

Significance
Yeast species in the subphylum Saccharomycotina are crucial to research, industry, and human health, but very little is known about what governs their diversity and distributions in the wild. We addressed this by predicting range maps for 186 species representative of yeast biodiversity using machine learning. We uncovered several hotspots of yeast diversity in mixed montane forests. Unlike many other eukaryotes, yeast diversity was higher outside the tropics. Additionally, variables that traditionally scale with species richness, such as temperature and area, appeared to be uncorrelated in yeasts. Our predictions can be used to guide future sampling efforts and assess how yeast species are affected by past, present, and future climate scenarios.
Keywords: macroecology, fungi, AI, biogeography, latitudinal species gradient
Abstract
The Saccharomycotina yeasts (“yeasts” hereafter) are a fungal clade of scientific, economic, and medical significance. Yeasts are highly ecologically diverse, found across a broad range of environments in every biome and continent on earth; however, little is known about what rules govern the macroecology of yeast species and their range limits in the wild. Here, we trained machine learning models on 12,816 terrestrial occurrence records and 96 environmental variables to infer global distribution maps at ~1 km2 resolution for 186 yeast species (~15% of described species from 75% of orders) and to test environmental drivers of yeast biogeography and macroecology. We found that predicted yeast diversity hotspots occur in mixed montane forests in temperate climates. Diversity in vegetation type and topography were some of the greatest predictors of yeast species richness, suggesting that microhabitats and environmental clines are key to yeast diversity. We further found that range limits in yeasts are significantly influenced by carbon niche breadth and range overlap with other yeast species, with carbon specialists and species in high-diversity environments exhibiting reduced geographic ranges. Finally, yeasts contravene many long-standing macroecological principles, including the latitudinal diversity gradient, temperature-dependent species richness, and a positive relationship between latitude and range size (Rapoport’s rule). These results unveil how the environment governs the global diversity and distribution of species in the yeast subphylum. These high-resolution models of yeast species distributions will facilitate the prediction of economically relevant and emerging pathogenic species under current and future climate scenarios.
Saccharomycotina is a fungal subphylum as genetically diverse as plants and animals (1) that occurs across a broad range of environments and metabolic modalities (2). Saccharomycotina yeasts (sometimes called the “true” or “budding” yeasts) provide a plethora of crucial ecosystem functions, acting as mutualists, parasites, and decomposers (3). Some yeasts are used as biological pest control while others are pathogens of important crop species (4). This subphylum contains the genus Saccharomyces, whose members are responsible for baking, brewing, and winemaking industries, which total over a trillion-dollar annual market revenue (5–7). Along with the popular model organism Saccharomyces cerevisiae, other emerging yeast models, such as Komagataella (Pichia) pastoris, Lipomyces starkeyi, Yarrowia lipolytica, and Zygosaccharomyces spp., are being developed with applications for pharmaceuticals, biofuels, cosmetics, and other biotechnologies (8–12). Seven of 19 priority fungal pathogens (13) recently identified by the World Health Organization occur in Saccharomycotina. These include members of the polyphyletic genus Candida, which are responsible for over 400,000 life-threatening infections annually with 46 to 75% mortality (14).
Despite their relevance to science, technology, industry, and human health, very little is known about the natural distribution of yeast diversity and the factors that govern it (15). The pathogen Candida auris was only described in 2009 but has since been found in 30 countries globally within a decade for unknown reasons (16). The yeast Saccharomyces eubayanus, one of the parental species that gave rise to the lager brewing hybrid S. pastorianus, was identified in the wild in 2011 (17), and European populations were only discovered in 2022 (18). Fungi more generally have been traditionally excluded from macroecological studies and are notably absent from seminal studies on which current theory is based (19–21). What large-scale studies do exist are concentrated in soil fungi, where yeasts accounted for only 0.4% of species (22). While yeasts can be isolated from soil, their environmental range is far broader, and they are commonly found in a variety of substrates and microbiomes across plants, animals, and other fungi (23). Yeasts have been isolated from locations as diverse as sterile hospital environments (24) to penguin feces (25) and can metabolize alcohols, ketones, organic acids, and more (4). Due to the unique and exceptional diversity of their fundamental niche space, the macroecology of yeasts may differ significantly from other eukaryotic clades. To identify global patterns in yeast diversity and distributions, we used public records from environmental metabarcodes and individual isolate samples to predict distribution maps for 186 species and test drivers across 96 environmental variables.
Results and Discussion
The full filtered search (Methods) resulted in 22,443 Saccharomycotina occurrence records, representing every biome on earth and 49.7% of terrestrial ecoregions. Of the 77.3% of occurrences with substrate sampling information, the majority were collected from soil samples (66.8%), followed by plant stem (9.9%), and root (6.8%). To explore the global distribution of yeast species diversity, we used machine learning to infer the full geographic ranges of each described species with at least five unique occurrence records. Of these 233 species, 47 with a true positive or true negative rate less than 75% were removed, yielding a total of 186 species representing 9 of 12 Saccharomycotina orders (12) (SI Appendix, Fig. S1).
Taxonomic bias is known to confound geographic analyses of species richness (26). Care must be taken to ensure diversity hotspots are indeed areas of increased species richness, and not just areas of increased taxonomic scrutiny. To assess this possible bias, we compared the observed geographic species richness of the training data defined by the taxonomy used by this study (based on conventional taxonomic standards) to species hypotheses defined by the UNITE database (27) (based on genetic clustering). Diversity patterns were highly congruent globally between both taxonomies (P < 2.2E-16, r2 = 0.798) (SI Appendix, Fig. S2), which indicates that the species richness estimates used by this study reflect true biological patterns.
Sampling bias is another factor that can significantly influence biogeographic analyses. To address this, we performed additional analyses on 19,626 environmental samples provided to us from the GlobalFungi (28) database. Species richness for these analyses was defined as the number of unique species hypothesis barcodes per gram of soil sample as a way to ground our predictions in empirical observations. Large scale patterns were largely congruent between the two analyses (SI Appendix, Fig. S3 and Dataset S1). Additionally, the relationship between sampling density was much weaker for predicted diversity estimates (P = 6.5E-7, r2 = 0.028) than empirical observations in our training dataset (P = 2.2E-16, r2 = 0.402), demonstrating the power of the machine learning approach used by this study to disentangle meaningful phenomena from false signal produced by sampling artifacts (SI Appendix, Fig. S4).
Distribution maps predicted through machine learning revealed several distinct hotspots of yeast diversity (Fig. 1 and SI Appendix, Fig. S5A), particularly in temperate forests (Fig. 2). Of the 11 most species-rich ecoregions, all were extratropical forests. Eight were classified as mixed forests and another eight were montane, associated with mountain ranges such as the Alps, Pyrenees, Caucasus, and the Appalachians. This trend was consistent across all major clades (SI Appendix, Fig. S6). Mixed forests harbor the greatest higher-level taxonomic plant diversity, which is thought to contribute heavily to the biodiversity of other fungal groups like ectomycorrhizal mycobionts (29, 30). Similarly, montane ecosystems are known to be exceptionally diverse (31, 32), with radically different assemblages of plants and animals occurring in close proximity along elevational clines.
Fig. 1.

Global yeast diversity. (A) Heat map of the distributions of 186 yeast species inferred through random forest machine learning models. (B) Average species richness per grid cell for each latitude band or line.
Fig. 2.

Yeast species richness is concentrated in temperate, mixed forests. Average species richness per grid cell for each Köppen-Geiger climate class (A) and biome (B).
Predicted yeast species richness was highest in mixed, montane forests. To explore which environmental drivers contribute most to yeast diversity in these regions, regression models were performed for 96 variables (Dataset S2). The heterogeneity in vegetation and topography across montane mixed forests provides a plethora of microhabitats and ecological niches for yeasts to occupy, which may contribute to their high diversity in these environments. This hypothesis is supported by our environmental regression analysis. Two of the variables with 100% relative importance in predicting species richness are enhanced vegetation index diversity and the topography principal component (Fig. 3 and Dataset S3). Plant species richness and geomorphic class diversity were also highly significant, with 98 and 99% relative importance, respectively. By contrast, relative importance for the principal component including forest biomass was just 39%, and altitude was not significant at all [false discovery rate (FDR) = 0.33]. This result suggests that it is not the forests and mountainous regions per se that are conducive to yeast diversity, but rather the heterogeneity of hosts and landscapes these environments often provide. However, it is worth mentioning that species richness estimates are highly variable within predicted diversity hotspots, which increased sampling efforts may help resolve.
Fig. 3.

Traditional predictors of species diversity are poor indicators of yeast species diversity. (A) Variables that scale with diversity in other clades, such as tropical climates (Left), temperature (Center), and area (Right), did not scale with yeast species diversity. (B) Three select variables that were among the best predictors of yeast species diversity: temperate climates (Left), vegetation diversity (Center), and geomorphic class diversity (Right). All graphs represent the same regression analysis with the following summary statistics; FDR: false discovery rate of the negative binomial regression. m: scaled slope of linear regression. Black curves represent locally estimated scatterplot smoothing.
Yeast species show extensive variation in their species ranges. For example, Metschnikowia gruessii (order Serinales) was predicted to occur in just nine ecoregions while Kockiozyma suomiensis (order Lipomycetales) was predicted to occur in 338, covering over a quarter of the earth’s terrestrial surface (Fig. 4A). We tested three variables that are expected to influence species range size: niche breadth, species richness, and absolute latitude. Niche breadth was obtained from a recent study (2) that generated experimental growth curves across 18 carbon sources for every species in our dataset. We found that the number of different carbon sources a species was able to metabolize had a significant impact on range size (Fig. 4A). Carbon specialists, which can only grow on a limited number of carbon sources (2), had significantly (P = 0.02) smaller geographic ranges compared to nonspecialist species. Range size was also significantly negatively correlated with species richness (P ≈ 0) (Fig. 4B). Species who occupied environments with high numbers of other yeast species were more likely to have smaller ranges. Last, absolute latitude had a negative effect (P ≈ 0) on species range size such that yeast species occupying ecoregions closer to the equator had larger range sizes than more temperate species (Fig. 4C), with species ranges becoming smaller with distance from the equator.
Fig. 4.
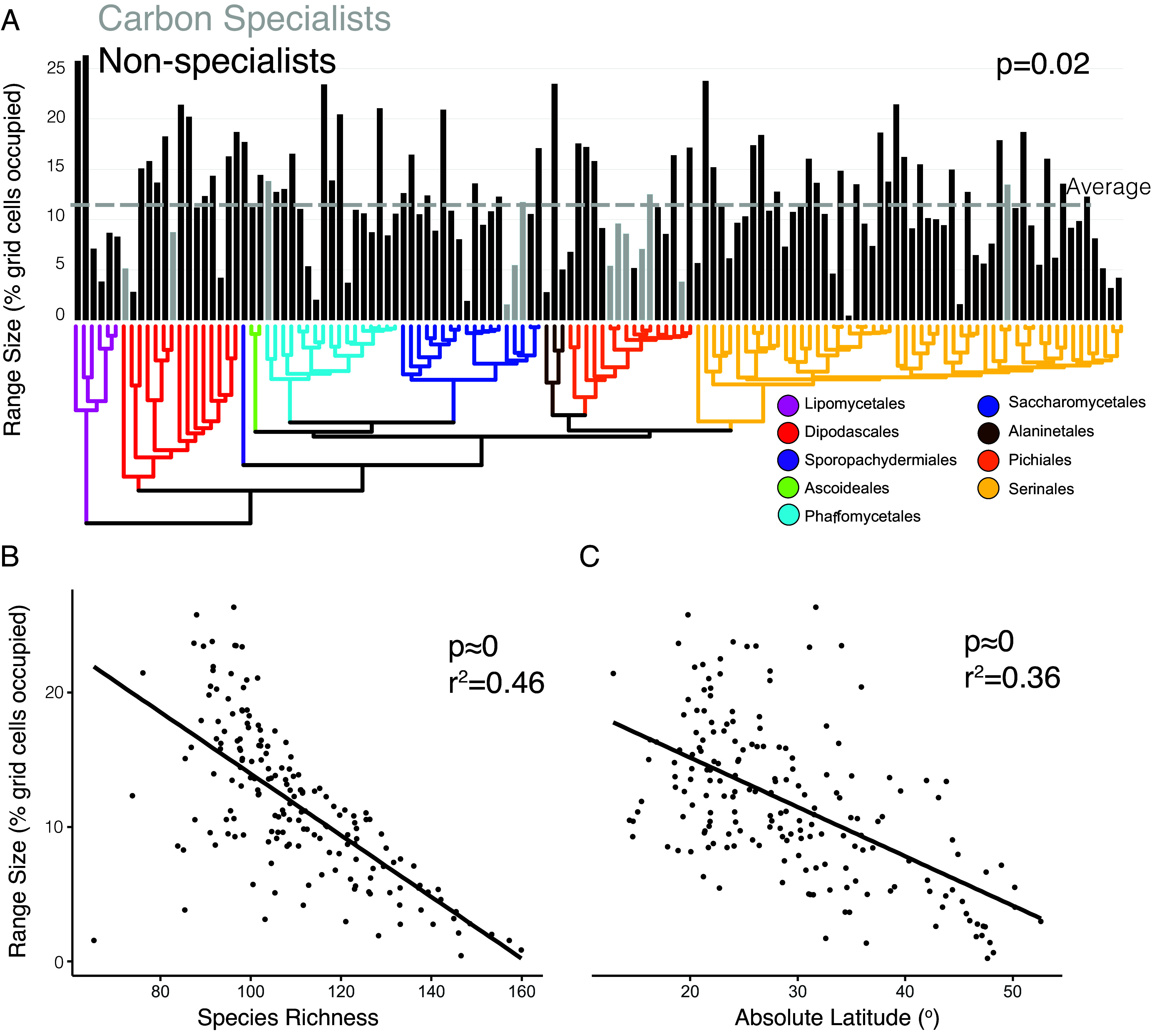
Yeast species range size scales negatively with species richness and latitude, positively with carbon niche breadth. (A) Specialist species that grew on only a few carbon sources had significantly smaller geographic ranges than nonspecialists. P-values represent a phylogenetic ANOVA test. Size of inferred ranges for each species included in this study, compared to (B) species richness and (C) absolute latitude. Summary statistics represented phylogenetic generalized least squares tests. All tests use the same underlying time-calibrated tree from Opulente et al. (2).
Yeast species ranges are negatively correlated with high species richness and positively correlated with carbon niche breadth, which may suggest that niche partitioning plays an important role in yeast biogeography. Species in high-diversity areas have restricted ranges, possibly implying that interspecific competition could limit geographic expansion. The limited range of specialists demonstrates that the fundamental niche space available to a species can have macroecological consequences. Positive relationships between range size and metabolic plasticity have also been observed in bacterial studies (33), further suggesting that niche breadth and range size are tightly linked in microbial taxa.
Comparisons of the macroecology of yeasts to other eukaryotic clades reveal several additional similarities. For example, richness peaks in montane forests (31, 32) and a positive association between niche breadth and range size (19, 34) are general patterns found in many other groups. Nevertheless, we also identified three major respects in which yeast macroecology deviates substantially from that of many other eukaryotic groups.
First, it is generally expected that species richness scales with available energy and resources, usually represented with proxy variables, such as area, temperature, or productivity (35). However, of these traditional predictors, only productivity emerged as a driver of yeast diversity. Net primary productivity (NPP) had a strong, significant relationship with species richness (FDR = 1.1E-57, m = 24.60) (Dataset S3). After highly correlated variables were decomposed into principal components (Methods), the resulting productivity principal component constructed from net primary productivity, growing season, and soil respiration was similarly predictive, with 100% relative importance (SI Appendix, Fig. S7 and Dataset S3). Notably, while the linear trend between productivity and richness was positive, the relationship more closely resembled the nonlinear humped-back model commonly reported in plants (36–38) (but see ref. 39), wherein species richness peaks at intermediate productivity but declines in either extremely high or extremely low productivity environments (SI Appendix, Fig. S7).
Neither temperature nor area had a positive effect on yeast species richness. Area size had a significant effect on richness (FDR = 6.8E-11, m = −4.92), but the hump-shaped relationship was weakly negative (Fig. 3). Mean annual air temperature had no significant relationship with yeast diversity (FDR = 0.078, m = 2.84). The temperature-associated principal component constructed from snow cover and energy from the sun was also insignificant (FDR = 0.23, m = −1.63). However, in our supplementary analysis of soil metabarcodes, temperature had a small but significant (FDR = 7.29E-33, m = 1.18) positive relationship with diversity (Dataset S1).
The metabolic theory of ecology predicts that biodiversity increases with both productivity and temperature. Productivity represents the amount of available resources and therefore the number of species a given environment can support. Temperature meanwhile represents the biochemical kinetic energy of an environment, positively influencing growth rates and metabolism in ectotherms and therefore generation times, mutation rates, and, hypothetically, speciation (40–42) (but see refs. 43 and 44). As productivity and temperature are tightly correlated, distinguishing between these hypotheses has historically been challenging. However, we find evidence only for the productivity hypothesis, recovering little support for the idea that temperature regulates species diversity in yeasts.
Temperature was previously identified as an important factor influencing the range of Saccharomyces species (45, 46), which has important implications as the ranges of many fungal pathogens are predicted to expand due to climate change (47). Our analysis suggests that this association between temperature and species range is also true throughout the subphylum since temperature range and temperature mean were the 7th and 9th most important continuous variables in our distribution models, respectively (Dataset S4). However, while temperature is an important determinant of yeast species distributions, it is likely not predictive of yeast species diversity globally.
Second, the latitudinal diversity gradient, or the tendency for species richness to peak in tropical climates, is arguably the most widely observed macroecological trend (20). We partially recover this trend; species richness exhibits a tropical peak due to elevated diversity from rainforests in South America, Africa, and Southeast Asian islands (Fig. 1). However, temperate regions held the most diversity with an average species richness of 73.6 species per grid cell, a value 2.6× higher than that of tropical regions (Fig. 2). Additionally, while temperate regions held significantly more richness per grid cell than nontemperate regions (FDR = 3.9E-13, m = 22.45), the species richness of tropical regions did not significantly differ from the richness of nontropical regions (FDR = 0.051, m = 5.38) (Fig. 3). Basidiomycete yeasts also exhibit an inverse latitudinal diversity gradient, but many other non-Saccharomycotina yeasts do not (22). In other fungal clades, the presence of an inverse latitudinal gradient on local scales has been attributed to negative relationships between fungal diversity and plant richness (48) or temperature (49) as potential drivers. However, as mentioned above, we found that yeast species diversity was positively correlated with plant species richness (FDR = 5.9E-43, m = 23.87) and uncorrelated with temperature (FDR = 0.078, m = 2.84).
The relative dearth of tropical diversity in certain fungal clades could also be due to historical biogeographical factors (29). In ectomycorrhizal fungi, for example, there are no known obligately tropical lineages (50) and most species are thought to have originated in temperate climates (51, 52). However, as has been reported in other clades (53, 54), diversity and diversification appear to be only weakly correlated in yeasts, suggesting that historical hotspots of diversification are not necessarily current hotspots of diversity (SI Appendix, Fig. S5). Additionally, variables tracking climate changes since the last glacial maximum were largely insignificant and had some of the smallest effect sizes measured. It is possible, that due to the short generation times and widespread dispersal capabilities of many yeast species (55, 56), historical processes that operate over thousands to millions of years have had minimal impact on modern distributions. Such a scenario may also help to explain the absence of a latitudinal diversity gradient. If yeast species can rapidly colonize and saturate environmental niches that were previously unavailable due to climate shifts or glacial cycles, it may explain why species richness is not concentrated in the more stable tropics.
Another explanation for the absence of a strong latitudinal gradient is water content. Both precipitation and moisture were strongly associated with species richness (relative importance >99%). However, these relationships appear to be logarithmic, scaling with richness until an inflection point after which additional water content appears to have little effect (SI Appendix, Fig. S8). This may explain why diversity peaks in both tropical and temperate climates (Fig. 1). Yeast diversity requires a certain baseline amount of water to be maintained, but does not benefit from any additional surplus past this threshold. A logarithmic relationship with species richness was also observed in other factors specific to the tropics like high temperatures and plant species richness (Datasets S2 and S3), which may suggest similar phenomena.
Third, Rapoport’s rule (57), or the positive relationship between species range size and latitude, was also found to be reversed in yeasts. As mentioned above, distance from the equator had a significant (P ≈ 0), negative relationship with species range size. Though the generality of Rapoport’s rule has been extensively questioned (58, 59), it has been identified as a major factor in the distribution of soil fungi, particularly in Agaricomycetes (22). Rapoport’s rule was originally postulated in order to explain the latitudinal diversity gradient since the smaller ranges of species in the tropics would enable more species to coexist. If Rapoport’s rule and latitudinal diversity gradients are indeed connected, it would explain the observed trend of both of them being inverted in yeasts. In microbes, metabolic niche theory predicts a positive relationship between temperature, niche breadth, and diversity (60, 61). We found evidence for a positive relationship between niche breadth and temperature (using latitude as a proxy, Fig. 4C), but diversity was uncorrelated with temperature (Fig. 3A) and negatively correlated with niche breadth (Fig. 4B).
In conclusion, we sought to uncover the global diversity and distribution of the Saccharomycotina yeasts. As single-celled organisms, the life history and lifestyle of yeasts are markedly different from many other eukaryotic clades. This divergence is reflected in their macroecology, which sets them apart from other fungi and even other yeasts (22). We did not find evidence of many commonly observed ecological patterns. Predicted yeast diversity is concentrated in temperate climates, not the tropics. Similarly, species range size decreases with distance from the equator, an inverse of Rapaport’s rule. Additionally, neither temperature, nor area, scale with species richness. These surprising findings emphasize the need in macroecology to study a variety of underexplored clades, especially those with unique life history traits.
The distribution models used by this study are reliant on environmental sampling. Wild yeasts are severely undersampled, which could influence the accuracy of our machine learning predictive models. Additionally, the majority of microbial sampling occurs in soil, whereas yeasts are expected to thrive across a wide range of different microhabitats and substrates (4). As such, much of yeast diversity, distribution, and life history remains obscured. Nevertheless, our estimates of yeast species richness are not strongly correlated with sampling density (SI Appendix, Fig. S4B); they are also consistent with current knowledge. For example, while biodiversity hotspots in Appalachia, Western/Southern Europe, and East Asia may appear at first glance to be artifacts from inflated research effort, they are also consistent with the known trend of yeast diversity peaks in temperate broadleaf and mixed forests (46), a biome common to all three regions. In fact, many diversity hotspots are actually undersampled in our dataset. The most heavily sampled ecoregion was the Samartic mixed forests in Eastern Europe, with 2.59× more sample sites in our training data than the Western European Broadleaf ecoregion, despite having 31.0% less estimated richness. Ecoregions around the Mediterranean and Black seas such as the north Turkish coast and montane forests along the Apennine and Rhodope mountain ranges were in the 98th percentile for yeast species richness, despite having zero samples in our training data. The Appalachian Mountains in the United States might similarly be an underappreciated biodiversity hotspot for yeasts (62), with species richness estimates rivaling that of western Europe despite having less than 3% of their sampling. For understudied and undersampled clades like the yeasts, employing a computational predictive framework, such as the one developed in this study, can guide future sampling efforts. Guiding both geographic and taxonomic sampling of this important clade toward specific poorly sampled ecoregions and substrates will likely greatly increase the resolution and power of future studies.
While the distribution patterns of yeast diversity are distinct from many other eukaryotes, the threats yeast face may be largely the same. We found that yeast diversity hotspots are characterized by temperate, montane, mixed forests. Notably, these ecosystems are some of the most impacted by human activities and climate change. Forests in central Europe, east Asia, and southwest Brazil, where yeast diversity is high, are dominated by secondary growth (63), having previously been disturbed by human activities. Similarly, montane environments are particularly impacted by climate change as communities shift upslope in response to rising temperatures, altering species ranges in the process (31, 64). As temperate ecosystems are forced to retreat to higher latitude and altitudes in a warming world, yeast diversity hotspots will need to adapt with them or face extinction. The methodology used by this study is readily adjustable to an array of future climate scenarios, and it may prove useful in assessing how yeast diversity, including economically relevant and pathogenic species, is affected by past, present, and future anthropogenic transformations.
Methods
Dataset.
To obtain a comprehensive record of Saccharomycotina biogeographic distribution, several data sources were queried. The majority (73%) of occurrence records were provided from the GlobalFungi (28) database (release 4). We also performed a query of all Saccharomycotina occurrence records without flagged geospatial issues from the Global Biodiversity Information Facility on December 14, 2022 (65), the details of which can be found at https://doi.org/10.15468/dl.n4fkqs. These data were further filtered by removing any record with a reported coordinate uncertainty of 1 km or greater. Saccharomycotina records were also taken from two published studies (15, 66). In Peris et al. (2022), records marked with the “anthropic” flag were removed, as this study is primarily interested in the diversity and distribution of naturally occurring yeasts. Similarly, the industrial hybrid species Saccharomyces bayanus and Saccharomyces pastorianus were excluded from analysis. Though now considered a naturally occurring species distinct from S. bayanus, Saccharomyces uvarum records were also removed as a conservative measure. Only known species were considered, as defined by The Yeasts Database (4), and species names were reconciled with the most recently published higher-level taxonomy (12) (Dataset S5). After synonymous species were merged, two additional filtering steps were applied. First, coordinate resolution needed to be at least two decimal places. Second, the R package CoordinateCleaner was employed to remove suspicious records, such as those with equal latitude and longitude coordinates, zero coordinates, or coordinates matching the centroid of counties/provinces or biodiversity institutions. A full record of all filtered coordinates can be found in Figshare (67) https://figshare.com/s/389e3f47e2d9f6ae242c.
Each occurrence record was associated with 96 environmental variables describing the climate, history, soil, vegetation, and anthropogenic inputs of the region. All variables were taken from publicly available sources and projected onto the WGS84 coordinate system at 30″ (~1 km2) resolution. Further details for each variable are available at Dataset S6. To avoid overfitting or overrepresentation of specific sampling sites in the training data, records with identical environmental variables of the same species within the same hundredth degree of latitude or longitude were aggregated into one. Finally, records with any missing data were also removed, resulting in a training dataset of 12,816 presences.
Species Distribution Modeling with Machine Learning.
To infer species occurrences in areas of limited sampling, machine learning random forest models were used. 233 models were constructed, one for every species with at least five occurrence records. 100,000 environmental data points were randomly sampled as pseudo-absences. Modeling was performed using the R package “randomforest.” A downsampling approach was used for training, which has been shown to reduce overfitting and significantly improve results in species distribution modeling (68). Each random forest model consisted of 100 decision trees. Otherwise, default parameters were used. A leave-one-out strategy was used for validation, and 186 models with at least a 75% true positive rate and 75% true negative rate were retained for downstream analysis. On average, models for these 186 species had an area under the receiver operating characteristic curve of 0.92, a true positive rate of 87%, and a true negative rate of 90% (SI Appendix, Fig. S9). Of the 96 environmental variables used in training, Köppen-Geiger climate classifications were the most predictive, followed by ecofloristic zones, biomes, and soil classifications. Together these four categorical variables represented almost a quarter of all variable importance, with 24.7% of the total mean decrease in Gini index across all variables. We also note that variables that are important for the binary classification task of random forest models are not necessarily those that are the most predictive of overall richness. For example, mean annual air temperature was the 9th most important continuous variable for distribution modeling but had an insignificant (FDR = 0.078) effect on richness. Conversely, geomorphic class diversity was the 3rd least important continuous variable for distribution modeling but had 100% relative importance to richness regressions.
Diversity and Diversification Estimation.
To reduce computational costs and to increase interpretability of results, terrestrial ecoregions were selected as the fundamental unit for environmental regression analysis. Ecoregions are defined by the World Wildlife Fund as “a large unit of land containing a geographically distinct assemblage of species, natural communities, and environmental conditions” (69). While environmental heterogeneity exists within ecoregions, they have been shown to accurately delimit biodiversity patterns in fungi (70). To accomplish this analysis, environmental variables and species richness estimates were aggregated into ecoregions. For the 90 continuous environmental variables in our training dataset, we simply took the mean value of all grid cells in a given ecoregion (Dataset S7). Select categorical variables were also encoded into six binary variables, which were based on the majority class within each ecoregion (Dataset S8). Species were said to be found in a particular ecoregion if they were predicted to occur in at least 10% of that ecoregion’s grid cells according to the random forest model. Speciation rates were inferred from the DR statistic (71, 72) calculated from the inverse equal splits method (73), using a recently published time-calibrated phylogeny (2). Ecoregion specific rates were calculated using a weighted mean of speciation rates for all species found in a given ecoregion. Weights represented the inverse of the number of ecoregions in which a given species occurred, such that species endemic to a specific ecoregion contributed more to that ecoregion’s estimate than a cosmopolitan species (72).
Environmental Analysis.
To determine environmental drivers of yeast diversity, regression models were constructed for each of the 96 quantitative variables, with yeast species richness as the dependent variable in each case. As species richness is always represented by a non-negative integer, negative binomial regressions were used, which are thought to be more appropriate for count data and, in practice, had consistently better Akaike information criterion scores than linear models. To increase interpretability of summary statistics, scaled linear regressions were also performed, taking the slope (m) as a measure of effect size. 16 variables whose negative binomial regressions had false discovery rates >0.05 were removed from downstream analysis. To reduce correlations between environmental variables, highly correlated variables were decomposed into single principal components. Effort was made to preserve the interpretation of principal components wherever possible. Each principal component explained at least 83% of the total variance (μ = 93%); further details can be found at Dataset S9. After highly correlated variables were decomposed, the greatest r2 between variables was 0.71 (μ = 0.11) (SI Appendix, Fig. S10). To estimate the contribution of the most predictive environmental variables and principal components, relative importance analysis was used. Negative binomial regression models were constructed from every combination of the 16 variables and principal components whose linear relationship with species richness had r2 > 0.15 and m > 0.20; species richness was the dependent variable. This strategy resulted in 65,535 total models. Akaike weights were then calculated and used to estimate relative importance for each predictor (74).
Species Range Size Analysis.
Several estimates were measured to test drivers of species range size. Species range size itself was estimated as the total fraction of grid cells predicted to be occupied by a given species. Latitude and species range overlaps were estimated for each species as the average value across every ecoregion in which a given species was predicted to occur (Dataset S10). Carbon niche-breadth classifications of specialists were taken from Opulente et al. 2023 (2), which inferred niche-breadth through experimental quantitative growth assays on 18 carbon sources. The positive relationship between niche-breadth and geographic range size has been identified as a major macroecological pattern in plants and animals (19, 21). However, this consensus has also attracted controversy for two main reasons. First, niche-breadth is a broadly defined concept often measured along multiple axes, such as diet, habitat, and tolerance, which are not necessarily correlated (19). Second, as range size and niche-breadth are typically inferred from the same underlying data (occurrence records), sampling artifacts can produce spurious correlations (34, 75, 76). The yeast dataset utilized by this study circumvents both these issues. The external absorption mode of feeding in yeasts (77) means that diet and habitat are one and the same, providing a convenient and unique lens through which to measure niche-breadth. Additionally, as this study defines niche-breadth independently through experimental growth assays conducted in a laboratory (2), there is no autocorrelation between niche-breadth and range size. Associations between species range size and diversity/latitude were tested with phylogenetic generalized least squares models implemented in the R package nlme (78) and niche breadth using phylogenetic ANOVAs implemented in the package Geiger (79).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Acknowledgments
We thank members of the Rokas Lab and the Y1000+ Project team for helpful discussions throughout the duration of this project. We would also like to thank the Society for the Protection of Underground Networks (SPUN) and the GlobalFungi database for their assistance, in particular Toby Kiers, Michael Van Nuland, Anastasiia Gromyko, and Tomas Vetrovsky. This work was performed using resources contained within the Advanced Computing Center for research and Education at Vanderbilt University in Nashville, TN. This work was partially supported by the NSF (grants DBI-1906759 to K.T.D., DEB-2110403 to C.T.H., DEB-2110404 to A.R.). X.-X.S. was supported by the NSF for Distinguished Young Scholars of Zhejiang Province (LR23C140001) and the Fundamental Research Funds for the Central Universities (226-2023-00021). M.P. is supported by award R35GM151348 from the NIGMS. Research in the Hittinger Lab is also supported by the USDA National Institute of Food and Agriculture (Hatch Projects 1020204 and 7005101), in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE–SC0018409, and an H. I. Romnes Faculty Fellowship (Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation). Research in the Rokas lab is also supported by the NIH/National Institute of Allergy and Infectious Diseases (R01 AI153356) and the Burroughs Welcome Fund.
Author contributions
K.T.D. and A.R. designed research; K.T.D. performed research; K.T.D., M.-C.H., D.A.O., A.L.L., J.F.W., X.Z., X.-X.S., M.G., M.P., C.T.H., and A.R. contributed new reagents/analytic tools; K.T.D. analyzed data; K.T.D. and A.R. wrote the paper; and all authors provided comments and edits to the paper.
Competing interests
A.R. is a scientific consultant of LifeMine Therapeutics, Inc. The authors declare no other competing interests.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All code required to run the species distribution models presented in this paper and replicate primary analyses as well as supplementary data files, including distribution maps and raster files for all 186 species, have been deposited online and will be made publicly accessible upon publication. Readers may access the code repository at https://github.com/KyleTDavid/YeastMacroecology2023 (80) and data files at https://figshare.com/articles/dataset/Yeast_Macroecology_2023/25145819 (67).
Supporting Information
References
- 1.Shen X.-X., et al. , Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175, 1533–1545.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opulente D. A., et al. , Genomic and ecological factors shaping specialism and generalism across an entire subphylum. BioRxiv [Preprint] (2023). 10.1101/2023.06.19.545611 (Accessed 8 September 2023). [DOI]
- 3.Starmer W. T., Lachance M.-A., “Yeast ecology” in The yeasts (2011), pp. 65–83. [Google Scholar]
- 4.Kurtzman C. P., Fell J. W., Boekhout T., The Yeasts: A Taxonomic Study (Elsevier, 2011). [Google Scholar]
- 5.Bread–Worldwide | Statista Market Forecast, Statista. https://www.statista.com/outlook/cmo/food/bread-cereal-products/bread/worldwide. Accessed 8 November 2023. [Google Scholar]
- 6.Beer–Worldwide | Statista Market Forecast, Statista. https://www.statista.com/outlook/cmo/alcoholic-drinks/beer/worldwide. Accessed 8 November 2023. [Google Scholar]
- 7.Wine–Worldwide | Statista Market Forecast, Statista. https://www.statista.com/outlook/cmo/alcoholic-drinks/wine/worldwide. Accessed 8 November 2023. [Google Scholar]
- 8.Spagnuolo M., Yaguchi A., Blenner M., Oleaginous yeast for biofuel and oleochemical production. Curr. Opin. Biotechnol. 57, 73–81 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Gassler T., et al. , The industrial yeast Pichia pastoris is converted from a heterotroph into an autotroph capable of growth on CO2. Nat. Biotechnol. 38, 210–216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan P., Smolke C. D., Biosynthesis of medicinal tropane alkaloids in yeast. Nature 585, 614–619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solieri L., The revenge of Zygosaccharomyces yeasts in food biotechnology and applied microbiology. World J. Microbiol. Biotechnol. 37, 96 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Groenewald M., et al. , A genome-informed higher rank classification of the biotechnologically important fungal subphylum Saccharomycotina. Stud. Mycol. 105, 1–22 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coordination G., Alastruey-Izquierdo A., “WHO fungal priority pathogens list to guide research, development and public health action” (Organización Mundial de la Salud, 2022). [Google Scholar]
- 14.Brown G. D., et al. , Hidden killers: Human fungal infections. Sci. Trans. Med. 4, 165rv13 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Spurley W. J., et al. , Substrate, temperature, and geographical patterns among nearly 2000 natural yeast isolates. Yeast 39, 55–68 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhodes J., Fisher M. C., Global epidemiology of emerging Candida auris. Curr. Opin. Microbiol. 52, 84–89 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Libkind D., et al. , Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc. Natl. Acad. Sci. U.S.A. 108, 14539–14544 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergin S. A., et al. , Identification of European isolates of the lager yeast parent Saccharomyces eubayanus. FEMS Yeast Res. 22, foac053 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slatyer R. A., Hirst M., Sexton J. P., Niche breadth predicts geographical range size: A general ecological pattern. Ecology Letters 16, 1104–1114 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Hillebrand H., On the generality of the latitudinal diversity gradient. Am. Nat. 163, 192–211 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Gaston K. J., Global patterns in biodiversity. Nature 405, 220–227 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Tedersoo L., et al. , Global diversity and geography of soil fungi. Science 346, 1256688 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Péter G., Takashima M., Čadež N., “Yeast habitats: Different but Global” in Yeasts in Natural Ecosystems: Ecology, Buzzini P., Lachance M.-A., Yurkov A., Eds. (Springer International Publishing, 2017), pp. 39–71. [Google Scholar]
- 24.Vallabhaneni S., et al. , Investigation of the first seven reported cases of Candida auris, a globally emerging invasive, multidrug-resistant fungus—United States, May 2013–August 2016. Morbidity Mortality Weekly Rep. 65, 1234–1237 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Goto S., Sugiyama J., Iizuka H., A taxonomic study of Antarctic yeasts. Mycologia 61, 748–774 (1969). [PubMed] [Google Scholar]
- 26.Freeman B. G., Pennell M. W., The latitudinal taxonomy gradient. Trends Ecol. Evol. 36, 778–786 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Nilsson R. H., et al. , The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Větrovský T., et al. , GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci. Data 7, 228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tedersoo L., Nara K., General latitudinal gradient of biodiversity is reversed in ectomycorrhizal fungi. New Phytol. 185, 351–354 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Ishida T. A., Nara K., Hogetsu T., Host effects on ectomycorrhizal fungal communities: Insight from eight host species in mixed conifer–broadleaf forests. New Phytol. 174, 430–440 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Rahbek C., et al. , Humboldt’s enigma: What causes global patterns of mountain biodiversity? Science 365, 1108–1113 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Rahbek C., et al. , Building mountain biodiversity: Geological and evolutionary processes. Science 365, 1114–1119 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Barberán A., et al. , Why are some microbes more ubiquitous than others? Predicting the habitat breadth of soil bacteria. Ecol. Lett. 17, 794–802 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Cardillo M., Dinnage R., McAlister W., The relationship between environmental niche breadth and geographic range size across plant species. J. Biogeogr. 46, 97–109 (2019). [Google Scholar]
- 35.Rabosky D. L., Hurlbert A. H., Species richness at continental scales is dominated by ecological limits. Am. Nat. 185, 572–583 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Fraser L. H., et al. , Worldwide evidence of a unimodal relationship between productivity and plant species richness. Science 349, 302–305 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Wang Y., et al. , Unimodal productivity–biodiversity relationship along the gradient of multidimensional resources across Chinese grasslands. Natl. Sci. Rev. 9, nwac165 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mittelbach G. G., et al. , What is the observed relationship between species richness and productivity? Ecology 82, 2381–2396 (2001). [Google Scholar]
- 39.Gillman L. N., et al. , Latitude, productivity and species richness. Global Ecol. Biogeogr. 24, 107–117 (2015). [Google Scholar]
- 40.Allen A. P., Brown J. H., Gillooly J. F., Global biodiversity, biochemical kinetics, and the energetic-equivalence rule. Science 297, 1545–1548 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Allen A. P., Gillooly J. F., Savage V. M., Brown J. H., Kinetic effects of temperature on rates of genetic divergence and speciation. Proc. Natl. Acad. Sci. U.S.A. 103, 9130–9135 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown J. H., Gillooly J. F., Allen A. P., Savage V. M., West G. B., Toward a metabolic theory of ecology. Ecology 85, 1771–1789 (2004). [Google Scholar]
- 43.Schluter D., Pennell M. W., Speciation gradients and the distribution of biodiversity. Nature 546, 48–55 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Liu H., Sun M., Zhang J., Genomic estimates of mutation and substitution rates contradict the evolutionary speed hypothesis of the latitudinal diversity gradient. Proc. R. Soc. B: Biol. Sci. 290, 20231787 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sweeney J. Y., Kuehne H. A., Sniegowski P. D., Sympatric natural Saccharomyces cerevisiae and S. paradoxus populations have different thermal growth profiles. FEMS Yeast Res. 4, 521–525 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Mozzachiodi S., et al. , Yeasts from temperate forests. Yeast 39, 4–24 (2022). [DOI] [PubMed] [Google Scholar]
- 47.Nnadi N. E., Carter D. A., Climate change and the emergence of fungal pathogens. PLoS Pathog. 17, e1009503 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi L.-L., et al. , Variation in forest soil fungal diversity along a latitudinal gradient. Fungal Diversity 64, 305–315 (2014). [Google Scholar]
- 49.Seena S., et al. , Biodiversity of leaf litter fungi in streams along a latitudinal gradient. Sci. Total Environ. 661, 306–315 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Tedersoo L., May T. W., Smith M. E., Ectomycorrhizal lifestyle in fungi: Global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 20, 217–263 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Corrales A., et al. , Diversity and distribution of tropical ectomycorrhizal fungi. Mycologia 114, 919–933 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Corrales A., Henkel T. W., Smith M. E., Ectomycorrhizal associations in the tropics—biogeography, diversity patterns and ecosystem roles. New Phytol. 220, 1076–1091 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Tietje M., et al. , Global variation in diversification rate and species richness are unlinked in plants. Proc. Natl. Acad. Sci. U.S.A. 119, e2120662119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Economo E. P., Narula N., Friedman N. R., Weiser M. D., Guénard B., Macroecology and macroevolution of the latitudinal diversity gradient in ants. Nat. Commun. 9, 1778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madden A. A., et al. , The ecology of insect–yeast relationships and its relevance to human industry. Proc. R. Soc. B: Biol. Sci. 285, 20172733 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Golan J. J., Pringle A., Long-distance dispersal of fungi. Microbiol. Spectr. 5, FUNK-0047-2016 (2017), 10.1128/microbiolspec.funk-0047-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stevens G. C., The latitudinal gradient in geographical range: How so many species coexist in the tropics. Am. Nat. 133, 240–256 (1989). [Google Scholar]
- 58.Gaston K. J., Blackburn T. M., Spicer J. I., Rapoport’s rule: Time for an epitaph? Trends Ecol. Evol. 13, 70–74 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Gaston K. J., Chown S. L., Why Rapoport’s rule does not generalise. Oikos 84, 309–312 (1999). [Google Scholar]
- 60.Okie J. G., et al. , Niche and metabolic principles explain patterns of diversity and distribution: Theory and a case study with soil bacterial communities. Proc. Biol. Sci. 282, 20142630 (2015), 10.1098/rspb.2014.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu B., et al. , Temperature determines the diversity and structure of N2O-reducing microbial assemblages. Funct. Ecol. 32, 1867–1878 (2018). [Google Scholar]
- 62.Barney J. B., Winans M. J., Blackwood C. B., Pupo A., Gallagher J. E. G., The Yeast Atlas of appalachia: Species and phenotypic diversity of herbicide resistance in wild yeast. Diversity 12, 139 (2020). [Google Scholar]
- 63.Potapov P., et al. , Mapping the world’s intact forest landscapes by remote sensing. Ecol. Soc. 13, 51 (2008). [Google Scholar]
- 64.Elsen P. R., Tingley M. W., Global mountain topography and the fate of montane species under climate change. Nat. Clim. Change 5, 772–776 (2015). [Google Scholar]
- 65.Global Biodiversity Information Facility, What is GBIF? https://www.gbif.org/what-is-gbif. Accessed 2 September 2022.
- 66.Peris D., et al. , Macroevolutionary diversity of traits and genomes in the model yeast genus Saccharomyces. Nat. Commun. 14, 690 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.David K. T., Yeast Macroecology 2023. Figshare. https://figshare.com/articles/dataset/Yeast_Macroecology_2023/25145819. Accessed 5 February 2024. [Google Scholar]
- 68.Valavi R., Elith J., Lahoz-Monfort J. J., Guillera-Arroita G., Modelling species presence-only data with random forests. Ecography 44, 1731–1742 (2021). [Google Scholar]
- 69.Olson D. M., et al. , Terrestrial Ecoregions of the World: A New Map of Life on EarthA new global map of terrestrial ecoregions provides an innovative tool for conserving biodiversity. BioScience 51, 933–938 (2001). [Google Scholar]
- 70.Smith J. R., et al. , A global test of ecoregions. Nat. Ecol. Evol. 2, 1889–1896 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Title P. O., Rabosky D. L., Tip rates, phylogenies and diversification: What are we estimating, and how good are the estimates? Methods Ecol. Evol. 10, 821–834 (2019). [Google Scholar]
- 72.Jetz W., Thomas G. H., Joy J. B., Hartmann K., Mooers A. O., The global diversity of birds in space and time. Nature 491, 444–448 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Redding D. W., Mooers A. Ø., Incorporating evolutionary measures into conservation prioritization. Conserv. Biol. 20, 1670–1678 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Anderson D., Burnham K., Model Selection and Multi-model Inference (Springer-Verlag, NY, ed. 2, 2004), vol. 63, p. 10. [Google Scholar]
- 75.Burgman M. A., The habitat volumes of scarce and ubiquitous plants: A test of the model of environmental control. Am. Nat. 133, 228–239 (1989). [Google Scholar]
- 76.Gregory R. D., Gaston K. J., Explanations of commonness and rarity in British breeding birds: Separating resource use and resource availability. Oikos 88, 515–526 (2000). [Google Scholar]
- 77.Whittaker R. H., New concepts of kingdoms of organisms: Evolutionary relations are better represented by new classifications than by the traditional two kingdoms. Science 163, 150–160 (1969). [DOI] [PubMed] [Google Scholar]
- 78.Pinheiro J., et al. , Package ‘nlme’: Linear and nonlinear mixed effects models (Version 3, 2017), p. 336.
- 79.Pennell M. W., et al. , geiger v2. 0: An expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics 30, 2216–2218 (2014). [DOI] [PubMed] [Google Scholar]
- 80.David K. T., Yeast Macroecology2023. Github. https://github.com/KyleTDavid/YeastMacroecology2023. Accessed 10 February 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Data Availability Statement
All code required to run the species distribution models presented in this paper and replicate primary analyses as well as supplementary data files, including distribution maps and raster files for all 186 species, have been deposited online and will be made publicly accessible upon publication. Readers may access the code repository at https://github.com/KyleTDavid/YeastMacroecology2023 (80) and data files at https://figshare.com/articles/dataset/Yeast_Macroecology_2023/25145819 (67).