

Abstract
Agents which increase the intracellular cyclic GMP (cGMP) concentration and cGMP analogs inhibit cell growth in several different cell types, but it is not known which of the intracellular target proteins of cGMP is (are) responsible for the growth-suppressive effects of cGMP. Using baby hamster kidney (BHK) cells, which are deficient in cGMP-dependent protein kinase (G-kinase), we show that 8-(4-chlorophenylthio)guanosine-3′,5′-cyclic monophosphate and 8-bromoguanosine-3′,5′-cyclic monophosphate inhibit cell growth in cells stably transfected with a G-kinase Iβ expression vector but not in untransfected cells or in cells transfected with a catalytically inactive G-kinase. We found that the cGMP analogs inhibited epidermal growth factor (EGF)-induced activation of mitogen-activated protein (MAP) kinase and nuclear translocation of MAP kinase in G-kinase-expressing cells but not in G-kinase-deficient cells. Ras activation by EGF was not impaired in G-kinase-expressing cells treated with cGMP analogs. We show that activation of G-kinase inhibited c-Raf kinase activation and that G-kinase phosphorylated c-Raf kinase on Ser43, both in vitro and in vivo; phosphorylation of c-Raf kinase on Ser43 uncouples the Ras-Raf kinase interaction. A mutant c-Raf kinase with an Ala substitution for Ser43 was insensitive to inhibition by cGMP and G-kinase, and expression of this mutant kinase protected cells from inhibition of EGF-induced MAP kinase activity by cGMP and G-kinase, suggesting that Ser43 in c-Raf is the major target for regulation by G-kinase. Similarly, B-Raf kinase was not inhibited by G-kinase; the Ser43 phosphorylation site of c-Raf is not conserved in B-Raf. Activation of G-kinase induced MAP kinase phosphatase 1 expression, but this occurred later than the inhibition of MAP kinase activation. Thus, in BHK cells, inhibition of cell growth by cGMP analogs is strictly dependent on G-kinase and G-kinase activation inhibits the Ras/MAP kinase pathway (i) by phosphorylating c-Raf kinase on Ser43 and thereby inhibiting its activation and (ii) by inducing MAP kinase phosphatase 1 expression.
Many mitogenic stimuli, including growth factors like epidermal growth factor (EGF), cytokines such as granulocyte-monocyte colony-stimulating factor, and hormones which bind to G-protein-coupled receptors, activate the Ras/mitogen-activated protein (MAP) kinase pathway (42). Through a series of protein-protein interactions, Ras is converted to its active, GTP-bound form and promotes translocation of Raf kinase to the plasma membrane, where the latter is activated (29, 42). This initiates a cascade of sequential phosphorylation events leading to phosphorylation of tyrosine and threonine regulatory sites in the MAP kinase isozymes p44 and p42, which are then activated and can translocate to the nucleus (34, 43). Important MAP kinase substrates include the transcription factors c-Myc, c-Jun, ATF-2, and p62TCF, which are involved in triggering G1-S phase transition and initiation of cell proliferation (23, 42).
Nitric oxide (NO) is synthesized by several types of enzymes, two constitutively expressed and activated by Ca2+ and one transcriptionally regulated by a variety of inflammatory cytokines (41). One of the major intracellular targets of NO is soluble guanylate cyclase, a heme-containing enzyme which converts GTP to cyclic GMP (cGMP) and is activated several hundred-fold by NO (41, 45). In addition, cGMP is synthesized by membrane-bound receptor guanylate cyclases which are activated by peptide hormones such as atrial natriuretic factor (45). cGMP has several intracellular targets, including gated ion channels, cGMP-dependent protein kinase (G-kinase), a cGMP-activated phosphodiesterase, and a cGMP-inhibited phosphodiesterase (5, 41, 45).
NO-generating agents and natriuretic peptides inhibit the proliferation of several cell types, including endothelial, smooth muscle, and glomerular mesangial cells; membrane-permeable cGMP analogs mimic these effects (1, 17, 21, 22, 37, 44, 48, 53). To determine whether the growth-inhibitory effects of cGMP were mediated by G-kinase, we transfected baby hamster kidney (BHK) cells, which express very little detectable G-kinase activity, with an expression vector for G-kinase Iβ. We found that growth inhibition by cGMP analogs correlated with inhibition of growth factor signaling through the Ras/MAP kinase pathway and occurred in G-kinase-expressing but not in parental BHK cells; cells expressing a catalytically inactive G-kinase behaved similarly to parental cells. Moreover, we found that G-kinase inhibits the Ras/MAP kinase pathway at the level of c-Raf kinase by phosphorylating c-Raf kinase on Ser43 and that G-kinase induced MAP kinase phosphatase 1 (MKP-1) expression, but this occurred later than the G-kinase-induced inhibition of MAP kinase activation.
MATERIALS AND METHODS
Materials.
In stably transfected cells, wild-type G-kinase Iβ was expressed under control of the mouse metallothionein promoter (31); catalytically inactive G-kinase was expressed from the cytomegalovirus (CMV) early promoter and has a single amino acid mutation, Asp516 to Ala, in the catalytic domain of the enzyme (15). In transiently transfected cells, G-kinase Iβ was expressed from the CMV promoter (31). Mammalian expression vectors for wild-type c-Raf kinase and for c-Raf kinase with a mutation of Ser43 to Ala [Raf(S43-A)] were from H. Mischak (32); the expression vector for B-Raf kinase was from P. Stork (49). Bacterial expression vectors containing the first 149 amino acids of c-Raf kinase were from P. Worley (52), and the vectors containing the BXB mutant c-Raf kinase lacking amino acids 26 to 303 were from H. Mischak (32); the produced proteins are fused to glutathione S-transferase (GST). An expression vector for the influenza virus hemagglutinin (HA) epitope-tagged MAP kinase p44 was from M. Karin, University of California, San Diego (UCSD), and the monoclonal antibody used to immunoprecipitate HA-tagged MAP kinase was from BAbCO. The polyclonal G-kinase antibody was raised in rabbits (28); the monoclonal anti-Ras antibodies Y13-259 and Y13-238 and the polyclonal anti-c-Raf kinase, anti-B-Raf kinase, and anti-MAP kinase antibodies were from Santa Cruz Biotechnology, except for the polyclonal antibody against the dually phosphorylated, active form of MAP kinase, which was from Promega. The heptapeptide Kemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly) and protein kinase inhibitor (PKI), a specific cAMP-dependent protein kinase (A-kinase) inhibitor, were from S. Taylor, UCSD. G-kinase Iβ was expressed in baculovirus-infected insect cells and purified as described previously (36). Lipofectamine was from Life Technologies, myelin basic protein (MBP) and the Raf kinase cascade assay kit were from Upstate Biotechnology Incorporated, recombinant MAP kinase kinase (MEK-1) was from Santa Cruz Biotechnology, 8-(4-chlorophenylthio)guanosine-3′,5′-cyclic monophosphate (8-pCPT-cGMP) and 8-bromoguanosine-3′,5′-cyclic monophosphate (8-Br-cGMP) were from BioLog, and [γ-32PO4]ATP (3,000 Ci/mmol) was from New England Nuclear.
Cell culture.
Wild-type (parental) BHK cells were obtained from I. Scheffler, UCSD, and were routinely grown in Dulbecco’s modified eagle’s (DME) medium supplemented with 10% fetal bovine serum (FBS) unless stated otherwise (14). Cells were quantitated in a model ZM Coulter Counter after being removed from plates by trypsinization.
Generation of BHK cells stably expressing G-kinase.
Approximately 1.5 × 105 parental BHK cells were plated in wells of a six-well cluster dish and were transfected by using Lipofectamine with 2 μg of plasmid DNA, pMM11 for wild-type G-kinase (31) or pCB6-GK(D516-A) for catalytically inactive G-kinase (15). After 24 h, the cells were split 1:100; 24 h later, G418 was added at a final concentration of 1 mg/ml. The cells were incubated for an additional 15 days, at which time individual clones were assayed for G-kinase expression and activity. Clones were maintained in G418 at a concentration of 0.4 mg/ml and were used in experiments within 10 to 14 passages.
Assessment of G-kinase expression and measurement of G-kinase activity in cell extracts.
Cells were extracted, and G-kinase expression and activity were measured in 10,000 × g supernatants as described previously (14). Briefly, G-kinase expression was assessed by immunoblotting using a G-kinase I-specific polyclonal antibody and detection by enhanced chemiluminescence with a horseradish peroxidase-coupled secondary antibody; G-kinase activity was measured with Kemptide and [γ-32PO4]ATP as substrates in the presence of PKI to inhibit A-kinase activity. To determine the time course of G-kinase activation in intact cells, cells were incubated in the presence or absence of 250 μM 8-pCPT-cGMP for the indicated time and then rapidly rinsed five times in ice-cold phosphate-buffered saline before cells were extracted and Kemptide phosphorylation was measured for 10 min at 7°C to minimize dissociation of cGMP from the enzyme. Kemptide phosphorylation observed in BHK cells lacking G-kinase expression reflected nonspecific protein kinase activity and was subtracted (14).
Stimulation of BHK cells with EGF.
Parental BHK cells and stably transfected cells were placed at a density of 104 cells/cm2; 24 h later the cells were placed in starvation medium consisting of DME with 0.2% bovine serum albumin and 0.1% FBS. The cells were starved for 36 h; where indicated, 80 μM ZnCl2 was present during the last 24 h of starvation. Cells were treated with 10 to 100 ng of EGF per ml for 5 min prior to harvest (10 ng/ml provided near-maximal stimulation of MAP kinase activity). The indicated concentrations of 8-pCPT-cGMP or 8-Br-cGMP were added 30 min before the EGF except as indicated in the legend to Fig. 6.
FIG. 6.

Time course of EGF-induced MAP kinase activation and effect of preincubation time with 8-pCPT-cGMP. G-kinase-expressing clone C11 cells were serum starved in the presence of zinc. (A) Cells were incubated with 100 ng of EGF per ml for the indicated times prior to extraction (filled circles); to half of the cultures, 250 μM 8-pCPT-cGMP was added 30 min prior to addition of EGF (open circles). MAP kinase activation was measured as described for Fig. 3. (B) Cells were preincubated with 250 μM 8-pCPT-cGMP for the indicated times and then incubated for 5 min with 100 ng of EGF per ml prior to extraction; MAP kinase activation was measured as described above. Thus, for 0-min preincubation, cells received 8-pCPT-cGMP and EGF together for 5 min prior to harvesting. (C) Cells were incubated with 250 μM 8-pCPT-cGMP for the indicated times and then were washed and extracted rapidly to measure the amount of G-kinase activated in vivo as described in Materials and Methods. To minimize 8-pCPT-cGMP dissociation from G-kinase, protein kinase activity was measured at 7°C immediately after cell extraction (14). The data in panels A and B were obtained from scanning autoradiographs as described in Materials and Methods and are the means of duplicates from two experiments; data in panel C represent the means ± SD of three experiments.
Transient transfection of BHK cells.
BHK cells were transiently transfected in six-well cluster dishes by using Lipofectamine as previously described (14). After transfection in serum-free medium, cells were allowed to recover for 1 h in full serum-containing medium before being transferred to starvation medium and treated with EGF and cGMP analogs as described above.
Assessment of MAP kinase activation. (i) Assessment of MAP kinase phosphorylation by a gel shift assay.
Cells stimulated with EGF were harvested directly into sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sample buffer by adding the buffer to culture plates and scraping the cells with a rubber policeman. The lysates were heated immediately to 100°C for 5 min and clarified by centrifugation, and the particulate-free extracts were resolved on 9% polyacrylamide gels. Western blots were generated as described previously (39), using an antibody specific for p44 and p42 MAP kinase; in the phosphorylated activated state, both isoforms of MAP kinase migrate on gels with apparent higher molecular masses than the enzymes in their nonphosphorylated inactive state (10).
(ii) Assessment of MAP kinase phosphorylation by using an antibody specific for dually phosphorylated MAP kinase.
Western blots were generated as described above but developed with an affinity-purified rabbit polyclonal antibody which specifically recognizes the dually phosphorylated, active form of MAP kinase and was raised against a dually phosphorylated peptide sequence corresponding to Thr183 and Tyr185 of p42 (34). Equal loadings of protein were verified by reprobing the blot with the pan-MAP kinase antibody described above.
(iii) Measurement of MAP kinase activity.
MAP kinase activity was measured as described elsewhere (50). Briefly, cells treated as described above were extracted in a HEPES-based lysis buffer containing 1% Triton X-100 and protease and phosphatase inhibitors. After clarification by centrifugation, a MAP kinase-specific antibody was added to the extracts and the immunoprecipitates were collected on protein G-agarose beads (39). The immunoprecipitates were washed three times with lysis buffer and once with lysis buffer lacking the Triton X-100. The immunoprecipitates were incubated with 15 μg of MBP and [γ-32PO4]ATP (0.2 Ci/mmol), and half were applied to SDS-polyacrylamide gels, with phosphorylated MBP detected by autoradiography; immunoprecipitation of equal amounts of MAP kinase was verified by immunoblotting. The other half of the reaction products were applied to squares of P81 phosphocellulose paper which were washed four times in 75 mM phosphoric acid, and radioactivity was measured by liquid scintillation counting. The assay was linear with time and with protein concentration.
(iv) Assessment of nuclear translocation of MAP kinase.
When phosphorylated and activated, MAP kinase can translocate to the nucleus (43). We assessed the cellular location of MAP kinase by immunofluorescent staining using a MAP kinase-specific antibody and a fluorescein 5-isothiocyanate-conjugated goat anti-rabbit immunoglobulin; cells were fixed and stained as described previously for assessing the cellular location of G-kinase (15). Cells were visualized with a Bio-Rad MRC-1024 laser scanning confocal system equipped with a krypton-argon laser.
Measurement of Ras-bound GTP and GDP.
Ras-bound GTP and GDP were measured as described previously after immunoprecipitation of Ras from cell extracts with antibody Y13-259 (40). This method uses a coupled enzymatic assay and measures absolute amounts of GTP and GDP bound to Ras; the data are expressed as femtomoles of GTP or GDP per milligram of cell protein. Antibody Y13-259 is directed against the Ras effector domain; it prevents the action of Ras GTPase-activating protein (Ras GAP) on Ras (19). Theoretically, antibody Y13-259 may not immunoprecipitate Ras-GTP with proteins bound to the effector domain, whereas antibody Y13-238 directed against an epitope outside the effector domain can immunoprecipitate Ras-GTP complexed with effector molecules (18). However, immunoprecipitation of Ras with a combination of Y13-259 and Y13-238 did not increase the amount of Ras-GTP recovered. Moreover, immunoprecipitation of Ras with Y13-238 alone resulted in substantially lower amounts of Ras-GTP recovery, indicating the need for Ras GAP blockade by Y13-259 for optimal results.
Measurement of Raf kinase activity. (i) Raf kinase cascade assay.
c-Raf kinase activity was measured similarly to MAP kinase activity except that a c-Raf kinase-specific antibody was substituted for the MAP kinase-specific antibody in the immunoprecipitation procedure, and nonactivated nonphosphorylated MEK and MAP kinase were added to the assay system as recommended by the manufacturer. Control reactions, in which either MEK or MAP kinase was omitted, showed no MBP phosphorylation.
(ii) MEK-1 phosphorylation.
To determine MEK phosphorylation by Raf kinase directly, c-Raf or B-Raf kinase immunoprecipitates were incubated in the presence of 400 ng of recombinant MEK-1 and 125 μM [γ-32PO4]ATP (1 Ci/mmol) at 30°C for 10 to 20 min as indicated previously (46). The reaction product was analyzed by SDS-PAGE and autoradiography. Immunoprecipitation of equal amounts of Raf kinase was verified by immunoblotting. Both Raf kinase assays were linear with time and with protein concentration.
Assessment of c-Raf kinase phosphorylation by G-kinase. (i) In vitro studies.
GST fusion proteins containing the N-terminal 149 amino acids of c-Raf kinase or the BXB mutant protein of c-Raf kinase were immobilized on glutathione-agarose and were incubated with 20 μM [γ-32PO4]ATP (0.4 μCi/μmol) for 30 min at 30°C in the presence of 200 ng of purified G-kinase and 10 μM 8-Br-cGMP. Control incubations contained the GST fusion proteins in the absence of G-kinase. Reaction products were separated by SDS-PAGE, and the gels were stained with Coomassie blue 250 and exposed to X-ray film.
(ii) In vivo studies.
Parental BHK cells and BHK cells stably expressing G-kinase were placed at a density of 104 cells/cm2 and were transfected in six-well cluster dishes by using Lipofectamine with 1.5 μg of plasmid DNA containing wild-type c-Raf or Raf(S43-A). Cells were incubated for 1 h in DME medium with 10% FBS and then placed for 24 h in the previously described starvation medium. To radioactively label the cells with 32PO4, cells were incubated in phosphate-free medium for 1 h and then 32PO4 was added at a concentration of 0.2 mCi/ml for 4 h. Where indicated, 500 μM 8-pCPT-cGMP was present during the last 20 min of the radioactive labeling period. c-Raf kinase was immunoprecipitated from cell extracts as described above, and the washed immunoprecipitates were resuspended in SDS sample buffer and subjected to SDS-PAGE. Proteins were transferred to polyvinylidene fluoride membranes, which were exposed to X-ray film. Immunoprecipitation of equal amounts of c-Raf kinase was verified by immunoblotting.
Assessment of MKP-1 expression.
Total cytoplasmic RNA was extracted from cells, electrophoresed on denaturing formaldehyde-agarose gels, blotted onto nitrocellulose membranes, and hybridized to radioactively labeled probes as described previously (39). The probe for MKP-1 was generated by PCR using primers encoding nucleotides 140 to 161 and complementary to nucleotides 691 to 710 of the MKP-1 mRNA sequence (25); identity of the probe with MKP-1 was verified by DNA sequencing.
Data analysis.
Results are expressed as means ± standard deviations (SD) of results of three independent experiments performed in duplicate. The significance of differences in mean values was determined by the two-tailed Student t test. Densitometric scanning of autoradiographs for Fig. 6 and 11 was performed with an Alpha Innotech IS-1000 digital imaging system.
FIG. 11.

In vivo phosphorylation of Ser43 of c-Raf kinase by G-kinase. Parental BHK cells were transiently transfected with either empty vector (lanes 1 and 2) or wild-type c-Raf (Raf, wt) vector (lanes 3 and 4), and G-kinase-expressing clone C11 cells were transfected with either wild-type c-Raf (lanes 5 and 6) or Raf(S43-A) (lanes 7 and 8) as described in Materials and Methods. Approximately 2 × 105 cells were radioactively labeled with 32PO4 for 4 h, with 500 μM 8-pCPT-cGMP added to some of the cultures during the last 20 min of the labeling period. c-Raf kinase was immunoprecipitated, and the immunoprecipitates were subjected to SDS-PAGE. Proteins were transferred to polyvinylidene fluoride membranes, and autoradiographs were generated (upper panel). Equal amounts of c-Raf kinase were present in the immunoprecipitates, as demonstrated by immunoblotting (lower panel).
RESULTS
G-kinase expression in stably transfected BHK cells.
As we described previously (14), wild-type (parental) BHK cells express very low amounts of G-kinase, either when measured by an enzymatic activity assay (Fig. 1A) or when assessed by Western blotting (Fig. 1B). Of 24 G418-resistant clones obtained from transfection of a vector encoding wild-type G-kinase Iβ, 23 clones expressed various amounts of the enzyme; results for two of these clones, C11 and C18, are shown in Fig. 1. The metallothionein promoter was leaky, leading to G-kinase expression even in the absence of zinc, but addition of zinc increased G-kinase expression in all of these clones (Fig. 1). One clone, C21, showed very little G-kinase expression and served as a control (Fig. 1). The amount of G-kinase expressed in clone C11, in the presence of zinc, corresponds to 7 pmol/mg of protein, which is in the range of physiological G-kinase concentrations found in smooth muscle cells, neuronal cells, and platelets (1 to 9 pmol/mg of protein) (5).
FIG. 1.

G-kinase expression in stably transfected BHK cells. BHK cells were transfected with vectors encoding either wild-type G-kinase Iβ under control of the mouse metallothionein promoter or catalytically inactive G-kinase under control of the CMV promoter. Individual G418-resistant colonies were grown in the presence or absence of 80 μM ZnCl2 for 24 h prior to harvesting. (A) G-kinase activity was measured as described in Materials and Methods, using the synthetic peptide substrate Kemptide in the presence of the specific A-kinase inhibitor PKI; G-kinase activity was calculated as the difference in Kemptide phosphorylation in the presence and absence of 10 μM cGMP (14). (B) Whole-cell lysates (104 cells/lane) were fractionated by SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and probed with a G-kinase I-specific antibody. Cell line designations: Wt, wild-type (parental) BHK cells; C21, a G418-resistant BHK clone expressing minimal amounts of G-kinase; C18 and C11, BHK clones expressing significant amounts of wild-type G-kinase; D19, a BHK clone expressing catalytically inactive G-kinase containing a point mutation of aspartate to alanine at amino acid residue 516.
Of 17 G418-resistant clones that had been transfected with a vector encoding catalytically inactive G-kinase, 12 showed significant G-kinase protein expression as assessed by immunoblotting (Fig. 1B, D19). As expected, clone D19 showed the same minimal amount of G-kinase activity as parental cells (Fig. 1A).
G-kinase activation inhibits proliferation of BHK cells.
cGMP analogs inhibit the growth of several different types of cells (1, 17, 21, 22, 37, 44, 48, 53, 54). When we added either of two membrane-permeable cGMP analogs, 8-pCPT-cGMP and 8-Br-cGMP, to the culture media of BHK cells, there was no growth inhibition observed with parental BHK cells, which are G-kinase deficient, clone C21 cells, which express extremely low amounts of G-kinase, and clone D19 cells, which express catalytically inactive G-kinase (Fig. 2; in this and all subsequent figures only data obtained with 8-pCPT-cGMP are shown, but similar results were obtained with 8-Br-cGMP). In the absence of cGMP analogs, the growth rate of several clones expressing catalytically active G-kinase was similar to the rates observed in G-kinase-deficient cells, and the addition of zinc to the culture media did not affect growth significantly (Fig. 2, clones C18, C11, and C8). However, the addition of cGMP analogs to the culture media of these G-kinase-expressing clones resulted in significant growth inhibition: 8-pCPT-cGMP inhibited cell doubling rates by about 20 to 30% in the absence of zinc and by about 50 to 70% in the presence of zinc (Fig. 2, C18, C11, and C8). Since zinc did not affect cell growth in the absence of cGMP analogs or in G-kinase-deficient cells, these results suggest that the degree of growth inhibition correlated with the amount of G-kinase expressed. Thus, BHK cell growth was inhibited by cGMP analogs only when the cells contained significant amounts of G-kinase activity.
FIG. 2.

Effect of 8-pCPT-cGMP on the proliferation of BHK cells. BHK cells were cultured in media containing 5% heat-inactivated FBS in the absence (open and striped bars) or presence (stippled and solid bars) of 80 μM ZnCl2. Cells were cultured in the absence (open and stippled bars) or presence (striped and solid bars) of 500 μM 8-pCPT-cGMP. After 24 and 48 h, individual wells were trypsinized and cell counts were determined. The data are presented as the number of population doublings in 24 h. Cell line designations are as for Fig. 1; C8 is an additional clone expressing catalytically active G-kinase at levels similar to those expressed by C11.
G-kinase inhibits EGF-induced MAP kinase activation.
Since the Ras/MAP kinase pathway transmits growth-promoting signals from growth factor receptors to the nucleus (42), it seemed possible that G-kinase might exert its antiproliferative effect by inhibiting this key mitogenic pathway. It is well established that activation of A-kinase inhibits growth factor-induced activation of the Ras/MAP kinase pathway in several different types of cells (8, 13, 20, 51). Since activation of MAP kinase is the last step in the Ras/MAP kinase pathway, we assessed the effects of cGMP analogs on EGF-induced phosphorylation and activation of MAP kinase in BHK cells expressing various amounts of G-kinase.
(i) G-kinase inhibits MAP kinase phosphorylation.
EGF induced the phosphorylation of >90% of both the p42 and p44 isoforms of MAP kinase in parental cells and in all of the clones tested (Fig. 3A; note that the EGF-induced phosphorylation of p42 and p44 resulted in a shift of the two proteins to an apparently higher molecular mass on SDS-polyacrylamide gels). In the absence of zinc, 8-pCPT-cGMP inhibited the EGF-induced activation of MAP kinase by approximately 30 and 50% in clones C18 and C11, respectively (Fig. 3A). There was no effect of 8-pCPT-cGMP on EGF-induced activation of MAP kinase in parental cells and clone D19 cells, which express catalytically inactive G-kinase, and only a minimal effect of 8-pCPT-cGMP in clone C21 cells, which express very small amounts of G-kinase (Fig. 3A). When the cells were pretreated with ZnCl2 to increase the amount of G-kinase expressed, 8-pCPT-cGMP almost completely inhibited EGF-induced MAP kinase phosphorylation in clone C11 (Fig. 3B). Zinc had no effect on the EGF-induced MAP kinase phosphorylation in parental cells or clone C21 (not shown). As shown in Fig. 3C, the effect of 8-pCPT-cGMP on EGF-induced MAP kinase phosphorylation was dose dependent, with a maximal effect occurring at 500 μM drug.
FIG. 3.

Effect of 8-pCPT-cGMP on EGF-induced MAP kinase phosphorylation. Cells were serum starved for 36 h as described in Materials and Methods; 80 μM ZnCl2 was present for the last 24 h (B and C) or was not present (A). The cells were extracted, the extracts were resolved by SDS-PAGE, and MAP kinase isozymes p42 and p44 were detected by Western blotting using an antibody which recognizes both isozymes irrespective of their phosphorylation state. EGF induces a mobility shift which causes ≥90% of p42 and p44 to migrate with a higher apparent molecular mass indicative of the phosphorylated state of p42 and p44. Cell line designations are as for Fig. 1. (A) EGF (100 ng/ml; lanes 3 to 6) was added for the last 5 min prior to harvesting, and 8-pCPT-cGMP (250 μM; lanes 5 and 6) was added 30 min prior to addition of EGF. (B) Clone C11 cells were cultured in the absence (lanes 1 to 3) or presence (lanes 4 to 6) of zinc chloride and were treated with 500 μM 8-pCPT-cGMP (lanes 3 and 6) for 30 min prior to addition of EGF (lanes 2, 3, 5 and 6) for 5 min as described above. (C) Clone C11 cells were cultured in the presence of zinc and were treated with the indicated concentrations of 8-pCPT-cGMP (lanes 3 to 6) for 30 min prior to addition EGF (lanes 2 to 6) for 5 min as described above.
We also examined the phosphorylation state of MAP kinase by using an antibody specific for activated MAP kinase dually phosphorylated on Thr and Tyr (34). In serum-starved cells, the signal was weak and detectable only after prolonged exposure of the Western blots (compare lane 1 of Fig. 4A to lane 1 of Fig. 4B). EGF treatment increased the signal dramatically and to similar extents in all cell lines examined (Fig. 4A; only results for clone C8 cells in the absence of EGF are shown, but similar results were observed in the other cell lines). Preincubation with 8-pCPT-cGMP inhibited the EGF-induced MAP kinase phosphorylation in all clones expressing significant amounts of G-kinase activity but not in G-kinase-deficient C21 cells (Fig. 4A). In the absence of EGF, the addition of 8-pCPT-cGMP resulted in some inhibition of the low, basal MAP kinase phosphorylation in serum-starved G-kinase-expressing cells (Fig. 4B; compare lanes 2 to 4 to lane 1).
FIG. 4.

Assessment of MAP kinase phosphorylation by using a phospho-MAP kinase-specific antibody. Cells were serum starved in the absence of zinc and treated with 8-pCPT-cGMP and/or EGF as indicated. Western blots were prepared as described for Fig. 3 but were developed with an antibody specific for the dually phosphorylated, active form of MAP kinase as described in Materials and Methods. (A) EGF (10 ng/ml; lanes 2 to 9) was added for the last 5 min prior to harvesting, and 8-pCPT-cGMP (500 μM; lanes 3, 5, 7, and 9) was added 30 min prior to addition of EGF. Clone C8, C18, and C11 cells express significant amounts of G-kinase activity, whereas C21 cells are G-kinase deficient. (B) Clone C11 cells were left untreated (lane 1) or were treated with 500 μM 8-pCPT-cGMP for 15 (lane 2), 30 (lane 3), or 60 (lane 4) min prior to harvesting. Lane 5, cells treated with EGF (10 ng/ml) for 5 min. Exposure times were 15 s (A) and 5 min (B).
(ii) G-kinase inhibits MAP kinase activity.
When the EGF-induced activation of MAP kinase was assessed by measuring MBP phosphorylation in MAP kinase immunoprecipitates, we found that EGF induced a 14.3 ± 2.8- and a 16.0 ± 3.5-fold increase in MAP kinase activity in G-kinase-deficient and G-kinase-expressing cells, respectively (data are means ± SD of results of two experiments performed with three G-kinase-deficient and four G-kinase-expressing cell lines; there is no statistically significant difference between the G-kinase-deficient and G-kinase-expressing cell lines). When the cells were pretreated with 8-pCPT-cGMP, MAP kinase activation was unaffected in the G-kinase-deficient cells but was reduced by 60 to 70% in G-kinase-expressing cells (Fig. 5A shows results obtained with the previously described clones). Since there was some clonal variation in the cells’ response to EGF, we repeated the experiment in transiently transfected BHK cells. There was no significant effect of 8-pCPT-cGMP in cells transfected with the control vector, but there was a 65% inhibition of EGF-induced MAP kinase activity by 8-pCPT-cGMP in cells transfected with the G-kinase expression vector (Fig. 5B). The data are consistent with the results shown in Fig. 3 and 4 and indicate that significant inhibition of growth factor-induced MAP kinase activation by cGMP required the presence of G-kinase.
FIG. 5.

Effect of 8-pCPT-cGMP on EGF-induced MAP kinase activation. Cells were serum starved in the absence of zinc and treated with 10 ng of EGF per ml for 5 min with or without a 30-min preincubation with 500 μM 8-pCPT-cGMP as indicated. Cells were extracted in the presence of phosphatase and protease inhibitors, and MAP kinase was immunoprecipitated from the extracts as described in Materials and Methods; washed immunoprecipitates were incubated with MBP and [γ-32PO4]ATP for 10 min to determine MAP kinase activity. Half of the samples were analyzed by SDS-PAGE, with phosphorylated MBP visualized by autoradiography (upper panels); in the other half, phosphorylated MBP was separated from residual [γ-32PO4]ATP by differential binding to phosphocellulose paper and quantitated by scintillation counting (the lower panels summarize the means ± SD of three independent experiments). MAP kinase activity is expressed as a percentage of the EGF-induced MAP kinase activity of G-kinase-deficient C21 cells (A) or control vector-transfected BHK cells (B). An immunoblot developed with a pan-MAP kinase antibody demonstrated equal amounts of MAP kinase present in the immunoprecipitates (not shown). (A) Individual clones of stably transfected BHK cells expressing very low G-kinase activity (C21) or expressing high amounts of catalytically active G-kinase (C18, C11, and C8) or catalytically inactive G-kinase (D19) were analyzed as described above. (B) Wild-type BHK cells were transiently transfected with either empty vector (control vector) or a G-kinase expression vector (GK vector) as described in Materials and Methods. After transfection, cells were serum starved and treated as described above. Since the transfection efficiency of BHK cells was >75%, we measured MAP kinase activity in the entire cell population.
(iii) Time course of G-kinase activation and inhibition of MAP kinase phosphorylation.
In Chinese hamster lung fibroblasts, cAMP only transiently inhibits growth factor-stimulated MAP kinase activation: the maximal effect of cAMP was observed 5 min after growth factor was added, and by 10 min there was no difference between untreated and cAMP-treated cells (30). We therefore examined a time course of EGF-induced MAP kinase activation and found that for as long as 1 h after addition of EGF, growth factor-induced MAP kinase activation was inhibited in 8-pCPT-cGMP-treated G-kinase-expressing cells (Fig. 6A). As found by other workers (4), the activation of MAP kinase by EGF was maximal between 5 and 10 min and then declined; note that 8-pCPT-cGMP inhibited MAP kinase activation at every time point by approximately 50% and did not alter the kinetics. Like other workers who have studied A-kinase inhibition of growth factor-induced MAP kinase activation (30), we found that G-kinase had to be activated prior to adding the growth factor; in the case of 8-pCPT-cGMP, preincubating the cells with the drug for 10 to 15 min yielded essentially maximal inhibition of MAP kinase activation (Fig. 6B). When we measured in vivo activation of G-kinase by 8-pCPT-cGMP, we found that a 15-min exposure to the drug resulted in maximal activation of the kinase, correlating with the time required for maximal effects of 8-pCPT-cGMP on MAP kinase (Fig. 6C).
(iv) G-kinase inhibits EGF-induced nuclear translocation of MAP kinase.
In the phosphorylated activated state, MAP kinase translocates to the nucleus, where it phosphorylates several transcription factors involved in regulating cell growth and differentiation (9, 43). We found that 8-pCPT-cGMP inhibited EGF-induced nuclear translocation of MAP kinase in G-kinase-expressing BHK cells but not in G-kinase-deficient cells (compare Fig. 7C and F).
FIG. 7.

Effect of 8-pCPT-cGMP on EGF-induced MAP kinase nuclear translocation. Clone C11 (A to C) or clone C21 (D to F) cells were serum starved in the presence of zinc as described in Materials and Methods. Cells were incubated without additives (A and D) or with EGF (100 ng/ml) added for the last 10 min prior to fixing (B, C, E, and F); in panels C and F, 250 μM 8-pCPT-cGMP was added 30 min before the EGF. Cells were immunostained with a MAP kinase-specific antibody followed by a fluorescamine-labeled secondary antibody and were analyzed by confocal laser microscopy as described in Materials and Methods.
G-kinase does not inhibit EGF-induced Ras activation.
The data thus far demonstrate that activating G-kinase inhibits the terminal step in the Ras/MAP kinase pathway. To determine whether G-kinase was acting upstream or downstream of Ras, we assessed the effect of G-kinase activation on EGF-induced Ras activation. We found that EGF increased Ras activation (defined as the ratio of Ras-GTP over Ras-GTP plus Ras-GDP) approximately eightfold in parental BHK cells and in clone C11 cells and that pretreating the cells with 8-pCPT-cGMP had no effect on the degree of Ras activation (Table 1; only the data for clone C11 cells, which express high G-kinase activity, are shown). Thus, G-kinase was affecting the Ras/MAP kinase pathway distal to Ras activation.
TABLE 1.
Effects of EGF and 8-pCPT-cGMP on the activation state of Ras
| Culture conditiona | Concn (fmol/mg of protein)b
|
GTP 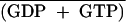 (%) (%) |
|
|---|---|---|---|
| GDP | GTP | ||
| Control | 866 ± 205 | 35 ± 11 | 4 |
| EGF | 841 ± 189 | 259 ± 55 | 31 |
| EGF + 8-pCPT-cGMP | 762 ± 184 | 265 ± 62 | 35 |
G-kinase-expressing BHK cells (clone C11) were treated with EGF (100 ng/ml) with or without 250 μM 8-pCPT-cGMP as described in Materials and Methods.
Ras was immunoprecipitated from 4 × 106 cells by using the monoclonal antibody Y13-259, and the amount of GTP and GDP bound to Ras was measured by the luciferase-based assay described in Materials and Methods. Data are means ± SD of three independent experiments.
G-kinase inhibits EGF-induced c-Raf kinase activation.
Because c-Raf kinase is immediately distal to Ras in the Ras/MAP kinase pathway and because A-kinase phosphorylates c-Raf kinase and thereby inhibits c-Raf kinase activity (8, 11, 16, 32, 51), we assessed the affect of G-kinase on EGF-induced c-Raf kinase activation by two methods. First, we examined MEK phosphorylation in c-Raf immunoprecipitates of stably transfected BHK cells expressing either very little G-kinase (C21) or significant amounts of G-kinase activity (C11 and C18). In untreated cells, MEK phosphorylation was barely detectable, but EGF increased MEK phosphorylation to similar degrees in all clones examined (Fig. 8A shows a short exposure; MEK phosphorylation in untreated cells was detectable only on longer exposures). While 8-pCPT-cGMP had no effect on EGF-induced MEK phosphorylation in Raf immunoprecipitates from the G-kinase-deficient cells (C21), it inhibited EGF-induced MEK phosphorylation in immunoprecipitates from cells containing G-kinase activity (C11 and C18). Similar amounts of Raf kinase were present in all immunoprecipitates (Fig. 8A, lower panel).
FIG. 8.

Effect of 8-pCPT-cGMP on EGF-induced c-Raf kinase activation measuring MEK phosphorylation. Cells were cultured as described for Fig. 5, and cells were extracted in the presence of phosphatase and protease inhibitors. c-Raf kinase was immunoprecipitated from the extracts, and washed immunoprecipitates were incubated with MEK-1 and [γ-32PO4]ATP for 10 min to determine Raf kinase activity as described in Materials and Methods. Control immunoprecipitates obtained with nonimmune rabbit serum were incubated with MEK-1 in parallel reactions (lane 10 in panel A and lane 7 in panel B). Samples were split in half and analyzed by SDS-PAGE, with proteins of one of the gels electroblotted onto membranes. Phosphorylated MEK-1 was visualized by autoradiography (upper panels); incubation of the blot with a c-Raf antibody demonstrated similar amounts of c-Raf kinase present in the immunoprecipitates (lower panels). (A) Individual clones of stably transfected BHK cells (approximately 107 cells) expressing very low G-kinase activity (C21) or expressing significant amounts of G-kinase activity (C11 and C18) were analyzed as described above. MEK-1 phosphorylation by c-Raf kinase immunoprecipitated from untreated cells was detectable on longer exposures (not shown). (B) Approximately 106 wild-type BHK cells were transiently transfected with either control vector or G-kinase vector as described for Fig. 5C and analyzed as described above.
Second, we quantitated c-Raf kinase activity in a coupled enzymatic assay (the Raf kinase cascade assay described in Materials and Methods). With this assay, we found that EGF increased c-Raf kinase activity 11.1 ± 2.1- and 7.3 ± 1.0-fold in parental and clone C11 cells, respectively (data are means ± SD of values from three independent experiments) (Fig. 9). This degree of c-Raf kinase activation is similar to what we observed for EGF-induced MAP kinase activation (Fig. 5), with clone C11 cells showing somewhat less c-Raf kinase activation than parental BHK cells. When the cells were treated with 8-pCPT-cGMP, the degrees of c-Raf kinase activation observed were 9.5 ± 1.9- and 3.0 ± 0.7-fold in parental and clone C11 cells, respectively (Fig. 9). Thus, 8-pCPT-cGMP inhibited EGF-induced c-Raf kinase activation by 14% in parental cells, a difference which did not reach statistical significance (P < 0.1), and by 59% in clone C11 cells, a difference which was statistically significant (P < 0.01). In parental BHK cells, the small degree of inhibition of c-Raf kinase by 8-pCPT-cGMP could be secondary to the drug cross-activating endogenous A-kinase (16, 51), even though 8-pCPT-cGMP is a relatively poor A-kinase activator (6). In G-kinase-expressing cells, the amount of inhibition of c-Raf kinase activation by 8-pCPT-cGMP is similar to what we found for 8-pCPT-cGMP inhibition of MAP kinase activation (Fig. 5) and again indicates that cGMP analogs require G-kinase activity for their effects.
FIG. 9.

Effect of 8-pCPT-cGMP on EGF-induced c-Raf kinase activation measured by a coupled enzymatic assay. Wild-type BHK (A) and clone C11 (B) cells were treated as described in the legend to Fig. 5, and c-Raf kinase was immunoprecipitated as described in Materials and Methods. Washed immunoprecipitates were sequentially incubated with recombinant, nonactivated MEK and MAP kinase in the presence of nonradioactive ATP; for the last 10 min of the reaction, MBP and [γ-32PO4]ATP were added, and 32PO4 incorporation into MBP was assessed as described for Fig. 5. The middle panel is a Western blot, probed for c-Raf kinase, of the gel shown in the top panel and demonstrates equal amounts of c-Raf kinase present in the immunoprecipitates. The values in the lower panel represent the means ± SD of three independent experiments.
To exclude the effect of clonal variation on our results, we examined MEK phosphorylation in Raf kinase immunoprecipitates from transiently transfected BHK cells. There was no significant effect of 8-pCPT-cGMP in parental BHK cells transfected with control vector, but there was significant inhibition of EGF-induced MEK phosphorylation by 8-pCPT-cGMP in cells transfected with the G-kinase expression vector (Fig. 8B).
G-kinase phosphorylates c-Raf kinase on Ser43.
A-kinase phosphorylates c-Raf kinase on Ser43, and phosphorylation of this residue has been shown to inhibit c-Raf kinase binding to Ras, thus effectively uncoupling the Ras/MAP kinase pathway at this step (7, 16, 51). We found that G-kinase efficiently phosphorylates Ser43 of c-Raf kinase both in vitro and in vivo.
(i) In vitro studies.
Purified G-kinase and [γ-32PO4]ATP were incubated with GST fusion constructs containing (i) the N-terminal 149 amino acids of c-Raf kinase [GST(1-149)Raf]; (ii) amino acids 51 to 149 of c-Raf kinase [GST(51-149)Raf]; and (iii) the N-terminal 149 amino acids of c-Raf kinase with Ser43 replaced by Asp [GST(1-149)Raf(S43-D)] (Fig. 10). The products of the reaction were analyzed on duplicate SDS-polyacrylamide gels. In Fig. 10A is shown Coomassie blue staining of one of the gels; the G-kinase monomer has an approximate mass of 74 kDa, GST(1-149)Raf and GST(1-149)Raf(S43-D) have approximate masses of 40 kDa, GST(51-149)Raf has an approximate mass of 32 kDa, and GST alone has an approximate mass of 27 kDa. In Fig. 10B is shown an autoradiograph of the duplicate gel; note that G-kinase highly phosphorylated the GST(1-149)Raf construct but that G-kinase did not phosphorylate the same construct with Ser43 mutated to Asp, nor did G-kinase phosphorylate the GST(51-149)Raf construct or GST alone. These studies indicate that Ser43 is the only residue within the first 149 amino acids of c-Raf kinase which is phosphorylated by G-kinase in vitro.
FIG. 10.

In vitro phosphorylation of Ser43 of c-Raf kinase by G-kinase. N-terminal fragments of c-Raf kinase fused to GST were purified from bacteria, immobilized on glutathione-agarose, and incubated for 30 min with 20 μM [γ-32PO4]ATP in the presence of purified G-kinase and 8-Br-cGMP as described in Materials and Methods. Reaction products were analyzed on duplicate SDS-polyacrylamide gels; one gel was stained with Coomassie blue R250 (A), and the other was exposed to X-ray film (B). Approximate masses of the proteins tested: GST(51-149)Raf, 32 kDa; GST(1-149), 40 kDa; GST(1-149)Raf(S43-D), 40 kDa; GST alone, 27 kDa. G-kinase (GK) added to the reaction can be seen on the Coomassie blue stain and, because of autophosphorylation, on the autoradiograph.
(ii) In vivo studies.
To determine if Ser43 of c-Raf kinase was also phosphorylated by G-kinase in vivo, we performed 32PO4-labeling experiments of parental and clone C11 BHK cells transfected with wild-type c-Raf kinase and Raf(S43-A) (Fig. 11). We found no increase in phosphorylation of wild-type c-Raf when parental cells were treated with 8-pCPT-cGMP, but we found greater than a threefold increase in c-Raf kinase phosphorylation when clone C11 cells were treated with 8-pCPT-cGMP (Fig. 11; compare lanes 3 and 4 to lanes 5 and 6). Thus, G-kinase clearly phosphorylated c-Raf kinase in vivo, and Ser43 appeared to be the only site phosphorylated by G-kinase because there was no increase in c-Raf kinase phosphorylation when clone C11 cells transfected with c-Raf(S43-A) were treated with 8-pCPT-cGMP (Fig. 11; compare lanes 7 and 8, to lanes 5 and 6). We confirmed the same amount of c-Raf kinase immunoprecipitated under each condition by probing the blot with an anti-c-Raf kinase antibody (Fig. 11, lower panels). Under the conditions shown, 32PO4 incorporation into endogenous Raf was below the limit of detection (Fig. 11, lanes 1 and 2).
The c-Raf kinase BXB mutant protein is a poor G-kinase substrate.
The BXB protein is a mutant of c-Raf kinase in which most of the regulatory domain, including the Ras binding domain, has been removed by deletion of amino acids 26 to 303 (38). It has been shown previously that A-kinase phosphorylates the BXB mutant protein, and the site of phosphorylation was localized to Ser621 (32). We therefore studied phosphorylation of the BXB mutant protein by G-kinase in vitro (Fig. 12). In the presence of G-kinase, a small amount of BXB phosphorylation was observed which was not observed in the absence of G-kinase; the phosphorylation of BXB by G-kinase was far less than the phosphorylation of GST(1-149)Raf and considerably less than G-kinase autophosphorylation (Fig. 12B). When Ser621 or Ser619 of the BXB protein were mutated to Ala, G-kinase phosphorylation of BXB was reduced but not eliminated, indicating that the small amount of G-kinase phosphorylation of BXB was not only on Ser621, the previously identified A-kinase phosphorylation site (32) (Fig. 12A). Thus, one can conclude that the BXB protein is a very poor in vitro G-kinase substrate.
FIG. 12.

Phosphorylation of c-Raf kinase BXB mutant protein by G-kinase. The BXB mutant protein, which is lacking amino acids 26 to 303 of c-Raf kinase, was purified from bacteria as a GST fusion protein and incubated with G-kinase and [γ-32PO4]ATP as described in the legend to Fig. 10. Reaction products were analyzed by SDS-PAGE, and the gel was stained with Coomassie blue R250 (left panels) and exposed to X-ray film (right panels); the BXB protein has an approximate mass of 60 kDa. (A) Lane 1, BXB protein; lane 2, BXB protein with Ser 619 mutated to alanine [BXB(S619-A)]; lane 3, BXB protein with Ser 621 mutated to alanine [BSB(S621-A)]. (B) For direct comparison, similar amounts of GST(1-149)Raf (lane 1) and BXB protein (lane 3) were incubated with G-kinase as described above. Lane 2, G-kinase (GK) only. As in Fig. 10, G-kinase added to the reaction can be seen both on the Coomassie blue stain and on the autoradiograph.
Raf(S43-A) and B-Raf kinase are insensitive to inhibition by G-kinase.
To determine whether the G-kinase inhibition of EGF-stimulated c-Raf kinase activity was mediated by phosphorylation of Ser43, we examined the effect of G-kinase on the activity of the mutant Raf(S43-A). To this end, G-kinase-expressing C11 cells were transiently transfected with expression vectors encoding either wild-type c-Raf or Raf(S43-A). The amount of wild-type or mutant c-Raf kinase expressed from these vectors was about 5- to 10-fold more than the amount of endogenous c-Raf kinase immunoprecipitated from cells transfected with the control vector (Fig. 13A, lower panel). In C11 cells transfected with wild-type c-Raf, the EGF-induced increase in MEK phosphorylation was inhibited in the presence of 8-pCPT-cGMP, as observed with endogenous Raf activity (Fig. 13A, upper panel; compare lanes 5 and 6). However, in C11 cells transfected with Raf(S43-A), there was little effect of 8-pCPT-cGMP on EGF-induced MEK phosphorylation (Fig. 13A; compare lanes 8 and 9). Endogenous Raf kinase activity, measured in cells transfected with the control vector, contributed little to the MEK phosphorylation measured in cells transfected with the wild-type or mutant Raf kinase expression vector (Fig. 13A; compare lanes 5 and 8 to lane 2). Thus, mutant Raf(S43-A) activity was insensitive to inhibition by G-kinase, suggesting that Ser43 is the major target for regulation by G-kinase.
FIG. 13.

Effects of G-kinase on the activities of mutant Raf(S43-A) and B-Raf kinase. G-kinase-expressing C11 cells were transiently transfected as described for Fig. 11; cells were serum starved and treated with 8-pCPT-cGMP and/or EGF as described for Fig. 5. Immunoprecipitates of c-Raf kinase (A) or B-Raf kinase (B) were incubated with MEK-1 and [γ-32PO4]ATP for 20 min as described in Materials and Methods. Autoradiographs of phosphorylated MEK-1 are shown in the upper panels; the immunoprecipitates were also analyzed by immunoblotting with c-Raf or B-Raf kinase antibody as indicated in the lower panels. (A) C 11 cells were transfected with empty vector (control vector) or with expression vectors encoding either wild-type c-Raf kinase [Raf (WT)] or Raf(S43-A). c-Raf kinase activity was determined as described above. Lane 10, MEK-1 incubated in the absence of immunoprecipitates. (B) C11 cells were transfected with control vector or with an expression vector for B-Raf kinase; B-Raf kinase activity and the amount of B-Raf in the immunoprecipitates were determined as described above. The amount of MEK-1 phosphorylation seen with immunoprecipitates from cells transfected with empty vector was little above the background MEK-1 phosphorylation observed with control immunoprecipitates obtained with nonimmune rabbit serum (lane 9).
Since Ser43 of c-Raf kinase is not conserved in B-Raf kinase, we next examined the effect of G-kinase on the activity of B-Raf kinase. BHK cells express very little if any endogenous B-Raf kinase, but transient transfection of a B-Raf kinase expression vector into C11 cells resulted in high levels of B-Raf expression (Fig. 13B, lower panel). Consistently, we observed high basal B-Raf kinase activity in serum-starved cells, and EGF treatment resulted only in a small increase in B-Raf kinase activity; similar findings have been reported by others (26). There was no significant effect of 8-pCPT-cGMP on basal or EGF-stimulated B-Raf kinase activity (Fig. 13B, upper panel). Thus, B-Raf kinase activity does not appear to be regulated by G-kinase.
Expression of Raf(S43-A) or B-Raf protects cells from MAP kinase inhibition by cGMP and G-kinase.
To test whether the G-kinase inhibition of EGF-stimulated MAP kinase activity was primarily mediated by the inhibition of c-Raf by G-kinase, we transfected G-kinase-expressing clone C11 cells with mutant Raf(S43-A) or B-Raf, which we had found to be insensitive to inhibition by G-kinase. Cells were cotransfected with HA-tagged MAP kinase, and MAP kinase activity was measured in immunoprecipitates obtained with anti-HA antibody from cells treated with EGF for 5 min with or without a 30-min preincubation with 8-pCPT-cGMP. In cells transfected with wild-type c-Raf, 8-pCPT-cGMP inhibited EGF-induced MAP kinase activity by 50.0% ± 11.3%; the effect of 8-pCPT-cGMP was less than observed previously in clone C11 cells (Fig. 5A), possibly because of the larger amounts of c-Raf kinase expressed in the present experiments. However, in cells transfected with mutant Raf(S43-A) and B-Raf, the levels of inhibition by 8-pCPT-cGMP were only 20.2% ± 8.6% and 14.7 ± 6.4%, respectively (data are the means ± SD of values from three independent experiments). Thus, expression of mutant Raf(S43-A) or B-Raf kinase significantly protected cells from inhibition of EGF-induced MAP kinase activity by cGMP and G-kinase. These results suggest that phosphorylation of c-Raf Ser43 by G-kinase is a major, albeit not the only, mechanism by which G-kinase regulates the MAP kinase pathway.
G-kinase induces MKP-1 expression.
MKP-1 is a dual-specificity protein phosphatase capable of removing the phosphates from both the phosphotyrosine and phosphothreonine residues of activated MAP kinase (24). cAMP analogs induce MKP-1 expression in Chinese hamster lung fibroblasts, and 8-Br-cGMP induces MKP-1 expression in rat glomerular mesangial cells which contain endogenous G-kinase; MKP-1 expression was maximal 1 h after drug exposure in the former case and 30 min after drug exposure in the latter case (3, 44). We found that both 8-pCPT-cGMP and 8-Br-cGMP induced MKP-1 mRNA expression in G-kinase-expressing BHK cells but not in parental cells and that the maximal effect occurred between 30 min and 1 h of drug exposure.
DISCUSSION
We found that cGMP analogs inhibited the Ras/MAP kinase pathway in BHK cells in a strictly G-kinase-dependent manner and that in cells expressing high amounts of G-kinase the cGMP analogs inhibited cell growth. Thus, the inhibition of cell growth by cGMP analogs noted by other workers in several cell types (1, 17, 21, 22, 37, 44, 48, 53) may be at least partially mediated by G-kinase inhibition of the Ras/MAP kinase pathway. This notion was postulated recently by a group of investigators for the cGMP-mediated inhibition of vascular smooth muscle cell growth since these workers found that 8-Br-cGMP inhibits the Ras/MAP kinase pathway in these cells and that the effect of 8-Br-cGMP was blocked by KT5822, an inhibitor of protein kinases (53). However, as discussed in an editorial that accompanied the paper, KT5822 shows only partial selectivity for G-kinase and the authors provided no evidence for its membrane permeability or efficacy in intact cells (33). By using BHK cells which are G-kinase deficient, we have been able to demonstrate that cGMP analogs clearly require G-kinase expression to inhibit the Ras/MAP kinase pathway. Our results do not exclude the possibility that cGMP inhibits cell growth through mechanisms other than inhibition of the Ras/MAP kinase pathway, but at least in BHK cells, these other mechanisms seem to require G-kinase since we found no inhibition of cell growth in cells lacking G-kinase activity.
The loss of G-kinase expression in many cultured cell lines may explain why some investigators found no effect of 8-Br-cGMP on EGF- or lysophosphatidic acid-induced activation of MAP kinase in Rat-1 fibroblasts (20). Similarly, low G-kinase activity may explain the lack of an effect of cGMP analogs on serum-induced mitogenesis of BALB/3T3 fibroblasts (12). In cultured vascular smooth muscle cells transfected with G-kinase, Boerth et al. (2) found only transient growth inhibition by 8-pCPT-cGMP, and the cells later reached the same saturation density as control cells transfected with an empty vector; the small effect of 8-pCPT-cGMP on the growth of these cells may be explained by the use of only 20 μM 8-pCPT-cGMP, which is generally a suboptimal concentration of this cGMP analog for intact cells (6).
There was no change in EGF-induced Ras activation in G-kinase-expressing cells treated with cGMP analogs, which indicated that the series of steps from EGF binding to its receptor to Ras activation was not affected by G-kinase. Thus, G-kinase inhibition of MAP kinase activation had to be at the level of either c-Raf kinase or MEK. We found that G-kinase inhibited EGF-induced c-Raf kinase activation and that the degree of c-Raf kinase inhibition was similar to the degree of MAP kinase inhibition (compare Fig. 5 and 9). Furthermore, we found that G-kinase phosphorylated Ser43 of c-Raf kinase efficiently both in vitro and in vivo; Ser43 is next to the Ras-binding domain of c-Raf kinase (47). Several previous groups of workers studying A-kinase inhibition of MAP kinase activation have shown that A-kinase phosphorylates c-Raf kinase on Ser43, also both in vitro and in vivo, and that phosphorylation at this site inhibits c-Raf kinase binding to Ras (7, 16, 51). (An abstract presenting results that are in disagreement with the previous work concerning the mechanism for A-kinase inhibition of Raf kinase has appeared recently [43a].) Thus, phosphorylation at Ser43 effectively uncouples the Ras-Raf kinase interaction.
In addition to Ser43, A-kinase phosphorylates Ser621 of c-Raf kinase in vitro and in vivo; this residue is in the catalytic domain of c-Raf kinase, and its phosphorylation reduces the enzyme’s catalytic activity (32, 50). We could not demonstrate efficient phosphorylation of Ser621 by G-kinase under optimal conditions in vitro, and it is therefore unlikely that G-kinase phosphorylates Ser621 in vivo. Moreover, the in vivo phosphorylation studies using wild-type c-Raf kinase and Raf(Ser43-A) suggest that Ser43 of c-Raf kinase is the only site phosphorylated significantly by G-kinase in vivo (Fig. 11). Since the mutant Raf(S43-A) was insensitive to inhibition by G-kinase, our results indicate that Ser43 in c-Raf is the major target for G-kinase regulation (Fig. 13A).
B-Raf kinase does not contain any potential A-kinase or G-kinase phosphorylation sites near its N-terminal Ras-binding domain corresponding to Ser43 of c-Raf, although there are two other potential A-kinase or G-kinase phosphorylation sites near the C-terminal catalytic domain and A-kinase has been reported to phosphorylate B-Raf kinase in vitro (35). We found no effect of cGMP and G-kinase on the activity of B-Raf kinase in serum-starved and EGF-treated cells (Fig. 13B). The effect of cAMP and A-kinase on B-Raf kinase activity is controversial: one group reported activation (49), whereas others found no effect or an inhibitory effect (11, 26, 35). Ectopic expression of B-Raf kinase in Rat-1 cells was reported to protect the cells from cAMP-mediated inhibition of growth factor-stimulated MAP kinase activity (11). We found that expression of Raf(S43-A) or B-Raf kinase in G-kinase-expressing BHK cells protected the cells from cGMP-mediated inhibition of EGF-stimulated MAP kinase activity. Since these two Raf kinases were refractory to inhibition by G-kinase (Fig. 13), we conclude that inhibition of wild-type c-Raf through phosphorylation of Ser43 by G-kinase is a major (albeit not the only) mechanism for rapid inhibition of the Raf/MAP kinase pathway by cGMP.
Part of the mechanism by which G-kinase inhibits growth factor-induced MAP kinase activation could be through G-kinase increasing the expression of MKP-1. However, the G-kinase-dependent induction of MKP-1 mRNA expression occurred with a delay of 30 min, much later than the G-kinase-mediated inhibition of EGF-induced MAP kinase activation that we observed in BHK cells. Thus, increased MKP-1 expression could contribute to prolonged effects of G-kinase activation, for example, to the inhibition of cell growth that we observed, but not to the rapid inhibitory effects of G-kinase on MAP kinase activation which we observed when cells were pretreated for only 5 to 15 min with 8-pCPT-cGMP prior to addition of EGF (Fig. 6B).
ACKNOWLEDGMENTS
We thank P. Worley and H. Mischak for the c-Raf kinase expression vectors, M. Karin for HA-tagged MAP kinase, and S. Taylor for PKI and Kemptide.
This work was supported in part by NSF grant MCB-9506327 and a University of California Cancer Research Coordinating Committee grant to R.B.P. and USPHS grant GM4960 and an American Heart Association grant to G.R.B. The Bio-Rad MRC-1024 laser scanning confocal system is part of the San Diego Microscopy and Imaging Resource at UCSD supported by NIH grant RR04050 (principal investigator, M. H. Ellisman); we thank N. Alinejad for technical assistance.
REFERENCES
- 1.Appel R G. Growth-regulatory properties of atrial natriuretic factor. Am J Physiol. 1992;262:F911–F918. doi: 10.1152/ajprenal.1992.262.6.F911. [DOI] [PubMed] [Google Scholar]
- 2.Boerth N J, Dey N B, Cornwell T L, Lincoln T M. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res. 1997;34:245–259. doi: 10.1159/000159231. [DOI] [PubMed] [Google Scholar]
- 3.Brondello J-M, Brunet A, Pouyssègur J, McKenzie F R. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J Biol Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- 4.Burgering B M, Pronk G J, vanWeeren P C, Chardin P, Bos J L. cAMP antagonizes p21ras-directed activation of extracellular signal-regulated kinase 2 and phosphorylation of mSos nucleotide exchange factor. EMBO J. 1993;12:4211–4220. doi: 10.1002/j.1460-2075.1993.tb06105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butt E, Geiger J, Jarchau T, Lohmann S M, Walter U. The cGMP-dependent protein kinase—gene, protein, and function. Neurochem Res. 1993;18:27–42. doi: 10.1007/BF00966920. [DOI] [PubMed] [Google Scholar]
- 6.Butt E, Nolte C, Schulz S, Beltman J, Beavo J A, Jastorff B, Walter U. Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochem Pharmacol. 1992;43:2591–2600. doi: 10.1016/0006-2952(92)90148-c. [DOI] [PubMed] [Google Scholar]
- 7.Chuang E, Barnard D, Hettich L, Zhang X-F, Avruch J, Marshall M S. Critical binding and regulatory interactions between Ras and Raf occur through a small, stable N-terminal domain of Raf and specific Ras effector residues. Mol Cell Biol. 1994;14:5318–5325. doi: 10.1128/mcb.14.8.5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cook S J, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–1071. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- 9.Davis R J. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 10.de Vries-Smits A M M, Burgering B M T, Leevers S J, Marshall C J, Bos J L. Involvement of p21ras in activation of extracellular signal-regulated kinase 2. Nature. 1992;357:802–805. doi: 10.1038/357602a0. [DOI] [PubMed] [Google Scholar]
- 11.Erhardt P, Troppmair J, Rapp U R, Cooper G M. Differential regulation of Raf-1 and B-Raf and Ras-dependent activation of mitogen-activated protein kinase by cyclic AMP in PC12 cells. Mol Cell Biol. 1995;15:5524–5530. doi: 10.1128/mcb.15.10.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg U C, Hassid A. Nitric oxide generating vasodilators inhibit mitogenesis and proliferation on BALB/c 3T3 fibroblasts by a cyclic GMP-independent mechanism. Biochem Biophys Res Commun. 1990;171:474–479. doi: 10.1016/0006-291x(90)91417-q. [DOI] [PubMed] [Google Scholar]
- 13.Graves L M, Bornfeldt K E, Raines E W, Potts B C, Macdonald S G, Ross R, Krebs E G. Protein kinase A antagonizes platelet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proc Natl Acad Sci USA. 1993;90:10300–10304. doi: 10.1073/pnas.90.21.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gudi T, Huvar I, Meinecke M, Lohmann S M, Boss G R, Pilz R B. Regulation of gene expression by cGMP-dependent protein kinase. J Biol Chem. 1996;271:4597–4600. doi: 10.1074/jbc.271.9.4597. [DOI] [PubMed] [Google Scholar]
- 15.Gudi T, Lohmann S M, Pilz R B. Regulation of gene expression by cyclic GMP-dependent protein kinase requires nuclear translocation of the kinase: identification of a nuclear localization signal. Mol Cell Biol. 1997;17:5244–5254. doi: 10.1128/mcb.17.9.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Häfner S, Adler H S, Mischak H, Janosch P, Heidecker G, Wolfman A, Fippig S, Lohse M, Ueffing M, Kolch W. Mechanism of inhibition of Raf-1 by protein kinase A. Mol Cell Biol. 1994;14:6696–6703. doi: 10.1128/mcb.14.10.6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagiwara H, Inoue A, Yamaguchi A, Yokose S, Furuya M, Tanaka S, Hirose S. cGMP produced in response to ANP and CNP regulates proliferation and differentiation of osteoblastic cells. Am J Physiol. 1996;270:C1311–C1318. doi: 10.1152/ajpcell.1996.270.5.C1311. [DOI] [PubMed] [Google Scholar]
- 18.Hallberg B, Rayter S I, Downward J. Interaction of Ras and Raf in intact mammalian cells upon extracellular stimulation. J Biol Chem. 1994;269:3913–3916. [PubMed] [Google Scholar]
- 19.Hattori S, Clanton D J, Satoh T, Nakamura S, Kaziro Y, Kawakita M, Shih T Y. Neutralizing monoclonal antibody against ras oncogene product p21 which impairs guanine nucleotide exchange. Mol Cell Biol. 1987;7:1999–2002. doi: 10.1128/mcb.7.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hordijk P L, Verlaan I, Jalink K, vanCorven E J, Moolenaar W H. cAMP abrogates the p21ras-mitogen-activated protein kinase pathway in fibroblasts. J Biol Chem. 1994;269:3534–3538. [PubMed] [Google Scholar]
- 21.Hutchinson H G, Trindade P T, Cunanan D B, Wu C F, Pratt R E. Mechanisms of natriuretic-eptide-induced growth inhibition of vascular smooth muscle cells. Cardiovasc Res. 1997;35:158–167. doi: 10.1016/s0008-6363(97)00086-2. [DOI] [PubMed] [Google Scholar]
- 22.Itoh H, Pratt R E, Ohno M, Dzau V J. Atrial natriuretic polypeptide as a novel antigrowth factor of endothelial cells. Hypertension. 1992;19:758–760. doi: 10.1161/01.hyp.19.6.758. [DOI] [PubMed] [Google Scholar]
- 23.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 24.Keyse S M. An emerging family of dual specificity MAP kinase phosphatases. Biochim Biophys Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- 25.Kwak S P, Hakes D J, Martell K J, Dixon J E. Isolation and characterization of a human dual specificity protein-tyrosine phosphatase gene. J Biol Chem. 1994;269:3596–3604. [PubMed] [Google Scholar]
- 26.Lange-Carter C A, Johnson G L. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science. 1994;265:1458–1461. doi: 10.1126/science.8073291. [DOI] [PubMed] [Google Scholar]
- 27.Lohmann S M, Vaandrager A B, Smolenski A, Walter U, De Jonge H R. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem Sci. 1997;22:307–312. doi: 10.1016/s0968-0004(97)01086-4. [DOI] [PubMed] [Google Scholar]
- 28.Markert T, Vaandrager A B, Gambaryan S, Poehler D, Haeusler C, Walter U, De Jonge H R, Jarchau T, Lohmann S M. Endogenous expression of type II cGMP-dependent protein kinase mRNA and protein in rat intestine. J Clin Investig. 1995;96:822–830. doi: 10.1172/JCI118128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marshall M. Ras target proteins in eukaryotic cells. FASEB J. 1995;9:1311–1318. doi: 10.1096/fasebj.9.13.7557021. [DOI] [PubMed] [Google Scholar]
- 30.McKenzie F R, Pouyssegur J. cAMP-mediated growth inhibition in fibroblasts is not mediated via mitogen-activated protein (MAP) kinase (ERK) inhibition. cAMP-dependent protein kinase induces a temporal shift in growth factor-stimualted MAP kinases. J Biol Chem. 1996;271:13476–13483. doi: 10.1074/jbc.271.23.13476. [DOI] [PubMed] [Google Scholar]
- 31.Meinecke M, Geiger J, Butt E, Sandberg M, Jahnsen T, Chakraborty T, Walter U, Jarchau T, Lohmann S. Human cyclic GMP-dependent protein kinase Iβ overexpression increases phosphorylation of an endogenous focal contact-associated vasodilator-stimulated phosphoprotein without altering the thrombin-evoked calcium response. Mol Pharmacol. 1994;46:283–290. [PubMed] [Google Scholar]
- 32.Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M, Philipp A, Kolch W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol Cell Biol. 1996;16:5409–5418. doi: 10.1128/mcb.16.10.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murad F. What are the molecular mechanisms for the antiproliferative effects of nitric oxide and cGMP in vascular smooth muscle? Circulation. 1997;95:1101–1103. doi: 10.1161/01.cir.95.5.1101. [DOI] [PubMed] [Google Scholar]
- 34.Payne D M, Rossomando A J, Martino P, Erickson A K, Her J-H, Shabanowitz J, Hunt D F, Weber M J, Sturgill T W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase) EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peraldi P, Frödin F, Barnier J V, Calleja V, Scimeca J-C, Filloux C, Calothy G, van Obberghen E. Regulation of the MAP kinase cascade in PC12 cells: B-Raf activates MEK-1 (MAP kinase or ERK kinase) and is inhibited by cAMP. FEBS Lett. 1995;357:290–296. doi: 10.1016/0014-5793(94)01376-c. [DOI] [PubMed] [Google Scholar]
- 36.Pöhler D, Butt E, Meissner J, Müller S, Lohse M, Walter U, Lohmann S M, Jarchau T. Expression, purification and characterization of the cGMP-dependent protein kinase Iβ and II using the baculovirus system. FEBS Lett. 1995;374:419–425. doi: 10.1016/0014-5793(95)01168-e. [DOI] [PubMed] [Google Scholar]
- 37.Porter J G, Catalano R, McEnroe G, Lewicki J A, Protter A A. C-type natriuretic peptide inhibits growth factor-dependent DNA synthesis in smooth muscle cells. Am J Physiol. 1992;263:C1001–C1006. doi: 10.1152/ajpcell.1992.263.5.C1001. [DOI] [PubMed] [Google Scholar]
- 38.Ruder T, Heidecker G, Rapp U R. Serum-, TPA-, and Ras-induced expression from Ap-1/Ets-driven promoters requires Raf-1 kinase. Genes Dev. 1992;6:545–556. doi: 10.1101/gad.6.4.545. [DOI] [PubMed] [Google Scholar]
- 39.Scheele J S, Pilz R B, Quilliam L A, Boss G R. Identification of a Ras-related protein in murine erythroleukemia cells that is a cAMP-dependent protein kinase substrate and is phosphorylated during chemically induced differentiation. J Biol Chem. 1994;269:18599–18606. [PubMed] [Google Scholar]
- 40.Scheele J S, Rhee J M, Boss G R. Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: comparison of parental and Ras-overproducing NIH 3T3 fibroblasts. Proc Natl Acad Sci USA. 1995;92:1097–1100. doi: 10.1073/pnas.92.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt H H H W, Lohmann S M, Walter U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta. 1993;1178:153–175. doi: 10.1016/0167-4889(93)90006-b. [DOI] [PubMed] [Google Scholar]
- 42.Seger R, Krebs E. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 43.Seth A, Gonzalet F A, Gupta S, Raden D L, Davis R J. Signal transduction within the nucleus by mitogen-activated protein kinase. J Biol Chem. 1992;267:24796–24804. [PubMed] [Google Scholar]
- 43a.Sidovar M F, He Y, Graves L M. Cellular regulation by protein phosphorylation: forty years of progress. Seattle: University of Washington School of Medicine; 1997. Phosphorylation of serine 43 is not required for the inhibition of c-Raf-1 kinase by cAMP, abstr. P5A-14; p. 54. [Google Scholar]
- 44.Sugimoto T, Haneda M, Togawa M, Isono M, Shikano T, Araki S, Nakagawa T, Kashiwagi A, Guan K-L, Kikkawa R. Atrial natriuretic peptide induces the expression of MKP-1, a mitogen-activated protein kinase phosphatase, in glomerular mesangial cells. J Biol Chem. 1996;271:544–547. doi: 10.1074/jbc.271.1.544. [DOI] [PubMed] [Google Scholar]
- 45.Vaandrager A B, De Jonge H R. Signaling by cGMP-dependent protein kinases. Mol Cell Biochem. 1996;157:23–30. doi: 10.1007/BF00227877. [DOI] [PubMed] [Google Scholar]
- 46.Vaillancourt R R, Gardner A M, Johnson G L. B-Raf-dependent regulation of the MEK-1/mitogen-activated protein kinase pathway in PC12 cells and regulation by cyclic AMP. Mol Cell Biol. 1994;14:6522–6530. doi: 10.1128/mcb.14.10.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vojtik A B, Hollenberg S M, Cooper J A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:204–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 48.Vollmar A M, Schmidt K-N, Schulz R. Natriuretic peptide receptors on rat thymocytes: inhibition of proliferation by atrial natriuretic peptide. Endocrinology. 1996;137:1706–1713. doi: 10.1210/endo.137.5.8612505. [DOI] [PubMed] [Google Scholar]
- 49.Vossler M R, Yao H, York R D, Pan M-G, Rim C S, Stork P J S. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Raf1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 50.Whitehurst C E, Owaki H, Bruder J T, Rapp U R, Gepper T D. The MAP kinase activity of the catalytic domain of RAF-1 is regulated independently of Ras binding in T cells. J Biol Chem. 1995;270:5594–5599. doi: 10.1074/jbc.270.10.5594. [DOI] [PubMed] [Google Scholar]
- 51.Wu J, Dent P, Jelinek T, Wolfman A, Weber M J, Sturgill T W. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science. 1993;262:1065–1068. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- 52.Yee W M, Worley P F. Rheb interacts with Raf-1 kinase and may function to integrate growth factor- and protein kinase A-dependent signals. Mol Cell Biol. 1997;17:921–933. doi: 10.1128/mcb.17.2.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu S-M, Hung L-M, Lin C-C. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway. Circulation. 1997;5:1269–1277. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]
- 54.Yu S M, Cheng Z J, Guh J H, Lee F Y, Kuo S C. Mechanism of anti-proliferation caused by YC-1, an indazole derivative, in cultured rat A10 vascular smooth-muscle cells. Biochem J. 1995;306:787–792. doi: 10.1042/bj3060787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]