GRKs. For example, at the GPCR CCR7, the ligand CCL19 leads to GRK3- and GRK6-dependent receptor phosphorylation, whereas the ligand CCL21 only activates GRK6 (103). Similarly, at the AT1R, knockdown of GRK2 and GRK3 lead to decreased receptor phosphorylation, β-arrestin recruitment, and endocytosis, whereas knockdown of GRK5 and GRK6 abolish β-arrestin–mediated ERK activation (109). Additionally, we did not investigate the role of β-arrestin 1 in this system, and it is possible that the GRKs may have different effects on β-arrestin 1 recruitment and functionality as compared to β-arrestin 2, as others have recently reported (86, 107). Further insight is needed into understanding how the GRKs modulate receptor internalization and signaling through β-arrestin–dependent, and possibly β-arrestin–independent, mechanisms.
Our data demonstrated that the GRK location and ligand identify both impacted the ability of a GPCR to modulate ERK activity. We also did not expect to find that the regulation of ERK activation also depended on which subcellular pool of ERK was being investigated (cytosolic versus nuclear ERK). There is burgeoning evidence demonstrating that many GPCR second messengers or effector proteins including cAMP (110), ERK (45, 111), JNK (112), and G proteins (113) exhibit location-specific effects. However, the physiologic importance and molecular determinants of this phenomenon are incompletely understood. It will be important to determine the functional consequences of location-specific GPCR signaling and how GRKs modulate and contribute to this activity.
Receptor internalization and recycling were initially considered a mechanism of signal attenuation and re-sensitization (114, 115). Specifically, β-arrestins sterically inhibit GPCRs to prevent further G protein activation while simultaneously promoting receptor internalization to prevent further ligand binding. However, subsequent studies have demonstrated that many GPCRs can continue to signal through both G proteins and β-arrestins from endosomes. Previous work on the β2AR showed that endosomal cAMP production is required to generate a complete transcriptional response (39). Subsequent work has shown that signaling from endosomes also impacts the global phosphoproteome (38). Many studies support the paradigm of location bias, and it is now appreciated that endocytosis does not solely serve as a mechanism to attenuate GPCR signaling; rather, it also can be a critical component to achieving maximal GPCR signaling (36, 44, 116–118). Additionally, prior work demonstrates the therapeutic potential of pharmacologically targeting signaling from endosomes by the neurokinin 1 receptor, MOR, and other GPCRs (119–122). Recent studies have observed that a specific ligand:GPCR complex can activate signaling pathways from endosomes that are distinct from those activated at the plasma membrane (37, 39, 118). It is likely that signaling from endosomes and other subcellular locations is one mechanism by which a GPCR can generate diverse signaling outputs in response to biased ligands. We previously demonstrated that the biased agonists of CXCR3 demonstrate different relative amounts of G protein and β-arrestin signaling at the plasma membrane and endosome, providing evidence that the degree of biased agonism observed for a GPCR depends on where the signaling output is measured (45).
Notably, our data demonstrate that changes in a given readout of CXCR3 signaling were not necessarily predictive of the alterations for other signaling events canonically viewed as sequential. As mentioned above, although differential GRK6 localization had little effect on the ability of CXCR3 to engage β-arrestin 2, GRK6 increased CXCR3 internalization across all ligands. These results are consistent with GRKs demonstrating function beyond receptor phosphorylation and increasing the affinity of β-arrestin for a GPCR. Other studies demonstrate that the GRKs can modulate β-arrestin conformation, which is tightly associated with its ability to scaffold other signaling effectors (86, 108, 123, 124). Additionally, GRKs are known to phosphorylate other non-GPCR receptors, such as receptor tyrosine kinases and toll-like receptors, transcription factors, and more (23). GRKs are also known to have kinase-independent functionality. For example, a reduction in GRK2 leads to impairment in zebrafish development, but a kinase-dead mutant form of GRK2 restores normal development (125). This led to the discovery that GRK2 interacts with the cyclin B1 regulator patched homolog 1 (PTCH1) in manner that is independent of its kinase activity, and the interaction is necessary for normal embryogenesis (125). Similar work has demonstrated that GRK5 can inhibit nuclear factor κB (NF-κB) signaling by promoting the nuclear accumulation of the NF-κB inhibitor IκBα independently of its catalytic activity (126).
Biased agonism at GPCRs is traditionally characterized by a ligand’s relative ability to activate G proteins and recruit β-arrestins. However, we now appreciate that there are many other dimensions of biased agonism including location bias, receptor bias, kinetic bias, and more (4). In this manuscript, we determined that VUF10661 was the only CXCR3 ligand to appreciably recruit GRKs to endosomes, even though all chemokines except CXCL9 could recruit GRKs to the plasma membrane. Although previous studies demonstrate that VUF10661 and VUF11418 have similar biased signaling profiles regarding G protein activation and β-arrestin recruitment (127), these ligands differed substantially in their abilities to recruit GRKs to the endosome. Notably, we did not observe a relationship between G protein activation, β-arrestin recruitment, or GRK recruitment to the plasma membrane and a ligand’s ability to recruit GRKs to the endosome. Together, our findings suggest another functionally distinct dimension of biased GPCR signaling, one that incorporates ligand and location bias.
Our findings highlight the importance of the GRKs in initiating biased responses at GPCRs and demonstrate the complexity of downstream signaling that can be achieved through a single GPCR. These data support the phosphorylation barcode hypothesis by demonstrating that differential GPCR engagement with the GRKs can generate multiple agonist-specific, signaling pathways. This complex signaling exists at different compartments within the cell. Together, our data demonstrate the therapeutic promise and simultaneous complexity of drugging GPCRs and GPCR effectors given the vast diversity of signaling that can be achieved within this receptor family. The molecular determinants that promote GRK translocation to endosomes are still unclear at this time, and the complex interconnection between ligand and GRK subtype bias has not been fully resolved. It will be important to determine the mechanisms by which individual GRKs modulate signaling at different locations. Deconvoluting the structural elements that exist between a GPCR and GRK are critical to understanding the biochemical basis for biased signaling observed across different cellular compartments.
MATERIALS AND METHODS
Bacterial Strains
XL-10 Gold ultracompetent E. coli (Agilent) were used to express all constructs used in this manuscript.
Cell Lines
Human Embryonic Kidney (HEK293) cells were grown in minimum essential media (MEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C and 5% CO2. ΔGRK2/3, ΔGRK5/6, ΔG6, and ΔG7 CRISPR/Cas9 KO HEK293 cells were provided by Asuka Inoue, Tohoku University, Japan, and validated as previously described (Table S1) (55, 79, 82).
Generation of Constructs
Construct cloning was performed using conventional techniques such as restriction enzyme/ligation methods. Linkers between the fluorescent proteins or luciferases and the cDNAs for receptors, transducers, or other proteins were flexible and ranged between 2 and 18 amino acids. Fluorescence resonance energy transfer (FRET) based ERK1/2 biosensors previously published (89) were used to generate BRET versions of these sensors by removing the N-terminal mCerulean through restriction digest and inserting a nanoluciferase. We utilized the 2x-Fyve targeting sequence (RKHHCRACG) from the hepatocyte growth factor-regulated tyrosine kinase substrate to target labeled constructs to endosomes. We utilized the membrane targeting sequence (MGCIKSKGKDS) from Lyn kinase to target labeled constructs to the plasma membrane. Location tags were attached to the N-terminus of all constructs, followed by a flexible amino acid linker. CD8α-smBiT and CD8α-mCherry were cloned using human CD8α isoform 1. Human EEa1-dsRed was a gift from the Nina Tsvetanova Laboratory. Bovine GRK2, human GRK3 Isoform 1, human GRK5, and human GRK6 Isoform B were used for all confocal microscopy experiments (GRK-YFP), and all experiments involving expression of either the WT or location-specific mutant GRKs (WT-GRK, Mem-GRK, Endo-GRK). GRK-LgBiT constructs were provide by Asuka Inoue (55). Specifically, we used human GRK2-LgBiT that is codon optimized, human GRK3-LgBiT that is codon optimized, human GRK5-LgBiT, and human GRK6 Isoform B-LgBiT. Human Gαi1 was used in recue experiments in ΔG7 cells. Rat β-arrestin 2-mKO was used for recruitment BRET assays. Human CXCR3A, AT1R, V2R isoform 1, B2AR, MOR, and ACKR3 were used in this study.
Cell Culture and Transfection
For luminescence-based assays, HEK293 cells were transiently transfected with an optimized calcium phosphate protocol as previously described unless otherwise indicated (77). In the calcium phosphate transfection method, cell culture media was replaced 30 minutes prior to transfection. Plasmid constructs were suspended in water to a final volume of 90μL. 10μL of 2.5 M calcium chloride was added to the plasmid constructs and mixed. 100μL of 2x HEPES-buffered saline solution (10mM D-Glucose, 40mM HEPES, 10 mM potassium chloride, 270 mM sodium chloride, 1.5 mM disodium hydrogen phosphate dihydrate) was added to the solution, allowed to incubate for two minutes, and subsequently added to the cells. For BRET based assays, luminescence-based assays in G protein KO cell lines, and confocal microscopy, cells were transiently transfected using polyethylenimine (PEI). In the PEI transfection method, cell culture media was replaced 30 minutes prior to transfection. Plasmid constructs were suspended in Opti-MEM (Gibco) to a final volume of 100μL and, in a separate tube, PEI at a concentration of 1 mg/mL was added to Opti-MEM to a final volume of 100μL. For experiments in this manuscript, 3μL of PEI was used per 1μg of plasmid DNA. After 5 minutes, the 100μL PEI solution was added to the 100μL DNA solution, gently mixed, and allowed to incubate at room temperature for 10–15 minutes, after which the mixture was added to the cells.
BRET and Split Luciferase Assays
For all BRET and Split Luciferase assays, HEK293 cells seeded in 6 well plates (~750,000 cells/well) were transiently transfected with the appropriate constructs using the calcium phosphate or PEI method previously described. GRK recruitment was assessed using a NanoBiT complementation assay, where GRK2, GRK3, GRK5, or GRK6 were tagged with a C-terminal LgBiT and transfected with either CXCR3-smBiT (receptor), wild-type CXCR3 and 2x-Fyve-SmBiT (endosome), or wild-type CXCR3 and CD8α-SmBiT (plasma membrane). The role of G protein activation in GRK2/3 recruitment was assessed using PEI transfection of CXCR3-SmBiT and the indicated GRK-LgBiT, with rescue of Gαi1 in G7 GKO cell lines and pertussis toxin (PTX) treatment at a final concentration of 200ng/mL in G6 GKO cells.
To examine role of GRK subcellular localization on effector engagement and downstream signaling, we assessed β-arrestin 2 recruitment and CXCR3 internalization using the wild-type GRKs, membrane-bound GRK2 or 3 constructs, or endosome-bound GRK2, GRK3, GRK5, or GRK6. Rescue of GRK2 or GRK3 was performed in GRK2/3 KO cells while rescue of GRK5 or GRK6 was performed in GRK 5/6 KO cells. For β-arrestin 2 recruitment assays, cells were transfected with the indicated WT- or mutant GRK, CXCR3-RLuc2, and β-arrestin 2-mKO. CXCR3 internalization assays used the indicated WT- or mutant GRK with CXCR3-RLuc2 and a 2x-Fyve-mVenus to assess proximity to the early endosome.
Location-specific BRET-biosensors of ERK activity were transfected using PEI. This ERK biosensor consists of a target ERK substrate that, following phosphorylation by activated phosphorylated ERK, binds to a phosphorylation binding domain, causing a conformational change in the biosensor and subsequent change in BRET efficiency (45, 89).
Twenty-four hours after transfection, cells were washed with phosphate buffered saline (PBS), collected with trypsin, and plated onto clear-bottomed, white-walled, Costar 96-well plates at 50,000 to 100,000 cells/well in BRET medium (clear minimum essential medium (Gibco) supplemented with 2% FBS, 10 mM HEPES, 1x GlutaMax (Gibco), and 1x Antibiotic-Antimycotic (Gibco)).
The following day, media was removed, and cells were incubated at 37°C with 80μL of HBSS supplemented with 20mM HEPES, and 3μM coelenterazine h for all BRET or NanoBiT complementation assays (Cayman Chemical, Ann Arbor, MI and Nanolight Technology, Pinetop, AZ) for 10 to 15 minutes. For all BRET assays, a standard 480nm RLuc emission filter and 530nm (for experiment using mVenus) or custom 542-nm (for experiments using mKO) long pass filter was utilized (Chroma Technology Co., Bellows Falls, VT). Cells were stimulated with either vehicle control (HBSS with 20 mM HEPES) or the indicated concentration of ligand.
For NanoBiT complementation and BRET experiments, three initial reads were taken prior to the addition of ligand to quantify baseline luminescence or BRET signal before adding ligand. The change in luminescence after ligand stimulation was subsequently normalized to vehicle treatment. For BRET experiments, the BRET ratio was calculated by dividing the acceptor signal by the luminescent signal, and a net BRET ratio was calculated by normalizing to vehicle treatment. Plates were read with a BioTek Synergy Neo2 plate reader or Berthold Mithras LB 940 set at 37°C. All readings were performed using a kinetic protocol.
Confocal Microscopy
HEK293 cells were plated on 35 mm glass bottomed dishes (MatTek Corporation, Ashland, MA) and transiently transfected using PEI with the listed constructs. To evaluate the baseline expression of wild-type GRK2-YFP, GRK3-YFP, GRK5-YFP, and GRK6-YFP, or the Lyn-tagged or 2xFyve-tagged indicated GRK-YFP, cells were imaged forty-eight hours after transfection. To assess ligand-induced GRK localization, forty-eight hours after transfection, the cells were washed once with PBS and then serum starved for one hour. The cells were subsequently treated with a control of serum free media or the listed chemokine at 100nM or listed VUF compound at 10μM for the indicated time at 37°C. Cells were then imaged with a Zeiss CSU-X1 spinning disk confocal microscope using the corresponding lasers for YFP (480nm excitation), mCerulean (433nm excitation), dsRed (561nm excitation). Images were analyzed using ImageJ (NIH, Bethesda, MD) and false colors were applied for clarity.
Immunoblotting
Immunoblotting was performed as described previously (51). ΔGRK2/3 and ΔGRK5/6 cells seeded in 6 well plates were transiently transfected with increasing amounts of GRK using the calcium phosphate transfection method. 24 hours after transfection, cells were serum starved in minimum essential medium supplemented with 0.01% bovine serum albumin (BSA) and 1% penicillin/streptomycin for 16 hours. The cells were then washed with ice cold PBS and lysed in ice cold RIPA buffer supplemented with protease inhibitors (cOmplete EDTA free (Sigma)). The samples were rotated at 4°C for forty-five minutes and cleared of insoluble debris by centrifugation at 17,000g at 4°C for 15 minutes, after which the supernatant was collected. Protein was resolved on SDS-10% polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with the indicated primary antibody overnight at 4°C. GRK2 (Santa Cruz, #sc-13143), GRK3 (Cell Signaling Technology, #80362), GRK5 (Santa Cruz, #sc-518005), and GRK6 (Cell Signaling Technology, #5878) antibodies were used to compare rescued GRK expression level in ΔGRK2/3 and ΔGRK5/6 cells to GRK expression levels WT HEK293 cells (128). Immunoblots were normalized using an alpha-tubulin antibody (Sigma, #T5168). Horseradish peroxidase-conjugated anti-rabbit-IgG or anti-mouse-IgG were used as secondary antibodies. The nitrocellulose membranes were imaged by SuperSignal enhanced chemiluminescent substrate (Thermo Fisher) using a ChemiDoc MP Imaging System (Bio-Rad). To determine the amount of GRK to rescue to match the expression levels in wild-type HEK293 cells, we performed a linear regression analysis of the tubulin normalized signal intensity versus the amount of GRK transfected. We then used these derived equations to solve for the amount of GRK plasmid needed to generate a western blot signal comparable to that seen in the wild-type HEK293 cells.
CXCR3 Ligands
Recombinant Human CXCL9, CXCL10, and CXCL11 (PeproTech) were diluted according to the manufacturer’s specifications, and aliquots were stored at −80°C until needed for use. VUF10661 (Sigma-Aldrich) and VUF11418 (Aobius) were reconstituted in dimethyl sulfoxide (DMSO) and stored at −20°C in a desiccator cabinet.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed in Excel (Microsoft, Redmond, WA) and graphed in Prism 9.0 (GraphPad, San Diego, CA). Dose-response curves were fitted to a log agonist versus stimulus with three parameters (span, baseline, and EC50), with the minimum baseline corrected to zero using Prism 9.0. Statistical tests were performed using a one or two-way ANOVA followed by Tukey’s multiple comparison’s test when comparing treatment conditions. When comparing ligands or treatment conditions in concentration-response assays or time-response assays, a two-way ANOVA of ligand and concentration or ligand and AUC, respectively, was conducted. For experiments using mutant GRKs, a two-way ANOVA was performed followed by Tukey’s multiple comparison’s test when comparing transfection conditions within a ligand. If a significant interaction effect was observed (P < 0.05), then comparative two-way ANOVAs between individual experimental conditions were performed. Further details of statistical analysis and replicates are included in the figure legends. Lines represent the mean, and error bars signify the SEM, unless otherwise noted. Experiments were not randomized, and investigators were not blinded to treatment conditions. Critical plate-based experiments were independently replicated by at least two different investigators when feasible.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Asuka Inoue (Tohoku University, Japan), Robert J. Lefkowitz (Duke University, USA), Christopher Cole Honeycutt (National Institutes of Health, USA), and Noelia Boldizsar (National Institutes of Health, USA) for reagents, guidance, and thoughtful feedback throughout this work; N. Nazo for laboratory assistance. Graphical figures were created with BioRender.
FUNDING
Duke Cardiovascular Research Center Undergraduate Research Fellowship (C.H.)
Duke University Deans Summer Research Fellowship (C.H.)
National Institutes of Health grant T32GM007171 (D.S.E.)
Duke Medical Scientist Training Program (D.S.E.)
American Heart Association Predoctoral Fellowship 20PRE35120592 (D.S.E.)
National Institutes of Health grant 1R01GM122798 (S.R.)
Burroughs Wellcome Career Award for Medical Scientists (S.R.)
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
DATA AND MATERIALS AVAILABILITY
All data needed to evalulate the conclusions in the paper are present in the paper or the Supplementary Materials. All plasmids generated in this study will be distributed upon reasonable request and completion of a materials transfer agreement with Duke University. All data reported in this paper will be shared by the lead contact upon request.
REFERENCES AND NOTES
- 1.Bockaert J, Pin JP, Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J 18, 1723–1729 (1999); published online EpubApr 1 ( 10.1093/emboj/18.7.1723). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP, A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16, 19–34 (2017); published online EpubJan ( 10.1038/nrd.2016.230). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Devries ME, Skolnick J, Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol 2, e13 (2006); published online EpubFeb ( 10.1371/journal.pcbi.0020013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eiger DS, Pham U, Gardner J, Hicks C, Rajagopal S, GPCR systems pharmacology: a different perspective on the development of biased therapeutics. Am J Physiol Cell Physiol 322, C887–C895 (2022); published online EpubMay 1 ( 10.1152/ajpcell.00449.2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reiter E, Lefkowitz RJ, GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab 17, 159–165 (2006); published online EpubMay-Jun ( 10.1016/j.tem.2006.03.008). [DOI] [PubMed] [Google Scholar]
- 6.Eishingdrelo H, Kongsamut S, Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Curr Chem Genom Transl Med 7, 9–15 (2013) 10.2174/2213988501307010009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian X, Kang DS, Benovic JL, β-arrestins and G protein-coupled receptor trafficking. Handb Exp Pharmacol 219, 173–186 (2014) 10.1007/978-3-642-41199-1_9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caron MG, Barak LS, A Brief History of the beta-Arrestins. Methods Mol Biol 1957, 3–8 (2019) 10.1007/978-1-4939-9158-7_1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenakin T, Christopoulos A, Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12, 205–216 (2013); published online EpubMar ( 10.1038/nrd3954). [DOI] [PubMed] [Google Scholar]
- 10.Smith JS, Lefkowitz RJ, Rajagopal S, Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17, 243–260 (2018); published online Epub04 ( 10.1038/nrd.2017.229). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eiger DS, Boldizsar N, Honeycutt CC, Gardner J, Rajagopal S, Biased agonism at chemokine receptors. Cell Signal 78, 109862 (2021); published online EpubFeb ( 10.1016/j.cellsig.2020.109862). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z, Yang F, Zhang D, Liu Z, Lin A, Liu C, Xiao P, Yu X, Sun JP, Phosphorylation of G Protein-Coupled Receptors: From the Barcode Hypothesis to the Flute Model. Mol Pharmacol 92, 201–210 (2017); published online EpubSep ( 10.1124/mol.116.107839). [DOI] [PubMed] [Google Scholar]
- 13.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ, Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal 4, ra51 (2011); published online EpubAug 9 ( 10.1126/scisignal.2001707). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villen J, Haas W, Kovacs JJ, Shukla AK, Hara MR, Hernandez M, Lachmann A, Zhao S, Lin Y, Cheng Y, Mizuno K, Ma’ayan A, Gygi SP, Lefkowitz RJ, Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci U S A 107, 15299–15304 (2010); published online EpubAug 24 ( 10.1073/pnas.1008461107). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaya AI, Perry NA, Gurevich VV, Iverson TM, Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proceedings of the National Academy of Sciences 117, 14139–14149 (2020) 10.1073/pnas.1918736117). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latorraca NR, Masureel M, Hollingsworth SA, Heydenreich FM, Suomivuori CM, Brinton C, Townshend RJL, Bouvier M, Kobilka BK, Dror RO, How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 183, 1813–1825 e1818 (2020); published online EpubDec 23 ( 10.1016/j.cell.2020.11.014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dwivedi-Agnihotri H, Chaturvedi M, Baidya M, Stepniewski TM, Pandey S, Maharana J, Srivastava A, Caengprasath N, Hanyaloglu AC, Selent J, Shukla AK, Distinct phosphorylation sites in a prototypical GPCR differently orchestrate beta-arrestin interaction, trafficking, and signaling. Sci Adv 6, (2020); published online EpubSep ( 10.1126/sciadv.abb8368). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liggett SB, Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci Signal 4, pe36 (2011); published online EpubAug 9 ( 10.1126/scisignal.2002331). [DOI] [PubMed] [Google Scholar]
- 19.Premont RT, Macrae AD, Aparicio SA, Kendall HE, Welch JE, Lefkowitz RJ, The GRK4 subfamily of G protein-coupled receptor kinases. Alternative splicing, gene organization, and sequence conservation. J Biol Chem 274, 29381–29389 (1999); published online EpubOct 8 ( [DOI] [PubMed] [Google Scholar]
- 20.Lodowski DT, Tesmer VM, Benovic JL, Tesmer JJ, The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem 281, 16785–16793 (2006); published online EpubJun 16 ( 10.1074/jbc.M601327200). [DOI] [PubMed] [Google Scholar]
- 21.Packiriswamy N, Parameswaran N, G-protein-coupled receptor kinases in inflammation and disease. Genes Immun 16, 367–377 (2015); published online EpubSep ( 10.1038/gene.2015.26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaudhary PK, Kim S, The GRKs Reactome: Role in Cell Biology and Pathology. Int J Mol Sci 22, (2021); published online EpubMar 25 ( 10.3390/ijms22073375). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV, G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther 133, 40–69 (2012); published online EpubJan ( 10.1016/j.pharmthera.2011.08.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthees ESF, Haider RS, Hoffmann C, Drube J, Differential Regulation of GPCRs-Are GRK Expression Levels the Key? Front Cell Dev Biol 9, 687489 (2021) 10.3389/fcell.2021.687489). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin TY, Liberatore K, Cizin DM, Cohen BM, Vasan N, Ma Y, Krismer K, Robles JT, van de Kooij B, van Vlimmeren AE, Andree-Busch N, Kaufer NF, Dorovkov MV, Ryazanov AG, Takagi Y, Kastenhuber ER, Goncalves MD, Hopkins BD, Elemento O, Taatjes DJ, Maucuer A, Yamashita A, Degterev A, Uduman M, Lu J, Landry SD, Zhang B, Cossentino I, Linding R, Blenis J, Hornbeck PV, Turk BE, Yaffe MB, Cantley LC, An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766 (2023); published online EpubJan ( 10.1038/s41586-022-05575-3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Touhara K, Inglese J, Pitcher JA, Shaw G, Lefkowitz RJ, Binding of G protein beta gamma-subunits to pleckstrin homology domains. J Biol Chem 269, 10217–10220 (1994); published online EpubApr 8 ( [PubMed] [Google Scholar]
- 27.Thiyagarajan MM, Stracquatanio RP, Pronin AN, Evanko DS, Benovic JL, Wedegaertner PB, A predicted amphipathic helix mediates plasma membrane localization of GRK5. J Biol Chem 279, 17989–17995 (2004); published online EpubApr 23 ( 10.1074/jbc.M310738200). [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Benovic JL, Wedegaertner PB, Plasma membrane and nuclear localization of G protein coupled receptor kinase 6A. Mol Biol Cell 18, 2960–2969 (2007); published online EpubAug ( 10.1091/mbc.e07-01-0013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katritch V, Cherezov V, Stevens RC, Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33, 17–27 (2012); published online EpubJan ( 10.1016/j.tips.2011.09.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komolov KE, Benovic JL, G protein-coupled receptor kinases: Past, present and future. Cell Signal 41, 17–24 (2018); published online Epub01 ( 10.1016/j.cellsig.2017.07.004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doll C, Poll F, Peuker K, Loktev A, Gluck L, Schulz S, Deciphering micro-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol 167, 1259–1270 (2012); published online EpubNov ( 10.1111/j.1476-5381.2012.02080.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, Mistry S, Tobin AB, Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J Biol Chem 286, 11506–11518 (2011); published online EpubApr ( 10.1074/jbc.M110.154526). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin FT, Daaka Y, Lefkowitz RJ, beta-arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem 273, 31640–31643 (1998); published online EpubNov 27 ( [DOI] [PubMed] [Google Scholar]
- 34.Jensen DD, Lieu T, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Herenbrink CK, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, Geppetti P, Hicks GA, Christie MJ, Porter CJH, Canals M, Bunnett NW, Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med 9, (2017); published online EpubMay 31 ( 10.1126/scitranslmed.aal3447). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakamoto H, Matsuda K, Hosokawa K, Nishi M, Morris JF, Prossnitz ER, Kawata M, Expression of G protein-coupled receptor-30, a G protein-coupled membrane estrogen receptor, in oxytocin neurons of the rat paraventricular and supraoptic nuclei. Endocrinology 148, 5842–5850 (2007); published online EpubDec ( 10.1210/en.2007-0436). [DOI] [PubMed] [Google Scholar]
- 36.Irannejad R, Tsvetanova NG, Lobingier BT, von Zastrow M, Effects of endocytosis on receptor-mediated signaling. Curr Opin Cell Biol 35, 137–143 (2015); published online EpubAug ( 10.1016/j.ceb.2015.05.005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsvetanova NG, Irannejad R, von Zastrow M, G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem 290, 6689–6696 (2015); published online EpubMar 13 ( 10.1074/jbc.R114.617951). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsvetanova NG, Trester-Zedlitz M, Newton BW, Peng GE, Johnson JR, Jimenez-Morales D, Kurland AP, Krogan NJ, von Zastrow M, Endosomal cAMP production broadly impacts the cellular phosphoproteome. J Biol Chem 297, 100907 (2021); published online EpubJul ( 10.1016/j.jbc.2021.100907). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsvetanova NG, von Zastrow M, Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10, 1061–1065 (2014); published online EpubDec ( 10.1038/nchembio.1665). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ, Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7, e1000172 (2009); published online EpubAug ( 10.1371/journal.pbio.1000172). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calebiro D, Nikolaev VO, Persani L, Lohse MJ, Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci 31, 221–228 (2010); published online EpubMay ( 10.1016/j.tips.2010.02.002). [DOI] [PubMed] [Google Scholar]
- 42.Godbole A, Lyga S, Lohse MJ, Calebiro D, Internalized TSH receptors en route to the TGN induce local G(s)-protein signaling and gene transcription. Nat Commun 8, 443 (2017); published online EpubSep 5 ( 10.1038/s41467-017-00357-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyga S, Volpe S, Werthmann RC, Gotz K, Sungkaworn T, Lohse MJ, Calebiro D, Persistent cAMP Signaling by Internalized LH Receptors in Ovarian Follicles. Endocrinology 157, 1613–1621 (2016); published online EpubApr ( 10.1210/en.2015-1945). [DOI] [PubMed] [Google Scholar]
- 44.Vilardaga JP, Jean-Alphonse FG, Gardella TJ, Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10, 700–706 (2014); published online EpubSep ( 10.1038/nchembio.1611). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eiger DS, Boldizsar N, Honeycutt CC, Gardner J, Kirchner S, Hicks C, Choi I, Pham U, Zheng K, Warman A, Smith JS, Zhang JY, Rajagopal S, Location bias contributes to functionally selective responses of biased CXCR3 agonists. Nat Commun 13, 5846 (2022); published online EpubOct 4 ( 10.1038/s41467-022-33569-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Higuera I, Penela P, Murga C, Egea G, Bonay P, Benovic JL, Mayor F Jr., Association of the regulatory beta-adrenergic receptor kinase with rat liver microsomal membranes. J Biol Chem 269, 1348–1355 (1994); published online EpubJan 14 ( [PubMed] [Google Scholar]
- 47.Murga C, Ruiz-Gomez A, Garcia-Higuera I, Kim CM, Benovic JL, Mayor F Jr., High affinity binding of beta-adrenergic receptor kinase to microsomal membranes. Modulation of the activity of bound kinase by heterotrimeric G protein activation. J Biol Chem 271, 985–994 (1996); published online EpubJan 12 ( 10.1074/jbc.271.2.985). [DOI] [PubMed] [Google Scholar]
- 48.Bychkov E, Zurkovsky L, Garret MB, Ahmed MR, Gurevich EV, Distinct cellular and subcellular distributions of G protein-coupled receptor kinase and arrestin isoforms in the striatum. PLoS One 7, e48912 (2012) 10.1371/journal.pone.0048912). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed MR, Bychkov E, Gurevich VV, Benovic JL, Gurevich EV, Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by l-DOPA treatment. J Neurochem 104, 1622–1636 (2008); published online EpubMar ( 10.1111/j.1471-4159.2007.05104.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz-Gomez A, Mayor F Jr., Beta-adrenergic receptor kinase (GRK2) colocalizes with beta-adrenergic receptors during agonist-induced receptor internalization. J Biol Chem 272, 9601–9604 (1997); published online EpubApr 11 ( 10.1074/jbc.272.15.9601). [DOI] [PubMed] [Google Scholar]
- 51.Smith JS, Nicholson LT, Suwanpradid J, Glenn RA, Knape NM, Alagesan P, Gundry JN, Wehrman TS, Atwater AR, Gunn MD, MacLeod AS, Rajagopal S, Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation. Sci Signal 11, (2018); published online EpubNov 6 ( 10.1126/scisignal.aaq1075). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng K, Smith JS, Eiger DS, Warman A, Choi I, Honeycutt CC, Boldizsar N, Gundry JN, Pack TF, Inoue A, Caron MG, Rajagopal S, Biased agonists of the chemokine receptor CXCR3 differentially signal through Galphai:beta-arrestin complexes. Sci Signal 15, eabg5203 (2022); published online EpubMar 22 ( 10.1126/scisignal.abg5203). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colvin RA, Campanella GS, Manice LA, Luster AD, CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol 26, 5838–5849 (2006); published online EpubAug ( 10.1128/MCB.00556-06). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groom JR, Luster AD, CXCR3 in T cell function. Exp Cell Res 317, 620–631 (2011); published online EpubMar 10 ( 10.1016/j.yexcr.2010.12.017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawakami K, Yanagawa M, Hiratsuka S, Yoshida M, Ono Y, Hiroshima M, Ueda M, Aoki J, Sako Y, Inoue A, Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying beta-arrestin transducer bias. Nat Commun 13, 487 (2022); published online EpubJan 25 ( 10.1038/s41467-022-28056-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scholten DJ, Canals M, Wijtmans M, de Munnik S, Nguyen P, Verzijl D, de Esch IJ, Vischer HF, Smit MJ, Leurs R, Pharmacological characterization of a small-molecule agonist for the chemokine receptor CXCR3. Br J Pharmacol 166, 898–911 (2012); published online EpubJun ( 10.1111/j.1476-5381.2011.01648.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith JS, Nicholson LT, Suwanpradid J, Glenn RA, Knape NM, Alagesan P, Gundry JN, Wehrman TS, Atwater AR, Gunn MD, Macleod AS, Rajagopal S, Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation. Science Signaling 11, eaaq1075 (2018) 10.1126/scisignal.aaq1075). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stoffel RH, Randall RR, Premont RT, Lefkowitz RJ, Inglese J, Palmitoylation of G protein-coupled receptor kinase, GRK6. Lipid modification diversity in the GRK family. J Biol Chem 269, 27791–27794 (1994); published online EpubNov 11 ( [PubMed] [Google Scholar]
- 59.Premont RT, Inglese J, Lefkowitz RJ, Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J 9, 175–182 (1995); published online EpubFeb ( [DOI] [PubMed] [Google Scholar]
- 60.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ, Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. Science 300, 1256–1262 (2003); published online EpubMay 23 ( 10.1126/science.1082348300/5623/1256 [pii]). [DOI] [PubMed] [Google Scholar]
- 61.Jean-Alphonse F, Bowersox S, Chen S, Beard G, Puthenveedu MA, Hanyaloglu AC, Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem 289, 3960–3977 (2014); published online EpubFeb 14 ( 10.1074/jbc.M113.526350). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snyder JC, Rochelle LK, Ray C, Pack TF, Bock CB, Lubkov V, Lyerly HK, Waggoner AS, Barak LS, Caron MG, Inhibiting clathrin-mediated endocytosis of the leucine-rich G protein-coupled receptor-5 diminishes cell fitness. J Biol Chem 292, 7208–7222 (2017); published online EpubApr 28 ( 10.1074/jbc.M116.756635). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA, Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7, 12178 (2016); published online EpubJul 11 ( 10.1038/ncomms12178). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wright SC, Lukasheva V, Le Gouill C, Kobayashi H, Breton B, Mailhot-Larouche S, Blondel-Tepaz E, Antunes Vieira N, Costa-Neto C, Heroux M, Lambert NA, Parreiras ESLT, Bouvier M, BRET-based effector membrane translocation assay monitors GPCR-promoted and endocytosis-mediated Gq activation at early endosomes. Proc Natl Acad Sci U S A 118, (2021); published online EpubMay 18 ( 10.1073/pnas.2025846118). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Namkung Y, LeGouill C, Kumar S, Cao Y, Teixeira LB, Lukasheva V, Giubilaro J, Simoes SC, Longpre JM, Devost D, Hebert TE, Pineyro G, Leduc R, Costa-Neto CM, Bouvier M, Laporte SA, Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci Signal 11, (2018); published online EpubDec 4 ( 10.1126/scisignal.aat1631). [DOI] [PubMed] [Google Scholar]
- 66.Wright SC, Lukasheva V, Le Gouill C, Kobayashi H, Breton B, Mailhot-Larouche S, Blondel-Tepaz E, Antunes Vieira N, Costa-Neto C, Heroux M, Lambert NA, Parreiras ESLT, Bouvier M, BRET-based effector membrane translocation assay monitors GPCR-promoted and endocytosis-mediated G(q) activation at early endosomes. Proc Natl Acad Sci U S A 118, (2021); published online EpubMay 18 ( 10.1073/pnas.2025846118). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beautrait A, Paradis JS, Zimmerman B, Giubilaro J, Nikolajev L, Armando S, Kobayashi H, Yamani L, Namkung Y, Heydenreich FM, Khoury E, Audet M, Roux PP, Veprintsev DB, Laporte SA, Bouvier M, A new inhibitor of the beta-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat Commun 8, 15054 (2017); published online EpubApr 18 ( 10.1038/ncomms15054). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koch WJ, Inglese J, Stone WC, Lefkowitz RJ, The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J Biol Chem 268, 8256–8260 (1993); published online EpubApr 15 ( [PubMed] [Google Scholar]
- 69.Gurevich VV, Gurevich EV, GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front Pharmacol 10, 125 (2019) 10.3389/fphar.2019.00125). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Touhara K, Koch WJ, Hawes BE, Lefkowitz RJ, Mutational analysis of the pleckstrin homology domain of the beta-adrenergic receptor kinase. Differential effects on G beta gamma and phosphatidylinositol 4,5-bisphosphate binding. J Biol Chem 270, 17000–17005 (1995); published online EpubJul 14 ( 10.1074/jbc.270.28.17000). [DOI] [PubMed] [Google Scholar]
- 71.Zarca A, Perez C, van den Bor J, Bebelman JP, Heuninck J, de Jonker RJF, Durroux T, Vischer HF, Siderius M, Smit MJ, Differential Involvement of ACKR3 C-Tail in beta-Arrestin Recruitment, Trafficking and Internalization. Cells 10, (2021); published online EpubMar 11 ( 10.3390/cells10030618). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palczewski K, Buczylko J, Kaplan MW, Polans AS, Crabb JW, Mechanism of rhodopsin kinase activation. J Biol Chem 266, 12949–12955 (1991); published online EpubJul 15 ( [PubMed] [Google Scholar]
- 73.Premont RT, Koch WJ, Inglese J, Lefkowitz RJ, Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem 269, 6832–6841 (1994); published online EpubMar 4 ( [PubMed] [Google Scholar]
- 74.Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ, Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science 257, 1264–1267 (1992); published online EpubAug 28 ( 10.1126/science.1325672). [DOI] [PubMed] [Google Scholar]
- 75.Pitcher JA, Touhara K, Payne ES, Lefkowitz RJ, Pleckstrin homology domain-mediated membrane association and activation of the beta-adrenergic receptor kinase requires coordinate interaction with G beta gamma subunits and lipid. J Biol Chem 270, 11707–11710 (1995); published online EpubMay 19 ( 10.1074/jbc.270.20.11707). [DOI] [PubMed] [Google Scholar]
- 76.Fredericks ZL, Pitcher JA, Lefkowitz RJ, Identification of the G protein-coupled receptor kinase phosphorylation sites in the human beta2-adrenergic receptor. J Biol Chem 271, 13796–13803 (1996); published online EpubJun 7 ( 10.1074/jbc.271.23.13796). [DOI] [PubMed] [Google Scholar]
- 77.Pack TF, Orlen MI, Ray C, Peterson SM, Caron MG, The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J Biol Chem 293, 6161–6171 (2018); published online EpubApr 20 ( 10.1074/jbc.RA117.001300). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smit MJ, Verdijk P, van der Raaij-Helmer EM, Navis M, Hensbergen PJ, Leurs R, Tensen CP, CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 102, 1959–1965 (2003); published online EpubSep 15 ( 10.1182/blood-2002-12-3945). [DOI] [PubMed] [Google Scholar]
- 79.Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Ruttiger N, Ziegler N, Benkel T, Schmitt NK, Ishida S, Muller I, Reher R, Kawakami K, Inoue A, Rick U, Kuhl T, Imhof D, Aoki J, Konig GM, Hoffmann C, Gomeza J, Wess J, Kostenis E, Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 9, 341 (2018); published online EpubJan 23 ( 10.1038/s41467-017-02661-3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katada T, Ui M, ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J Biol Chem 257, 7210–7216 (1982); published online EpubJun 25 ( [PubMed] [Google Scholar]
- 81.Kurose H, Katada T, Amano T, Ui M, Specific uncoupling by islet-activating protein, pertussis toxin, of negative signal transduction via alpha-adrenergic, cholinergic, and opiate receptors in neuroblastoma x glioma hybrid cells. J Biol Chem 258, 4870–4875 (1983); published online EpubApr 25 ( [PubMed] [Google Scholar]
- 82.Hisano Y, Kono M, Cartier A, Engelbrecht E, Kano K, Kawakami K, Xiong Y, Piao W, Galvani S, Yanagida K, Kuo A, Ono Y, Ishida S, Aoki J, Proia RL, Bromberg JS, Inoue A, Hla T, Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J Exp Med 216, 1582–1598 (2019); published online EpubJul 1 ( 10.1084/jem.20181895). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Q, Plasencia M, Li Z, Mukherjee S, Patra D, Chen CL, Klose T, Yao XQ, Kossiakoff AA, Chang L, Andrews PC, Tesmer JJG, Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600–605 (2021); published online EpubJul ( 10.1038/s41586-021-03721-x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones Brunette AM, Sinha A, David L, Farrens DL, Evidence that the Rhodopsin Kinase (GRK1) N-Terminus and the Transducin Galpha C-Terminus Interact with the Same “Hydrophobic Patch” on Rhodopsin TM5. Biochemistry 55, 3123–3135 (2016); published online EpubJun 7 ( 10.1021/acs.biochem.6b00328). [DOI] [PubMed] [Google Scholar]
- 85.Moller TC, Pedersen MF, van Senten JR, Seiersen SD, Mathiesen JM, Bouvier M, Brauner-Osborne H, Dissecting the roles of GRK2 and GRK3 in mu-opioid receptor internalization and beta-arrestin2 recruitment using CRISPR/Cas9-edited HEK293 cells. Sci Rep 10, 17395 (2020); published online EpubOct 15 ( 10.1038/s41598-020-73674-0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Drube J, Haider RS, Matthees ESF, Reichel M, Zeiner J, Fritzwanker S, Ziegler C, Barz S, Klement L, Filor J, Weitzel V, Kliewer A, Miess-Tanneberg E, Kostenis E, Schulz S, Hoffmann C, GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat Commun 13, 540 (2022); published online EpubJan 27 ( 10.1038/s41467-022-28152-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ, Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem 279, 35518–35525 (2004); published online EpubAug 20 ( 10.1074/jbc.M405878200). [DOI] [PubMed] [Google Scholar]
- 88.Zheng H, Loh HH, Law PY, Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein-dependent ERK activation. Mol Pharmacol 73, 178–190 (2008); published online EpubJan ( 10.1124/mol.107.039842). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K, A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A 105, 19264–19269 (2008); published online EpubDec 9 ( 10.1073/pnas.0804598105). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheroutre H, Lambolez F, Doubting the TCR coreceptor function of CD8alphaalpha. Immunity 28, 149–159 (2008); published online EpubFeb ( 10.1016/j.immuni.2008.01.005). [DOI] [PubMed] [Google Scholar]
- 91.Cole DK, Laugel B, Clement M, Price DA, Wooldridge L, Sewell AK, The molecular determinants of CD8 co-receptor function. Immunology 137, 139–148 (2012); published online EpubOct ( 10.1111/j.1365-2567.2012.03625.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ, GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 166, 907–919 (2016); published online EpubAug 11 ( 10.1016/j.cell.2016.07.004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, Samaan F, Dandey VP, Tan YZ, Hong C, Mahoney JP, Triest S, Little J. t., Chen X, Sunahara R, Steyaert J, Molina H, Yu Z, des Georges A, Lefkowitz RJ, Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex. Nat Struct Mol Biol 26, 1123–1131 (2019); published online EpubDec ( 10.1038/s41594-019-0330-y). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cahill TJ, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J, Antar A, Blanc A, Qu CX, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun JP, Bouvier M, Skiniotis G, Lefkowitz RJ, Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A 114, 2562–2567 (2017); published online Epub03 ( 10.1073/pnas.1701529114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kavelaars A, Eijkelkamp N, Willemen HL, Wang H, Carbajal AG, Heijnen CJ, Microglial GRK2: a novel regulator of transition from acute to chronic pain. Brain Behav Immun 25, 1055–1060 (2011); published online EpubAug ( 10.1016/j.bbi.2011.03.019). [DOI] [PubMed] [Google Scholar]
- 96.Singhmar P, Huo X, Eijkelkamp N, Berciano SR, Baameur F, Mei FC, Zhu Y, Cheng X, Hawke D, Mayor F Jr., Murga C, Heijnen CJ, Kavelaars A, Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc Natl Acad Sci U S A 113, 3036–3041 (2016); published online EpubMar 15 ( 10.1073/pnas.1516036113). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kuai J, Han C, Wei W, Potential Regulatory Roles of GRK2 in Endothelial Cell Activity and Pathological Angiogenesis. Front Immunol 12, 698424 (2021) 10.3389/fimmu.2021.698424). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ, Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther 16, 302–307 (2008); published online EpubFeb ( 10.1038/sj.mt.6300371). [DOI] [PubMed] [Google Scholar]
- 99.Obrenovich ME, Smith MA, Siedlak SL, Chen SG, de la Torre JC, Perry G, Aliev G, Overexpression of GRK2 in Alzheimer disease and in a chronic hypoperfusion rat model is an early marker of brain mitochondrial lesions. Neurotox Res 10, 43–56 (2006); published online EpubAug ( 10.1007/BF03033333). [DOI] [PubMed] [Google Scholar]
- 100.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ, Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 87, 454–463 (1993); published online EpubFeb ( 10.1161/01.cir.87.2.454). [DOI] [PubMed] [Google Scholar]
- 101.Li J, Ge Y, Huang JX, Stromgaard K, Zhang X, Xiong XF, Heterotrimeric G Proteins as Therapeutic Targets in Drug Discovery. J Med Chem 63, 5013–5030 (2020); published online EpubMay 28 ( 10.1021/acs.jmedchem.9b01452). [DOI] [PubMed] [Google Scholar]
- 102.Nobles KN, Guan Z, Xiao K, Oas TG, Lefkowitz RJ, The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and −2. J Biol Chem 282, 21370–21381 (2007); published online EpubJul 20 ( 10.1074/jbc.M611483200). [DOI] [PubMed] [Google Scholar]
- 103.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ, Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci U S A 106, 9649–9654 (2009); published online EpubJun ( 10.1073/pnas.0904361106). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moo EV, van Senten JR, Brauner-Osborne H, Moller TC, Arrestin-Dependent and -Independent Internalization of G Protein-Coupled Receptors: Methods, Mechanisms, and Implications on Cell Signaling. Mol Pharmacol 99, 242–255 (2021); published online EpubApr ( 10.1124/molpharm.120.000192). [DOI] [PubMed] [Google Scholar]
- 105.Krilov L, Nguyen A, Miyazaki T, Unson CG, Williams R, Lee NH, Ceryak S, Bouscarel B, Dual mode of glucagon receptor internalization: role of PKCalpha, GRKs and beta-arrestins. Exp Cell Res 317, 2981–2994 (2011); published online EpubDec 10 ( 10.1016/j.yexcr.2011.10.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nishiyama K, Nishimura A, Shimoda K, Tanaka T, Kato Y, Shibata T, Tanaka H, Kurose H, Azuma YT, Ihara H, Kumagai Y, Akaike T, Eaton P, Uchida K, Nishida M, Redox-dependent internalization of the purinergic P2Y(6) receptor limits colitis progression. Sci Signal 15, eabj0644 (2022); published online EpubJan 11 ( 10.1126/scisignal.abj0644). [DOI] [PubMed] [Google Scholar]
- 107.Haider RS, Matthees ESF, Drube J, Reichel M, Zabel U, Inoue A, Chevigne A, Krasel C, Deupi X, Hoffmann C, beta-arrestin1 and 2 exhibit distinct phosphorylation-dependent conformations when coupling to the same GPCR in living cells. Nat Commun 13, 5638 (2022); published online EpubSep 26 ( 10.1038/s41467-022-33307-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J. t., Antar A, Blanc A, Qu CX, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun JP, Bouvier M, Skiniotis G, Lefkowitz RJ, Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A 114, 2562–2567 (2017); published online EpubMar 7 ( 10.1073/pnas.1701529114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ, Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A 102, 1442–1447 (2005); published online EpubFeb 1 ( 10.1073/pnas.0409532102). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anton SE, Kayser C, Maiellaro I, Nemec K, Moller J, Koschinski A, Zaccolo M, Annibale P, Falcke M, Lohse MJ, Bock A, Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell 185, 1130–1142 e1111 (2022); published online EpubMar 31 ( 10.1016/j.cell.2022.02.011). [DOI] [PubMed] [Google Scholar]
- 111.Kwon Y, Mehta S, Clark M, Walters G, Zhong Y, Lee HN, Sunahara RK, Zhang J, Non-canonical beta-adrenergic activation of ERK at endosomes. Nature 611, 173–179 (2022); published online EpubNov ( 10.1038/s41586-022-05343-3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ, Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 (2000); published online EpubNov 24 ( 10.1126/science.290.5496.1574). [DOI] [PubMed] [Google Scholar]
- 113.Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M, Endocytosis promotes rapid dopaminergic signaling. Neuron 71, 278–290 (2011); published online EpubJul 28 ( 10.1016/j.neuron.2011.05.036). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crilly SE, Puthenveedu MA, Compartmentalized GPCR Signaling from Intracellular Membranes. J Membr Biol 254, 259–271 (2021); published online EpubJun ( 10.1007/s00232-020-00158-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Calebiro D, Godbole A, Internalization of G-protein-coupled receptors: Implication in receptor function, physiology and diseases. Best Pract Res Clin Endocrinol Metab 32, 83–91 (2018); published online EpubApr ( 10.1016/j.beem.2018.01.004). [DOI] [PubMed] [Google Scholar]
- 116.Irannejad R, von Zastrow M, GPCR signaling along the endocytic pathway. Curr Opin Cell Biol 27, 109–116 (2014); published online EpubApr ( 10.1016/j.ceb.2013.10.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pavlos NJ, Friedman PA, GPCR Signaling and Trafficking: The Long and Short of It. Trends Endocrinol Metab 28, 213–226 (2017); published online EpubMar ( 10.1016/j.tem.2016.10.007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mohammad Nezhady MA, Rivera JC, Chemtob S, Location Bias as Emerging Paradigm in GPCR Biology and Drug Discovery. iScience 23, 101643 (2020); published online EpubOct 23 ( 10.1016/j.isci.2020.101643). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thomsen ARB, Jensen DD, Hicks GA, Bunnett NW, Therapeutic Targeting of Endosomal G-Protein-Coupled Receptors. Trends Pharmacol Sci 39, 879–891 (2018); published online EpubOct ( 10.1016/j.tips.2018.08.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mai QN, Shenoy P, Quach T, Retamal JS, Gondin AB, Yeatman HR, Aurelio L, Conner JW, Poole DP, Canals M, Nowell CJ, Graham B, Davis TP, Briddon SJ, Hill SJ, Porter CJH, Bunnett NW, Halls ML, Veldhuis NA, A lipid-anchored neurokinin 1 receptor antagonist prolongs pain relief by a three-pronged mechanism of action targeting the receptor at the plasma membrane and in endosomes. J Biol Chem 296, 100345 (2021); published online EpubJan-Jun ( 10.1016/j.jbc.2021.100345). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jimenez-Vargas NN, Yu Y, Jensen DD, Bok DD, Wisdom M, Latorre R, Lopez C, Jaramillo-Polanco JO, Degro C, Guzman-Rodriguez M, Tsang Q, Snow Z, Schmidt BL, Reed DE, Lomax AE, Margolis KG, Stein C, Bunnett NW, Vanner SJ, Agonist that activates the micro-opioid receptor in acidified microenvironments inhibits colitis pain without side effects. Gut 71, 695–704 (2022); published online EpubApr ( 10.1136/gutjnl-2021-324070). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Latorre R, Ramirez-Garcia PD, Hegron A, Grace JL, Retamal JS, Shenoy P, Tran M, Aurelio L, Flynn B, Poole DP, Klein-Cloud R, Jensen DD, Davis TP, Schmidt BL, Quinn JF, Whittaker MR, Veldhuis NA, Bunnett NW, Sustained endosomal release of a neurokinin-1 receptor antagonist from nanostars provides long-lasting relief of chronic pain. Biomaterials 285, 121536 (2022); published online EpubJun ( 10.1016/j.biomaterials.2022.121536). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shukla AK, Xiao K, Lefkowitz RJ, Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36, 457–469 (2011); published online EpubSep ( 10.1016/j.tibs.2011.06.003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peterson YK, Luttrell LM, The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev 69, 256–297 (2017); published online EpubJul ( 10.1124/pr.116.013367). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jiang X, Yang P, Ma L, Kinase activity-independent regulation of cyclin pathway by GRK2 is essential for zebrafish early development. Proc Natl Acad Sci U S A 106, 10183–10188 (2009); published online EpubJun 23 ( 10.1073/pnas.0812105106). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sorriento D, Ciccarelli M, Santulli G, Campanile A, Altobelli GG, Cimini V, Galasso G, Astone D, Piscione F, Pastore L, Trimarco B, Iaccarino G, The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc Natl Acad Sci U S A 105, 17818–17823 (2008); published online EpubNov 18 ( 10.1073/pnas.0804446105). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zheng K, Smith JS, Eiger DS, Warman A, Choi I, Honeycutt CC, Boldizsar N, Gundry JN, Pack TF, Inoue A, Caron MG, Rajagopal S, Biased agonists of the chemokine receptor CXCR3 differentially signal through Galpha(i):beta-arrestin complexes. Sci Signal 15, eabg5203 (2022); published online EpubMar 22 ( 10.1126/scisignal.abg5203). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reichel M, Weitzel V, Klement L, Hoffmann C, Drube J, Suitability of GRK Antibodies for Individual Detection and Quantification of GRK Isoforms in Western Blots. Int J Mol Sci 23, (2022); published online EpubJan 21 ( 10.3390/ijms23031195). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evalulate the conclusions in the paper are present in the paper or the Supplementary Materials. All plasmids generated in this study will be distributed upon reasonable request and completion of a materials transfer agreement with Duke University. All data reported in this paper will be shared by the lead contact upon request.