

Significance
Although clonal hematopoiesis (CH) becomes a model in the study of aging and cancer, the genomic and transcriptomic alterations over aging have not been fully elucidated in acute myeloid leukemia (AML). Through analysis of a large cohort of AML, we revealed gene fusion (GF) events as main drivers in younger patients, whereas gene mutations constitute major genomic abnormalities in the elderly cases. Three mutational patterns are postulated among patients without GF, one likely transformed directly from CH (CH-AML), a second through or coupled with a stage of myelodysplasia syndrome (MDS) (CH-MDS-AML), and a third temporarily designated as the other GF- AML. This study provides insights into how aging shapes AML pathogenesis and helps to identify risk factors and molecular targets.
Keywords: acute myeloid leukemia, aging, clonal hematopoiesis, RNA-seq, molecular alterations
Abstract
Acute myeloid leukemia (AML) is an aging-related and heterogeneous hematopoietic malignancy. In this study, a total of 1,474 newly diagnosed AML patients with RNA sequencing data were enrolled, and targeted or whole exome sequencing data were obtained in 94% cases. The correlation of aging-related factors including age and clonal hematopoiesis (CH), gender, and genomic/transcriptomic profiles (gene fusions, genetic mutations, and gene expression networks or pathways) was systematically analyzed. Overall, AML patients aged 60 y and older showed an apparently dismal prognosis. Alongside age, the frequency of gene fusions defined in the World Health Organization classification decreased, while the positive rate of gene mutations, especially CH-related ones, increased. Additionally, the number of genetic mutations was higher in gene fusion–negative (GF-) patients than those with GF. Based on the status of CH- and myelodysplastic syndromes (MDS)-related mutations, three mutant subgroups were identified among the GF- AML cohort, namely, CH-AML, CH-MDS-AML, and other GF- AML. Notably, CH-MDS-AML demonstrated a predominance of elderly and male cases, cytopenia, and significantly adverse clinical outcomes. Besides, gene expression networks including HOXA/B, platelet factors, and inflammatory responses were most striking features associated with aging and poor prognosis in AML. Our work has thus unraveled the intricate regulatory circuitry of interactions among different age, gender, and molecular groups of AML.
Acute myeloid leukemia (AML) is a malignant blood cancer characterized by aggressive proliferation and blocked differentiation of hematopoietic stem/progenitor cells (HSPCs) (1, 2). The incidence of AML increases with age with a peak at an median of 68 y and slightly decreases in people aged over 75 y (3, 4). More importantly, aging is strongly associated with the leukemogenesis and poor prognosis of AML (5). With the global aging of population, it becomes more important to capture the molecular basis of aging in AML.
In recent decades, the common genomic abnormalities including gene fusions and sequence variants defined in the genomic classification of AML have spurred the implementation of precision medicine in this disease (6, 7). Cellular differentiation stages, transcriptomic subtypes, and their gene expression profiles have further improved the performance of molecular classification and even rationalized the tailored therapy in AML (7–10). On the other hand, clonal hematopoiesis (CH) with specific genetic mutations, mostly DNMT3A, TET2, and ASXL1 (DTA) mutations, has been identified based on large-scale genomic sequencing using peripheral blood from healthy populations (11). A recent work has further identified 24 loci, including 21 previously undefined ones, where germline genetic variation influences the predisposition to CH of indeterminate potential (CHIP) using the genome-wide association study (12). We conducted the survey of CHIP in the healthy Chinese population aged 40 to 110 y and identified the high incidence of CHIP in people aged 60 to 89 y (>40%) and older (>65%) (13). CH is not only closely associated with malignant blood diseases such as myelodysplastic syndromes (MDS), myeloproliferative neoplasms, and AML (14) but also shows the risk of development toward other systemic diseases (15). The dynamic monitoring of CH-related genetic mutations using peripheral blood at different ages can aid the early prediction of myeloid neoplasms (16). Of note, in the latest version of the World Health Organization (WHO) classification of AML, 8 secondary-type mutations (ASXL1, BCOR, EZH2, STAG2, SF3B1, SRSF2, U2AF1, and ZRSR2) associated with MDS and aging have been introduced into the definition of AML with myelodysplasia-related (AML-MR) subtype with or without a previous history of MDS. In addition to affecting the survival of HSPCs, CH may promote the tumor-dependent microenvironment and lead to the expansion of mutant clones, as exemplified by the inflammation resistance, which is one of the known risk factors for the generation of CH (17, 18).
Until now, relevant studies have mainly focused on the incidence and adverse significance of CH-related mutations in different age groups. In our previous work, we identified differences in the age and gender distribution of gene expression subgroups in AML that were strongly associated with specific genetic lesions (7). However, the aging models of AML patients and their clinical and biological significance remain largely unknown. In particular, the transformation patterns and their molecular characteristics between age-related AML including AML-MR and other CH-transformed AML still need to be compared in large-scale adulthood AML patients. Besides, the trends of age-related gene fusions, genetic mutations, and gene expression markers/pathways, such as various aging hallmarks and epigenetic factors, are rarely studied in an integrated manner. Meanwhile, key gender-related factors in AML that may influence the generation of different CH-related mutations also need to be investigated.
In this work, we established a discovery cohort with manually reviewed clinical and omics data from 1,474 newly diagnosed AML patients. First, we determined the prognosis of older age groups, which is closely related to the prevalence of CH-related mutations. Then, the common molecular features were systematically compared in different biological contexts, including age groups and gene fusion status. Some clinical and molecular features associated with age groups in AML were identified, such as different incidence of rare gene fusions and accumulation patterns of genetic mutations, as well as their potential gender distribution. These results may further improve the understanding of aging models and enrich the age-related molecular classification and prognostic stratification for adult AML.
Results
Age-Related Prognosis Stratification of AML.
First, we analyzed the age distribution of the entire cohort across age groups, mostly collected from January 2019 to December 2022 in Shanghai (n = 1,007), Hangzhou (n = 283), and Suzhou (n = 184) with RNA sequencing data. The median age of integrated cohorts of AML was 50 (IQR: 37, 62) y old, with the number of patients reaching a peak at the 50 to 59 y age group (Fig. 1A and Dataset S1). Gene fusion–positive (GF+) AMLs (n = 559) were slightly younger (median: 45, IQR: 31, 56) (Fig. 1B). In contrast, gene fusion–negative (GF-, n = 915) patients with CH-related genetic mutations were mostly prevalent in the 60 to 69 age group (median: 59, IQR: 48, 67) (Fig. 1C). Different age groups showed distinct prognostic stratification of overall survival (OS) and event-free survival (EFS) (Fig. 1D and SI Appendix, Fig. S1). Patients over 60 y of age showed the worst 3-y OS rates including the 60 to 69 y group (33.0, 95% CI: 23.9-45.6) and >=70 y group (24.5, 95% CI: 12.3-48.7) (Fig. 1D). Similar age-related risk stratification was observed in two independent validation groups, namely, The Cancer Genome Atlas (TCGA) LAML and Beat AML cohorts (SI Appendix, Fig. S2). In the multivariate analysis incorporating clinical features, we observed the independent prognostic significance in the two age groups [60 to 69 (P = 0.001) and >=70 y (P < 0.001)] (Fig. 1E). Of note, the number of patients eligible for hematopoietic stem cell transplantation (HSCT) decreased with age (SI Appendix, Fig. S3A). Nevertheless, HSCT can improve the prognosis of non-M3 AML patients for all age groups, including those aged 60 to 69 y (SI Appendix, Fig. S3B).
Fig. 1.

Distribution and prognostic stratification of age groups in AML. Histograms show all 1,474 AML patients (A), gene fusion–positive patients (B), and gene fusion–negative patients (C) from three centers. Common CH genetic mutations including DNMT3A, TET2, ASXL1, and secondary-type mutations suggestive of MDS transformation (BCOR, EZH2, STAG2, SF3B1, SRSF2, U2AF1, and ZRSR2) and other gene fusion–negative patients are shown in Left and Right panels (C), respectively. (D) Three-year OS analysis (KM curve) of AML patients from three centers based on age groups. The log-rank test is used to compare outcomes between age groups. (E) Multivariate analysis (Cox proportional hazard model) of basic clinical data including age groups, gender, BM blasts (%), WBC (×109/L), HGB (g/L), PLT (×109/L), FAB subtypes, and HSCT identified >60 y as having independent prognostic significance. The BM blasts, WBC, HGB, and PLT parameters are scaled using R function “scale.” BM, bone marrow. WBC, white cell count. HGB, hemoglobin. PLT, platelet. HSCT, hematopoietic stem cell transplantation.
Gene Fusions Decrease while Sequence Mutations Increase in the Older Age Group of AMLs.
Gene fusions and sequence mutations are common molecular alterations in AML, which showed obvious age correlation in this series. Potential gene fusion events were detected in 38% of all AML patients. Logistic regression indicated that the incidence of gene fusions in AML was significantly and negatively correlated with age, with occurring rates ranging from 62% in 20 to 29 y old to 16% in 70+ y old (P < 0.001) (Fig. 2A and SI Appendix, Fig. S4), while some rare gene fusions were more common in elderly AML patients (Fig. 2B). Among the 35 most frequent gene mutations including the FLT3-internal tandem duplication (ITD) and KMT2A-partial tandem duplication (PTD) (total positive rate >85% in this cohort), activated signaling pathways showed higher involvement rates in younger patients, while gene mutations in other pathways were more commonly seen in older patients, especially for spliceosome, tumor suppressor (TS), DNA methylation, and chromatin modifier genes (Fig. 2C, SI Appendix, Figs. S3–S5, and Dataset S2). Intriguingly, the mutation rates of several genes displayed a continuous increase after certain ages, as exemplified by DNMT3A and NPM1 above the age of 30 and ASXL1, TP53, and SRSF2 in 60 y or older (Fig. 2D and SI Appendix, Tables S1 and S2). In addition, several genetic mutations including TET2 and BCOR showed two potential increase points above 40 and 60 y of age. In total, 9 gene mutation terms tended to show higher median age and age correlation.
Fig. 2.
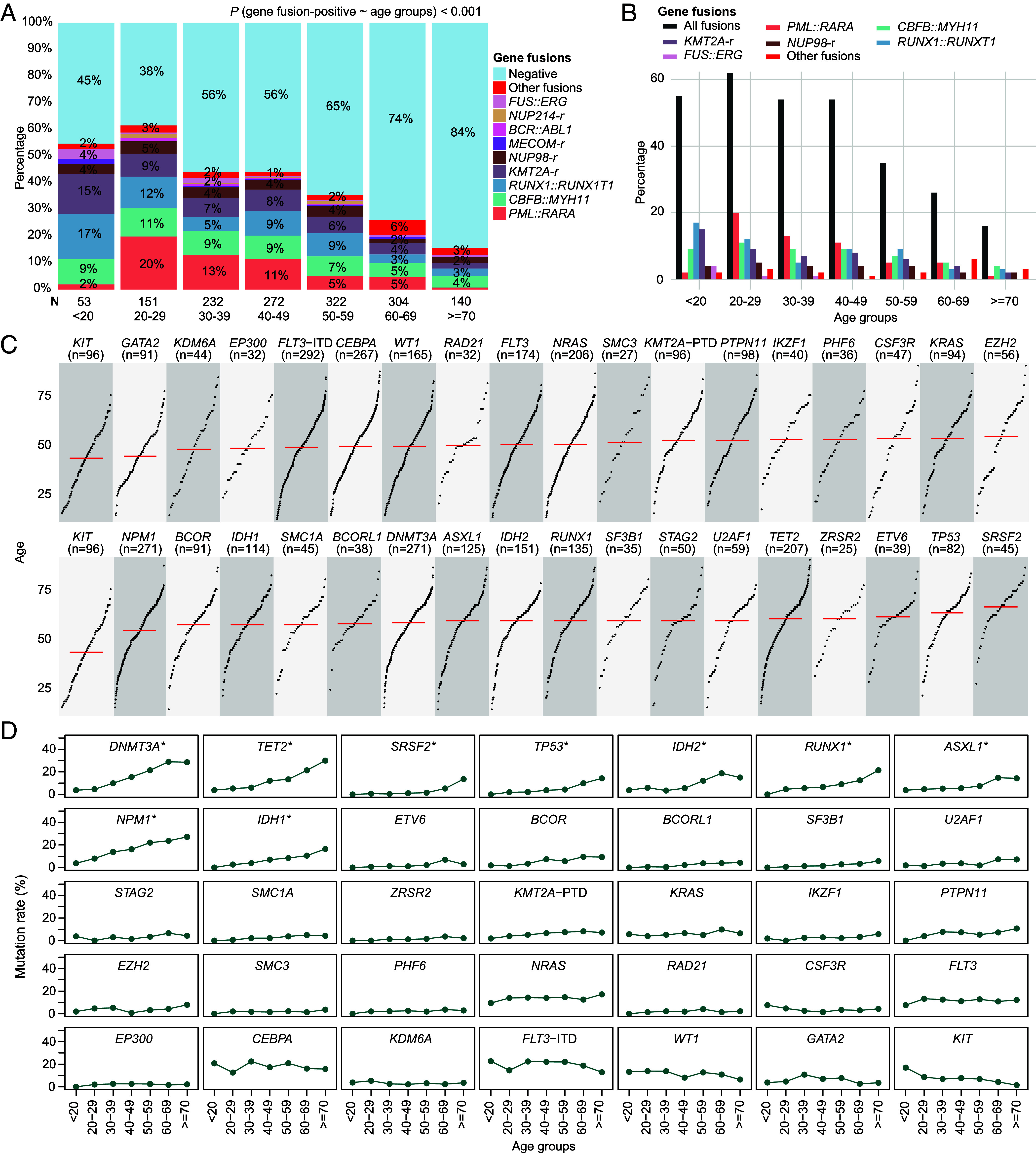
Incidence trend of gene fusions and common genetic mutations with age in AML. (A) and (B) show the positive rate of high-frequency and rare gene fusion events in different age groups. Most common gene fusions showed a negative correlation with age, PML::RARA, CBFB::MYH11, and RUNX1::RUNX1T1 in particular. Logistic regression indicates the decreased trend of gene fusions with age. (C) Scatter plots show the median age distribution (red line) of the 35 common gene mutation terms including FLT3-ITD and KMT2A-PTD. Each point represents a patient. The points are sorted by patient age from Left-Bottom to Right-Top. (D) Scatter plots show the mutation rates of common mutant events in different age groups. Top 9 genes with age-related mutations are marked with asterisks.
Different Mutational Patterns of Gene Fusion–Negative AML.
We then examined the interaction of gene fusion status and the number of genetic mutations in different age groups. Among the 35 frequently mutated genes in AML, about 30 to 60% GF- and 10 to 20% GF+ patients showed four or more mutations in all age groups (Fig. 3A), while the vast majority of healthy CH populations aged 60 y or older contained three or less mutations (13). It is noted that compared with GF+ patients, the proportion of patients harboring more than four mutations was 2 to 3 times higher in the GF- group (Fig. 3 A and B). The modest correlation between the number of genetic mutations and age was observed in GF- patients (Fig. 3C). These tendencies were more obvious when the top 9 gene mutation terms enriched in elderly patients were taken into consideration (Fig. 3 B and C, Lower). The whole exome sequencing-based TCGA LAML and Beat AML cohorts with lower sequencing depth could validate similar difference between GF- and GF+ groups (SI Appendix, Figs. S6 and S7) (1, 7, 8, 14).
Fig. 3.
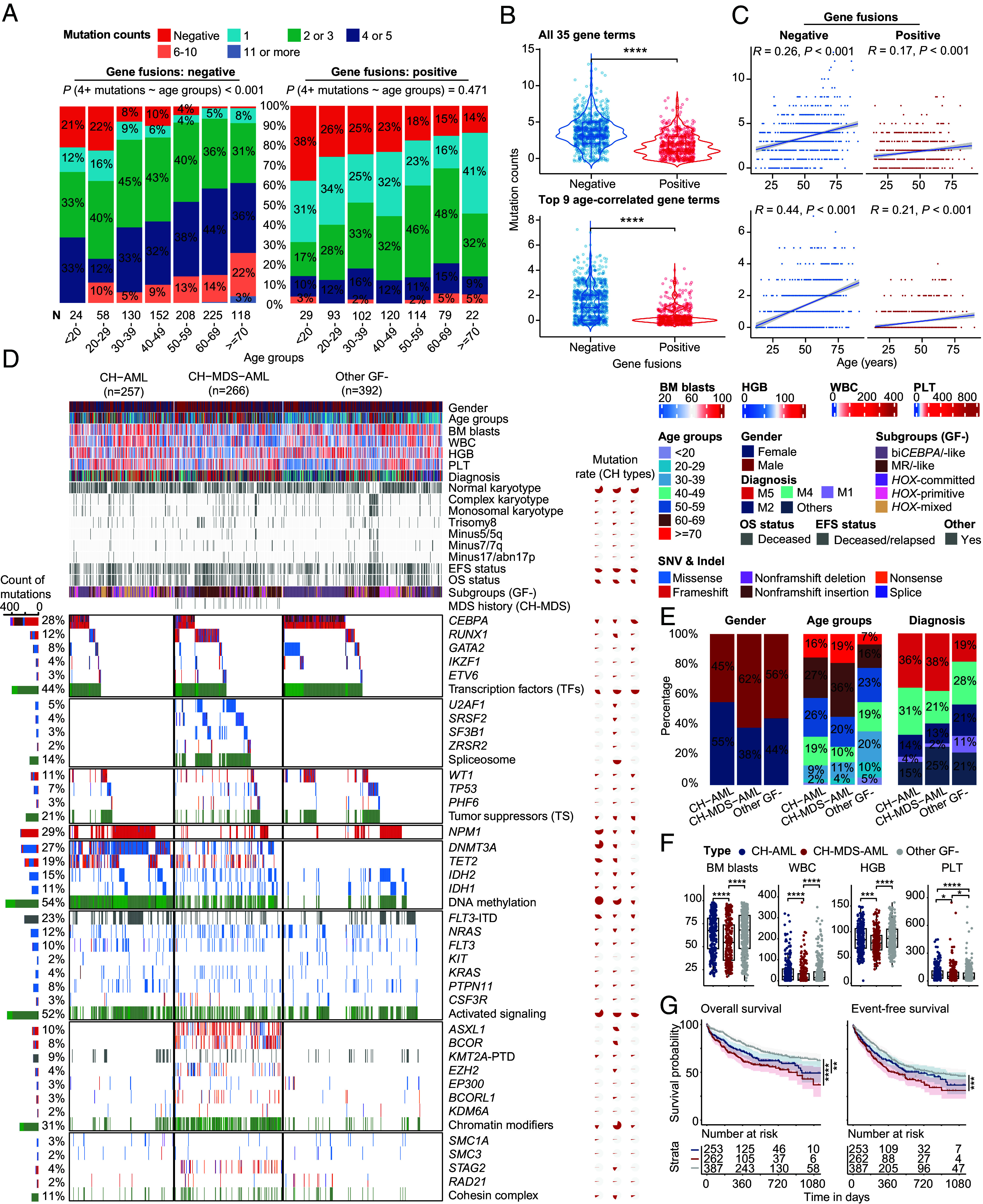
Genomic landscape of gene fusion–negative patients with AML. (A–C) compare the number of mutations between gene fusion–positive and gene fusion–negative patients with AML. (A) Percentage bar graphs show the classes of mutation numbers in gene fusion–negative (Left)/-positive (Right) groups. Gene fusion–negative patients have a higher rate of four or more mutations. (B) Violin plots show the difference in mutation numbers between gene fusion–negative/-positive groups. (C) Scatter plot of mutation number and age. Spearman correlations are shown in the Top. Regression line and the CI are shown. The Top and Bottom panels show all 35 gene mutation items and top 9 genes with age-related mutations, respectively. (D) Genomic landscape including clinical features, gene fusions, and genetic mutations of representative CH-related patients. CH-AML contains DNMT3A or TET2 genetic mutations, while the secondary mutations are negative. In contrast, patients in the CH-MDS-AML group contain at least one of the secondary-type mutations. Multicolored bars map the different types of gene mutations, representing the number of gene mutations. The percentage on the Left indicates the mutation rates of genes in all patients, while the pie chart on the Right shows the mutation rates of CH-AML, CH-MDS-AML and other GF- groups. (E) The bar plot shows the percentage difference of gender, age groups, diagnosis. (F) Boxplots show the difference of BM blasts, WBC, HGB, and PLT. (G) OS and EFS of CH-AML and CH-MDS-AML groups. Statistical significances of (B) and (F) were inferenced by the Wilcoxon signed-rank test. CH, clonal hematopoiesis. MDS, myelodysplastic syndromes. GF, gene fusion. BM, bone marrow. WBC, white cell count. HGB, hemoglobin. PLT, platelet. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we compared the genomic landscape and clinical significance of GF- patients, who enriched more aging-related CH markers. Currently, the DTA or other secondary-type mutations represent the most well-known and common genetic anomalies associated with CH. Among them, the eight secondary-type mutations that are part of the AML-MR definition in the 5th edition of WHO classification can facilitate the identification of MDS-related AML. According to the status of CH-related mutations, we assumed three mutant subgroups in GF- patients: CH-AML (DNMT3A or TET2 mutations without secondary-type mutations), CH-MDS-AML (secondary-type mutations-positive, with or without a previous MDS history), and other GF- AML (Fig. 3D and SI Appendix, Tables S3 and S4), respectively, accounting for 28%, 29%, and 43% of GF- AML. Compared with other GF- AML, the proportion of elderly patients (>=60) [43% (P < 0.001) and 55% (P < 0.001) vs. 23%] and FAB-M5 subtype [36% (P < 0.001) and 38% (P < 0.001) vs. 19%] was significantly higher in CH-AML and CH-MDS-AML. In contrast, other GF- AML showed a higher percentage of M1 [11% vs. 4% (P < 0.001) and 2% (P < 0.001)] and M2 subtype [21% vs. 14% (P = 0.043) and 13% (P = 0.013)] of AML. In addition, the other GF- patients can be further subdivided into distinct molecular subgroups with clinical significance, such as biCEBPA/-like, MR/-like and HOX-primitive AML, according to the gene expression profiles enriched for the CEBPA, RUNX1/TSs, and NPM1 mutations, respectively (7).
As compared to CH-AML, the CH-MDS-AML group showed a pattern of higher percentage of male (62% vs. 45%, P < 0.001) and elderly patients (55% vs. 43%, P = 0.004), while lower bone marrow (BM) blasts (P < 0.0001), white blood count (WBC, ×109/L, P < 0.0001), hemoglobin (HGB, g/L, P < 0.001), and platelet (PLT, ×109/L, P < 0.05) at diagnosis (Fig. 3 E and F). In terms of prognosis, a shorter OS and EFS could be observed in patients with CH-MDS-AML (Fig. 3G). Notably, the multivariate analysis indicated that age, -17/abn(17p), and the presence of genetic mutations of ETV6 and TP53 could independently predict a poor prognosis in CH-AML (SI Appendix, Fig. S8A). In contrast, male gender, WBC, mutations of RUNX1, IKZF1, TP53, FLT3, as well as Spliceosome genes, monosomal karyotype, and trisomy eight discriminated the outcome of CH-MDS-AML patients (SI Appendix, Fig. S8B). In other GF- patients, CEBPA mutations and monosomal karyotype could predict better prognosis, while age, complex karyotype, and FLT3-ITD mutations showed an independent negative impact on outcome (SI Appendix, Fig. S8C). In addition, HSCT could significantly improve the outcome of CH-MDS-AML patients (P < 0.001), while it showed borderline significance in the CH-AML (P = 0.056) and other GF- group (P = 0.067).
Transcriptome Correlation Analysis of Age Groups in AML.
Next, we identified potential age-related gene expression features among all patients, with expression levels of 1,867 and 1,247 genes positively and negatively correlated with age, respectively (adjusted P < 0.0001) (Fig. 4A, SI Appendix, Fig. S9, and Dataset S3). Most of these gene expression patterns showed statistical significance in merged differentially expressed genes (DEGs) analysis of age groups using the adjusted P < 0.05 threshold (Fig. 4A and Dataset S4). The intersection between stringent DEGs and age-correlated gene expression patterns might provide more robust subset for functional studies. Among them, the expression of PAWR, CPNE8, SYTL4, and MYCT1 were previously reported as subtype-related markers in older and high-risk AML (7). Pathways positively associated with older age were mainly involved in embryonic development (e.g., HOXA/B, RDH10, MMP16, PCGF2, EXT1, and ACVR2A), stem cell differentiation (e.g., GATA6, PTPRC, ESR1, HMGA2, and FN1), nervous system (e.g., HDGFL3, NAP1L2, and NECAB1), cell death (e.g., BCL2L11, BCL3, BCL6, HMOX1, BEX2, CYP1B1, IL7, IL10, LILRB1, and TSLP), cytokine production (e.g., GATA3, IL6, RORA, and FCGR3A), and some well-known cancer-related signaling abnormalities (e.g., TGF−beta, TNF, PI3K−Akt, and BMP signaling) (Fig. 4B and Dataset S5) (19–22).
Fig. 4.
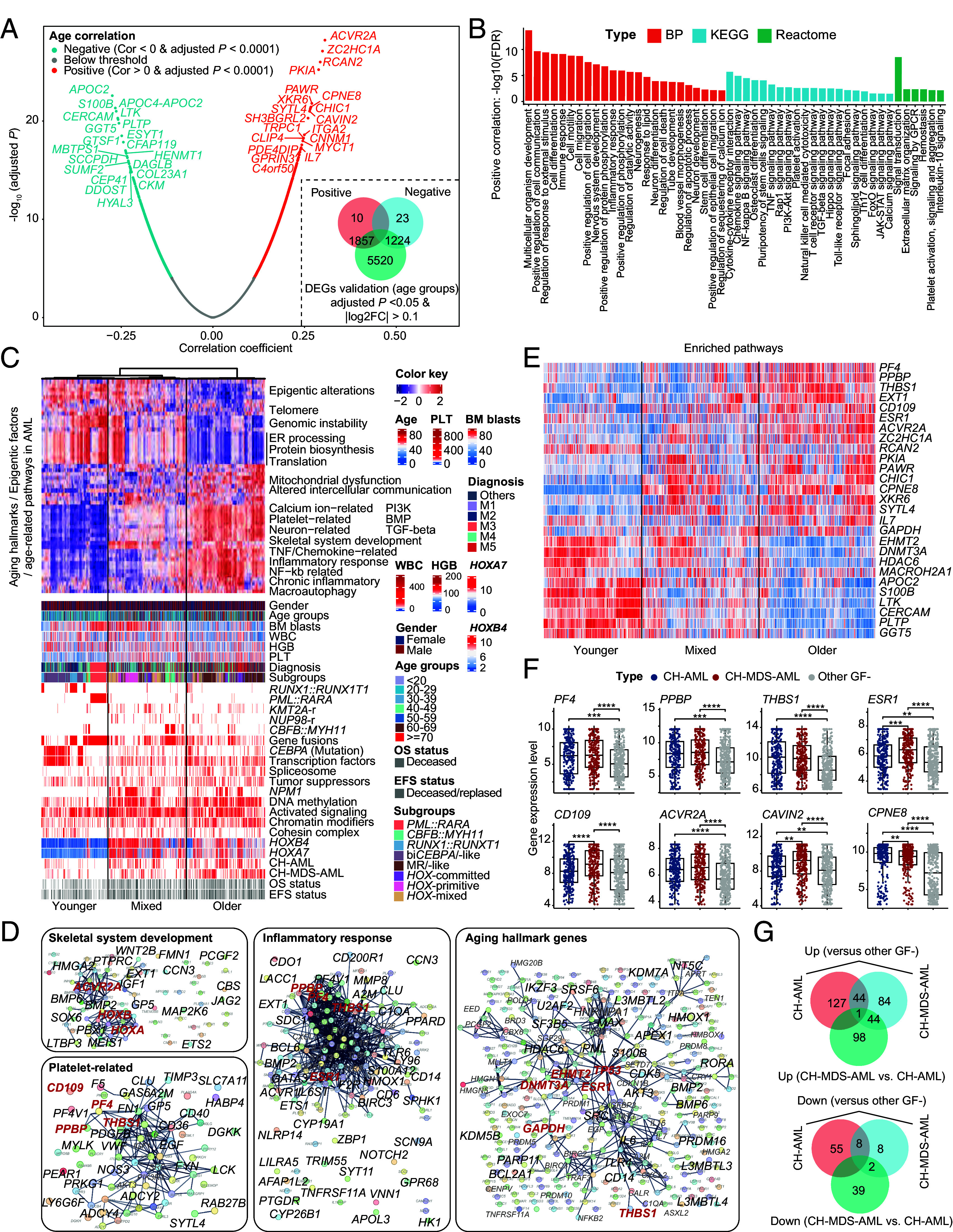
Transcriptomic screening of age-related genes and pathways in AML. (A) Volcano plots show the positive and negative correlation of gene expression patterns with age. The Pearson correlation coefficients with adjusted P < 0.0001 are colored red (>0) and cyan (<0) colors, respectively. The merged DEGs of age groups are calculated by comparing all possible combinations of age groups, such as >70 vs. 60 to 69 and 60 to 69 vs. 50 to 59. The <40 age groups are pooled in the DEGs analysis of age groups. (B) Functional enrichment analysis [GO BP (19), KEGG (21), and Reactome (22)] of positive-correlation gene expression patterns having positive correlation with age, performed on the STRING website (20). Only gene terms with false discovery rate (FDR)<0.05 are retained. (C) The integrative heatmap shows the age-related molecular and clinical features, including top heatmap (single-sample gene set enrichment scores of aging hallmarks, epigenetic gene sets, and enriched pathways based on positive and negative gene expression patterns with age in AML), gender, age, genetic mutations, gene fusions, CH-groups, gene expression subgroups, and survival data. Samples are sorted via the hierarchical clustering of pathways enrichment scores. At least three aging statuses can be found in AML patients, which show distinct patterns of aging hallmarks, epigenetic classes, and enriched pathways of age-related genes. Protein biosynthesis, DNA repair, and epigenetic factors (protein, reader, eraser, and bind eraser) show higher enrichment in younger patients, including PML::RARA, RUNX1::RUNX1T1, and CEBPA mutations with low HOXA/B gene expression. Epigenetic writer/binder writer, inflammation response, platelet- and neuron−related gene sets enriched in CH-related AML with higher stemness. CBFB::MYH11 and one branch of NPM-mutant patients show lower enrichment of inflammation response. (D) Selected gene networks show the known interactions of genes/proteins with age-related expression patterns. Each node represents one gene/protein. Some of key genes are labeled in red colors according to recurrence or priori knowledge, including the aberrant expression of PF4, THBS1, and PPBP as age- and prognosis-correlated genes in AML (7, 23–25). The CD109 can predict poor prognosis in the MR/-like subgroup. (E) Heatmap of partially representative age-related gene expression markers. (F) Boxplots show the normalized gene expression levels of a representative set of genes with age-correlated expression profiles. (G) Venn plots show the intersection of age-related gene expression markers between CH-AML and CH-MDS-AML groups. Statistical significances of (F) are inferenced by the Wilcoxon signed-rank test. GO, gene ontology. BP, biological process. KEGG, Kyoto Encyclopedia of Genes and Genomes. CH, clonal hematopoiesis. MDS, myelodysplastic syndromes. MR, myelodysplasia-related/-like. DEGs, differentially expressed genes. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In order to integrate different age-related features, we screened enriched pathways and different functional gene sets from aging hallmarks and epigenetic factors for conducting the supervised hierarchical clustering. The overall landscape of age-related features was displayed, and three distinct types of aging patterns were observed (Fig. 4C). The first type enriched PML::RARA, RUNX1::RUNX1T1, and CEBPA mutations, showing downmodulated HOXA/B gene family and up-regulated protein biosynthesis, endoplasmic reticulum processing, DNA repair, and epigenetic factors. The second type harbored one branch of CH-AML and most CH-MDS-AML patients, characterized by higher enrichment scores in loss of proteostasis, altered intercellular communication, mitochondrial dysfunction, BMP signaling pathway, platelet-, NF-κb-related networks as well as inflammatory response (Fig. 4D). The third type showed mixed features, including a subset of HOXA/B-related patients with NPM1 mutations, KMT2A-r, and NUP98-r displaying similar up-regulated protein biosynthesis to the first type, and CBFB::MYH11 exhibiting lower gene expression level of HOXA/B genes. Both higher blockage stages of hematopoietic stem cell differentiation (primitive signatures) or lower HOXA/B gene expression (CBFB::MYH11) indicative of monocytic differentiation might be associated with intermediate inflammatory microenvironment (7, 26). Similar results were validated in the pooled TCGA LAML and Beat AML cohorts (SI Appendix, Fig. S10 and Dataset S6).
In concordance with our previous work (7), the age relevance of HOXA/B and platelet-related gene network was prominent in AML, including THBS1, PF4, and PPBP genes (Fig. 4 D and E). In addition, we found a significant enrichment in the inflammatory response pathway, which also comprised aberrant expression of THBS1, PF4, and PPBP genes (Fig. 4F). Aging hallmarks-related network in AML was also extracted, with involvement of DNMT3A, GAPDH, HDAC6, EHMT2, TP53, etc. A recent work has reported that the high expression of epigenetic factor EHMT2 can increase the incidence of KMT2A rearrangements (27). We found that the expression level of the EHMT2 gene was significantly and negatively correlated with age (R = −0.14, P < 0.0001). More potential gene expression markers are provided in SI Appendix (Dataset S7). When narrowing down the analysis to patients in CH-related groups, we found that the number of genes with age-related expression patterns (adjusted P < 0.0001) in the CH-AML and CH-MDS-AML groups was small (Dataset S8). By comparing the DEGs between CH-AML, CH-MDS-AML, and other GF- AML patients, potential CH-related gene expression signatures were defined in the GF- AML group (Fig. 4G and Dataset S9).
Gender Difference of Age-Related Gene Fusions, Mutations, and Gene Expressions in AML.
Using Pearson’s Chi-squared test, the overall frequency of gene fusions tended to be higher in females (280/695, 40%, P = 0.077) compared to males (279/779, 36%) in this series (Fig. 5A and SI Appendix, Tables S5 and S6), especially the KMT2A (8.3% vs. 4.9%, P = 0.007) and NUP98 (4.2% vs. 2.7%, P = 0.12) gene fusions, proven to lead to the high expression of HOXA/B gene family (28, 29). We then compared the proportion of rare fusion genes and the common fusion entities in different age and gender groups. Intriguingly, above 30 y, the incidence of rare gene fusions in males began to increase with age. In the female counterpart, the increased incidence with age was impeded between 40 and 60 y (Fig. 5B).
Fig. 5.
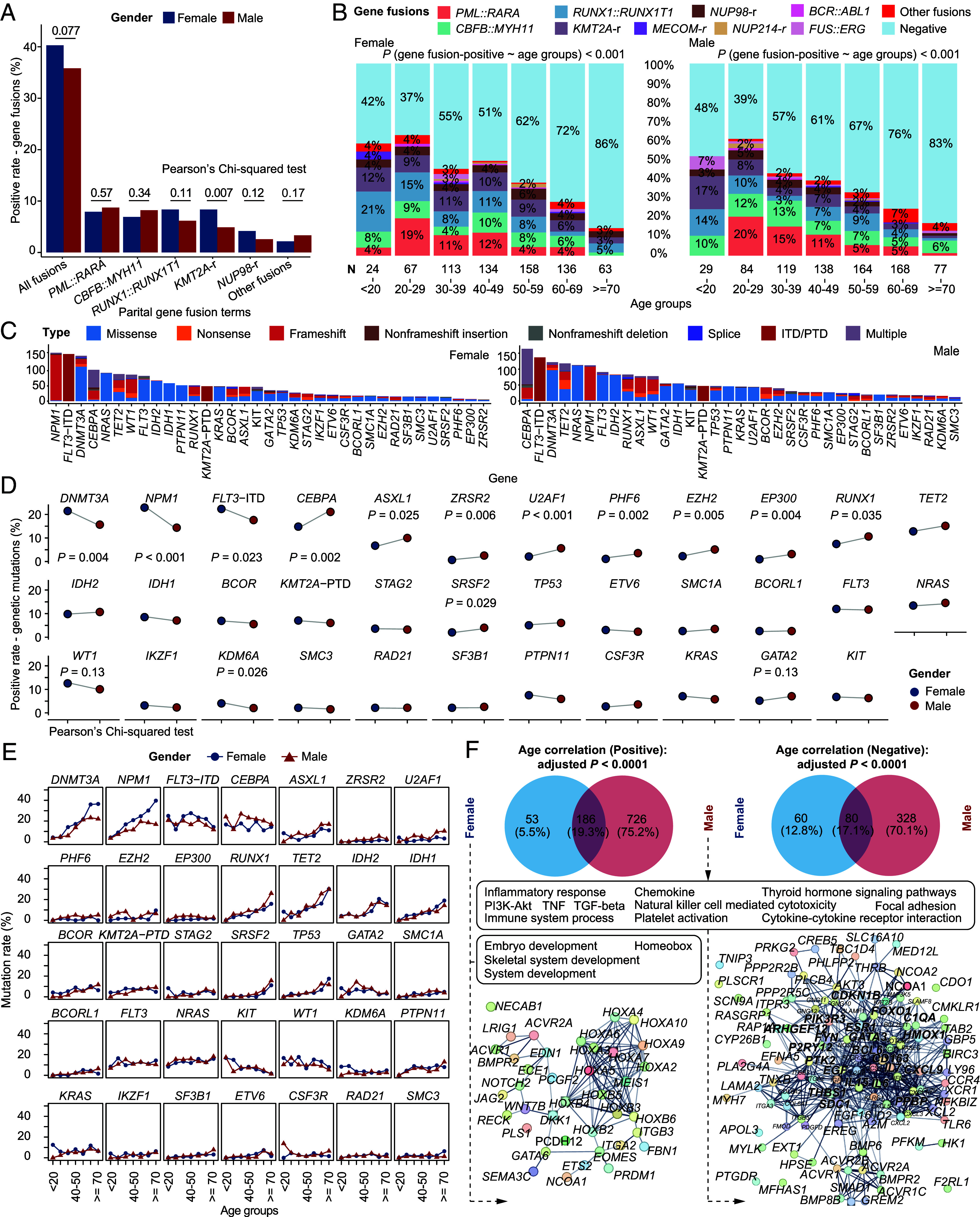
Gender differences in gene fusions, genetic mutations, and age-related gene expression markers in AML. (A) The bar chart shows the frequency of gene fusions between female (blue) and male (red). Females tend to have more gene fusions, mainly KMT2A and NUP98 gene fusions. (B) Positive rate of different classes of gene fusions in different age groups. High-frequency gene fusions decrease with age in both females and males, while rare gene fusions show higher incidence in older male AML patients. Logistic regressions indicate the decrease trend of gene fusions with age in male and female. (C–E) show the difference in the occurrence of gene mutations. (C) The bar chart shows the number of patients with common genetic mutations in female (Left) and male (Right). (D) Dot plots show the changes in proportions between females (blue dot) and males (red dot). (E) Dot plots show the sex difference of genetic mutations in different age groups. (F) Pathway enrichment analysis of genes with age-related expression profiles (adjusted P < 0.0001) in female and male highlighting the gender-related HOXA/B and inflammatory response pathways. Statistical significance of (A) and (D) is inferenced by the Pearson’s Chi-squared test.
Comparison of genetic mutations (Pearson’s Chi-squared test) showed that the positive percentage of CEBPA (P = 0.002), ASXL1 (P = 0.025), ZRSR2 (P = 0.006), U2AF1 (P < 0.001), PHF6 (P = 0.002), EZH2 (P = 0.005), and EP300 (P = 0.004) mutations was significantly higher in male patients (Fig. 5 C and D). In contrast, DNMT3A (P = 0.004), NPM1 (P < 0.001), and FLT3-ITD (P = 0.023) mutations were more frequently seen in female patients (Fig. 5D). Analysis of different age groups indicated that DNMT3A and NPM1 mutations showed higher difference between genders above 60 y (Fig. 5E), while mutations of CEBPA, ASXL1, etc., were more frequent in most males in >30 age groups. Focusing on age-correlated gene expression patterns and enriched pathways, we noticed that the HOXA/B-related gene network tended to be enriched in female, while the inflammatory response was significantly up-regulated in male patients with AML (Fig. 5F and Dataset S10). Therefore, we speculate that, apart from gene fusion status, both CH-AML and CH-MDS-AML groups, and gender may influence the accumulation of genetic mutations and aberrant gene expression regulatory circuitry in AML.
Combining Age-Correlated Genes and Clonal Hematopoietic Types to Uncover Gonadal-Related Immune Regulation in AML.
Different enrichment of gender and inflammatory pathways in CH-AML (female enriched) and CH-MDS-AML (male enriched) prompted us to investigate potential gonadal factors in the induction of different CH patterns through immune regulation. First, age-related gene expression patterns of CH-AML/CH-MDS-AML and female/male were compared (Fig. 6A). DEGs (|log2(fold change) > 1| and adjusted P < 0.05) between CH types and age groups were used to screen the candidate targets. Known gonadal-related gene sets from MsigDB were then used to narrow down the number of genes (Fig. 6A). A total of 68 genes, including AR (Androgen Receptor), THBS1, TRH (Thyrotropin Releasing Hormone), CYP1B1 (Cytochrome P450 Family 1 Subfamily B Member 1), CALCRL (Calcitonin Receptor Like Receptor), etc., were defined as potential gonadal-related genes potentially involved in immune regulation process (Fig. 6B).
Fig. 6.
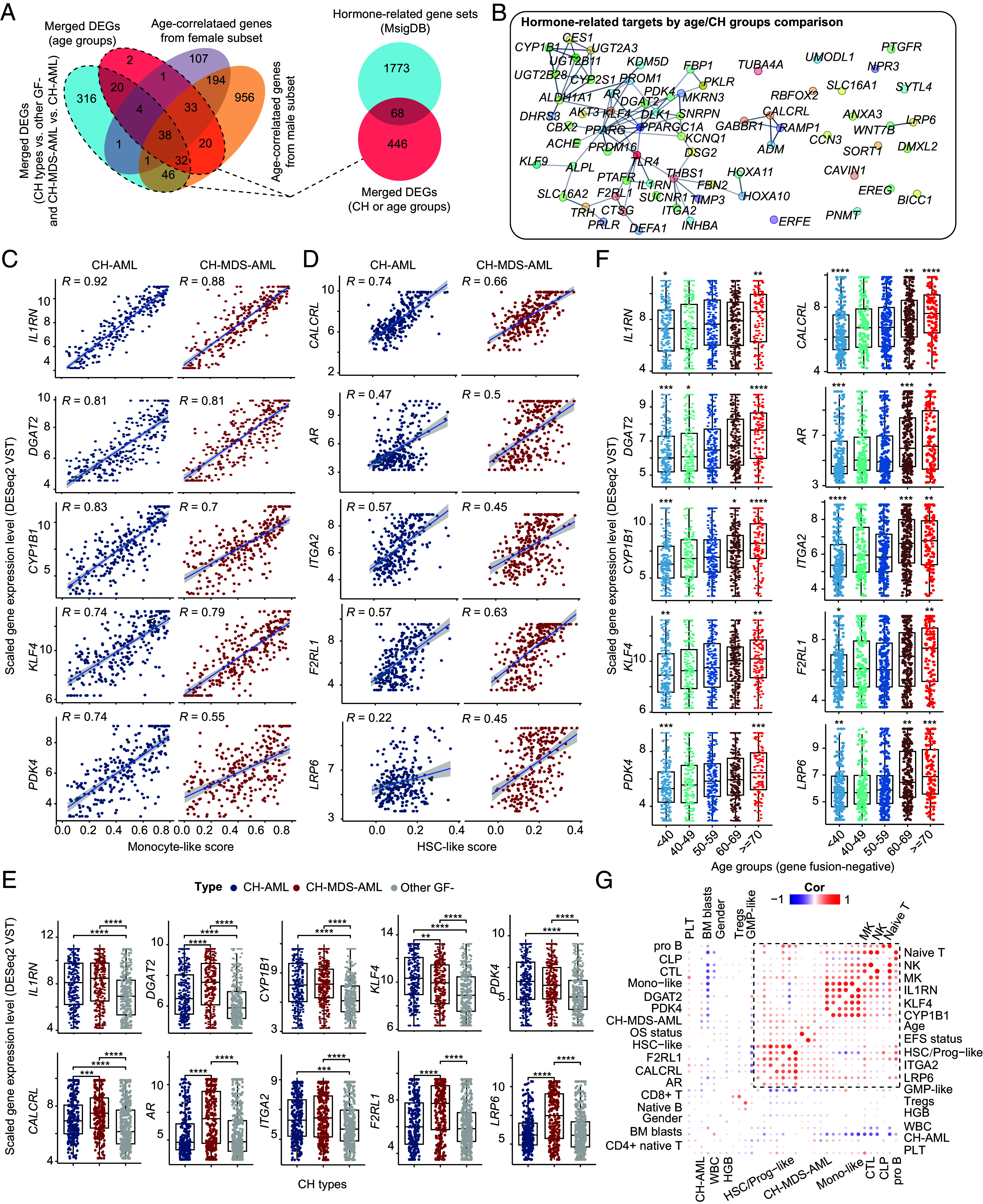
Correlation analysis of age-/gonadal-related gene expression patterns and immune signatures in CH groups. (A) The Venn plot displays the merged DEGs (|log2 (fold change)| > 1 and adjusted P < 0.05) of age/CH groups (Left). About 87% of merged DEGs in age groups can be found in the age-correlated gene sets (adjusted P < 0.0001) from female and male subset of AML patients. Intersections between hormone gene sets and merged DEGs of CH types and age groups (Right). (B) Network visualization of 68 genes using the intersection set. Scatter plots of the top 10 genes correlating with the tumor-derived monocyte-like (C) and HSC-like signatures (D). The expression levels (DESeq2 VST normalization) of IL1RN, CYP1B1, and DGAT2 are highly correlated with the monocyte-like score, while that of the CALCRL is most highly correlated with the HSC-like score. Boxplots of (E) and (F) represent the gene expression difference of screened age-/hormone-related gene sets between CH types and different age groups. Note that the age groups (<40) are combined in this comparison (F). Statistical significances of (E) and (F) are inferenced by the Wilcoxon signed-rank test. (G) Correlation heatmap of defined age-related targets and immune cells. Statistically insignificant pairs are filled with blank color. Both single-sample gene enrichment analysis and CIBERSORTx deconvolution of immune cells are used. HSC, hematopoietic stem cell. DEGs, differentially expressed genes. VST, variance-stabilizing transformation. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Then, we calculated enrichment scores of differentiation stages in leukemic/hematopoietic cells based on the published gene sets (Dataset S11) (26, 30). Deconvolution of immune cells was also performed, and several significant connections were identified. Correlation between genes and monocyte-like and HSC-like signatures, two known tumor-derived signatures associated with different immune environments, was given attention to further identify more robust interactions between gene expression and immune regulation. Here, we have shown the representative 10 genes of which the expression was highly and positively associated with monocyte-like (Fig. 6C) or HSC-like (Fig. 6D). The L1RN, CYP1B1, PDK4, and ITGA2 showed consistently high expression both in CH-AML and CH-MDS-AML (Fig. 6E), compared with other GF- AML. In contrast, the expression of DGAT2, CALCRL, AR, F2RL1, and LPR6 showed the highest level in the CH-MDS-AML group, while that of KLF4 was higher in the CH-AML group. The age correlation of the expression levels of these genes was double-checked in boxplots (Fig. 6F). The genes–immunocytes pairs could facilitate the identification of patterns of immune regulation where gonadal- and molecular subtype-related factors might interact (Fig. 6G and Dataset S11).
Discussion
Aging is a natural and inevitable process, which is associated with the elevated incidence and mortality rates of various types of cancers. A recent work has proposed twelve hallmarks of aging, such as genomic instability, epigenetic alterations, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, etc. (31), yet the specific impact of aging on AML remains largely unknown. Hence, gaining a comprehensive understanding of the underlying biologic and molecular mechanisms that link aging and AML is crucial for the management of the disease in an aging society.
It is currently recognized that somatic mutations accumulate in HSPCs during the natural aging process. By the age of 70, humans are expected to harbor between 350,000 and 1,400,000 coding mutations within the HSPCs pool (32). These mutations can provide a competitive growth advantage to cells and give rise to CH in healthy aging individuals (33). In parallel, we observed that among healthy individuals in the Chinese population, CH accounted for 6% and 14% in the 40 to 49 and 50 to 59 age groups, respectively. Remarkably, this proportion increased to more than 40 to 60% in the population aged 60 y and older (13).
Our large-scale study adds further clarity on the association of aging and AML. In general, gene fusions, primarily the WHO-defined entities and other high-frequency ones, are common in younger AML, while mutations are more frequently seen in older patients, which is in concordance with previous reports (34–37). Older adult AML is characterized by higher frequency of CH-related mutations of AML, including epigenetic factors (e.g., DNMT3A, TET2, and ASXL1), spliceosome (e.g., SRSF2, ZRSR2, and U2AF1), TP53, etc., while CEBPA mutations tend to occur in younger patients (38–40). In addition to differences in mutated genes, the number of mutations is another molecular parameter strongly associated with aging. Intriguingly, the correlation between age and the number of mutations is greater in gene fusion–negative patients. We postulate that the accumulation of genetic variants to a certain extent, such as the involvement of functionally critical genes, as well as sufficient number of mutations and clone size in a long period of time might lead to the eventual initiation and development of AML. This provides a plausible explanation for the high incidence of AML among the geriatric population. Collectively, these results, together with data we obtained from CHIP carriers in the healthy aging population (13), provide compelling evidence that CH-related mutations may serve as major driving force in the initiation of elderly AML.
Nevertheless, the sophisticated processes and mechanisms underlying the transition from CH to AML have not yet been fully elucidated. We posit that three distinct mutational patterns may exist in gene fusion–negative AML patients based on the status of CH-associated mutations, namely, CH-AML, CH-MDS-AML, and other GF- AML. CH-AML is likely to be developed directly from CHIP with eventual other factors, whereas CH-MDS-AML is distinguished by the presence of secondary-type mutations with markers recently incorporated into the WHO-defined MDS-related AML (41), and therefore is likely to be transformed through, or coupled with a stage of MDS. In comparison to CH-AML, the CH-MDS-AML group exhibits a higher proportion of elderly and male patients, and lower BM blasts, WBC, HGB, and PLT at diagnosis, which coincides with the feature of multilineage hypoplasia and cytopenia in MDS. Of particular note, the CH-MDS-AML group demonstrates the worst prognosis. These findings underscore the significant clinical and pathological implications of different patterns of mutation accumulation in AML.
It is noteworthy that gene expression profiles further shed light on the aging-related pathogenic mechanisms in AML. HOXA/B-, platelet factor-, and inflammatory response-related networks represent the most obvious enrichment characteristics correlated with aging and poor prognosis in AML. Of note, three research groups have recently reported the antiaging effect of platelet factor PF4 (23–25), which is also involved in the inflammatory response pathway, but its role in AML remains unknown. We speculate that platelet factors might be imperative for the maintenance of leukemic stem cells, which warrants additional mechanistic exploration. Besides, the aging hallmark gene sets of AML that defined in this work could provide a comprehensive reference resource for age-guided clinical trials, as exemplified by the HDAC6, and EHMT2. It has recently been reported that the hyperactivation of HDAC6 promotes the proliferation of HSPCs, whereas its inhibition can efficiently rescue the hematopoietic phenotype (42). EHMT2 (also known as G9a) acts as a histone methyltransferase and one of the molecular contributors to KMT2A rearrangements (27). Notably, EHMT2 can interact with DNMT1 to coordinate DNA and histone methylation, leading to the transcriptional silencing of target TS genes (43). Dual small-molecule inhibitors against G9a and DNA methyltransferases activity have been proved to inhibit cell proliferation and promote apoptosis in AML (44). Taken together, our work provides a pool of candidate targets for subsequent functional experiments and clinical research.
On the other hand, CH has been reported as a protective factor for Alzheimer’s disease (45), potentially via improving the survival and enhanced phagocytosis of marrow-derived microglia. Previous studies have suggested that the number of human HSPCs is limited, which may be affected by the aging process, leading to a deterioration in their potential, a process also termed stem cell exhaustion (31, 46). From an environmental adaptation perspective in the hematopoietic system, as the HSPC pool declines with age, CH may exert a positive effect by enhancing the survival advantage of HSPCs for necessary hematopoiesis (47). Pertinently, some neurologically relevant genes, such as HDGFL3, NAP1L2, and NECAB1, were found up-regulated in elderly AML in the present work. We suppose that in a positive way, certain degree of CH might serve as a protective mechanism against aging and play an important role in maintaining the physiological hematopoiesis and neurologic function. However, when mutations accumulate excessively, it may culminate in the development of MDS and AML. This consequence might represent the cost that humans have to pay in order to maintain fitness and essential body functions during the aging process, and further functional validation needs to be performed.
An intriguing finding of this study is that, in addition to age, gender bias appears to provide insights into the potential mechanisms underlying AML pathogenesis. For example, rare gene fusions are abundant in older male patients. Some genetic mutations also showed gender-related distributions, such as female-prone NPM1, DNMT3A, and FLT3-ITD, and preferential mutations of CEBPA, ASXL1, and U2AF1 in males. Besides, deregulated expression of HOXA/B-related and inflammatory response networks were enriched in female and male patients, respectively. We suppose that immune/autoimmune factors and abnormal expression of gonadal-related genes may contribute to these gender-related discrepancies. Further exploration of interaction patterns among age, gender, genetic lesions, and CH-related mutation accumulations will reveal the intricate complexity of AML pathogenesis, which is informative for screening potential molecular targets and establishing interventions to prevent or delay aging-driven malignant processes.
In summary, we performed a large-scale integrative analysis in 1,474 newly diagnosed AML patients. Potential correlations between molecular and clinical features, age, gender, and prognosis in particular, have been systematically explored. These results can shape a gross model of aging in AML, which may provide more optimized information on the molecular classification and prognostic stratification of adult AML in the future.
Materials and Methods
Patients and Samples.
This study incorporated 1,474 newly diagnosed AML patients from three centers in China, including 1007 from Shanghai Institute of Hematology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 283 from Zhejiang Institute of Hematology, the First Affiliated Hospital of Zhejiang University College of Medicine, and 184 from Jiangsu Institute of Hematology, the First Affiliated Hospital of Soochow University, mostly over the period of January 2019 to December 2022. Among them, 639 cases were previously reported (7), and 835 were newly incorporated in this study. BM samples from all patients were subjected to RNA sequencing, and targeted or whole exome sequencing data were available in the majority of these patients (n = 1,390, 94%). Sample processing, nucleic acid extraction, and next-generation sequencing were performed as previously reported (7). Treatment protocols are provided in SI Appendix.
This study was approved by the Ethics Committee of Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, the First Affiliated Hospital of Zhejiang University College of Medicine, and the First Affiliated Hospital of Soochow University. All patients have given informed consent for both treatment and cryopreservation of BM and peripheral blood samples according to the Declaration of Helsinki.
Visualization and Statistical Analysis.
Categorical variables were compared by Pearson’s Chi-square or Fisher’s exact test and continuous data by the t test or Wilcoxon rank-sum test. The R package survival (v3.5-7) and survminer (v0.4.9) was used to construct the Kaplan–Meier (KM) model and draw the survival curve. Multivariate Cox analysis of OS was performed by ezcox (v1.0.4). Most statistical analyses were performed using the R 4.3.0 software package. Age-related gene expression markers were defined by Pearson correlations. Predefined eight gene expression subgroups of AML were predicted via using AutoML-based models (7). Detailed information and parameters related to the analysis of gene fusions, sequence mutations, and gene expression profiles are provided in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Acknowledgments
This work was supported by the State Key Laboratory of Medical Genomics, the Double First-Class Project (WF510162602) from the Ministry of Education, the Shanghai Collaborative Innovation Program on Regenerative Medicine and Stem Cell Research (2019CXJQ01), the Overseas Expertise Introduction Project for Discipline Innovation (111 Project; B17029), the National Natural Science Foundation of China (NSFC 81861148030, 82230006, 82270166, and 82300168), the Samuel Waxman Cancer Research Foundation, the Shanghai Clinical Research Center for Hematological disease (19MC1910700), the Shanghai Shenkang Hospital Development Center (SHDC2020CR5002), the Shanghai Major Project for Clinical Medicine (2017ZZ01002), the Shanghai Sailing Program (23YF1424400), the Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20161406), the Innovative Research Team of High-level Local Universities in Shanghai, and the Shanghai Guangci Translational Medical Research Development Foundation.
Author contributions
Z.C., J.J., D.-P.W., R.-B.R., S.-J.C., and Y.S. designed research; J.-F.L., W.-Y.C., Xiang-Jie Lin, L.-J.W., K.W., Y.-M.Z., H.-M.Z., X.-J.C., Y.-L.Z., W.Y., J.-N.Z., X.Y., H.-Y.W., J.-Q.R., Xiao-Jing Lin, N.Q., Y.-T.D., G.L., X.-Y.Y., and S.-N.C. performed research; F.Z., X.-Q.W., S.-Y.W., L.J., H.F., Y.T., and X.-J.S. contributed new reagents/analytic tools; J.-F.L., W.-Y.C., and K.W. analyzed data; Xiang-Jie Lin, L.-J.W., H.-M.Z., X.-J.C., Y.-L.Z., W.Y., J.-N.Z., X.Y., F.Z., H.-Y.W., J.-Q.R., Xiao-Jing Lin, N.Q., Y.-T.D., G.L., X.-Y.Y., and S.-N.C. collected clinical data; Y.-M.Z., S.-Y.W., and L.J. performed DNA/RNA sequencing; and J.-F.L. and W.-Y.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: J.L., University of Florida Cancer and Genetics Research Center; and P.L., NIH.
Contributor Information
Zhu Chen, Email: zchen@stn.sh.cn.
Jie Jin, Email: jiej0503@zju.edu.cn.
De-Pei Wu, Email: wudepei@suda.edu.cn.
Rui-Bao Ren, Email: rbren@sjtu.edu.cn.
Sai-Juan Chen, Email: sjchen@stn.sh.cn.
Yang Shen, Email: yang_shen@sjtu.edu.cn.
Data, Materials, and Software Availability
Anonymized RNA sequencing data have been deposited in [The Genome Sequence Archive for Human (GSA-Human, https://ngdc.cncb.ac.cn/gsa-human)] (HRA002693 and HRA006117) (48, 49). Universal visualization codes of web-based plugins were deposited at the HIPLOT (ORG) website (50). Previously published data were used for this work (1, 7, 8, 35, 51, 52).
Supporting Information
References
- 1.Tyner J. W., et al. , Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dufva O., et al. , Immunogenomic landscape of hematological malignancies. Cancer Cell 38, 380–399.e13 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Miranda-Filho A., et al. , Epidemiological patterns of leukaemia in 184 countries: A population-based study. Lancet Haematol. 5, e14–e24 (2018). [DOI] [PubMed] [Google Scholar]
- 4.DiNardo C. D., Erba H. P., Freeman S. D., Wei A. H., Acute myeloid leukaemia. Lancet 401, 2073–2086 (2023). [DOI] [PubMed] [Google Scholar]
- 5.Newell L. F., Cook R. J., Advances in acute myeloid leukemia. BMJ 375, n2026 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Arber D. A., et al. , International consensus classification of myeloid neoplasms and acute leukemias: Integrating morphologic, clinical, and genomic data. Blood 140, 1200–1228 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng W. Y., et al. , Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia. Proc. Natl. Acad. Sci. U.S.A. 119, e2211429119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bottomly D., et al. , Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell 40, 850–864.e9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Docking T. R., et al. , A clinical transcriptome approach to patient stratification and therapy selection in acute myeloid leukemia. Nat. Commun. 12, 2474 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mer A. S., et al. , Biological and therapeutic implications of a unique subtype of NPM1 mutated AML. Nat. Commun. 12, 1054 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zink F., et al. , Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kessler M. D., et al. , Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., et al. , The impact of age and number of mutations on the size of clonal hematopoiesis. Proc. Natl. Acad. Sci. U.S.A. 121, e2319364121 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal S., Ebert B. L., Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaiswal S., et al. , Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu M., et al. , Multiparameter prediction of myeloid neoplasia risk. Nat. Genet. 55, 1523–1530 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avagyan S., et al. , Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 374, 768–772 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Meira A., et al. , Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat. Genet. 55, 1531–1541 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gene Ontology Consortium, The Gene Ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szklarczyk D., et al. , The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M., Furumichi M., Sato Y., Kawashima M., Ishiguro-Watanabe M., KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillespie M., et al. , The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroer A. B., et al. , Platelet factors attenuate inflammation and rescue cognition in ageing. Nature 620, 1071–1079 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leiter O., et al. , Platelet-derived exerkine CXCL4/platelet factor 4 rejuvenates hippocampal neurogenesis and restores cognitive function in aged mice. Nat. Commun. 14, 4375 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park C., et al. , Platelet factors are induced by longevity factor klotho and enhance cognition in young and aging mice. Nat. Aging 3, 1067–1078 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Galen P., et al. , Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265–1281.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray Z. H., et al. , Epigenetic balance ensures mechanistic control of MLL amplification and rearrangement. Cell 186, 4528–4545.e18 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heikamp E. B., et al. , The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 139, 894–906 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libbrecht C., et al. , Menin is necessary for long term maintenance of meningioma-1 driven leukemia. Leukemia 35, 1405–1417 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., et al. , Single-cell transcriptomics reveals multiple chemoresistant properties in leukemic stem and progenitor cells in pediatric AML. Genome Biol. 24, 199 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023). [DOI] [PubMed] [Google Scholar]
- 32.Jaiswal S., Clonal hematopoiesis and nonhematologic disorders. Blood 136, 1606–1614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fabre M. A., et al. , The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 606, 335–342 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cappelli L. V., et al. , DNMT3A mutations are over-represented in young adults with NPM1 mutated AML and prompt a distinct co-mutational pattern. Leukemia 33, 2741–2746 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Ley T. J., et al. ; Cancer Genome Atlas Research Network, Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metzeler K. H., et al. , Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 128, 686–698 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Burd A., et al. , Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat. Med. 26, 1852–1858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genovese G., et al. , Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abelson S., et al. , Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie M., et al. , Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khoury J. D., et al. , The 5th edition of the World Health Organization classification of haematolymphoid tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia 36, 1703–1719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pezzotta A., et al. , HDAC6 inhibition decreases leukemic stem cell expansion driven by Hedgehog hyperactivation by restoring primary ciliogenesis. Pharmacol Res. 183, 106378 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Estève P. O., et al. , Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.San José-Enériz E., et al. , Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 8, 15424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouzid H., et al. , Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nat. Med. 29, 1662–1670 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mejia-Ramirez E., Florian M. C., Understanding intrinsic hematopoietic stem cell aging. Haematologica 105, 22–37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Köhnke T., Majeti R., Clonal hematopoiesis: From mechanisms to clinical intervention. Cancer Discov. 11, 2987–2997 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng W. Y., et al. , Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia. GSA-Human. https://ngdc.cncb.ac.cn/gsa-human/browse/HRA002693. Deposited 14 July 2022. [DOI] [PMC free article] [PubMed]
- 49.Li J. F., et al. , Aging and comprehensive molecular profiling in acute myeloid leukemia. GSA-Human. https://ngdc.cncb.ac.cn/gsa-human/browse/HRA006117. Deposited 7 December 2023.
- 50.Li J., et al. , Hiplot: A comprehensive and easy-to-use web service for boosting publication-ready biomedical data visualization. Brief. Bioinform. 23, bbac261 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Lin X., et al. , Integration of genomic and transcriptomic markers improves the prognosis prediction of acute promyelocytic leukemia. Clin. Cancer. Res. 27, 3683–3694 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Jin P., et al. , Large-scale in vitro and in vivo CRISPR-Cas9 knockout screens identify a 16-gene fitness score for improved risk assessment in acute myeloid leukemia. Clin. Cancer Res. 28, 4033–4044 (2022), 10.1158/1078-0432.Ccr-22-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Data Availability Statement
Anonymized RNA sequencing data have been deposited in [The Genome Sequence Archive for Human (GSA-Human, https://ngdc.cncb.ac.cn/gsa-human)] (HRA002693 and HRA006117) (48, 49). Universal visualization codes of web-based plugins were deposited at the HIPLOT (ORG) website (50). Previously published data were used for this work (1, 7, 8, 35, 51, 52).