

Abstract
Background
Young‐onset multiple system atrophy (YOMSA) is defined as the onset of multiple system atrophy (MSA) before the age of 40 years old. YOMSA is rare and there is much uncertainty of the phenotype and natural history in patients with YOMSA.
Objective
The objective is to evaluate the characteristics and disease course of patients with YOMSA.
Methods
We retrospectively reviewed medical records of patients with MSA who were evaluated at all Mayo Clinic sites from 1998 to 2021. We identified patients with YOMSA and evaluated clinical characteristics, autonomic function testing results, and disease course.
Results
Of 1496 patients with a diagnosis of clinically probable or clinically established MSA, 20 patients had YOMSA. The median age of onset was 39.1 (interquartile range [IQR] = 37.1, 40.1) years; 13 patients (65%) were male. MSA‐parkinsonism was the most common subtype (65%). The median duration of symptom onset to YOMSA diagnosis was 4.9 (IQR = 3.7, 9) years. At the time of medical record review, 17 patients were deceased with a median survival of 8.3 (IQR = 7, 10.9) years. Univariate analysis showed that initial onset of autonomic failure predicted unfavorable survival (hazard ratio = 2.89, P = 0.04) compared to those who presented with motor impairment only at onset. At the time of YOMSA diagnosis, composite autonomic severity score was available in 19 patients with a median of 5 (IQR = 4, 6.5).
Conclusions
YOMSA resembles MSA in most aspects including phenotype and prognosis, although the diagnosis is usually delayed. The presence of autonomic failure at symptom onset may be a poor predictor for survival.
Keywords: multiple system atrophy, parkinsonism, ataxia, autonomic failure, synucleinopathies
Multiple system atrophy (MSA) is an α‐synucleinopathy that presents with autonomic failure and motor impairment. 1 The clinical phenotype is defined based on the predominant motor feature of parkinsonism (MSA‐P) or cerebellar ataxia (MSA‐C). 1 A definite diagnosis is possible only after brain autopsy, which can lead to potential for misdiagnosis throughout the disease course. 1 , 2 MSA usually has a mean age of onset of 63.4 years with an annual incidence of 3/100,000 among the United States (US) population. 3 , 4 Different types of MSA have been described based on age of onset: young onset‐MSA (YOMSA) is defined as onset before age 40 years, whereas later‐onset MSA (LOMSA) is defined as onset after the age of 75 years. 5 , 6 YOMSA has a frequency of 0.9% to 1.7% among patients who are clinically diagnosed with MSA, and 4.1% to 4.5% among patients with pathologically confirmed MSA, which suggests that the diagnosis of YOMSA can be missed during the disease course or that patients with YOMSA are more likely to undergo an autopsy to determine the final diagnosis. 5 , 6 , 7 The rarity of YOMSA in conjunction with similarities in clinical features with more common diseases, such as young onset Parkinson's disease (YOPD), may contribute to misdiagnosis. 5
Because there are very few studies on YOMSA, the clinical course, prognosis, and disease characteristics remain largely unknown. In the present study, we aim to describe the disease course and outcome of patients with YOMSA. We also compare disease characteristics of YOMSA with reported data of usual age onset of MSA, which is of importance as we seek to identify patients early in the disease course.
Materials and Methods
We retrospectively reviewed medical records of all patients with a diagnosis of possible or probable MSA based on the second consensus criteria for the diagnosis of MSA who were evaluated at Mayo Clinic in Rochester, Minnesota; Phoenix, Arizona; and Jacksonville, Florida from 1998 to 2021. We recorded age at symptom onset and identified patients with an onset at the age of 40 years and younger. We excluded patients if an alternative diagnosis to MSA was more likely based on the clinical, laboratory, and imaging findings.
We used the Movement Disorder Society (MDS) criteria for the diagnosis of MSA and defined the age at the time of symptom onset as the first time the patient reported symptoms of motor impairment (parkinsonism or cerebellar ataxia features) or autonomic failure (ie, bladder dysfunction, and orthostasis). 1 Parkinsonism features include bradykinesia, resting tremor, rigidity, gait abnormalities, and postural instability. Features of cerebellar ataxia include ataxic gait and dysarthria, limb ataxia, and persistent gaze‐evoked nystagmus. 1 Because of the low specificity of isolated erectile dysfunction, urinary failure was needed to accompany or occur within 1 year of erectile dysfunction to be considered because of autonomic failure. 1 We identified the age at the time of MSA diagnosis as the time that all requested evaluations by the physician were completed and the MSA diagnosis was concluded based on the clinical presentation, examination, and results of diagnostic evaluation. Further, we retrospectively applied the MDS criteria for the diagnosis of MSA, published in 2022, 1 to categorize these patients as clinically established or clinically probable MSA. We recorded demographic characteristics, clinical features, and course in all patients. We collected findings from neuroimaging and genetic studies, and autonomic function evaluations from autonomic reflex screen (ARS) and thermoregulatory sweat test (TST), when available, at the time of MSA diagnosis. We calculated the Composite Autonomic Severity Score (CASS) using findings from ARS with and without TST. CASS computes a score from 0 to 10, and scores >7 demonstrate severe autonomic failure. 8
We calculated levodopa equivalent daily dose (LEDD) based on the last available medical record, when applicable. 9
Statistical Analysis
BlueSky Statistics 7.1 (GA) was used to conduct statistical analyses. Categorical variables were reported as number and frequency, and interval variables as mean and standard deviation (SD) or median and first and third quartiles. To further investigate clinical characteristics of YOMSA, we divided patients based on the MSA subtype (MSA‐P or MSA‐C) and initial symptom at the time of symptom onset (presence of autonomic failure, or motor impairment only). For comparisons between groups, we used Fisher's exact test for categorical data, and Mann–Whitney U‐test for continuous variables. We performed survival analysis using Kaplan–Meier estimates and generated survival curves for the time from symptom onset to death (disease course). For living patients, we considered the last date of medical record review as the censor date. We used the log‐rank test to compare survival estimates of two groups. We used Cox regression hazards models to calculate univariate and multivariate hazard ratios for survival.
Considering the small sample size, all univariate factors that reached a P value of <0.10 were moved into a multivariate model. For all other analyses we considered a P value of <0.05 as statistically significant.
Results
Patient Characteristics
Of 1496 patients with a diagnosis of MSA, 20 (1.3%) patients had YOMSA; among them 13 (65%) patients had MSA‐P. The median age of onset was 39.1 (interquartile range [IQR] = 37.1, 40.1) years, and 13 (65%) patients were male. At the time of medical record review, 17 patients were deceased with a mean age of death of 47.7 (±3.7) years (Table 1), and the median duration of disease course of 8.3 (IQR = 7, 10.9) years. None of the patients underwent brain autopsy at our institution.
TABLE 1.
Patients' characteristics and comparison between groups
| Variable | All patients | Comparison based on MSA type | Comparison based on initial symptom | ||||
|---|---|---|---|---|---|---|---|
| Mean ± SD or frequency (n = 20) | MSA‐P (n = 13) | MSA‐C (n = 7) | P‐value | Motor impairment only (n = 10) | Autonomic failure (n = 10) | P‐value | |
| Demographics (y) | |||||||
| Age of onset (n = 20) | 38.58 ± 2.12 | 38.4 ± 2.3 | 38.9 ± 1.7 | 0.75 | 38.7 ± 1.6 | 38.47 ± 2.6 | 0.85 |
| Time from onset to diagnosis (n = 20) | 6.66 ± 4.53 | 6.9 ± 5.03 | 6.06 ± 3.7 | 0.64 | 8.28 ± 5.1 | 5.04 ± 3.3 | 0.07 |
| Time from diagnosis to death | 2.97 ± 1.99 | 3.1 ± 2.01 | 2.55 ± 2.09 | 0.72 | 3.2 ± 2.2 | 2.79 ± 1.9 | 0.88 |
| Time from onset to death | 9.06 ± 4.02 | 9.4 ± 4.6 | 8.1 ± 2.08 | 0.50 | 10.8 ± 4.02 | 7.8 ± 3.7 | 0.13 |
| Age of death | 47.77 ± 3.70 | 47.87 ± 4.2 | 47.5 ± 2.3 | 0.95 | 49.8 ± 4.5 | 46.3 ± 2.1 | 0.08 |
| Autonomic function testing | |||||||
| TST (%) | 66.57 ± 38.58 | 71.07 ± 34.6 | 55.7 ± 49.5 | 0.57 | 67.5 ± 39.8 | 65.5 ± 39.7 | 1 |
| CASS‐A | 4.78 ± 2.25 | 5.07 ± 2.2 | 4.1 ± 2.4 | 0.42 | 4.5 ± 2.3 | 5 ± 2.2 | 0.58 |
| CASS‐T | 5.93 ± 2.40 | 6.1 ± 2.08 | 5.25 ± 3.5 | 0.66 | 5.5 ± 2.4 | 6.3 ± 2.4 | 0.39 |
| Clinical features | |||||||
| LEDD (last visit) | 1013.33 ± 859.99 | 1083.33 ± 940.67 | 733.33 ± 404.14 | 0.94 | 986.33 ± 826.98 | 1053.83 ± 986.56 | 0.81 |
| Presence of dystonia | 8 (40%) | 7 (53.8%) | 1 (14.3%) | 0.15 | 6 (60%) | 2 (20%) | 0.16 |
| Positive Babinski | 5 (25%) | 2 (15.4%) | 3 (42.9%) | 0.28 | 2 (20%) | 3 (30%) | 1 |
| Hyperreflexia | 11 (55%) | 7 (53.8%) | 5 (71.4%) | 0.64 | 7 (70%) | 5 (50%) | 0.64 |
Abbreviations: MSA, multiple system atrophy; SD, standard deviation; MSA‐P, multiple system atrophy‐parkinsonism; MSA‐C, multiple system atrophy‐cerebellar ataxia; TST, thermoregulatory sweat test; CASS‐A, Composite Autonomic Severity Score using findings from autonomic reflex screen only; CASS‐T, Composite Autonomic Severity Score using findings from both autonomic reflex screen and thermoregulatory sweat test; LEDD, levodopa equivalent daily dose.
Regarding environmental exposures, a substantial history of exposure to trichloroethylene and xylene was reported in one patient because of his occupation. None of the patients reported a history of a neurological disorder in a first‐degree relative.
Initial Presentation
At the time of symptom onset, motor impairment (with or without autonomic failure) was the most common complaint and was reported by 13 (65%) patients; autonomic failure was present in 10 (50%) patients. At the first visit by a neurologist, Parkinson's disease was the most frequent diagnosis reported in eight (40%) patients, followed by parkinsonism and ataxia/sporadic cerebellar ataxia each in three (15%) patients (Table S1).
Disease Progression and YOMSA Diagnosis
Four patients (patients 3, 4, 7, and 8) fulfilled the MDS criteria for clinically established MSA and the rest were categorized as clinically probable MSA based on the findings at the time of MSA diagnosis. The median duration of symptom onset to a clinical diagnosis of YOMSA was 4.9 (IQR = 3.7, 9) years (Table 1). Broad genetic testing in four patients with MSA‐C and one patient with MSA‐P excluded other mimicking conditions (Table S1). The evaluated conditions include Huntington's disease, storage diseases, defects in 6‐pyruvoyl‐tetrahydropterin synthase gene, defects in ATPIA3 gene (responsible for rapid‐onset dystonia‐parkinsonism, cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss [CAPOS], and alternating hemiplegia of childhood), spinocerebellar ataxia, Friedrich's ataxia, and fragile X tremor ataxia syndrome (FXTAS).
Eight (40%) patients, including seven with MSA‐P and one with MSA‐C reported dystonia during their disease course. In 12 (60%) patients, at least one of the pyramidal signs of positive Babinski or hyperreflexia was found around the time of MSA diagnosis, with hyperreflexia being most common, found in 11 (55%) patients. Thirteen (65%) patients reported experiencing dream enactment behavior during their disease course. Seven (35%) patients reported at least one cognitive complaint. Depression was the most common mood disorder reported by eight (40%) patients, followed by anxiety that was present in four (20%) patients (Table S1).
Two patients underwent deep brain stimulation of bilateral subthalamic nuclei during their disease course with a diagnosis of YOPD and experienced only brief improvement followed by symptom recurrence. Levodopa was prescribed for 16 patients at some point in their disease course, with 12 (75%) patients reporting at least a partial response. In seven patients, levodopa was used for ≥2 years with a median of 5 (IQR = 4.5, 8) years. Levodopa‐induced dyskinesia (LID) was observed in six (37.5%) patients (Table S1). At the time of the last neurology visit, LEDD was able to be calculated in 15 patients with a mean of 1013.3 (±859.99).
Autonomic Function Testing
At the time of YOMSA diagnosis, TST was performed in 19 patients. From 17 with results available, the median percentage anhidrosis was 88% (IQR = 39, 87.5) (Table 1). CASS, based on the components of ARS, was available in 19 patients with a median of 5 (IQR = 4, 6.5).
Imaging
Brain imaging was performed in 19 patients (Table S1). Based on the last available magnetic resonance imaging (MRI), 11 (57.8%) patients had at least one MRI marker suggestive of MSA: atrophy of putamen, middle cerebellar peduncle, cerebellum, or pons (nine patients), hot cross bun sign (four patients), T2 hyperintensity in the middle cerebellar peduncles (three patients), and T2 hypointensity of the putamina (two patients).
Predictors of Survival
There was no statistically significant difference between MSA subtypes regarding disease duration, age of death, or presence of pyramidal features (Table 1). Disease duration was not statistically significantly different between individuals with autonomic failure at the time of symptom onset and those with motor impairment only (7.8 [±3.7] and 10.8 [±4.02] years, respectively; P = 0.13) (Table 1).
Regarding predictors of survival based on the univariate Cox regression analyses, patients with autonomic failure at the time of symptom onset had a hazard ratio (HR) of 2.89 as compared to those who presented with motor impairment only (95% confidence interval [CI] = 1.04–8.05; P = 0.04) (Fig. 1). There was an HR of 1.25 for each 1‐point increase in Composite Autonomic Severity Score using findings from autonomic reflex screen only (CASS‐A) (95% CI = 0.96–1.62; P = 0.09) (Table 2).
FIG. 1.
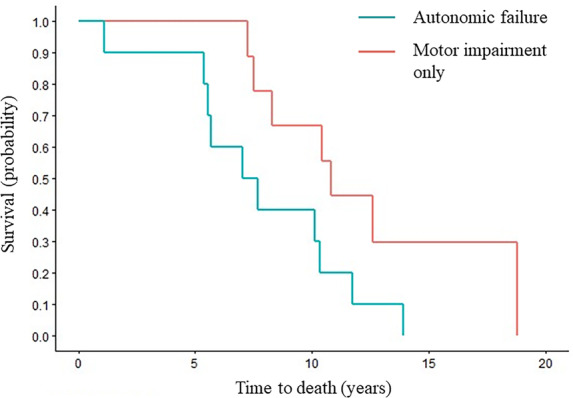
Symptom onset to death based on initial symptoms. Censored Kaplan–Meier survival curves for probability of death from symptom onset in all patients with onset of autonomic failure (blue) and motor impairment only (red).
TABLE 2.
Assessing predictors of death based on the univariate cox proportional hazards analysis for the disease course duration (time from symptom onset to death)
| Variable | HR | 95% Confidence interval | P‐value | |
|---|---|---|---|---|
| Lower bound | Upper bound | |||
| Gender (M) | 2.08 | 0.71 | 6.15 | 0.18 |
| Age of onset | 1.23 | 0.93 | 1.61 | 0.14 |
| MSA subtype (MSA‐C) | 1.36 | 0.45 | 4.14 | 0.58 |
| Initial symptom (autonomic failure) | 2.89 | 1.04 | 8.05 | 0.04* |
| TST | 0.99 | 0.98 | 1.01 | 0.91 |
| CASS‐A | 1.25 | 0.96 | 1.62 | 0.09* |
| CASS‐T | 1.12 | 0.88 | 1.44 | 0.35 |
| LEDD | 1 | 1 | 1 | 0.29 |
| Dystonia (yes) | 0.52 | 0.18 | 1.52 | 0.52 |
| Babinski (yes) | 1.04 | 0.36 | 3.03 | 1.04 |
| Hyperreflexia (yes) | 1 | 0.36 | 2.78 | 1 |
A P‐value <0.10 was considered as statistically significant to move into the multivariate model.
Abbreviations: HR, hazard ratio; M, male; MSA, multiple system atrophy; MSA‐C, multiple system atrophy‐cerebellar ataxia; TST, thermoregulatory sweat test; CASS‐A, Composite Autonomic Severity Score using findings from autonomic reflex screen only; CASS‐T, Composite Autonomic Severity Score using findings from both autonomic reflex screen and thermoregulatory sweat test; LEDD, levodopa equivalent daily dose.
Multivariate analysis with the presence of autonomic failure at the time of symptom onset and CASS‐A, revealed that autonomic failure at the time of symptom onset was associated with a worsened survival even after adjusting for the severity of autonomic failure using CASS (HR = 4.23, 95% CI = 1.29–13.91; P = 0.01) (Table S2).
Discussion
We present a large case series of patients diagnosed with YOMSA. Patients with YOMSA account for only 1.3% of all patients diagnosed with clinically established or clinically probable MSA at our institution during the past 23 years. Consistent with the rare nature, patients with YOMSA were frequently diagnosed later in the disease course than that reported for usual onset MSA, which suggests that the diagnosis of MSA is challenging in younger patients. It was also found that presenting with initial symptoms of autonomic failure led to worse prognosis, similar to previous studies.
Most patients received the YOMSA diagnosis in the middle to late stages of their disease course in our study, with a mean duration from symptom onset to YOMSA diagnosis of 6.6 years. A study by Batla and colleagues 5 examined a cohort of 22 YOMSA patients, eight with autopsy‐confirmation, and similarly reported a duration of 6.2 years from onset to diagnosis among patients who were clinically diagnosed with YOMSA. Similar to our cohort, none had a family history of a movement disorder.
Studying this cohort may help us better understand initial symptoms in YOMSA to allow for earlier and more accurate diagnosis for patients. Motor impairment (50%) followed by autonomic failure (35%) and a combination of both symptoms (15%) were the most common initial symptoms among our patients. Motor onset tends to be most common in MSA, particularly in LOMSA. 6 The initial presentation of autonomic‐only symptoms is important as the MDS MSA consensus criteria recently highlighted a new category of possible prodromal MSA. This category includes patients with autonomic failure and subtle motor signs. Although designated as research criteria, better clinical evaluation of this subset of patients, including in YOMSA, is indicated.
Identifying patients with early autonomic failure is crucial as we found a worse survival in patients with autonomic failure at the time of symptom onset compared with motor onset. This cohort had a moderate degree of autonomic failure at presentation, as shown by CASS and TST results, which was seen in both MSA‐C and MSA‐P subtypes. These findings are concordant with other studies showing that autonomic failure at the time of symptom onset or diagnosis is a negative prognostic indicator in MSA. 10 , 11 , 12 The underlying pathophysiology of poor prognosis in patients with MSA who have autonomic dysfunction is unclear. Prominent loss of medullary serotonergic neurons, which are involved in regulation of micturition reflex, thermoregulation, and respiratory chemosensitivity and rhythm, has been previously described in patients with MSA that suffer from autonomic failure. 11 , 13 Moreover, previous studies showed an association between loss of these neurons and sudden nocturnal death. 13 Consequently, patients with early autonomic dysfunction may have an increased risk of sudden death during sleep. 11
Clinical features may aid in the timely diagnosis of YOMSA. We found that 60% of our cases had at least one pyramidal sign such as hyperreflexia or positive Babinski at the time of diagnosis, which is higher than findings from a prospective North American study that found pyramidal signs in 33.7% of patients, 3 but is similar to other YOMSA cohorts. 5 Depression, both solely or in association with anxiety, was a common complaint reported by 40% of patients in our study and was higher than what was reported by Batla et al 5 (13.6%). Additionally, in contrast to the study by Batla et al, 5 which reported absence of any cognitive complaints among patients with YOMSA, 35% of the patients in our study reported at least one cognitive complaint, such as memory impairment. These differences suggest that neuropsychiatric and cognitive complaints may be ignored by both clinicians and patients with YOMSA because of the predominance of motor and autonomic symptoms.
Dystonia may be more common in YOMSA and was seen in 40% of our patients. In a prospective European cohort study, dystonia was evident in 30.5% of patients with MSA. 14 In the study by Batla et al, 76% of all patients who were clinically diagnosed with YOMSA showed dystonia, which may also be related to the higher proportion of MSA‐P in their study compared to our cohort. 5
One factor that can complicate diagnosis of MSA is response to dopaminergic agents and this study found that 75% of patients on levodopa showed at least a partial response. The response rate was almost similar in the Batla et al 5 study with 83% of patients reporting a moderate to good response to levodopa. Beneficial response to levodopa has been reported among 42.5% to 56.7% of patients with MSA‐P and 12.9% to 25% of patients with MSA‐C in previous cohort studies on MSA. 3 , 14 One possibility for the better response of patients with YOMSA to levodopa may be related to the higher neuronal reservoir in younger individuals. However, considering the differences between studies in the methodology of defining levodopa responsiveness, it may be hard to draw definitive conclusions. Nevertheless, these findings emphasize that levodopa responsiveness should not exclude the possibility of YOMSA in a young patient with parkinsonism. The presence of LID, seen in 37.5% of the patients with YOMSA, may also support a MSA diagnosis.
There are some limitations to the present study. First, it is a retrospective study that leads to the chance of recall bias regarding symptom onset and exposure history. Second, none of the patients underwent brain autopsy to receive a definite diagnosis of YOMSA. Third, although MDS criteria for MSA diagnosis have shown excellent diagnostic performance in validation studies, the diagnostic accuracy in patients with YOMSA has not been evaluated, leaving some degrees of diagnostic uncertainty in this group of patients. Fourth, in most of the patients, levodopa responsiveness was determined based on the physician's interpretation and not a levodopa challenge test.
In summary, this study provides valuable findings on the clinical characteristics of patients with YOMSA, especially those with MSA‐C subtype as previously little had been published on this group. Autonomic function testing can not only help in distinguishing YOMSA from mimicking conditions, but is a valuable tool in determining prognosis. Our findings suggest that patients with YOMSA frequently present with autonomic failure and may show a better response to levodopa compared to patients with MSA, although further studies are required. In line with previous studies, we found a higher prevalence of dystonia and pyramidal features among patients with YOMSA as compared to usual onset‐MSA. Additionally, we found that YOMSA resembles MSA in most aspects including phenotype and prognosis. These findings can help us in identifying patients with YOMSA earlier in their disease course, initiate treatment earlier in disease, and better understand the clinical course of YOMSA.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
N.B.: 1B, 1C, 2A, 2B, 3A.
R.S.: 1B, 2C, 3B.
C.H.A.: 1B, 2C, 3B.
Z.K.W.: 1B, 2C, 3B.
L.M.J.: 1B, 2C, 3B.
E.E.B.: 1B, 2C, 3B.
P.S.: 1B, 2C, 3B.
P.A.L.: 1A, 1B, 2C, 3B.
W.S.: 1A, 1B, 2C, 3B.
E.A.C.: 1A, 1B, 1C, 2A, 2B, 3A.
Disclosures
Ethical Compliance Statement: The study was approved by the Mayo Clinic Institutional Review Board and the informed consents were obtained from the patients. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This study was supported in part by National Institutes of Health (NIH) (P01NS44233, U54NS065736, K23NS075141, R01 FD004789, and R01 NS092625), Mayo CCaTS (UL1TR000135), Cure PSP Foundation, NIH (R01 NS092625, U19 AG71754, and UL1 TR000135), food and drug administration (FD‐R‐07290), The Michael J. Fox Foundation, Bishop Dr. Karl Golser Foundation, Sturm Foundation, Mayo Center for Regenerative Medicine, and Mayo Funds. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Financial Disclosures for the Previous 12 Months: N.B., C.H.A., L.M.J., E.E.B., P.S., P.A.L., W.S., and E.A.C. declare that there are no additional disclosures to report. R.S. receives research support from the National Center for Advancing Translational Sciences (NCATS), a component of the NIH, NIH, unrestricted grant from Acadia Pharmaceuticals, and The Michael J. Fox Foundation. Z.K.W. is partially supported by the NIH/NIA and NIH/The National Institute of Neurological Disorders and Stroke (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, the gifts from the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, The Albertson Parkinson's Research Foundation, and PPND Family Foundation. He serves as PI or Co‐PI on Biohaven Pharmaceuticals (BHV4157‐206) and Vigil Neuroscience (VGL101‐01.002, VGL101‐01.201, PET tracer development protocol, Csf1r biomarker and repository project, and ultra‐high field MRI in the diagnosis and management of CSF1R‐related adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia) projects/grants. He serves as Co‐PI of the Mayo Clinic APDA Center for Advanced Research and as an external advisory board member for the Vigil Neuroscience and as a consultant on neurodegenerative medical research for Eli Lilli and Company.
Supporting information
Table S1. Characteristics of each patient.
Table S2. Assessing predictors of death based on multivariate cox proportional hazards analysis for the disease course duration (time from symptom onset to death).
Relevant disclosures and conflict of interest are listed at the end of this article.
References
- 1. Wenning GK, Stankovic I, Vignatelli L, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord 2022;37(6):1131–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poewe W, Stankovic I, Halliday G, et al. Multiple system atrophy. Nat Rev Dis Primers 2022;8(1):56. [DOI] [PubMed] [Google Scholar]
- 3. Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14(7):710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 1997;49(5):1284–1288. [DOI] [PubMed] [Google Scholar]
- 5. Batla A, De Pablo‐Fernandez E, Erro R, et al. Young‐onset multiple system atrophy: clinical and pathological features. Mov Disord 2018;33(7):1099–1107. [DOI] [PubMed] [Google Scholar]
- 6. Sekiya H, Koga S, Otsuka Y, et al. Clinical and pathological characteristics of later onset multiple system atrophy. J Neurol 2022;269(8):4310–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jellinger KA. Young‐onset multiple system atrophy. Mov Disord 2018;33(12):1974–1975. [DOI] [PubMed] [Google Scholar]
- 8. Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc 1993;68(8):748–752. [DOI] [PubMed] [Google Scholar]
- 9. Schade S, Mollenhauer B, Trenkwalder C. Levodopa equivalent dose conversion factors: an updated proposal including opicapone and safinamide. Mov Disord Clin Pract 2020;7(3):343–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coon EA, Sletten DM, Suarez MD, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain 2015;138(Pt 12):3623–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Figueroa JJ, Singer W, Parsaik A, et al. Multiple system atrophy: prognostic indicators of survival. Mov Disord 2014;29(9):1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083. [DOI] [PubMed] [Google Scholar]
- 13. Tada M, Kakita A, Toyoshima Y, et al. Depletion of medullary serotonergic neurons in patients with multiple system atrophy who succumbed to sudden death. Brain 2009;132(Pt 7):1810–1819. [DOI] [PubMed] [Google Scholar]
- 14. Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12(3):264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of each patient.
Table S2. Assessing predictors of death based on multivariate cox proportional hazards analysis for the disease course duration (time from symptom onset to death).